

Abstract
Despite considerable efforts in modeling liver disease in vitro, it remains difficult to recapitulate the pathogenesis of the advanced phases of non-alcoholic fatty liver disease (NAFLD) with inflammation and fibrosis. Here, we developed a liver-on-a-chip platform with bioengineered multicellular liver microtissues composed of four major types of liver cells (hepatocytes, endothelial cells, Kupffer cells, and stellate cells) to implement a human hepatic fibrosis model driven by NAFLD: i) lipid accumulation in hepatocytes (steatosis), ii) neovascularization by endothelial cells, iii) inflammation by activated Kupffer cells (steatohepatitis), and iv) extracellular matrix (ECM) deposition by activated stellate cells (fibrosis). In our model, the presence of stellate cells in the liver-on-a-chip model with fat supplementation showed elevated inflammatory responses and fibrosis marker up-regulation. Compared to transforming growth factor-beta (TGFβ)-induced hepatic fibrosis models, our model includes the native pathological and chronological steps of NAFLD which shows (1) higher fibrotic phenotypes, (2) increased expression of fibrosis markers and (3) efficient drug transport and metabolism. Taken together, the proposed platform will enable a better understanding of the mechanisms underlying fibrosis progression in NAFLD as well as the identification of new drugs for the different stages of NAFLD.
Keywords: liver microtissues, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), liver fibrosis, co-culture
Graphical Abstract
1. Introduction
Fibrosis is characterized through the extreme accumulation of extracellular matrix (ECM) in an organ following an injury or trigger. Organ fibrosis, secondary to chronic injury, can eventually lead to organ dysfunction resulting from the remodeling process that accumulates ECM in the damaged parenchyma.[1] One of the health-threatening fibrotic disorders is non-alcoholic fatty liver disease (NAFLD) triggered via chronic abundant feeding of fat. Excessive food/calorie intake causes lipid accumulation in the liver (steatosis), followed by the release of proinflammatory cytokines that leads to tissue inflammation (fatty hepatitis), which can eventually lead to tissue fibrosis (cirrhosis).[2] The complex pathophysiology of end-stage liver disease involves multiple cell types across different disease stages that progress over several months to many years, varying for each individual patient. It starts with the accumulation of lipids (triglycerides) in hepatocytes which over time may trigger an inflammatory response of the tissue by the activation of macrophages (mainly Kupffer cells (KCs)). Immune cells as well as hepatocytes and liver sinusoidal cells release different signaling molecules such as cytokines that promote the activation of hepatic stellate cells (HSCs) into myofibroblasts.[3] Among those molecules, transforming growth factor-beta (TGFβ) is one of the most potent fibrogenic cytokines.[4] Myofibroblasts then remodel the ECM by depositing fibrosis-associated proteins, including collagen I and fibronectin, resulting in scar formation and cirrhosis.[5,6] With an increasing prevalence of roughly 25% in the United States, NAFLD is the foremost source of chronic liver disease, accruing large economic, emotional, and health tolls to patients. However despite its prevalence, it has yet to be properly studied due to difficulty in pathology modeling.[7]
Animal models have been used over the past decades for genetic-, chemical-, or diet-induced modeling of NAFLD. However, none of these can fully recapitulate the pathology and progression of the human NAFLD.[8,9] To overcome the limitation of animal models, various in vitro human NAFLD models have been suggested with varying levels of complexity, from simple two-dimensional (2D) monolayer cultures to advanced three-dimensional (3D) co-cultures.[10–13] However, an advanced 3D in vitro model that captures the structural complexities of the liver, which impact the physiological/pathological model, has not yet been achieved. To build improved pharmaceutical models, it is necessary to employ relevant hepatocellular models. For example, cytochromes P450 (CYP), which are a family of enzymes associated with the metabolism of many xenobiotics and drugs, exhibits varying metabolic capabilities in different cell types. In particular, culturing a human hepatoma cell line (HepaRG) and primary human hepatocytes (PHHs) in a 3D environment enhances both mRNA expression and CYP activities compared to hepatocellular carcinoma (HepG2) cells.[14,15]
To establish a clinically reliable human NAFLD model, it is essential to integrate non-parenchymal cells (HSCs, KCs, and endothelial cells comprising approximately 30–35% of the total liver cells) with hepatic parenchymal cells as each cell type plays specific roles in the subsequent pathological process of NAFLD. One study demonstrated this by developing a bioprinted human liver platform, incorporating PHHs with all the listed hepatic non-parenchymal cells, for studying hepatic fibrosis development.[16] However, although the TGFβ1 and methotrexate-based liver fibrosis model caused liver fibrosis, the pathophysiological progression pattern and mechanism of liver cirrhosis did not align with observations correlative with NAFLD. Another demonstration of this, human non-alcoholic steatohepatitis (NASH) model was developed using three major hepatic cell types (hepatocytes, KCs, and HSCs) as well as the sinusoidal flow in the lipotoxic environment to show the fibrosis development from lipid accumulations and inflammation.[17] However, these cells were cultured on 2D-like substrates only for 10 days without physical interaction between hepatocytes and hepatic non-parenchymal cells.
