

Abstract
In this study we developed a mesoporous silica nanoparticle (MSNP) based platform for high-dose loading of a range of activated platinum (Pt) chemo agents that could be attached to the porous interior through the use of electrostatic and coordination chemistry under weak-basic pH conditions. In addition to the design feature for improving drug delivery, the MSNP could also be encapsulated in a coated lipid bilayer (silicasome), to improve the colloidal stability after intravenous (IV) injection. We demonstrate improved pharmacokinetics and intratumor delivery of encapsulated activated oxaliplatin (DACHPt) over free drug in an orthotopic Kras-derived pancreatic cancer (PDAC) model. Not only did IV injection of the DACHPt silicasome provide more efficacious cytotoxic tumor cell killing, but could also demonstrate that chemotherapy-induced cell death is accompanied by the features of immunogenic cell death (ICD) as well as a dramatic reduction in bone marrow toxicity. The added ICD features were reflected by calreticulin and HMGB1 expression, along with increased CD8+/FoxP3+ T-cell ratios and evidence of perforin and granzyme B release at the tumor site. Subsequent performance of a survival experiment, demonstrated that the DACHPt silicasome generate a significant improvement in survival outcome, which could be extended by delayed administrated of the anti-PD-1 antibody.
Keywords: silicasome nanocarrier, platinum drug, pancreatic cancer (PDAC), immunogenic cell death (ICD), anti-PD-1 antibody
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal cancer with 5-year survival of ~8%[1]. As surgery is only suitable for ~15% of PDAC patients upon diagnosis, the best clinically available option is to use chemotherapy in the majority of PDAC patients[2]. This can be accomplished by the use of a gemcitabine (GEM)/Nab-paclitaxel combination or a four-drug regimen, FOLFIRINOX, which includes folinic acid, 5-fluorouracil, irinotecan, oxaliplatin[3]. While the recent breakthrough of a modified FOLFIRINOX regimen that reduces the irinotecan dose has yielded promising results in a phase 3 study, the alternative use option is still limited due to the occurrence of toxic side effects and chemo-resistance[3c]. In order to develop a nanocarrier with reduced irinotecan side effects, we have previously developed mesoporous silica nanoparticles (MSNP) coated with a lipid bilayer for safer and more efficacious irinotecan delivery[4]. The success of this carrier, also known as a “silicasome”, was attributed to the ability of a proton-generating trapping agent to allow remote import of the weak-basic molecule, irinotecan, into the porous packaging space in the particle interior[4]. This resulted in improved irinotecan delivery, which was accompanied by a significant improvement in treatment efficacy for PDAC, prolonged survival, and reduced toxicity compared to the free drug or a liposomal carrier, Onivyde®[4b].
Oxaliplatin is another active pharmaceutical ingredient (API) of the FOLFIRINOX regimen that is very potent but exhibits major, and dose-limiting toxicity (e.g., bone marrow)[5]. In addition, oxaliplatin is known to exert immunogenic effects that could be potentially useful to supplement its chemotherapeutic effects[6]. However, since Pt drugs are coordination compounds, it is not possible to perform remote loading to obtain MSNPs with high loading capacity, thereby limiting the utility of this carrier for Pt drugs. It would be of great advantage to develop MSNPs carriers for efficient Pt drug loading and delivery from the perspective that ~50% of all cancer patients undergoing chemotherapy receive at least one type of Pt-based treatment[7].
Pt-based antineoplastic molecules (Fig. S1) are coordination compounds, with the generalized chemical structure of cis-[PtA2X2] as shown in Fig. 1. A2 represents two monodentate or one bidentate ligands with a nitrogen donor atoms, while X2 are comprised of two monodentate or one bidentate anionic ligand(s)[8]. When delivered to a tumor site, Pt drugs provide DNA crosslinking through the formation of platinum-DNA adducts (a.k.a. alkylation)[9], in addition to the triggering of additional biological responses such as impacting the signal transducer and activator of transcription (STAT) pathway, induction of endoplasmic reticulum stress, and immunogenic cancer killing, etc[6]. This enables the Pt drugs, including encapsulated versions[10], to be useful to treat a variety of cancers, including PDAC[7b]. However, since remote loading of these drugs in MSNPs is not an option, alternative encapsulation attempts have been tried with limited success, yielding a Pt : MSNP with low (0.87 wt %) loading capacity (LC%) in an unmodified MCM-41 MSNP[11]. This triggered additional improvements to increase the LC% to 10~20 wt% by attaching a carboxy (-COOH) group to the MSNP surface, allowing coordination binding to cisplatin[12],[13]. Similar attempts were undertaken to improve oxaliplatin loading, providing an agent with a broader spectrum of activities and reduced cross-resistance compared to cisplatin[9]. Specifically, COOH-modified MSNP was used for complexation to cis-dichloro(1,2-diamminocyclohexane) platinum(II) (a.k.a. DACHPtCl2), which is an oxaliplatin precursor [14]. This resulted in improving the oxaliplatin loading capacity to 16.1 wt%[14]. The downside of a post-grafting carboxy modification, however, is that it interferes in coating with a lipid bilayer, which is critical for colloidal stability and systemic biodistribution[15]. This likely explains why the studies were only carried out under tissue culture conditions, rather than in vivo.
Fig. 1.
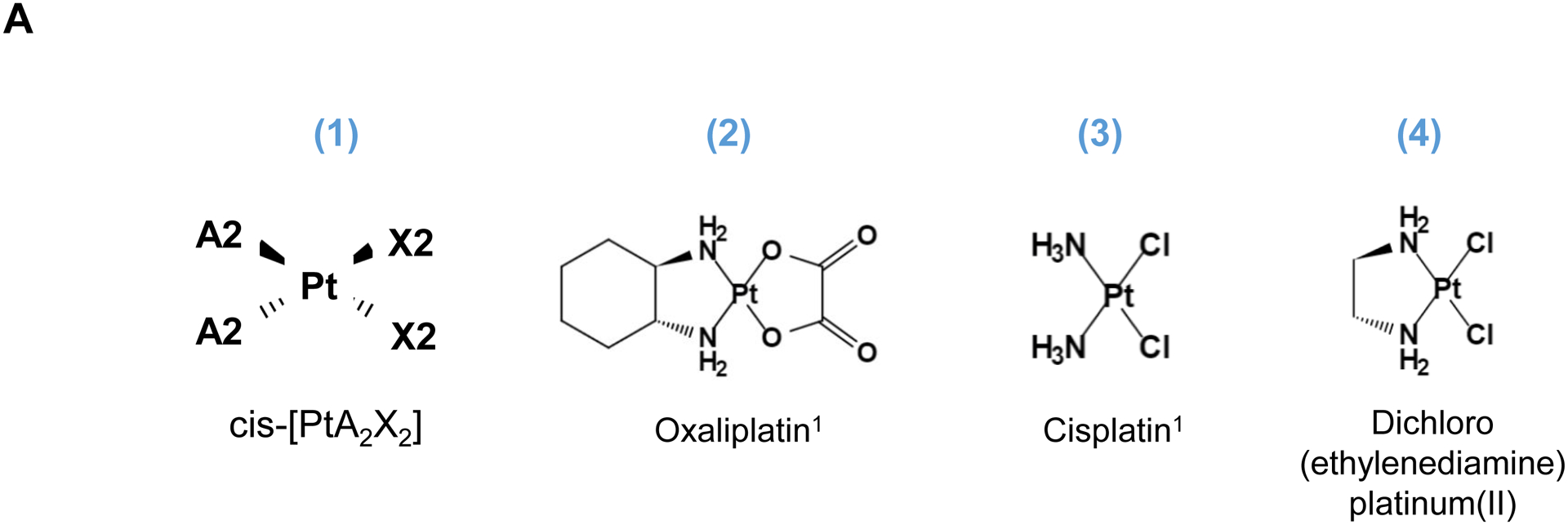
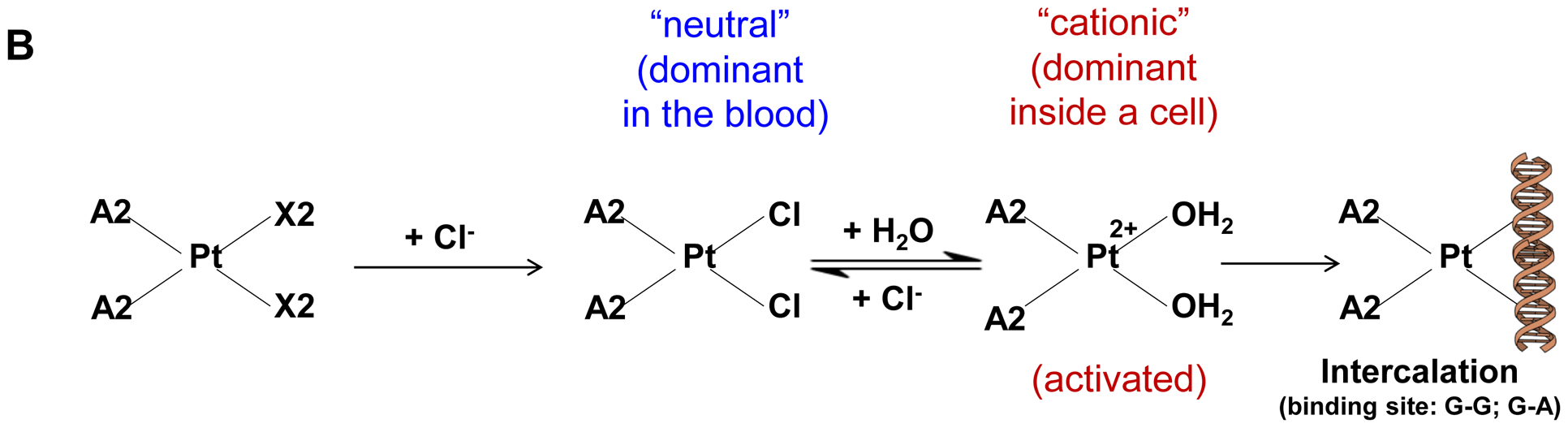
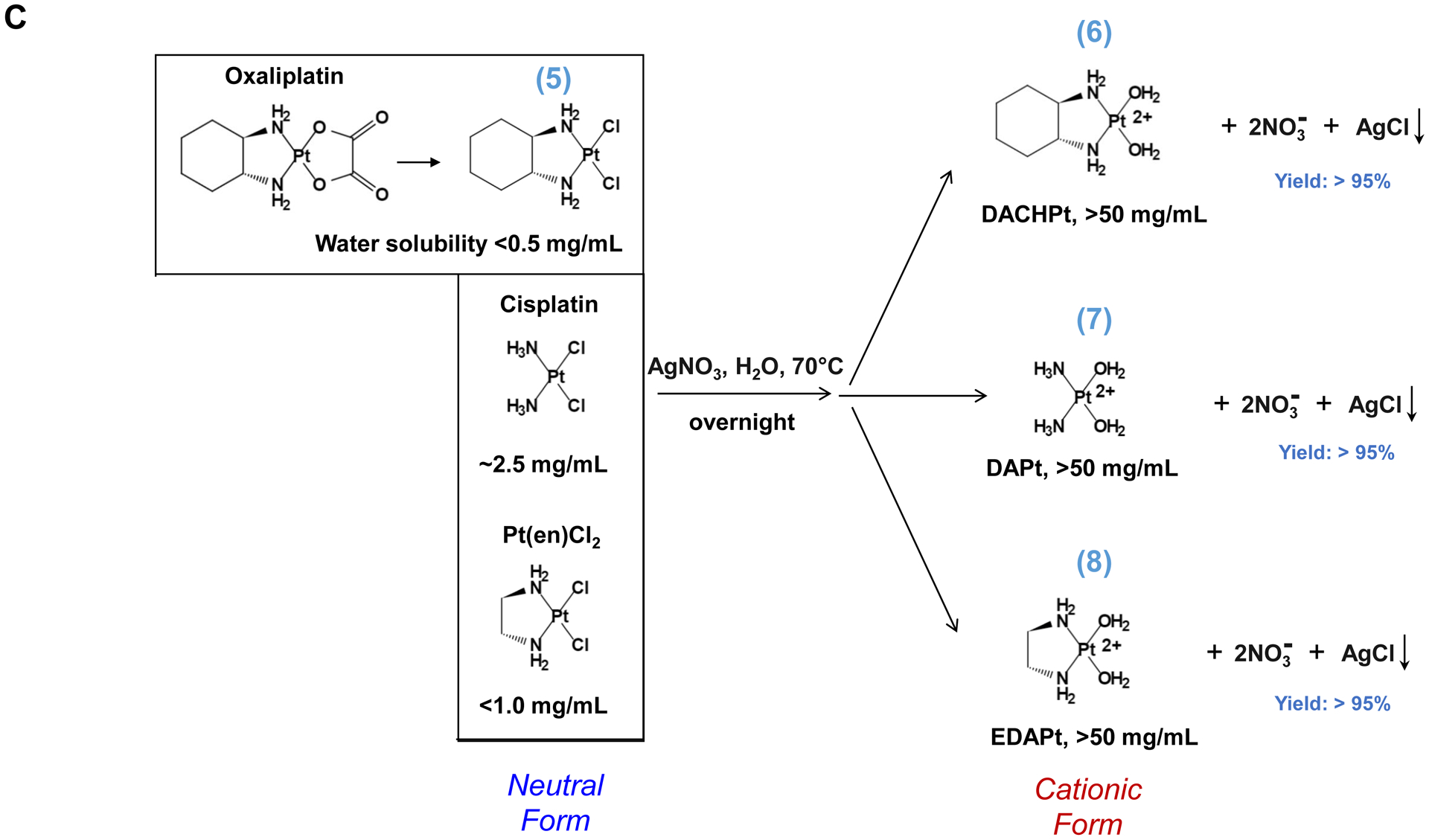
Synthesis of activated Pt drugs for the purpose of encapsulation by silicasomes. (A) Pt drugs are coordination compounds, with the basic structural composition of cis-[PtA2X2]. A2, i.e., two monodentate or one bidentate ligand with nitrogen donor atoms; X2: two monodentate or one bidentate anionic ligand(s). Representative Pt drugs studied in this project include oxaliplatin, cisplatin and Pt(en)Cl2. (B) Pt drugs in aqueous solution exist as equilibration species that can be depicted as “neutral” or “cationic”; the cationic variant is pharmaceutically active and capable of providing DNA cross linking. (C) Synthesis of cationic and active version of Pt drugs. This resulted in DACHPt (for oxaliplatin), DAPt (for cisplatin) and EDAPt (for Pt(en)Cl2), respectively. The yield of these reactions is high, i.e. ~95%.
Herein, we report an intravenously (IV) injectable tailored-designed silicasome carrier for Pt drug encapsulation. This was achieved by introducing active Pt drugs that can be efficiently loaded under a carefully designed complexation conditions, making use of weak basic pH conditions. Instead of using a post-grafting approach, we took advantage of the pH-dependent properties of the surface silanol groups for electrostatic and coordination binding of DACHPt, followed by applying a uniform lipid coat to seal the MSNP pores. This strategy, which is also adaptable for other Pt payloads, allowed the generation of carriers with colloidal stability and high loading capacity. The availability of a DACHPt silicasome allowed us to perform efficacy and safety studies in an orthotopic Kras PDAC model. An additional advantage proved to be the ability to use silicasome-encapsulated DACHPt for generating immunogenic cell death, and hence the development of a chemo-immunotherapy approach, where the carrier could be effectively combined with anti-PD-1 antibody for treatment of orthotopic PDAC tumors.
2. Results and Discussion
Three common Pt drug payloads were used in this study to develop an effective drug loading strategy, namely oxaliplatin, cisplatin and dichloro (ethylenediamine) platinum (Pt(en)Cl2) (Fig. 1A, structures 2–4)[7b]. It is generally agreed upon that Pt drugs exist as an equilibrium of “neutral” or “cationic” species in an aqueous solution (Fig. 1B)[16]. The binding equilibrium is dependent on the Cl− ion concentration (CCl−) as well as pH[16]. While the neutral drug version is dominant in the blood circulation due to a high CCl− (~150 mM), the formation of an intracellular cationic version is facilitated due to a lower CCl− (~30 mM)[17]. Moreover, the cationic formulation is regarded as pharmaceutically active due to the coordinated crosslinking to DNA, which is instrumental in preventing cancer growth[16]. These findings prompted us to consider loading cationic, activated Pt drugs into MSNP rather than experimenting with the pristine drugs. This requires the use of “neutral” Pt drugs where the X2 ligand is represented by Cl− ions. This required the purchasing of commercially available dichloro(1,2-diaminocyclohexane) platinum(II) (structure 5 in Fig. 1C) as the neutral version of oxaliplatin, in addition to the use of cisplatin and Pt(en)Cl2, which are already in the neutral form. This allowed us to proceed with the synthesis of 3 activated Pt drugs through the introduction of silver nitrate at 70°C, resulting in the formation of DACHPt (structure 6 in Fig. 1C), DAPt (structure 7 in Fig. 1C) and EDAPt (structure 8 in Fig. 1C). These represent the activated versions of oxaliplatin, cisplatin and (Pt(en)Cl2), respectively, and could be derived from high reaction yields (>95%). These reactions also generated a silver chloride precipitate, which can be easily removed through centrifugation and filtration.
The synthesis procedure for deriving the Pt-silicasomes nanocarriers is schematically outlined in Fig. 2A. The schematic illustrates the following steps: (1) synthesis of bare MSNP, (2) soak-in of the activated Pt drugs under controlled pH conditions, (3) lipid coating, and (4) purification and sterilization. In order to successfully implement the design process shown in Fig. 2A, it was necessary to develop a multi-parameter design process that takes into consideration pH, energy input, feed ratio and lipid coating (Fig. 2A, inserted table). Sixty-five nm bare MSNP was synthesized using a sol-gel reaction, as previously reported[4b]. MSNP is an amorphous silica nanocarrier that displays surface silanol groups[18]. While our previous studies have characterized the surface silanol groups on MSNPs[19], it is known that these silanols can be ionized at high pH condition, i.e. ≡Si–OH (low pH) ⇔ ≡Si–O− (high pH) (Fig. 2B1). Use of the ionized silanols, which exert most of the negative charge, is advantageous to accomplish attachment of the cationic Pt drugs via a combination of coordination and electrostatic interactions (Fig. 2B2). Thus, we asked whether pH-mediated fine-tuning could increase the abundance and efficiency of drug soak-in of the carrier. This demonstrated that incremental increase of the negative charge by pH adjustment from 5.5 to 8.5 could increase the drug binding efficiency of the carrier (Fig. 2C). During incubation of DACHPt with the MSNP to achieve a w/w coupling ratio of 1: 5 for Pt: MSNP, we observed a significant improvement in encapsulation efficiency (EE%), to achieve ~75% at pH 8.5. This was considerably higher than the EE% at a weak acidic (pH 5.5) or neutral pH (7.4), which could only accomplish values of 25% and 60%, respectively (Fig. 2C). In addition to pH, we also assessed the effect of stirring vs sonication, the soak-in time, and Pt: MSNP feed ratios to identify the optimal design parameters, as outlined in Fig. S2.
Fig. 2.
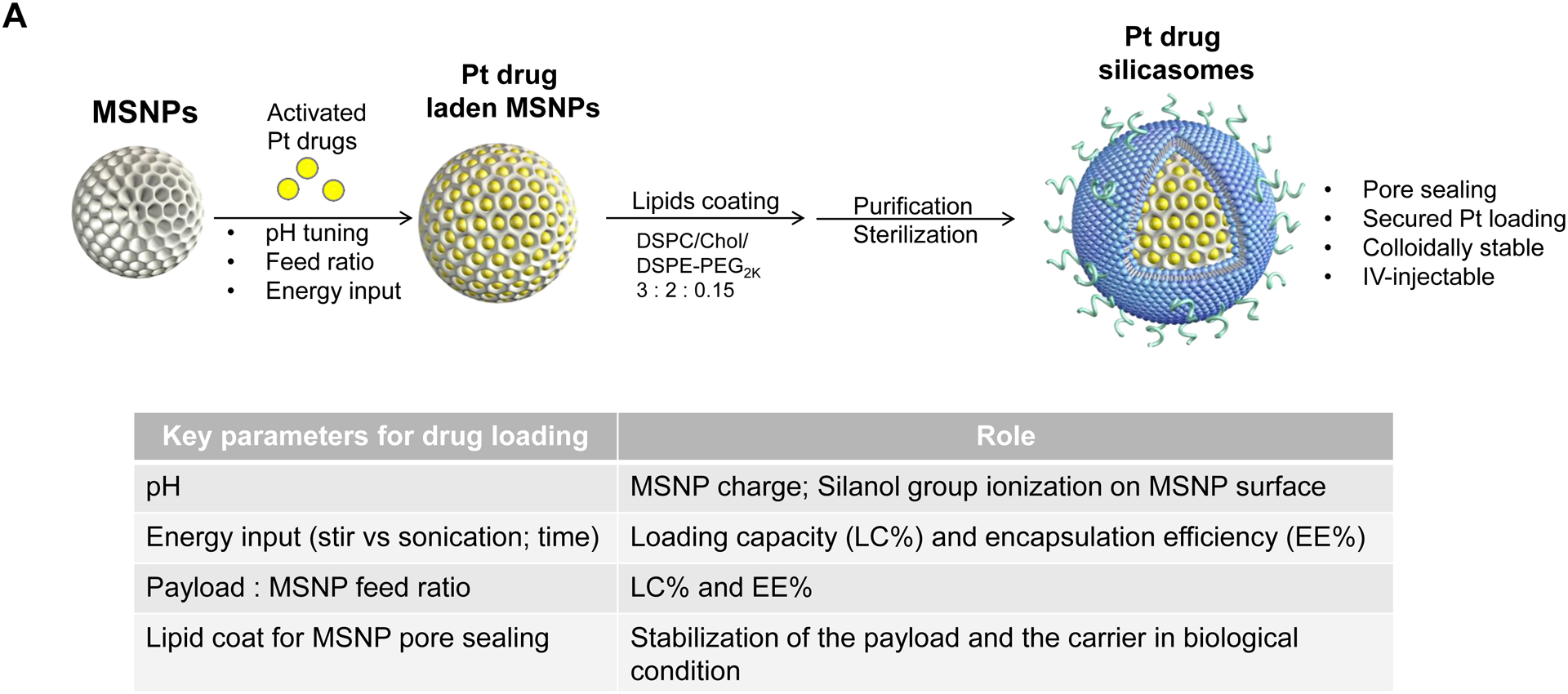
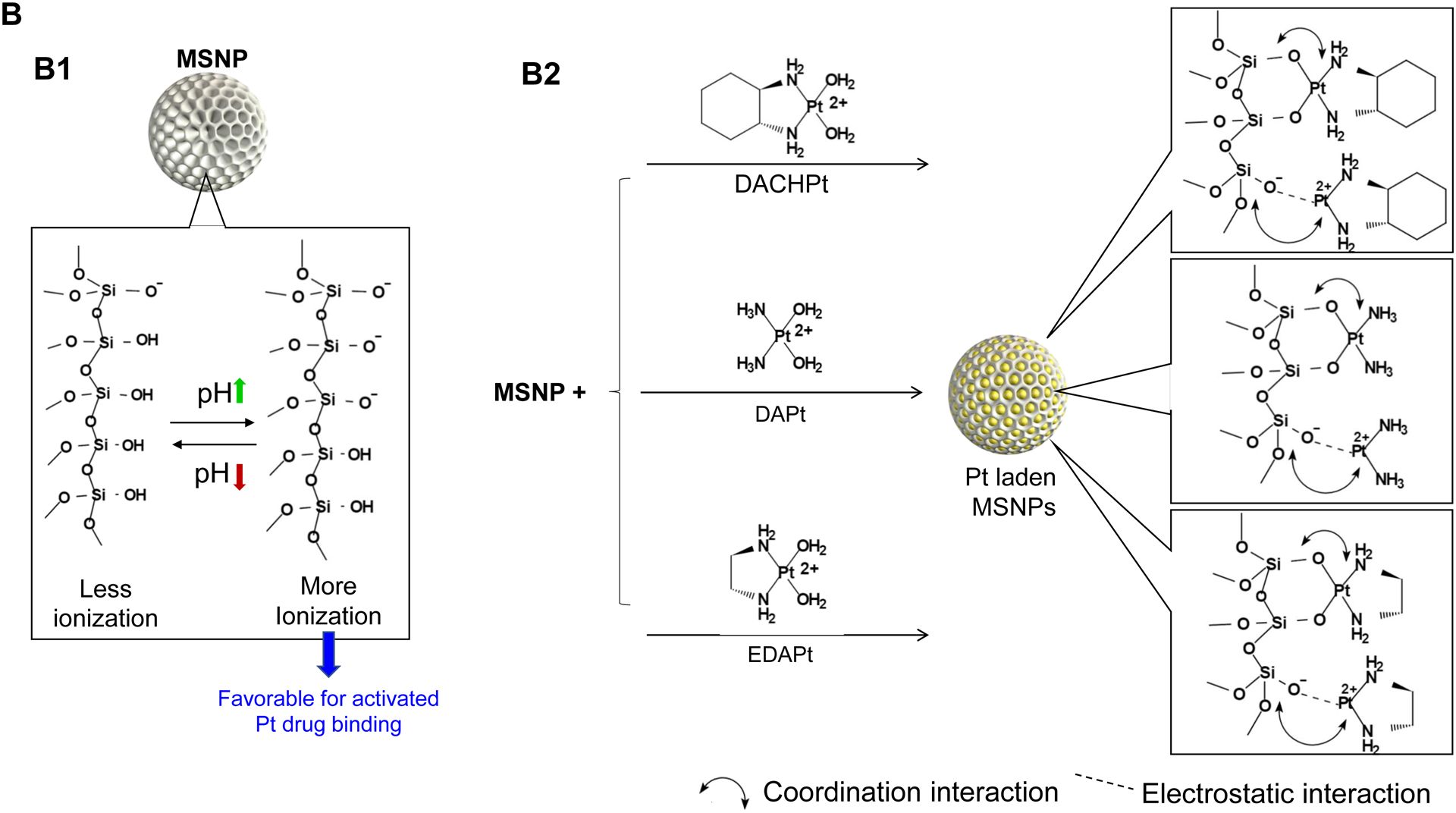
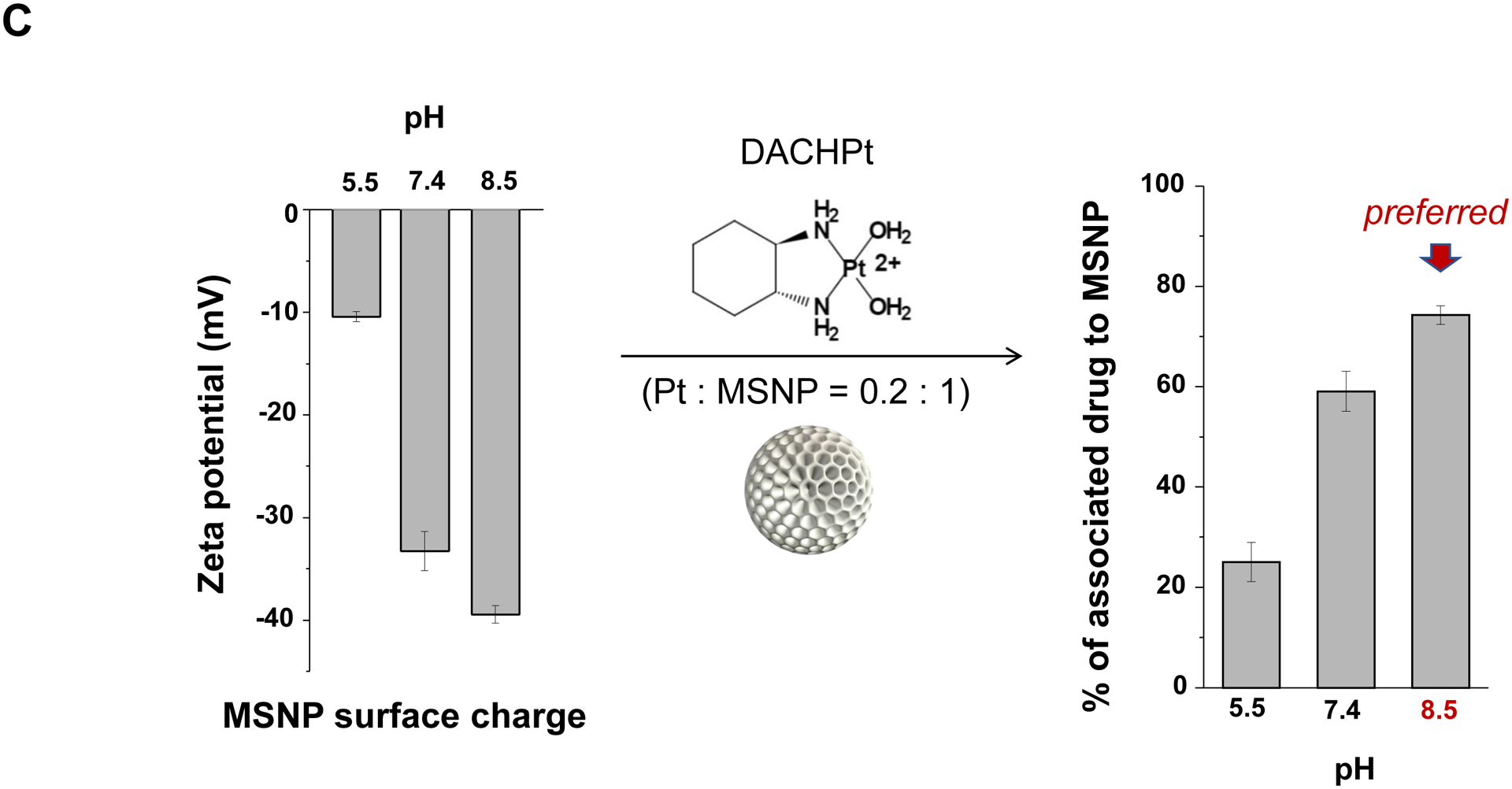
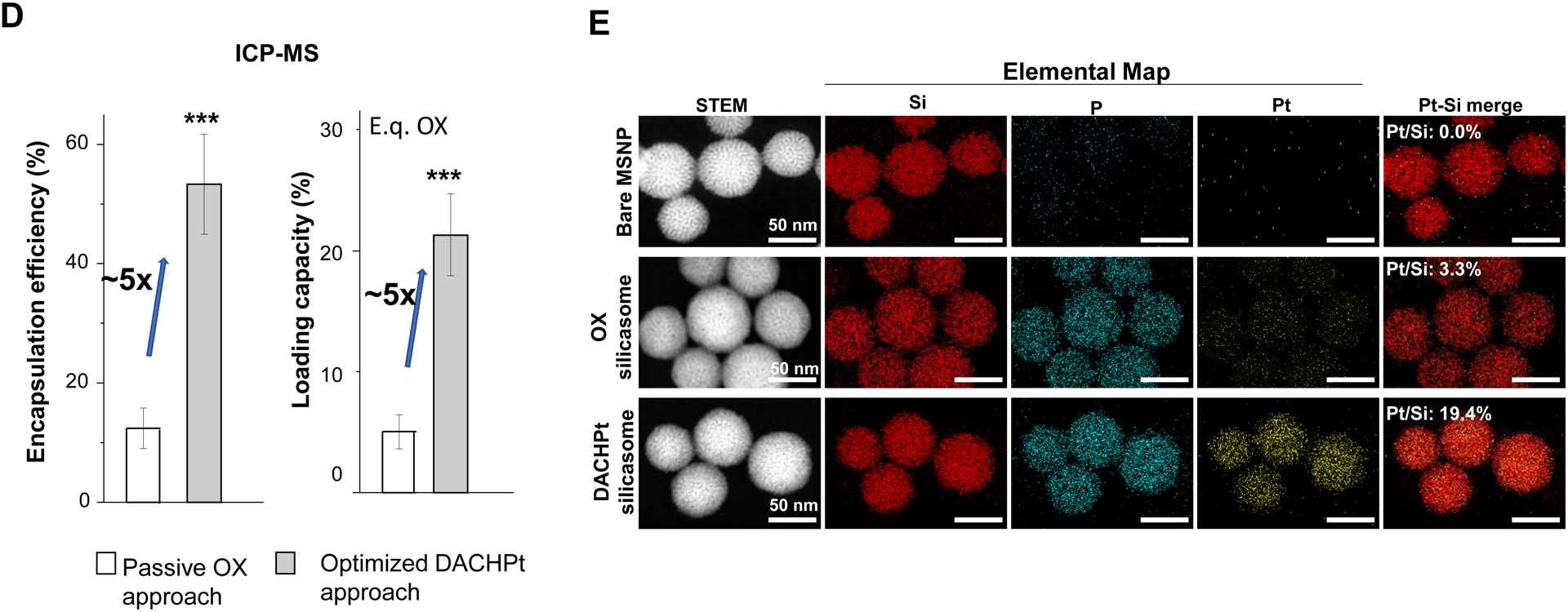
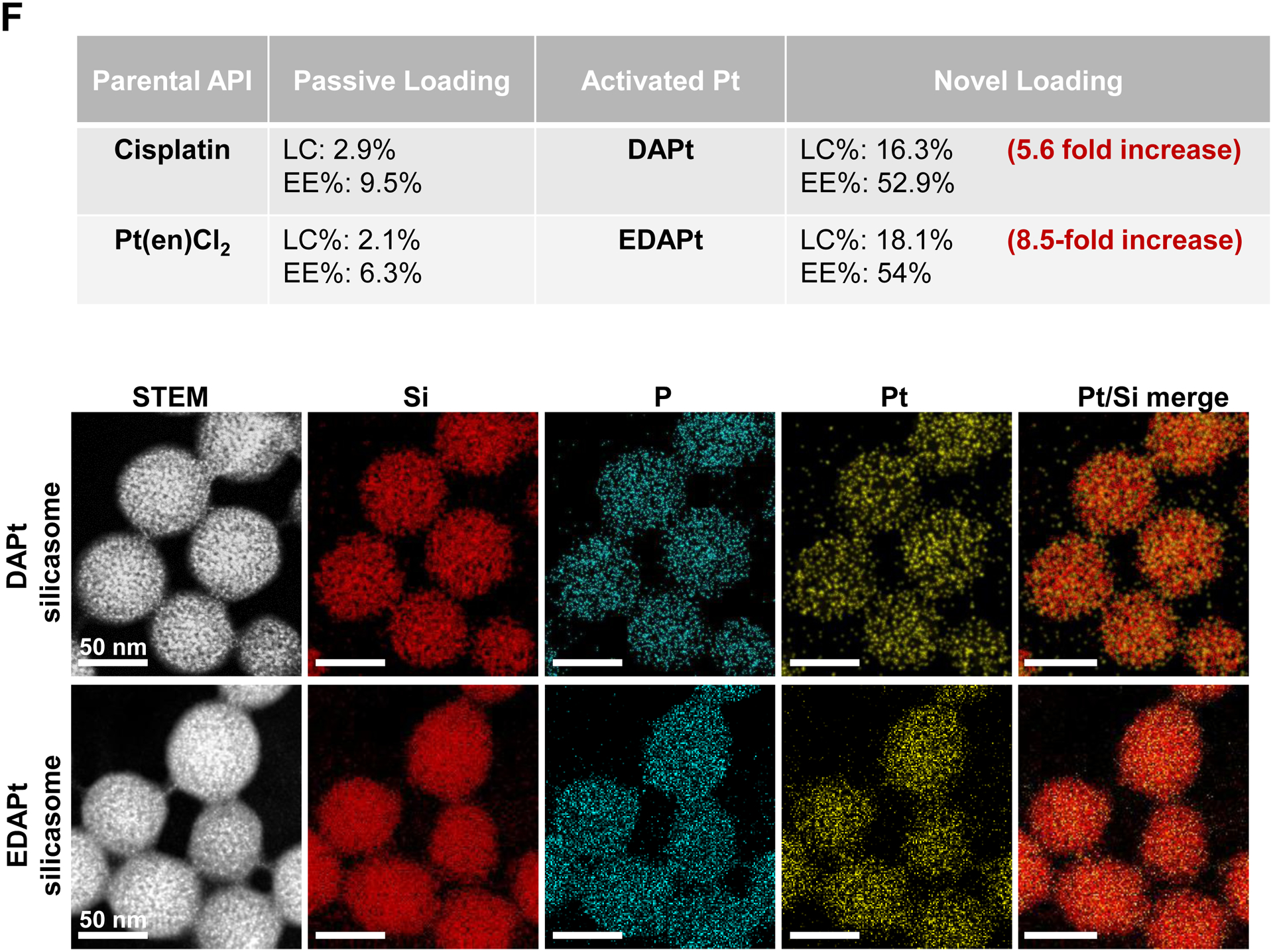
Design, synthesis and characterization of silicasomes, that contain the activated Pt drug. (A) The top panel provides a schematic that outlines the synthesis steps for tailored construction of silicasomes, incorporating the active Pt drug. The key parameters that govern successful drug loading are outlined in the table (lower panel). (B) pH adjustment to attain weak-basic conditions, favorable for drug loading. At basic pH, silanol groups can be ionized, i.e. ≡Si–OH ⇔ ≡Si–O− (B1), leading to a surface that allows efficient drug attachment via coordination chemistry and electrostatic binding (B2). (C) Zeta potential value of bare MSNP is pH-dependent. The development of a negative surface charge at pH 8.5 leads to the highest level of Pt drug binding, as exemplified by DACHPt. Other loading parameters, such the sonication condition, feed ratio, incubation time, etc., were systemically optimized, as illustrated online (Fig. S2). (D) After experimenting with multiple reaction conditions, it was possible to accomplish a ~5x increase in terms of EE% and LC%, as determined by ICP-MS analysis of Pt. The same binding level could not be achieved by a passive loading strategy. (E) Ultrastructural in situ STEM-EDS imaging confirmed the improved drug loading by coordination chemistry and electrostatic binding. The Pt/Si ratio (w/w) was determined to be 19.4% for the DACHPt silicasome, which is ~6x higher than a silicasome passively entrapping oxaliplatin. (F) And approximate 5–9x fold improvement in drug loading was also achieved for DAPt and EDAPt, according to ICP-MS analysis; this was also confirmed by STEM-EDS visualization. The hydrodynamic sizes of DAPt silicasome and EDAPt silicasome were 138.1 ± 1.5 nm (PDI: 0.113) and 137.8 ± 0.2 nm (PDI: 0.146).