To closely recapitulate the pathology and progression of human NAFLD, it is necessary to develop a highly organized, robust in vitro NAFLD model. These models need to mimic (1) the native 3D tissue architecture (such as spheroids/organoids), (2) physiological circulatory dynamics (nutrients/O2 gradients and hemodynamic transport) and milieu (nutrients and hormones, etc.), and (3) the relevant cellular composition of the native hepatic microenvironment.[18] Using spheroid-laden hydrogels as a 3D microtissue culture system provides an opportunity to construct in vitro models to recapitulate in vivo features, including nutrients/O2 gradients and direct cell-cell contacts between different cell types inside spheroids compared to 3D cultures of individual cells.[19] Recently, we demonstrated the role of vascular endothelial cells (HUVECs) co-cultured with HepG2 cells in spheroids in the modeling of steatosis by supplementing fat. The steatosis pathogenesis can be precisely modeled with the presence of around 20% of HUVECs in the liver spheroids.[20] In addition, it has been known that hepatocytes, endothelial cells, Kupffer cells, and stellate cells represent approximately 60–80%,[21] 15–20%,[22] 15%,[23] and 10%[24] of the total liver cell population, respectively. Based on the information from our previously developed platform and the ratio of liver cell population, we further integrated the role of KCs in inducing elevated levels of proinflammatory cytokine productions and reactive oxygen species (ROS) expression. Spehroids composed of hepatocytes (HepG2 or HepaRG cells), HUVECs, and KCs were encapsulated in gelatin methacryloyl (GelMA) hydrogels to demonstrate their role in developing steatohepatitis in the presence of inflammation.[25] GelMA can provide an excellent microenvironment for cells mimicking the native ECM and support their adhesion, proliferation, and growth.[26] However, further work must be done to capture the pathological progression of NAFLD, mainly hepatic fibrosis. In this study, we aim to create a hepatic fibrosis model by developing a NAFLD-on-a-chip platform with embedded bioengineered multicellular liver microtissues (BE-MLMs) composed of the four main liver cell types (hepatocytes (HepaRG or PHHs), HUVECs, KCs, and HSCs). The natural progression of NAFLD–steatosis followed by inflammation/steatohepatitis and, ultimately, ECM deposition/fibrosis–was replicated in our proposed NAFLD-on-a-chip platform to compare with TGFβ-induced fibrosis models in terms of levels of fibrosis marker expression and the efficiency of drug transport and metabolism. Furthermore, a microfluidic device for creating a precise gradient of a fibrotic drug was employed to verify our model for facilitating drug screening and development.