Following the synthesis of the DACHPt soaked-in bare particles, the MSNPs were coated by a lipid bilayer (LB), which was accomplished by an ethanol exchange method that resulted in the formation of a bilayer with a molar ratio of 3:2:0.15 for DSPC: cholesterol: PE-PEG2K, respectively[4b]. The LB provided rapid and uniform pore sealing, improved colloidal stability and capable of entrapping drug payloads of ~70% into the porous interior (Fig. S3). Following experimentation with multi-parameter adjustment, it was possible through the use of ICP-MS Pt elemental analysis, to accomplish an EE% of ~53% and a LC% of ~21 wt% (e.q. oxaliplatin) for the optimized DACHPt silicasome formulation. We also prepared a control silicasome carrier that encapsulated oxaliplatin by using a passive loading approach. This demonstrated a ~5x higher LC% and EE% values using the coordination approach, versus passive loading (Fig. 2D).
In order to confirm the LC% analysis, we conducted elemental mapping at nanostructural level by performing energy-dispersive X-ray spectroscopy (EDS) in combination with scanning transmission electron microscope (STEM) (Fig. 2E). While STEM allowed visualization of the silicasome morphology, complementary EDS spectrum imaging of the same regions of interest (ROI) allowed high resolution visualization of the compositional variations. EDS revealed a strong signature for Pt in the DACHPt silicasome compared to a weaker signal for the same element in the oxaliplatin silicasome (Fig. 2E). This is in agreement with the EDS spectrum that showed a Pt/Si element ratio (w/w) of 19.4% for the DACHPt silicasome compared to a ratio of 3.3% for the oxaliplatin silicasome. We also observed a phosphorus (P) signal in the EDS, which confirmed particle coating by a phospholipid containing bilayer.
In order to show the broader application of our loading approach, we also carried out drug loading studies for DAPt (activated cisplatin) and EDAPt (activated Pt(en)Cl2) at pH 8.5, followed by lipid coating. ICP-MS analysis confirmed that the LC% and EE% were improved 5.6 to 8.5-fold, respectively, compared to passive drug loading (Fig. 2F, upper panel). Improved drug loading was also confirmed by in situ STEM-EDS visualization (Fig. 2F, bottom panel).
To investigate the in vivo relevance of improved DACHPt delivery by silicasomes in an animal tumor model, a freshly prepared batch of DACHPt silicasome was fully characterized, as shown in Fig. 3A. This includes assessment of the primary and hydrodynamic particle sizes by cryoEM and DLS, allowing us to show a particle sizes of 82.4 ± 2.8 nm and 137.3 ± 1.1 nm (PDI 0.076), respectively. High-magnification cryoEM images confirmed that there was uniform coating on the MSNP surface by an intact 6–7 nm thick lipid bilayer (Fig. 3A). The particles were also used to determine drug release by incubation in H2O, acidified phagolysosomal simulation fluid (PSF) buffer (pH 4.5) and 100% serum (Fig. S4). This showed ~6% drug release from the carrier in H2O at 48 h, compared to ~20% and ~35% by serum and PSF buffer, respectively (Fig. S4). Before conducting animal experiment, we also assessed the impact of the carriers on the viability of human PANC-1 and murine KPC cells. The latter cell line was derived from a spontaneous KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-1-Cre PDAC tumor[20]. An MTS assay was used to demonstrate that the DACHPt silicasome resulted in roughly the same decrease in cell viability, as the free drug over 48 h (Fig. S5A and S5B). Since oxaliplatin is also useful for the treatment of colon cancer, similar cell viability experiments were carried out in MC38 and CT26 colon cancer cells. This demonstrated a dose-dependent cytotoxic effect (Fig. S5C and S5D). The characterization of the cell death response by the DACHPt silicasome as immunogenic cell death (ICD) will be discussed later.
Fig. 3.
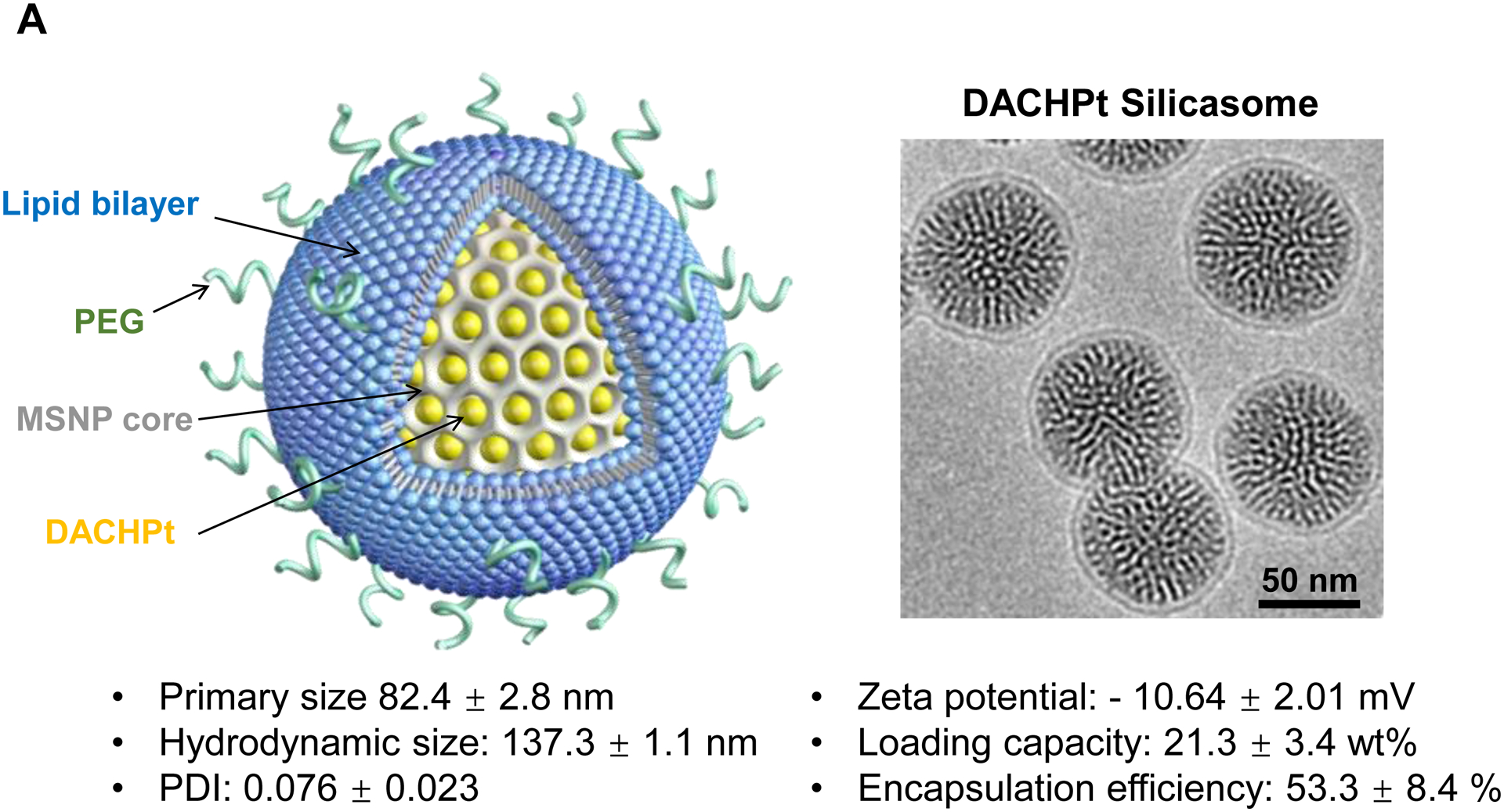
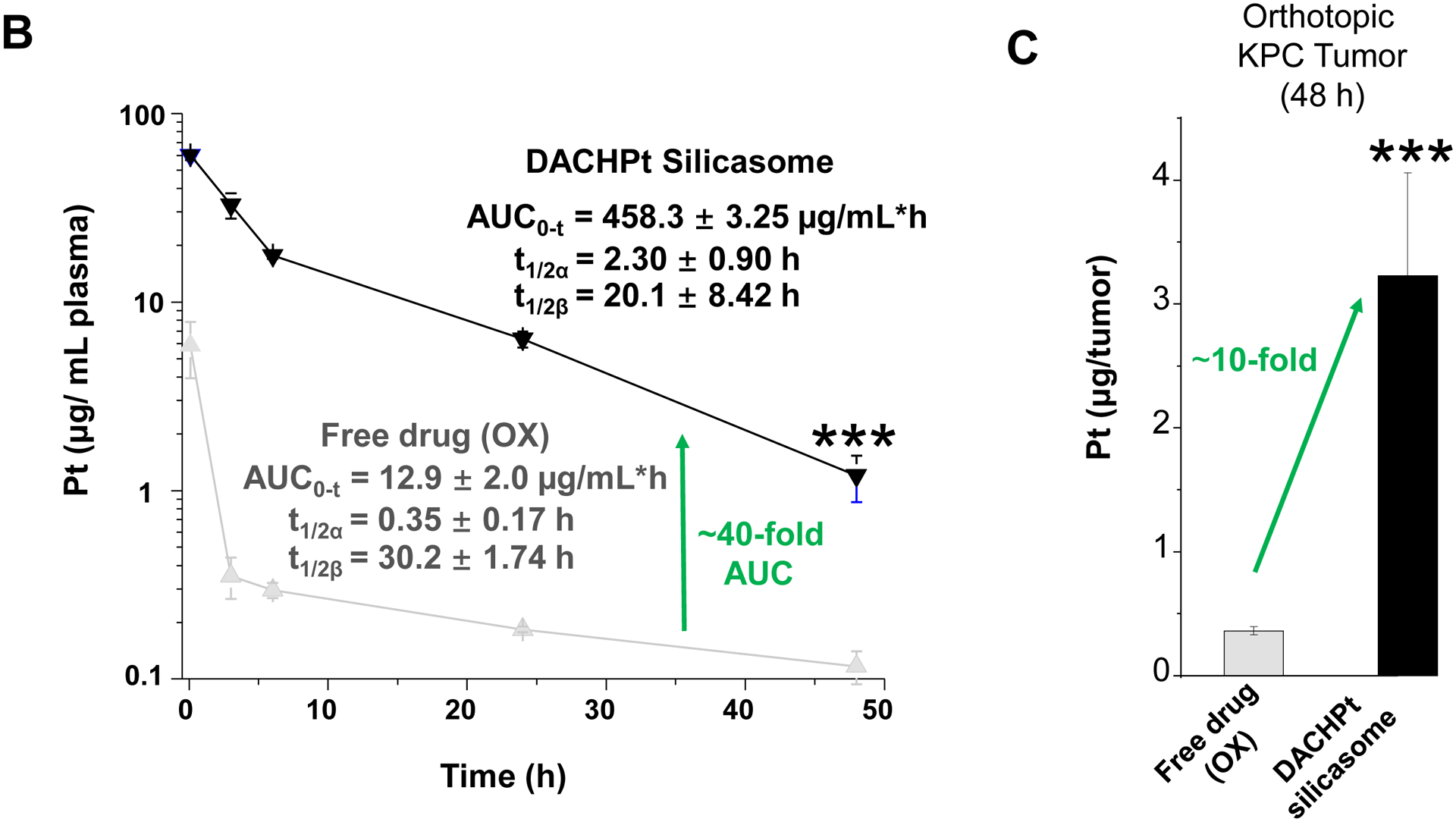
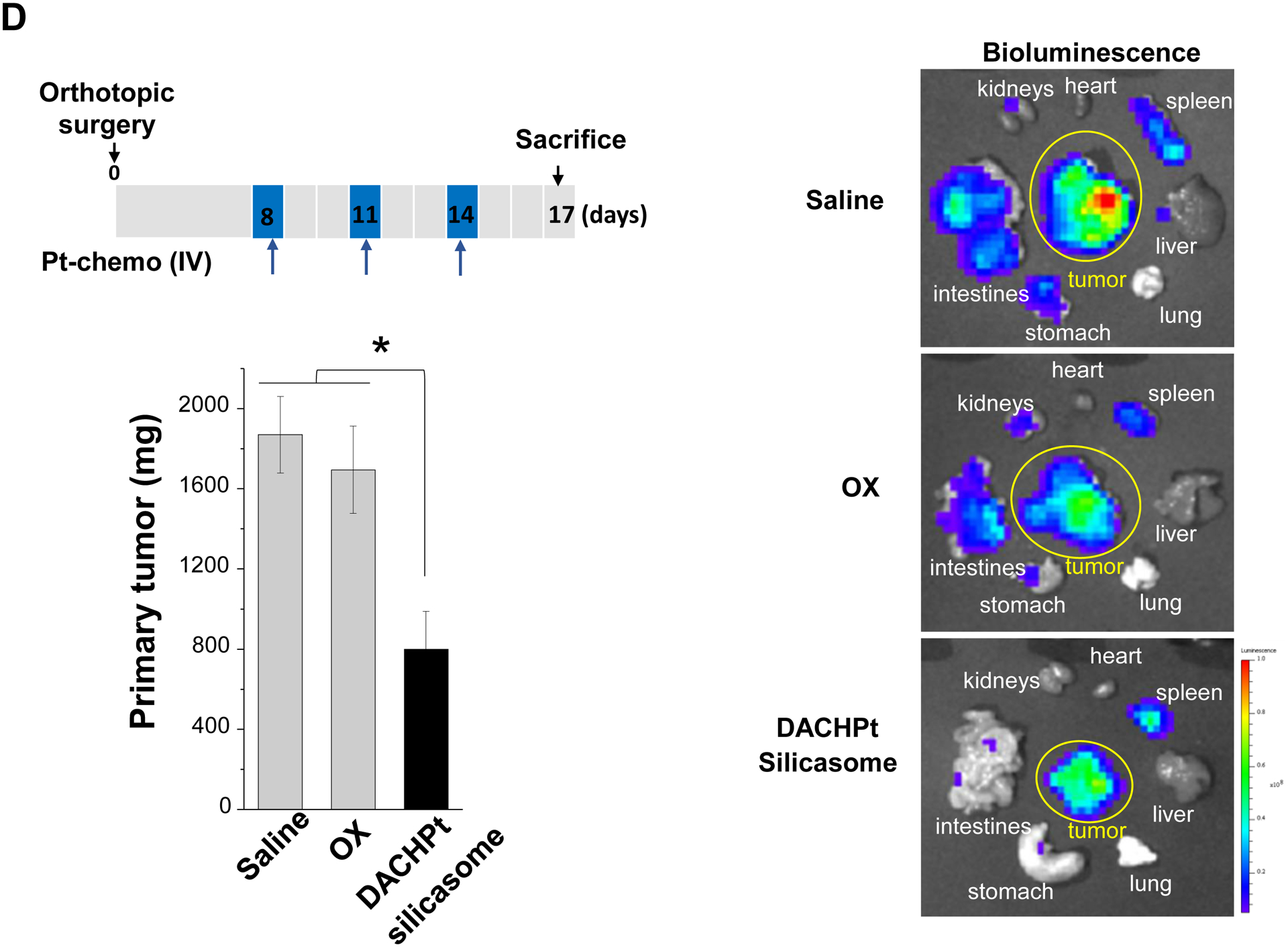
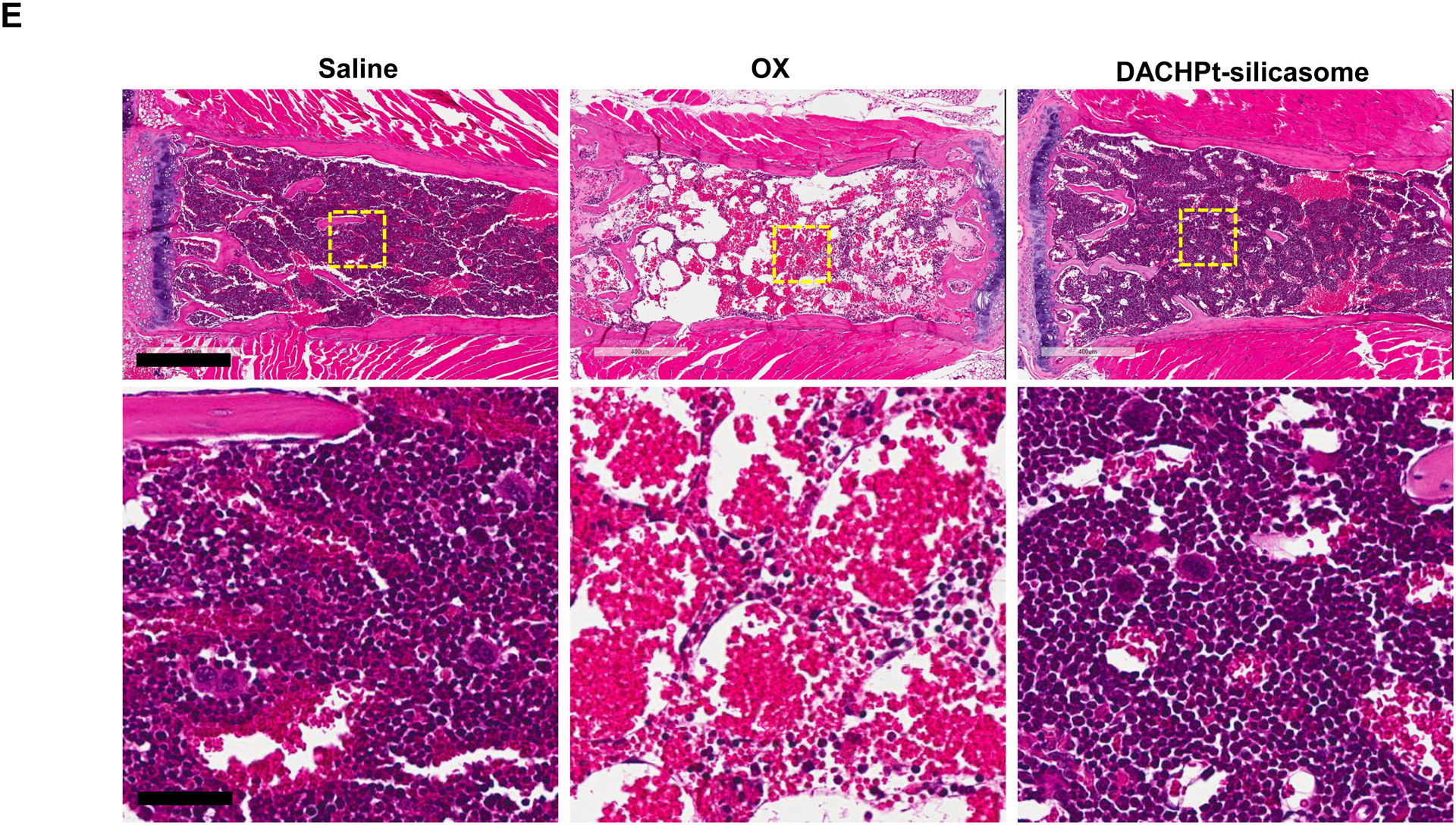
The DACHPt silicasome improves the PK, biodistribution and anti-cancer efficacy over the free drug in a KPC-derived orthotopic model. The encapsulated delivery also improves drug safety. (A) Before animal experimentation, the DACHPt silicasomes were fully characterized, including by cryoEM visualization. The physicochemical properties are summarized. (B-C) PK profile in healthy mice (B) and Pt drug content in orthotopic KPC tumor (C) after the animals received a single IV injection of free oxaliplatin or DACHPt silicasome at identical Pt dose, i.e. 4.95 mg/kg (n = 3). PK parameters were calculated by PKSolver software. Pt content was quantified by ICP-MS. Data represent mean ± SD; ***, p < 0.001 (two-tailed Student’s t-test). (D) Comparative efficacy testing of DACHPt silicasome vs free oxaliplatin in the orthotopic KPC model in B6129SF1/J mice. KPC-luc tumor-bearing mice received the free drug or DACHPt silicasomes 8 days after initial tumor implantation. A total of 3 IV administrations were performed. Saline was used as a negative control. In addition to assessing primary tumor size by weight, tumor size and metastases were also assessed by IVIS imaging as shown in the right-hand panel. Data represent mean ± SEM; *, p < 0.05 (one-way ANOVA followed by a Tukey’s test). (E) Histological analysis of bone marrow by H&E staining in the efficacy experiment in (D). Bar in upper panel is 400 μm and in lower panel is 50 μm. Additional histological analysis to show treatment safety in various organs are shown online (Fig. S7).
To further investigate the impact of the silicasome carrier in an orthotopic PDAC model, KPC cells were surgically implanted into the tail of the pancreas to establish a primary cancer that develops metastatic spread and resembles human PDAC in the expression of a robust dysplastic stroma and poor anti-PDAC immunity[20]. For ease of tumor visualization, the KPC cells were stably transfected with a luciferase vector, as previously described[4a, 21]. In order to assess the pharmacokinetics (PK) of the DACHPt silicasome, the plasma Pt content was quantitatively assessed by ICP-MS in animals receiving a single IV injection of 50 mg/kg MSNPs, which contain a Pt drug content equivalent to 10 mg/kg oxaliplatin. IV injection of non-encapsulated oxaliplatin (at identical Pt molar dose) served as the free drug control because free DACHPt leads to drug precipitates when interacting with Cl ions in the blood[17b]. Blood collection was performed at 5 mins, 3 h, 6 h, 24 h and 48 h after IV injection, followed by ICP-MS quantification. The PK profiling, using a two compartment PKSolver model, was calculated to reflect both the half-live (t1/2) in the circulation (α) as well as for the elimination (β) phase[22] (Fig. 3B). The area-under-the-curve (AUC) for the encapsulated DACHPt was determined to be 458.3 ± 3.25 μg/mL•h, which was ~40-fold higher than for the free drug (labeled as “OX”). To assess Pt drug content in tumor, orthotopic KPC tumor-bearing mice received a single IV injection of drug-laden particles as in the PK study, followed by animal sacrifice at 48 h. Intratumoral drug content improved ~10-fold for encapsulated vs. free drug delivery (Fig. 3C), which is highly significant (p < 0.001). The DACHPt silicasome also showed abundant distribution to the liver and spleen, while the kidney also showed a relatively high Pt level for the encapsulated vs. the free drug (Fig. S6).
An efficacy study was performed by IV injection of the silicasome carrier eight days after orthotopic KPC implantation in the pancreas of B6129SF1/J mice (Fig. 3D). This translates into starting treatment at a primary tumor size of 5~7 mm in the absence of macro-metastases[21]. IV injections were repeated every 3 days for a total of 3 administrations. The control groups included animals receiving IV saline or free oxaliplatin. Please notice that this experiment did not include free DACHPt because the drug is unstable in the systemic circulation due to the high concentration of chloride ions[17b, 23]. The tumor growth at the primary and metastatic sites was followed by IVIS bioluminescence imaging up to the time of concluding the experiment on day 17 (Fig. 3D, right). This demonstrated a lower tumor burden and metastatic spread in response to treatment with the DACHPt silicasome compared to saline and free drug (p < 0.05). We also assessed the primary tumor weight, which demonstrated ~60% tumor inhibition by the DACHPt-loaded silicasome compared to ~5% inhibition by the free drug (Fig. 3D, left). This difference was statistically significant (p < 0.05).
Overcoming chemotherapy side effects is an important objective of nano-based chemotherapy [24]. In order to determine if therapeutic safety also applies to the DACHPt silicasome, histological analysis was performed on all the main organs and the bone marrow. In contrast to the major reduction (>50%) in cellularity of the hemopoietic bone marrow cells by the free drug, there was no noticeable decrease in cellularity during treatment with the DACHPt silicasome, similar to saline administration (Fig. 3E). This finding is of significance from the perspective that PDAC patients frequently develop grade 3–4 neutropenia upon administration of oxaliplatin[5]. Histological examination of other major organs, such as liver, spleen, kidney, lung and heart, did not show any gross pathology in any of the treatment groups (Fig. S7). This is in agreement with our previous drug delivery studies using different mesoporous silica nanoparticle based formulations in vivo[4, 21, 25].