2. Results and Discussion
2.1. Establishment of BE-MLMs for Inducing Fibrosis Driven by NAFLD
NAFLD represents one of the most common chronic liver diseases and is closely associated with the development of liver fibrosis.[2] The degree of liver fibrosis in patients with NAFLD is associated with the risk of evolving end-stage liver diseases, including liver cirrhosis and hepatocellular carcinoma, which may lead to liver-specific mortality and morbidity. However, there is no approved remedy for patients with advanced stages of NAFLD. A healthy diet combined with regular exercise are the only options for preventing the progression of the disease. To build an effective pre-clinical NAFLD model which can be used for drug development, we developed a liver-on-a-chip platform using BE-MLMs composed of human hepatocytes (HepaRG or PHHs), HUVECs, KCs, and HSCs to reproduce the natural progression of NAFLD (steatosis → steatohepatitis → fibrosis) (Figure 1A). We hypothesized that the generated fibrosis model that closely follows the natural progression of NAFLD might differ from artificial fibrosis models induced by TGFβ1 to activate HSCs. To test our hypothesis, we first mixed HepaRG (cell line), HUVECs (human primary), KCs (human primary), and HSCs (human primary) at different ratios in inverse pyramidal microwells: (i) 100% HepaRG, (ii) 80% HepaRG + 20% HUVECs, (iii) 70% HepaRG + 15% HUVECs + 15% KCs, and (iv) 61% HepaRG + 13% HUVECs + 13% KCs + 13% HSCs. These cells were cultured for 4 days to generate BE-MLMs (Figure 1B and C). HepaRG, HUVECs, KCs, and HSCs were labeled with orange, green, blue, and deep red color dyes, respectively, for assessing their configuration and further tracking their movement in BE-MLMs. We employed a 3D spheroid model that recapitulates the native 3D tissue architecture as well as the oxygen and nutrients gradients similar to the hepatic lobules (zonation). A previous study which measured oxygen gradients in spheroid models demonstrated that below 150 μm diameter of spheroids could reduce the hypoxic/necrotic region (below 2%).