While clearly efficacious for chemotherapy delivery in the orthotopic PDAC model, we were also interested to see whether encapsulated DACHPt exerts an ICD effect, similar to what was described for oxaliplatin[26]. ICD represents a unique form of apoptotic cell death that is accompanied by a chemo-induced cell stress response, characterized by the expression of calreticulin (CRT) and the release of adjuvant stimuli[26a, 27]. While CRT expression on the dying tumor cell surface provides an “eat-me” signal to antigen-presenting cells (APC)[26], the subsequent release of high-mobility group box 1 (HMGB1) from the dying tumor cells provide an adjuvant stimulus for APC maturation (Fig. 4A)[26a, 26c–f, 28]. It is also highly feasible to consider combining the ICD stimulus with an immune checkpoint inhibitor (ICI) such as anti-PD-1 towards combination immunotherapy[28]. In order to investigate whether DACHPt induces CRT expression and HMGB1 release in KPC cells, an in vitro experiment was performed in cultured cells that were exposed to the DACHPt as well as oxaliplatin (Fig. S8). This demonstrated effective killing of 70% of the cells by both treatments after 24 h. Immunofluorescence staining with an anti-CRT primary antibody, followed by Alexa Fluor® 488 conjugated secondary antibody, demonstrated a significant increase in cell surface CRT expression during performance of flow cytometry (Fig. 4B, upper panel). We also performed an ELISA assay to measure HMGB1 release into the supernatant, demonstrating increased release of HMGB1 from treated cells (Fig. 4B, lower panel). In order to determine whether the vitro ICD biomarker expression translates into in vivo immunogenic effects according to a consensus ICD guideline[29], we also performed a vaccination study in a syngeneic KPC animal model. Suspensions of dying tumor cells, generated by oxaliplatin or DACHPt exposure for 24 h, were subcutaneously injected on 2 occasions (7 days apart) in left flank of immunocompetent B6129SF1/J mice. This was followed by tumor re-challenge on the contralateral side, using untreated KPC cells (Fig. 4C). The magnitude of the growth inhibition on the contralateral side demonstrated that both treatments could result in growth retardation, implying immunogenic effects (p < 0.05). Notably, 1 (out of 6) mouse in the DACHPt-treated group remained tumor free (see “spaghetti curves” in Fig. S9A). Tumor weight measurements (Fig. S9B) confirmed the reduction in tumor volume. Importantly, immunohistochemistry (IHC) analysis of the remove tumor tissue showed an increase in infiltration by CD8+ T-cells in parallel with a reduced number of Foxp3+ regulatory T cell (Treg) for both oxaliplatin and DACHPt treatment (Fig.4D). This amounted to a respective increase of 4.8- and 6-fold in the CD8+/Tregs cell ratios compared to PBS control (Fig. 4D).
Fig. 4.
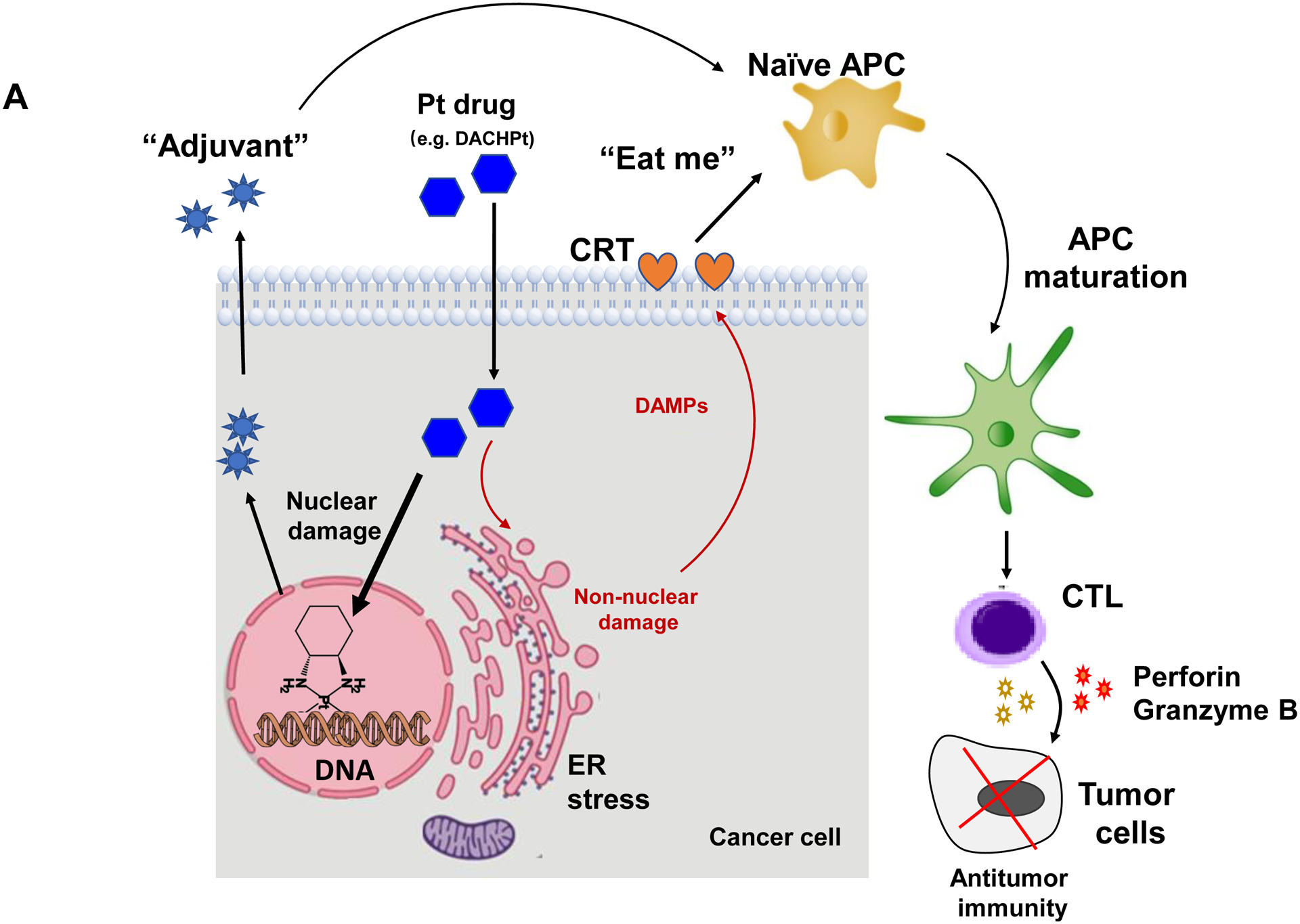
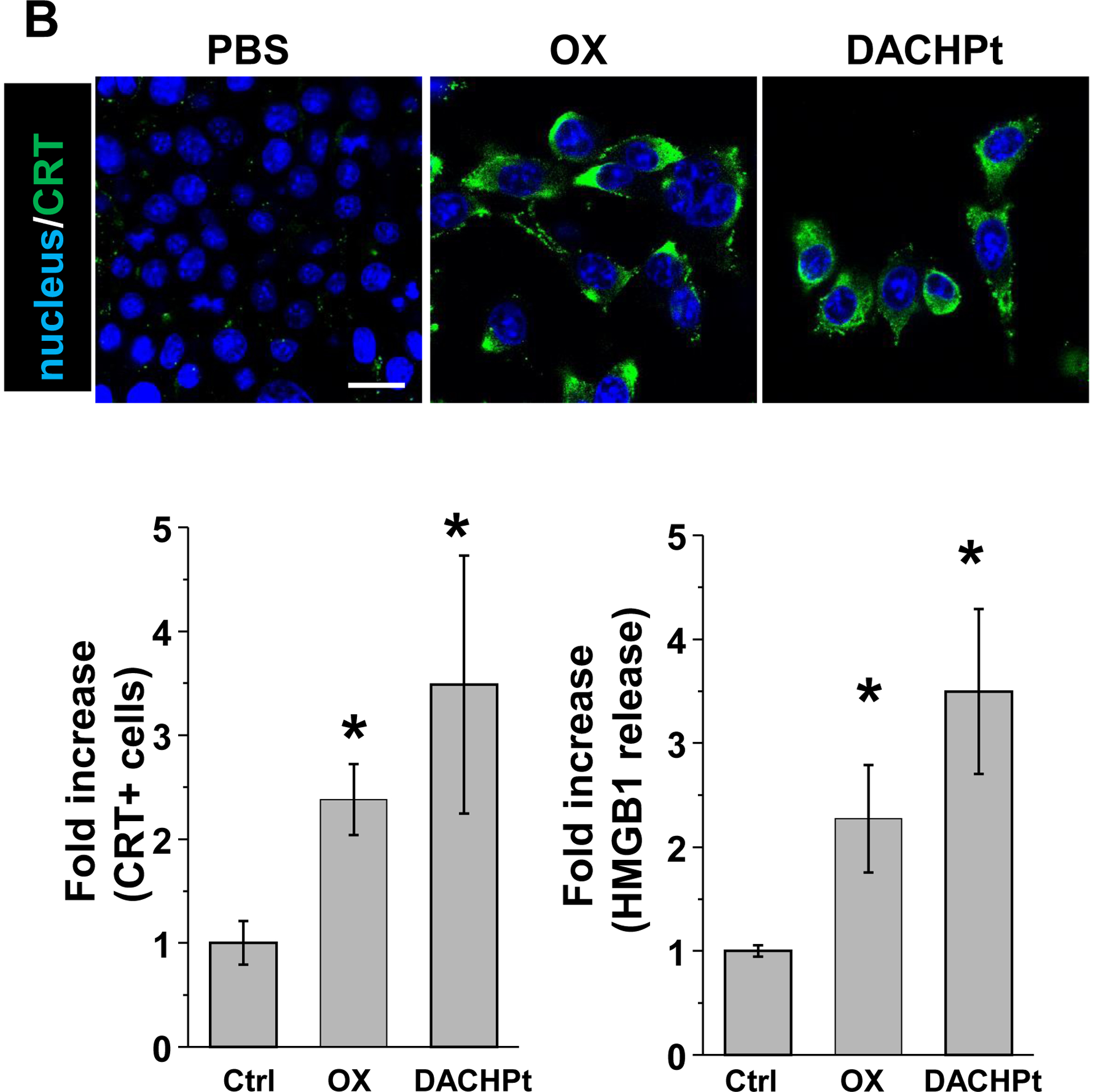
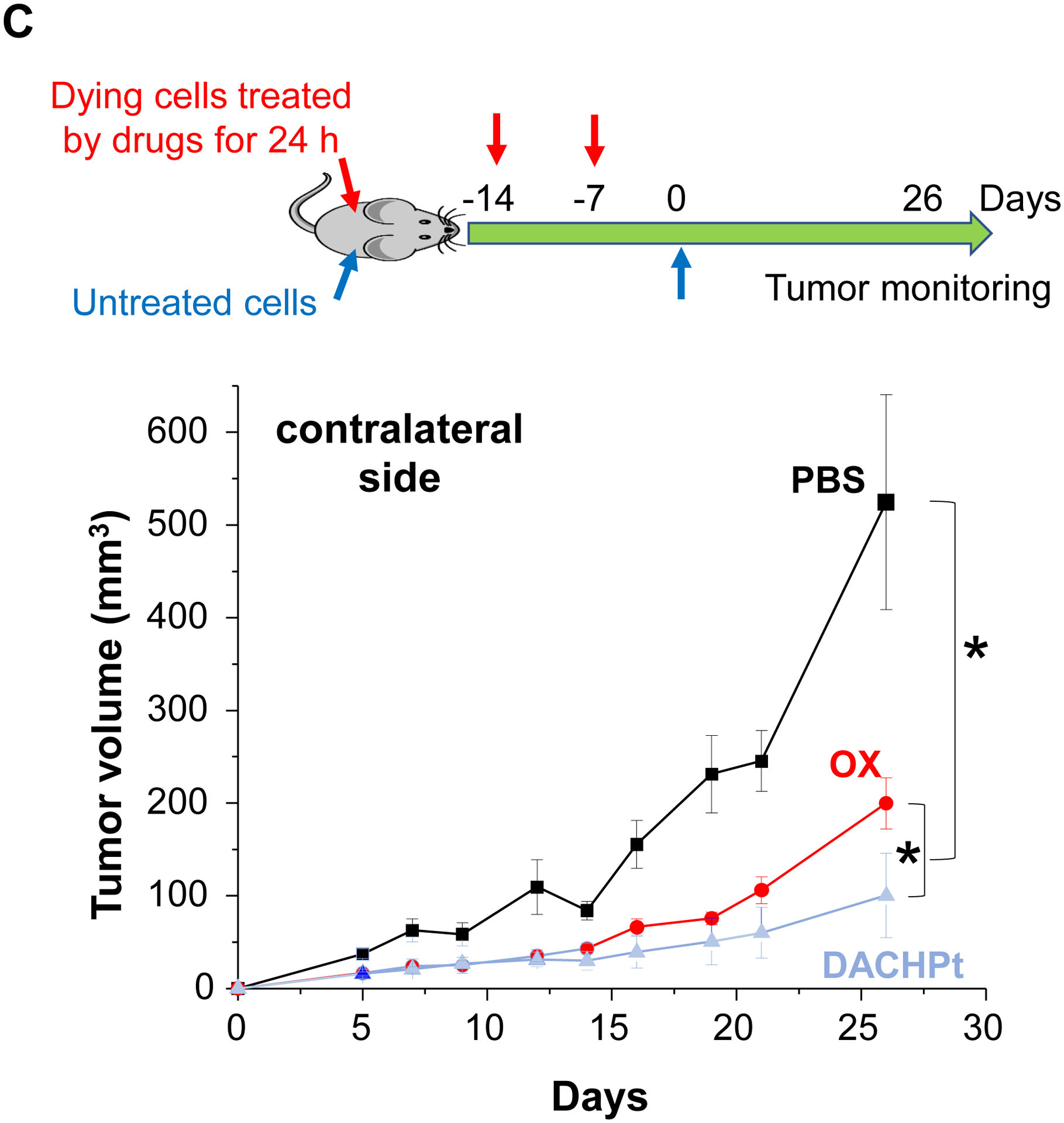
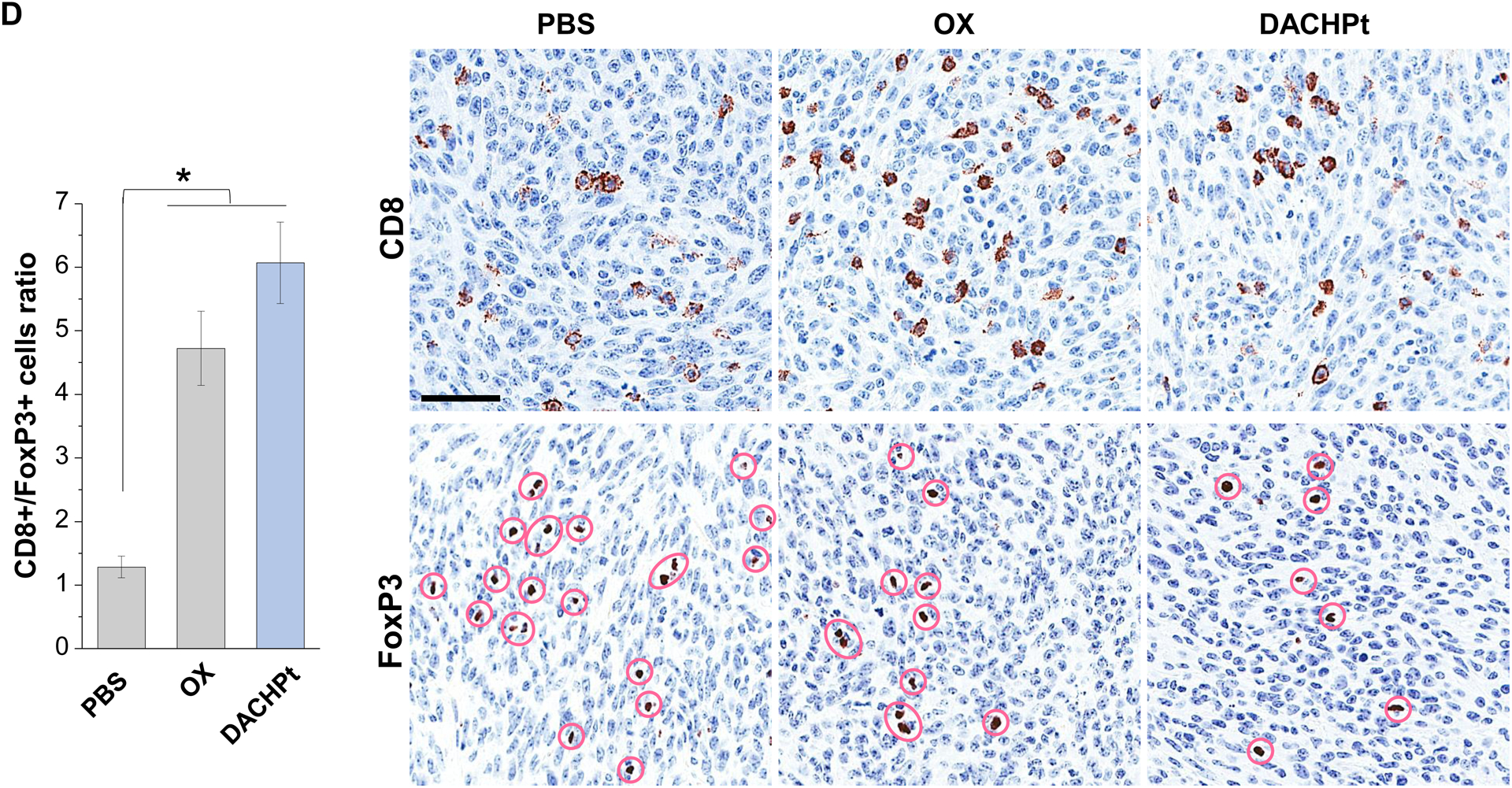
DACHPt induces immunogenic cell death (ICD) in the KPC tumor model. (A) Schematic to illustrate the action of DACHPt as an inducer of ICD. Select Pt chemo agents, such as DACHPt, induce and immunogenic response in which CRT expression on the dying cancer cell surface provides an “eat-me” signal for APC cells. The response is also accompanied by the release of adjuvant stimuli, such as HMGB1, which promote APC maturation and cross-presentation of endogenous tumor-associated antigens. This can lead to the activation and recruitment of CD8+ T cells capable of mediating cytotoxic cancer cell death by the release of perforin. (B) Upper panel: Confocal microscopy showing the appearance of CRT on the KPC cell surface treated with oxaliplatin or DACHPt (500 μM) for 24 h. Bar is 20 μm. Green: CRT; Blue: Nuclear. Lower panel: CRT expression was assessed by flow cytometer (left panel) and HMGB1 release was determined by ELSLA (right panel) in KPC cells exposed to oxaliplatin or DACHPt (500 μM) for 24 h. Data are expressed as mean ± SD, n=3. *, p < 0.05 compared to PBS control (one-way ANOVA followed by a Tukey’s test). (C) In vivo confirmation of the ICD effect by a vaccination study, in which healthy B6129SF1/J mice first received treatment with the chemo -induced dying KPC cells in one flank on two occasions one week apart, followed by injection of live KPC cells on the contralateral side. Tumors on the contralateral side were collected on day 26. (D) The tumor tissue was used for IHC analysis of CD8+ and Foxp3+ T cell appearance, allowing us to calculate a CD8+/Foxp3+ ratio. Data are expressed as mean ± SEM, n=6. *, p<0.05 (one-way ANOVA followed by a Tukey’s test). Bar is 50 μm.
Given this background, we proceeded to the performance of IHC staining for ICD markers, and immune activation in the tumor tissues harvested from the animals treated with the DACHPt silicasomes and oxaliplatin in Fig. 3D. This analysis demonstrated that encapsulated DACHPt was capable of inducing significantly higher CRT expression compared to animals treated with free drug (p < 0.05), or saline (p < 0.01) (Fig. 5A). Representative IHC pictures for CRT staining appear online (Fig. S10A). The in vivo CRT data are further supported by increased CRT expression in cultured KPC cells exposed to silicasome-encapsulated DACHPt or free drug controls (Fig. S10B). The DACHPt silicasome induced comparable CRT expression on the KPC surface as free OX and DACHPt in vitro. Similar findings were obtained during IHC staining to assess HMGB1 release (Fig. 5B); this required software analysis to quantify protein release from damaged nuclei (Fig. S11). Moreover, we also evaluated the number of CD8+ T cells and FoxP3+ Tregs at the tumor site. This resulted in a significant increase in the CD8+/Treg ratio compared to free drug (p < 0.05) or the saline control (p < 0.001) (Fig. 5C). Representative IHC images appear online (Fig. S12). Impressively, we observed significantly increased staining for perforin and granzyme B at the tumor site of animals treated with the DACHPt silicasome compared to free drug (p < 0.01) or the saline control (p < 0.001) (Figs. 5D and 5E). Representative IHC images appear on the right-side panels.
Fig. 5.
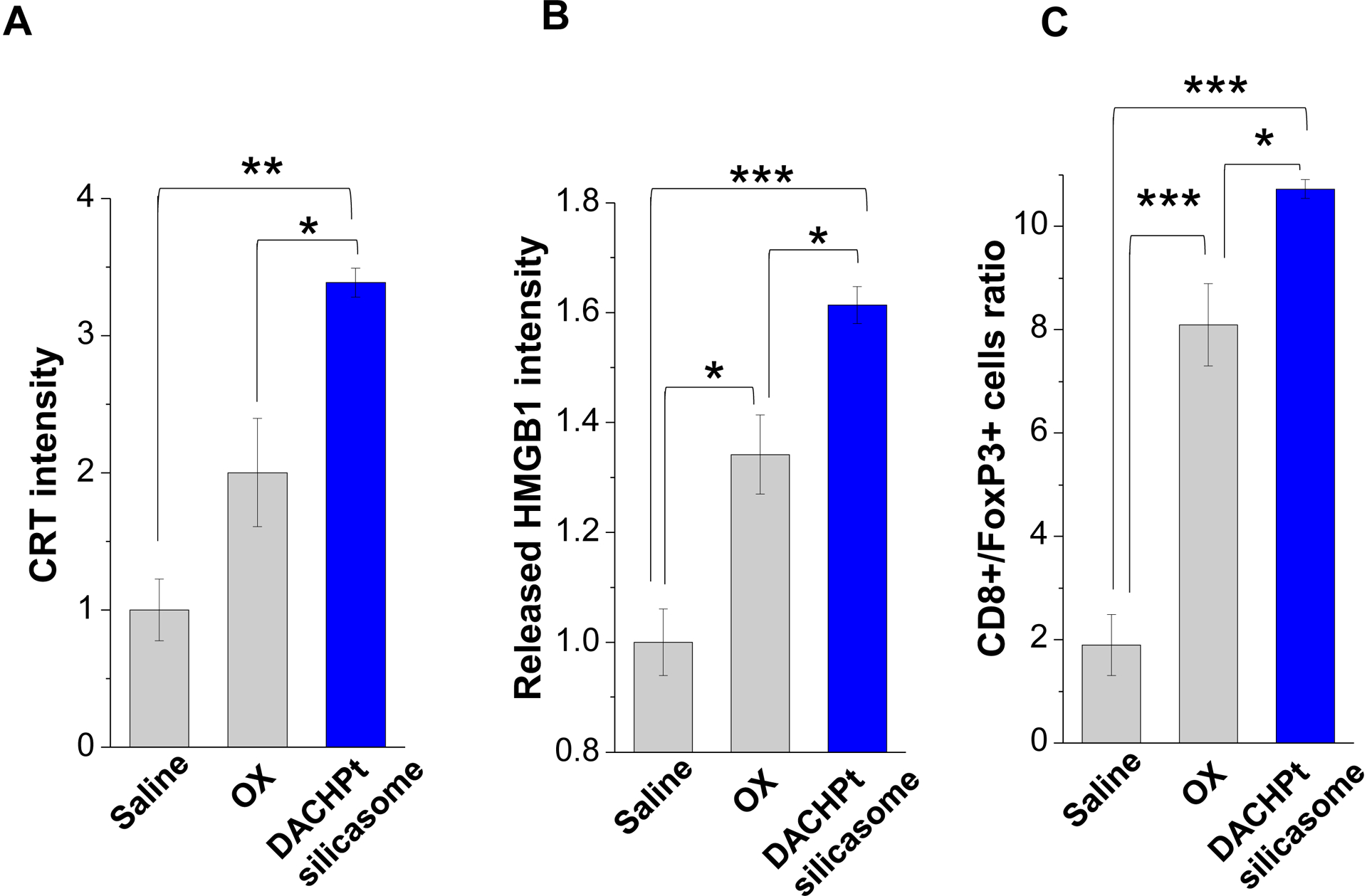
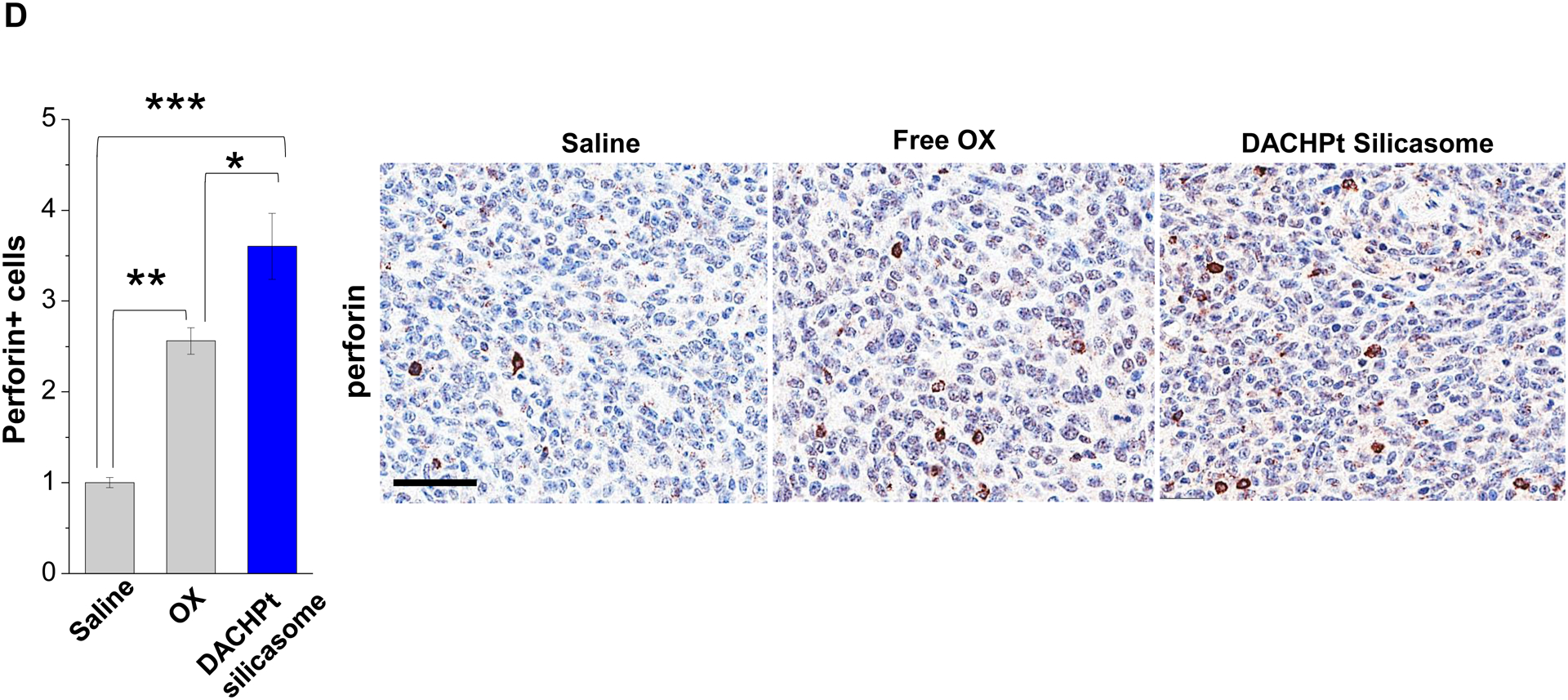
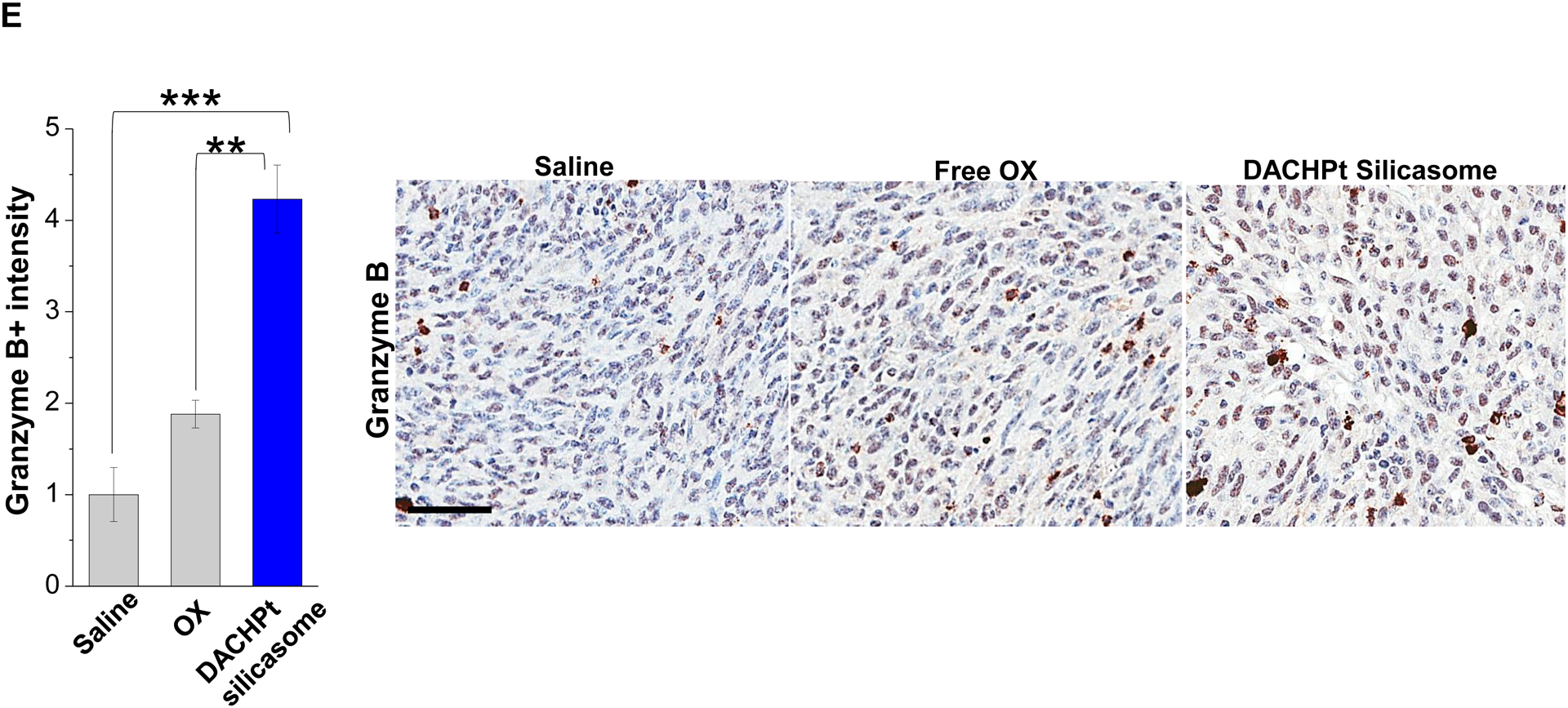
Immunogenic effects of the DACHPt silicasome in orthotopic PDAC tumors. The data were generated from the same efficacy study described in Fig. 3D. (A-C) IHC analysis shows the appearance of ICD biomarkers (CRT and HMGB1) as well as recruitment of activated CD8+ and Foxp3+ T-cells at the tumor site. A more comprehensive array of panels from the IHC staining data are shown in Supplementary Figure S10. (D-E) Assessment of perforin (D) and granzyme B (E) expression in the efficacy study. Data are expressed as mean ± SEM, n=3. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (one-way ANOVA followed by a Tukey’s test). Bars are 50 μm.