[27] However, comparing the structural organization between our model and native liver tissues as well as studying the oxygen and nutrient gradient of our spheroids remain to be confirmed to better understand the BE-MLMs. Further work will be conducted by histological assessment and oxygen/nutrient gradient analysis in the BE-MLMs for proving our liver model that recapitulates the native liver structural organization and oxygen/nutrient gradient, respectively. The size of BE-MLMs was controlled by the number of cells seeded in each microwell. A total of 200 cells were seeded per well (~100 μm in diameter on day 4) to avoid the formation of a necrotic core in the BE-MLMs, as previously described.[25] HepaRG cells, a human hepatoma-derived cell line potentially differentiating into hepatocyte-like and biliary-like cells, were employed as a substitute for PHHs.[28] Although the expression levels of CYP enzymes and drug transporters were slightly lower than PHHs, HepaRG cells have demonstrated higher cell viability, attachment efficacy to ECM, longevity in culture, and a lower cost compared to PHHs.[29] To take the advantages of HepaRG cells over PHHs, fibrotic BE-MLMs were first developed by employing HepaRG cells. Then, we employed PHHs to build a more physiologically and pathologically relevant fibrosis model of human NAFLD.
Figure 1. Establishment of BE-MLMs for inducing fibrosis driven by NAFLD.
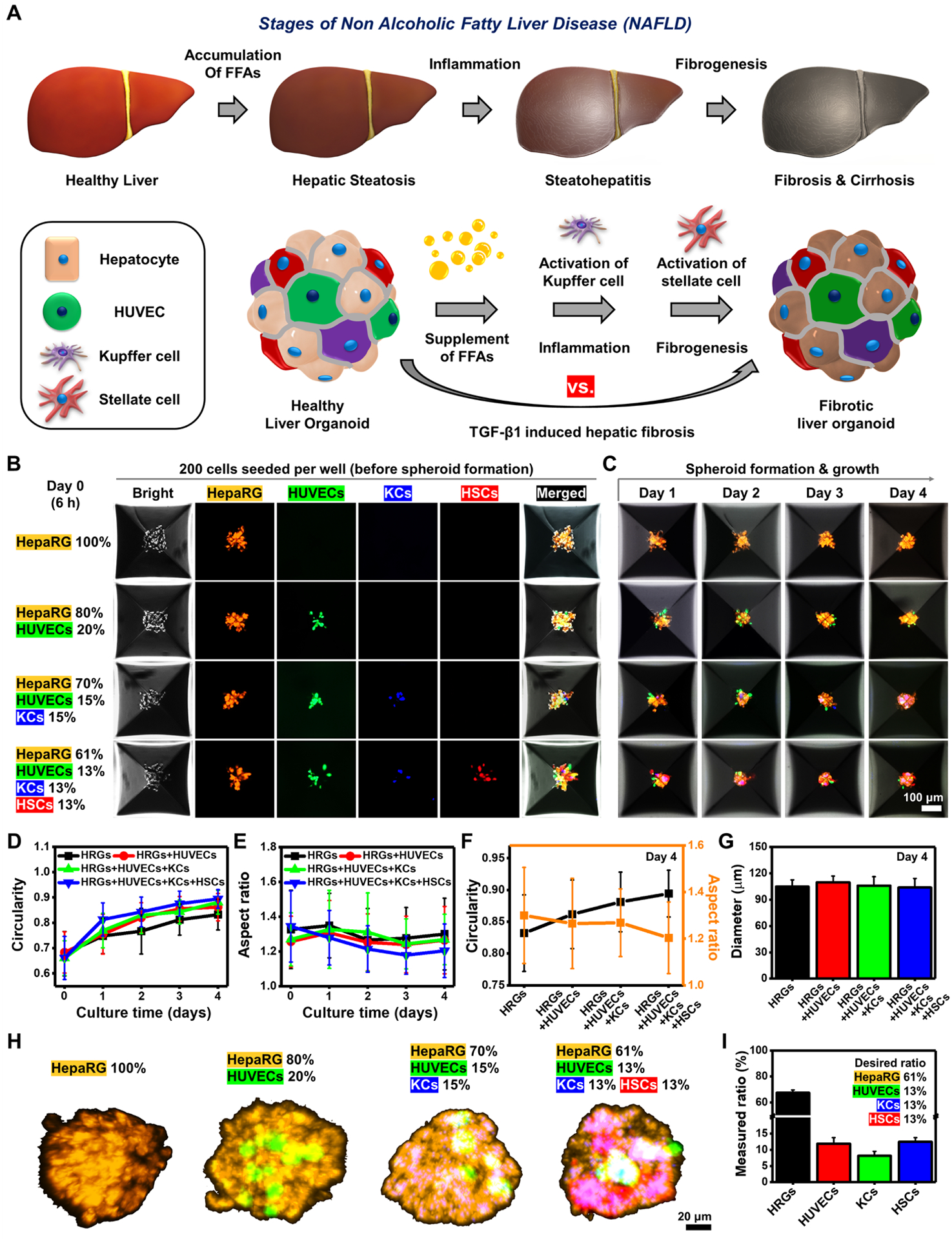
A) Schematic illustrations of liver fibrosis progression driven by NAFLD and BE-MLMs. B) Representative fluorescence images of hepatic cells with different combinations of parenchymal and non-parenchymal cells in inverse pyramidal microwells on day 0 (total number of cells in each well: 200 cells). C) Representative confocal images showing the formation of BE-MLMs in the microwells for 4 days. Profiles of D) circularity and E) aspect ratio for BE-MLMs culturing in the microwells for 4 days (n = 36 BE-MLMs in each condition). F) Comparison between BE-MLM circularity and aspect ratio in different combinations of parenchymal and non-parenchymal cells. G) Average diameters and H) representative confocal images of BE-MLMs across the groups on day 4 (n = 36 BE-MLMs in each condition). I) A measured fraction of each cell type included in BE-MLMs composed of HepaRG, HUVECs, KCs, and HSCs on day 4 (n = 6). Error bars indicate standard deviation.