To confirm the therapeutic benefit of the DACHPt silicasome in the efficacy study, we also performed a survival outcome study in the KPC orthotopic model, using the same dosimetry, and frequency of DACHPt silicasome administration as for Fig. 3D, with minor modifications (Fig. 6). We also included the use of the treatment with anti-PD-1 in this analysis to determine whether the generation of a “hot” immune environment (as a result of the ICD effect by encapsulated DACHPt) could affect the survival outcome through a chemo-immunotherapy effect. Consistent with the efficacy study, the DACHPt silicasome significantly improved the survival outcome compared to saline and free oxaliplatin in the syngeneic orthotopic KPC model (p < 0.05). Moreover, the performance of the already efficacious DACHPt silicasome was further enhanced by anti-PD-1 antibody, leading to a significant prolongation of the animal life span (p < 0.05). No obvious benefits were observed in the treatment using free oxaliplatin alone or anti-PD-1 alone. This likely reflects inadequate oxaliplatin access to the tumor site[23] as well as low immunogenicity of the PDAC tumor as a result of the low mutational load and a relatively “cold” tumor microenvironment in the absence of an immunogenic stimulus[30].
Fig. 6.
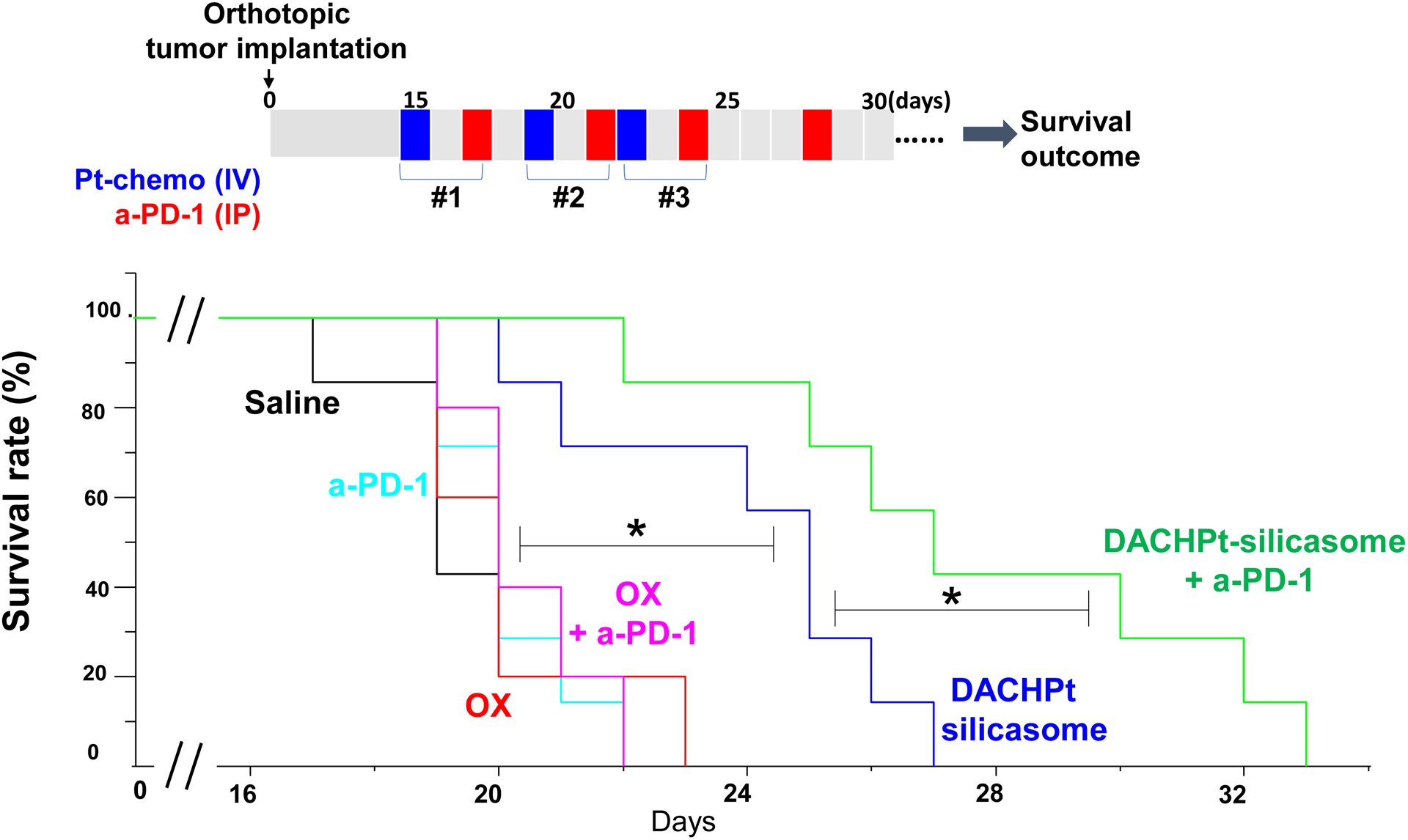
Animal survival study in the orthotopic KPC model, treated with DACHPt silicasome w/wo anti-PD-1 antibody. KPC tumor-bearing mice were treated with DACHPt silicasome at a Pt dose equivalent of 2 mg/kg IV every 3–4 days, with or without IP administration of 100 μg anti-PD-1 antibody. We also included free oxaliplatin with or without anti-PD-1 for comparison. While chemotherapeutic agent was administrated for 3 times, a total of 4 administrations was given for the antibody, as we outlined on the top panel. Saline and anti-PD-1 antibody alone were also used as controls. Kaplan-Meier plots were used to display the survival rate of the different animal groups (n=5–7, *p < 0.05, Log Rank test).
3. Conclusions
In this study, we developed a facile and effective drug loading approach to allow Pt drug delivery by a silicasome nanocarrier. This required the use of activated Pt drugs for efficient loading, and electrostatic/coordination attachment to the porous interior under weak basic conditions. Additional coating of the MSNPs surface by a lipid bilayer allowed secured entrapment of the Pt drug molecules, in addition to providing colloidal stability and effective systemic biodistribution. This was corroborated by the improved PK profile and intratumoral drug delivery by the DACHPt laden silicasome over free drug in the orthotopic KPC model. IV injection of DACHPt silicasome also demonstrated efficacy in the chemotherapy response of the tumor along with a significant reduction in bone marrow toxicity. In addition, the tumor killing response was associated with an ICD response that was reflected by increased biomarker expression for ICD and recruitment of cytotoxic T-cells. In a separate survival experiment, IV injection of DACHPt silicasome led to a significantly improved survival benefit compared to the free drug. Moreover, the efficacy of the chemo-immunotherapy response was further enhanced by the co-administration of anti-PD-1 antibody. Collectively, the successful development of a facile and versatile Pt silicasome offers great promise in improving the therapeutic index of Pt-based chemotherapy agents, both as a monotherapy as well as in combination with checkpoint inhibitors.
4. Experimental Section
Materials
Tetraethylorthosicate (TEOS), triethanolamine (TEA-ol), triethylamine (TEA) cetyltrimethylammonium chloride solution (CTAC, 25 wt% in water), silver nitrate(AgNO3), nitric acid (HNO3), 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), dextrose, dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPtCl2), cis-diammineplatinum(II) dichloride (cisplatin), and dichloro(ethylenediamine)platinum(II) (Pt(en)Cl2) were purchased from Sigma-Aldrich, USA. 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phospho-ethanol amine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000), and cholesterol (Chol) were purchased from Avanti Polar Lipids, USA. Oxaliplatin was purchased from LC Laboratories, USA. Murine anti-PD-1 antibody (#BE0146) and InVivoPure pH 7.0 dilution buffer (#IP0070) were purchased Bio X Cell (New Hampshire, USA). Penicillin, streptomycin, Dulbecco’s modified Eagle medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 Medium were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from Gemini Bio Products. Matrigel- Matrix Basement Membrane was purchased from BD Bioscience.
Preparation of cationic and activated Pt drugs: DACHPt, DAPt and EDAPt
To achieve efficient drug loading, a list of cationic and activated Pt drugs were prepared as detailed in the literature, with minor modification[31]. Taking DACHPt for example, DACHPtCl2 (506 mg, 1.33 mmol) and AgNO3 (406 mg, 2.39 mmol) were added in 9.37 mL DI H2O. The molar ratio of AgNO3 : DACHPtCl2 was 1.8 : 1. Subsequently, 93.7 μL of a 5% HNO3 solution was added to the mixture to achieve an acidic pH of < 2. The suspension was wrapped in aluminum foil and placed in a 70°C oil bath, with stirring overnight (~16 h). The mixture was cooled on ice and stored in a 4°C refrigerator overnight. The sample was spun down at 4,000 rpm for 10 min and the supernatant filtrated through a 0.22 μm syringe filter to obtain the final product, DACHPt. The DAPt and EDAPt aqueous complexes were prepared in similar fashion from commercially available cisplatin and Pt(en)Cl2, respectively. The ratio of AgNO3: cisplatin (or Pt(en)Cl2) remained the same (1.8 : 1), corresponding to 51 mg AgNO3 plus 50 mg cisplatin or 47 mg AgNO3 plus 50 mg Pt(en)Cl2, respectively. The Pt concentration was determined by ICP-MS (NexION 2000, PerkinElmer). We also used ICP-MS to measure the Ag ion concentration to ensure the removal of AgCl from our samples.
Synthesis, purification, and characterization of cationic and activated Pt drugs laden silicasomes
Sixty-five nm bare MSNPs were synthesized at 18 L scale and purified by extensive acidic ethanol washing to remove the CTAC detergent, as reported previously[4b]. In order to determine the optimal loading condition, we experimented with multiple rounds of drug loading to find the optimal pH, incubation time, sonication condition, and particle/drug feed ratio, etc. Details of the protocol optimization appear online (Fig. S2). In the case of DACHPt loading, purified MSNP (20 mg) from ethanol stock solution was spun at 15,000 rpm for 15 min, and resuspend in 1 mL HEPES buffer (200 mM, pH 8.5). The MSNP suspension was spun down and resuspend in 0.5 mL HEPES buffer (200 mM, pH 8.5). Then, 160 μL DACHPt solution (25 mg/mL, e.q. Pt) and 340 μL pure H2O were added to the MNSP suspension (40 mg/mL, 0.5 mL). The mixture was sonicated for 10 min. In a sonication water bath. The DACHPt-laden MSNPs were spun down at 15,000 rpm for 12 min, followed by collecting the supernatant for Pt elemental analysis by ICP-MS. This allowed us to calculate the amount of drug associated with MSNP. Drug-soaked MSNPs were further used to introduce surface lipid coating. Briefly, the DACHPt-laden MSNP pellet was resuspended in 1 mL 5% Dextrose 5 mM HEPES buffer (pH 7.4, absence of Cl− ion) and added to the lipid suspended in an ethanol solution (32 mg DSPC, 10.8 mg Chol and 5.6 mg PE-PEG2K, in 100 μL ethanol) at 65 °C. The mixture was sonicated by probe sonication (Ultrasonic Processor Model VCX130, 40% amplitude) at a 10s/5s on/off cycle for 5~10 mins. After sonication, the particles were purified by washing with 5% Dextrose 5 mM HEPES buffer (pH 7.4) through two rounds of centrifugation at 15,000 rpm for 15 mins. The washed DACHPt silicasome sample was re-suspended in 5% Dextrose 5 mM HEPES buffer (pH 7.4) and filtered across a 0.22 μm filter for sterilization. Similarly, the DAPt and EDAPt silicasomes were prepared by the similar procedure as above. In order to compare the complexation synthesis of Pt silicasomes to silicasomes that passively encapsulate oxaliplatin, cisplatin and Pt(en)Cl2, 20 mg bare MSNPs were mixed with a 4 mg equivalent Pt of the drugs dissolved in DI water during vigorously stirring for 24 h. The drug soaked MSNPs were subsequently used for applying the lipid coating, as described above.
The Pt drug content of the final synthesized products was determined by ICP-OES or ICP-MS by diluting the sample in 2% HNO3. The encapsulation efficiency was defined as EE% = [the total amount of encapsulated Pt drug (m1)]/[the total amount of Pt drug (m0)] × 100%. Drug loading capacity was defined as LC% = [the total amount of encapsulated Pt drug (mdrug)]/[the total amount of particle (mMSNP)] × 100%. Particle hydrodynamic size and zeta potential were measured by a ZETAPALS instrument (Brookhaven Instruments Corporation). The final product was visualized by cryoEM (TF20 FEI Tecnai-G2) to confirm the uniformity and integrity of the coated lipid bilayer. The energy-dispersive X-ray spectroscopy (EDS) and element mapping were performed by scanning transmission electron microscopy (STEM) in a FEI Titan 80–300 kV TEM.
Cell culture
The KPC pancreatic adenocarcinoma cell line, which was derived from a spontaneous tumor originating in a transgenic KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-1-Cre mouse (B6/129 background)[4, 20b, 21], was cultured in DMEM, containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine and 1 mM sodium pyruvate. To allow bioluminescence imaging, the KPC cells were permanently transfected with a luciferase-based lentiviral vector in the UCLA vector core facility, followed by a limiting dilution cloning as we previously described[4a].
Cytotoxicity MTS assay
Cytotoxicity testing of free Pt drugs or drug-laden silicasomes was performed by using a standard MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega). PDAC cells were plated at a density of 5×103 cells per well in a 96-well plate and cultured for 24 h before the medium was replaced with fresh medium containing free OX, free DACHPt or DACHPt laden silicasome at indicated concentrations. Non-treated cells were used as control. After treatment for 48 h, the medium was replaced with 100 μL of fresh medium containing MTS solution (5:1, v/v medium/ CellTiter 96® Aqueous stock solution), and the cells were further cultured at 37°C for 1 h. The absorbance of the culture wells at 490 nm was recorded by a microplate reader (M5e, Molecular Device, USA). Wells receiving the MTS solution without cells were used as blank. The relative cell viability (%) is [(the absorption of treated well - blank)/(the absorption of control well - blank)] ×100%.
Animal purchase and study permission
Female B6129SF1/J mice (JAX 101043) were purchased from The Jackson Laboratory, and maintained under pathogen-free conditions. All animal experiments were performed according to protocols (#2009–134) approved by the UCLA Animal Research Committee.
PK study
The PK study was performed on 10~12-week-old healthy female B6129SF1/J mice. The animals received a single IV injection of free OX or DACHPt silicasome at a Pt dose of 4.95 mg/kg (equal to oxaliplatin dose of 10 mg/kg), followed by collection of blood samples at 5 min, 3, 6, 24, and 48 hrs. After separation of the plasma fraction, the plasma samples were digested with HCl: HNO3 3:1, v/v) in a hot-block, before replenishment in 2% HNO3 for ICP-MS analysis of the Pt content. The PK data were analyzed by PKSolver software, using a two-compartment model[22].
Tumor drug content and biodistribution study
An orthotopic KPC tumor model in immunocompetent B6129SF1/J mouse was established as described previously[4, 21]. Briefly, 30 μL of DMEM/Matrigel (1:1 v/v), containing ~1×106 KPC-luc cells, was injected into the tail of the pancreas in female B6129SF1/J mice (8~10 weeks) by a sort surgical survival procedure[4, 21]. To determine the tumor drug content and biodistribution, tumor bearing mice received a single IV injection of free OX or DACHPt silicasome at a Pt dose of 4.95 mg/kg. Animals were sacrificed 48 h post-injection, followed by tumor and tissue collections. These samples were accurately weighted, and followed by digestion using aqua regia in a hot-block and reconstructed in 2% HNO3 for ICP-MS measurement to determine the Pt content.