Next, we assessed several characteristics (i.e., circularity, aspect ratio, and diameter) of BE-MLMs cultured in inverse pyramidal microwells. All the groups showed increased circularity and decreased aspect ratio for 4 days in culture (Figure 1D and E). However, when closely comparing the BE-MLMs with different configurations on day 4, the HepaRG + HUVECs + KCs + HSCs group showed the highest level of circularity (~0.9) and the lowest level of aspect ratio (~1.2), implying the better formation of spherical shape compared to all the other groups (Figure 1F). Both circularity and aspect ratio are important factors for generating a linear gradient of oxygen tension and nutrients inside BE-MLMs.[20] This indicates the HepaRG + HUVECs + KCs + HSCs group (the native configuration of hepatic tissue) may have more homogeneous gradients that mimic the native liver microarchitecture. The diameter of BE-MLMs (~105 μm) was not significantly different across the groups (Figure 1G). Moreover, a homogeneous distribution of different cell types was observed within the whole BE-MLMs. This suggests physical interactions between hepatocytes and hepatic non-parenchymal cells promote the formation of hepatic organoids similar to the native hepatic microenvironment (Figure 1H). Initial cell seeding ratio of HepaRG + HUVECs + KCs + HSCs group (61:13:13:13) corresponded to its actual composition ratio (67.4:11.9:8.2:12.5) on day 4 (Figure 1I). Although the slightly increased ratio of proliferative HepaRG and decreased ratio in non-proliferative KCs were observed relative to the initial feeding ratio on day 4, their composition seems to be nearly matched with the proportion of native hepatic tissues.[28]
2.2. Influences of HSCs on Hepatic Fibrosis Progression of BE-MLMs
In our previous study, the inclusion of KCs into HepaRG + HUVECs spheroids led to elevated levels of lipid accumulation and ROS expression.[25] To build a fibrosis model driven by NAFLD and study the role of HSCs in the liver fibrogenesis process, the BE-MLM (61% HepaRG + 13% HUVECs + 13% KCs + 13% HSCs) group was compared to that without the presence of HSCs (70% HepaRG + 15% HUVECs + 15% KCs) in terms of cell viability, lipid accumulation, and inflammatory responses. BE-MLMs with or without HSCs were generated after culturing different compositions of cell types in inverse pyramidal microwells for 4 days. The resulting BE-MLMs were then encapsulated in GelMA hydrogels to maintain their 3D conformation and mimic the native ECM.[26] The BE-MLMs with or without the presence of HSCs were cultured in mixed cell culture media (based on the ratio of different cell types) with or without free fatty acids (FFAs) for 21 days. Cell viability across the groups was obtained on days 3, 7, 14, and 21 (Figures S1 and S2). The results display higher numbers of dead cells for BE-MLMs with HSCs and FFAs on days 14 and 21 compared to those without HSCs and FFAs (Figure S2). This indicates that (1) accumulation of FFAs into BE-MLMs may accelerate inflammatory responses in the BE-MLMs, ultimately leading to cell death, and (2) cellular senescence and apoptosis mediated by the increasing activation of HSCs may also attribute to the increase in dead cell portion. Production levels of albumin and urea increased in all experimental groups over a 21-day culture period. This demonstrates the maintenance of normal liver functions of BE-MLMs in GelMA hydrogels (Figure S3). Although no notable change in albumin secretion was detected between all the groups, the urea production level of BE-MLMs with HSCs and FFAs supplement was notably higher than the other groups on day 14. In addition, BE-MLMs were cultured for 21 days and stained with CD31 to assess the formation of microvascular networks (Figure S4). Immunofluorescence (IF) images of BE-MLMs stained with CD31 showed notable differences in neovascularization between groups with and without the presence of HSCs on days 14 and 21. This result indicates that the interaction between HSCs and other hepatic cells in BE-MLMs may be involved in vascular development to promote the formation of functional microvascular networks. Furthermore, we found that different cell types in BE-MLMs exhibited a homogeneous distribution even after 21 days of culture. This suggests that our platform would be suitable for in vitro modeling of fibrosis driven by NAFLD through the period of long-term culture (Figure S5).
Next, degrees of lipid accumulation and ROS expression in BE-MLMs cultured for 21 days in GelMA hydrogels were assessed (Figure 2A and B). BE-MLMs supplemented with FFAs showed significantly elevated levels of lipid accumulations and ROS expression regardless of HSC presence. This leaded to the progression of NAFLD state and ROS-mediated inflammatory responses. However, the intensity ratios of accumulated lipid and ROS in the group with HSCs were only slightly higher than the group without HSCs. This indicates that HSCs in BE-MLMs may not play a critical role in lipid accumulation and ROS production.
Figure 2. Influences of the existence of HSCs on hepatic fibrosis progression of BE-MLMs.