Assessment of anti-PDAC efficacy by the DACHPt silicasome in the orthotopic KPC model described above
KPC-luc cells (~1×106) were orthotopically injected into the pancreas in mice. Eight-day post-surgery, the tumor-bearing mice received IV injections of DACHPt silicasome at Pt dose of 2 mg/kg. The control includes saline as well as free oxaliplatin. This dose arrangement is in agreement with the literature[23]. Tumor-bearing mice received IV injection of the indicated therapy every 3 days for a total of 3 administrations. Before animal sacrifice (72 h post the last IV injection), the mice received intraperitoneal injection of D-luciferin, followed by ex vivo bioluminescence imaging using an IVIS imaging system. Primary tumor and major organs (e.g. sternum, heart, liver, spleen, lung and kidneys) were harvested and fixed in 10% formalin, followed by paraffin embedding and sectioning to provide 4 μm slices for histological analysis in the UCLA Translational Pathology Core Laboratory (TPCL). H&E staining was performed to look at the pathological abnormality in mice receiving different treatments. The H&E slides for toxicity assessment were read in a blinded fashion by an experienced veterinary pathologist.
Identification of DACHPt as an ICD-inducing agent
Surface CRT expression was visualized by immunofluorescence (IF) staining using an anti-CRT primary antibody, followed by the incubation with Alex488 conjugated secondary antibody. Briefly, ~1.5×104 KPC cells were seeded into an 8-well confocal chamber slide. After 24 h, the cell culture medium was replaced with fresh medium containing the chemo agents, following which the cells were incubated for another 24 h. The cells were washed twice in cold PBS and fixed with 4% paraformaldehyde (PFA) at room temperature (r.t.) for 15 mins. After fixation, the cells were washed twice with cold PBS and blocked with 1% BSA in PBS for 0.5 h. The cells were incubated with anti-CRT primary antibody (ab2907, 1:200) in 200 μL blocking solution at 4 °C overnight, followed by washing with PBS and staining with secondary antibody (Alex488 conjugated goat anti-rabbit secondary antibody, A-11008, 1:1000) together with the nuclear dye, Hoechst 33342, at r.t. for 1 h. The cells were washed with PBS, then imaged by using a Leica SP8-MD confocal microscope under the 100× objective lens.
Surface CRT expression was measured by flow cytometry as previously described[27a]. Briefly, 7.5×104 KPC cells were seeded into 24-well plates. After cell attachment, KPC cells were treated with free oxaliplatin and DACHPt (500 μM), for 24 h. The loosely attached cells were combined with trypsin-treated at adherent cells. The cells were washed in cold PBS and then stained on ice with a primary anti-CRT antibody (Abcam, ab2907, 1: 140) in 200 μL BD staining buffer for 0.5 h. The cells were washed in cold PBS and stained with an Alexa Fluor® 680-conjugated secondary antibody (LifeScience Technologies #A21244) for 30 min on ice. After washing in cold PBS, the cells were assessed in a LSRII flow cytometer (BD Biosciences). In the same experiment, the cell culture media were spun down to collect the supernatants for HMGB1 detection by an ELISA kit (Catalog# ST51011, IBL International GmbH), according to the manufacture’s instruction.
Moreover, we also validated the ICD effect of DACHPt in a vaccination experiment, using a published protocol[29]. Briefly, eight million KPC cells were seeded in a tissue culture dish. After cellular attachment, DACHPt or free oxaliplatin (500 μM) were added for 24 h. Cells were collected and washed before being resuspended in 0.8 mL cold PBS. For vaccination, each mouse received subcutaneous injection (SC) of a 100 μL suspension of chemo-treated cells in the right flank. Control animals received PBS only. The vaccination was repeated after 7 days. Fourteen days after the 1st vaccination, the same mice received the SC injection of healthy KPC cells (1×106 cells) in the contralateral side. Tumor growth was measured by a digital caliper every 2–3 days. At the conclusion of the vaccination experiment (Day 26), animals were sacrificed and the tumors collected for IHC immunophenotyping of CD8+ T cells and FoxP3+ Treg cells. Primary antibodies to CD8 (#14-0808-82, 1: 100) and FoxP3 (#13-5773-82, 1: 200) were purchased from ThermoFisher. IHC staining was performed in the UCLA Translational Pathology Core Laboratory (TPCL). The slides were scanned and images were assessed by using Aperio ImageScope software (Leica).
Investigation of ICD and immune activation by silicasome encapsulated DACHPt in the orthotopic KPC model
The tumor tissues in the efficacy study (Fig. 3D) were used for further immunophenotyping with the focus of ICD induction and immune activation. Primary antibodies that recognizes ICD biomarkers include CRT (ab2907, 1: 200) and HMGB1 (ab18256, 1: 200). We also measured immune activation markers, such as CD8+ T cells (#14-0808-82, 1: 100), FoxP3+ Treg (#13-5773-82, 1: 200), granzyme B (ab4059, 1: 100), and perforin (ab16074, 1: 100) through IHC staining. The slides were scanned and images were analyzed by using Aperio ImageScope software (Leica).
Assessment of the survival outcome using DACHPt silicasome w/wo anti-PD-1 in an orthotopic KPC tumor model
Tumor-bearing mice were randomly assigned into 6 groups (n = 5–7) and received IV injection of Pt drug formulations twice per week as designed in Fig. 6. Anti-PD-1 antibody was injected at 100 μg/animal intraperitoneally two days later after each Pt chemo injection. To assess survival rate, animals were monitored daily up to the stage of spontaneous death or approaching moribund status based on the criteria of extensive abdominal ascites, severe dehydration, significant weight loss (>20%), extreme weakness or inactivity[4, 21, 32]. The survival data were plotted as Kaplan-Meier curves, followed by statistical analysis by Log Rank testing (Mantel-Cox), using GraphPad Prism 7.00 software.
Statistical analysis
Comparative analysis of differences between groups was performed using the 2-tailed Student’s t-test (Excel software, Microsoft) for two-group comparison. One-way ANOVA followed by a Tukey’s test (Origin software, OriginLab) was performed for multiple group comparisons. Data were expressed as mean ± SD or SEM, as stated in the figure legends. The survival analysis was performed by Log Rank testing (Mantel-Cox), using GraphPad Prism 7.00 software. A statistically significant difference was considered at *p < 0.05.
Supplementary Material
Acknowledgements
This study was supported by the U.S. Public Health Service Grants, 1U01CA198846 and 1R01CA247666-01A1. We acknowledge the use of the Preclinical Imaging Technology Center, the Translational Pathology Core Laboratory (TPCL), the Electron Imaging Center for Nanomachines (EICN), and the CNSI Advanced Light Microscopy/Spectroscopy (ALMS) Shared Facility at UCLA.
Footnotes
Competing financial interests: The authors declare the following competing financial interest(s): Andre E. Nel and Huan Meng are co-founders, board members and equity holders in Westwood Bioscience Inc. UCLA entered into a sponsored research agreement with Westwood Bioscience Inc. Andre E. Nel and Huan Meng are also co-founders and equity holders in NAMMI therapeutics. The remaining authors declared no conflict of interest.
Supporting Information Available: Additional figures, table, and methods as described in the text. This material is available free of charge online.
References
- [1].Siegel RL, Miller KD, Jemal A, CA Cancer J Clin 2019, 69, 7. [DOI] [PubMed] [Google Scholar]
- [2].a) Garrido-Laguna I, Hidalgo M, Nat Rev Clin Oncol 2015, 12, 319; [DOI] [PubMed] [Google Scholar]; b) Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH, Nature reviews. Gastroenterology & hepatology 2018, 15, 333. [DOI] [PubMed] [Google Scholar]
- [3].a) Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, U. Groupe Tumeurs Digestives of, P. Intergroup, N Engl J Med 2011, 364, 1817;21561347 [Google Scholar]; b) Suker M, Beumer BR, Sadot E, Marthey L, Faris JE, Mellon EA, El-Rayes BF, Wang-Gillam A, Lacy J, Hosein PJ, Moorcraft SY, Conroy T, Hohla F, Allen P, Taieb J, Hong TS, Shridhar R, Chau I, van Eijck CH, Koerkamp BG, The Lancet. Oncology 2016, 17, 801; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, Chone L, Francois E, Artru P, Biagi JJ, Lecomte T, Assenat E, Faroux R, Ychou M, Volet J, Sauvanet A, Breysacher G, Di Fiore F, Cripps C, Kavan P, Texereau P, Bouhier-Leporrier K, Khemissa-Akouz F, Legoux JL, Juzyna B, Gourgou S, O’Callaghan CJ, Jouffroy-Zeller C, Rat P, Malka D, Castan F, Bachet JB, G. Canadian Cancer Trials, G. I. P. G. the Unicancer, N Engl J Med 2018, 379, 2395. [DOI] [PubMed] [Google Scholar]
- [4].a) Liu XS, Situ A, Kang YA, Villabroza KR, Liao YP, Chang CH, Donahue T, Nel AE, Meng H, Acs Nano 2016, 10, 2702; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu X, Jiang J, Chan R, Ji Y, Lu J, Liao YP, Okene M, Lin J, Lin P, Chang CH, Wang X, Tang I, Zheng E, Qiu W, Wainberg ZA, Nel AE, Meng H, ACS Nano 2019, 13, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Cassidy J, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzeen F, Saltz L, Journal of Clinical Oncology 2008, 26, 2006; [DOI] [PubMed] [Google Scholar]; b) Haller DG, Oncology 2000, 14, 15. [PubMed] [Google Scholar]
- [6].Hato SV, Khong A, de Vries IJ, Lesterhuis WJ, Clin Cancer Res 2014, 20, 2831. [DOI] [PubMed] [Google Scholar]
- [7].a) Galanski M, Jakupec MA, Keppler BK, Curr Med Chem 2005, 12, 2075; [DOI] [PubMed] [Google Scholar]; b) Wheate NJ, Walker S, Craig GE, Oun R, Dalton Trans 2010, 39, 8113. [DOI] [PubMed] [Google Scholar]
- [8].Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK, Adv Drug Deliv Rev 2013, 65, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang D, Lippard SJ, Nat Rev Drug Discov 2005, 4, 307. [DOI] [PubMed] [Google Scholar]
- [10].a) Johnstone TC, Suntharalingam K, Lippard SJ, Chem Rev 2016, 116, 3436; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ma PA, Xiao HH, Li CX, Dai YL, Cheng ZY, Hou ZY, Lin J, Materials Today 2015, 18, 554; [Google Scholar]; c) Hang Z, Cooper MA, Ziora ZM, Biochemical Compounds 2016, 4, 1; [Google Scholar]; d) Xiao HH, Yan LS, Dempsey EM, Song WT, Qi RG, Li WL, Huang YB, Jing XB, Zhou DF, Ding JX, Chen XS, Progress in Polymer Science 2018, 87, 70; [Google Scholar]; e) Mochida Y, Cabral H, Kataoka K, Expert Opin Drug Deliv 2017, 14, 1423. [DOI] [PubMed] [Google Scholar]
- [11].Arean CO, Vesga MJ, Parra JB, Delgado MR, Ceramics International 2013, 39, 7407. [Google Scholar]
- [12].Gu J, Su S, Li Y, He Q, Zhong J, Shi J, The Journal of Physical Chemistry Letters 2010, 1, 3446. [Google Scholar]
- [13].Lin CH, Cheng SH, Liao WN, Wei PR, Sung PJ, Weng CF, Lee CH, Int J Pharm 2012, 429, 138. [DOI] [PubMed] [Google Scholar]
- [14].He H, Xiao H, Kuang H, Xie Z, Chen X, Jing X, Huang Y, Colloids and Surfaces B: Biointerfaces 2014, 117, 75. [DOI] [PubMed] [Google Scholar]
- [15].Zheng Y, Fahrenholtz CD, Hackett CL, Ding S, Day CS, Dhall R, Marrs GS, Gross MD, Singh R, Bierbach U, Chemistry–A European Journal 2017, 23, 3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Martinho N, Santos TCB, Florindo HF, Silva LC, Frontiers in Physiology 2019, 9, 1898; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E, Molecular cancer therapeutics 2002, 1, 227. [PubMed] [Google Scholar]
- [17].a) Salerno M, Yahia D, Dzamitika S, de Vries E, Pereira-Maia E, Garnier-Suillerot A, JBIC Journal of Biological Inorganic Chemistry 2009, 14, 123; [DOI] [PubMed] [Google Scholar]; b) Murakami M, Cabral H, Matsumoto Y, Wu S, Kano MR, Yamori T, Nishiyama N, Kataoka K, Sci Transl Med 2011, 3, 64ra2. [DOI] [PubMed] [Google Scholar]
- [18].Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ, Acc Chem Res 2013, 46, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang H, Dunphy DR, Jiang X, Meng H, Sun B, Tarn D, Xue M, Wang X, Lin S, Ji Z, Li R, Garcia FL, Yang J, Kirk ML, Xia T, Zink JI, Nel A, Brinker CJ, J Am Chem Soc 2012, 134, 15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA, Cancer Cell 2005, 7, 469; [DOI] [PubMed] [Google Scholar]; b) Tseng WW, Winer D, Kenkel JA, Choi O, Shain AH, Pollack JR, French R, Lowy AM, Engleman EG, Clin Cancer Res 2010, 16, 3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu X, Lin P, Perrett I, Lin J, Liao YP, Chang CH, Jiang J, Wu N, Donahue T, Wainberg Z, Nel AE, Meng H, J Clin Invest 2017, 127, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y, Huo M, Zhou J, Xie S, Comput Methods Programs Biomed 2010, 99, 306. [DOI] [PubMed] [Google Scholar]
- [23].Cabral H, Murakami M, Hojo H, Terada Y, Kano MR, Chung UI, Nishiyama N, Kataoka K, Proc Natl Acad Sci U S A 2013, 110, 11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R, Nature Nanotechnology 2007, 2, 751; [DOI] [PubMed] [Google Scholar]; b) Liu X, Tang I, Wainberg ZA, Meng H, Small 2020, 2000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Meng H, Xue M, Xia T, Ji Z, Tarn DY, Zink JI, Nel AE, ACS nano 2011, 5, 4131; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meng H, Mai WX, Zhang H, Xue M, Xia T, Lin S, Wang X, Zhao Y, Ji Z, Zink JI, ACS nano 2013, 7, 994; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng H, Wang MY, Liu HY, Liu XS, Situ A, Wu B, Ji ZX, Chang CH, Nel AE, Acs Nano 2015, 9, 3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P, Biochimica et biophysica acta 2010, 1805, 53; [DOI] [PubMed] [Google Scholar]; b) Zhao X, Yang K, Zhao R, Ji T, Wang X, Yang X, Zhang Y, Cheng K, Liu S, Hao J, Ren H, Leong KW, Nie G, Biomaterials 2016, 102, 187; [DOI] [PubMed] [Google Scholar]; c) Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, Sautes-Fridman C, Fucikova J, Galon J, Spisek R, Tartour E, Zitvogel L, Kroemer G, Galluzzi L, Oncoimmunology 2015, 4, e1008866; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G, Cancer Cell 2015, 28, 690; [DOI] [PubMed] [Google Scholar]; e) Bezu L, Gomes-da-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G, Frontiers in immunology 2015, 6, 187; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kroemer G, Galluzzi L, Kepp O, Zitvogel L, Annu Rev Immunol 2013, 31, 51. [DOI] [PubMed] [Google Scholar]
- [27].a) Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G, Nat Med 2007, 13, 54; [DOI] [PubMed] [Google Scholar]; b) Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G, Clin Cancer Res 2010, 16, 3100; [DOI] [PubMed] [Google Scholar]; c) Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, Nature reviews. Cancer 2012, 12, 860. [DOI] [PubMed] [Google Scholar]
- [28].Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ, Immunity 2016, 44, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, Oncoimmunology 2014, 3, e955691.25941621 [Google Scholar]
- [30].a) Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Proceedings of the National Academy of Sciences 2013, 110, 20212; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL, Current protocols in pharmacology 2016, 73, 14.39. 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, Vonderheide RH, Cancer Immunol Res 2015, 3, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sood P, Thurmond KB, Jacob JE, Waller LK, Silva GO, Stewart DR, Nowotnik DP, Bioconjugate Chemistry 2006, 17, 1270. [DOI] [PubMed] [Google Scholar]
- [32].Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA, Science 2009, 324, 1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.