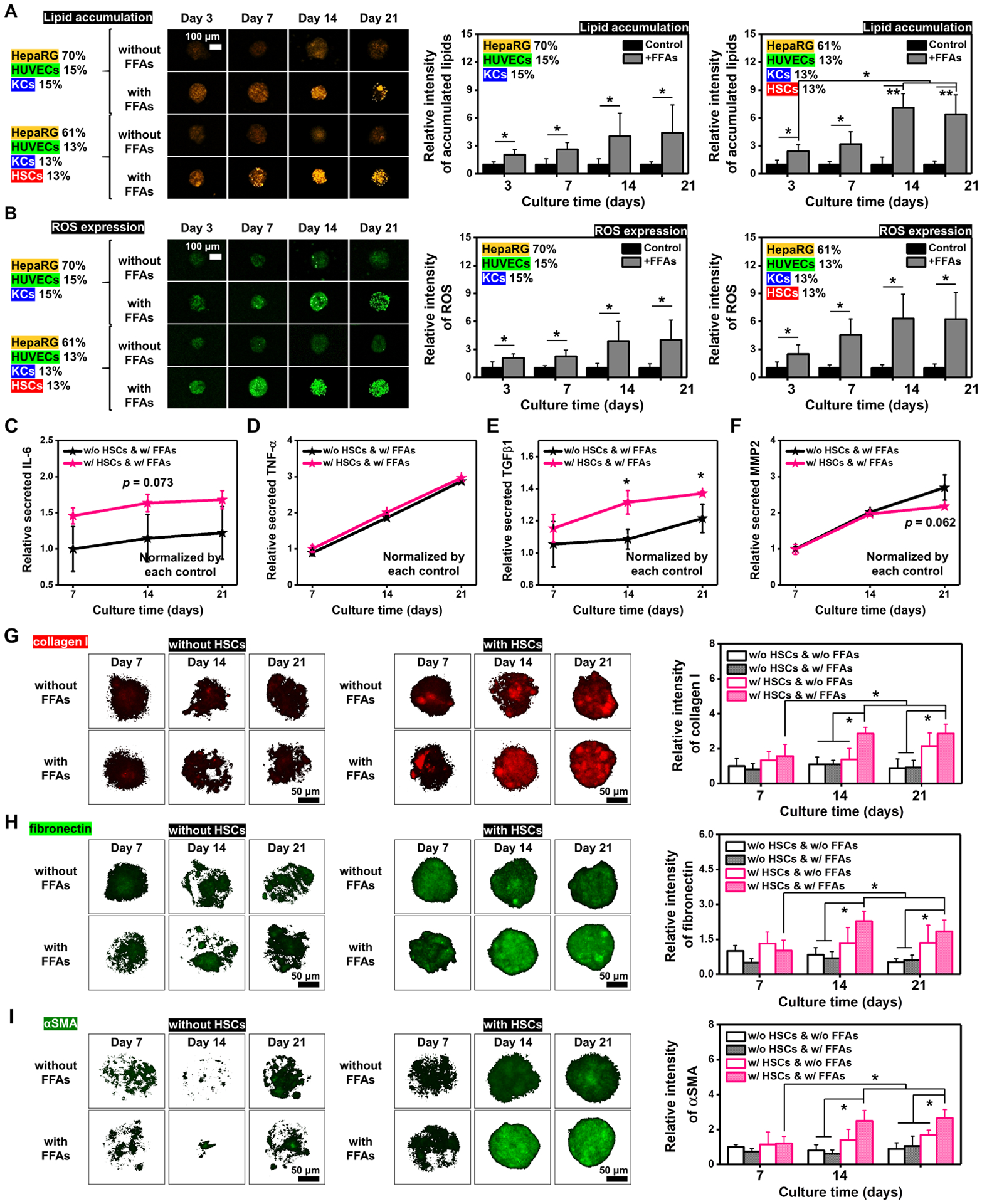
Representative confocal images and quantification of A) accumulated lipids and B) produced ROS in BE-MLMs with or without the HSC presence and with or without the FFAs supplement on days 3, 7, 14, and 21 (n = 4). Quantification of C) IL-6, D) TNF-α, E) TGFβ1, and F) MMP2 secretions from BE-MLMs supplemented with FFAs over 21 days (n = 3). Data were normalized according to each condition without FFAs supplement. Representative confocal images and relative intensity of G) collagen I, H) fibronectin, and I) αSMA in BE-MLMs with or without the HSC presence and with or without the FFAs supplement on days 7, 14, and 21 (n = 4). Error bars indicate standard deviation. ANOVA, *P < 0.05, **P < 0.005, between specified groups.
To uncover the role of HSCs in the subsequent pathological process of NAFLD, fat deposition-related inflammatory responses were compared between BE-MLMs with and without HSCs (Figure 2C–F). We observed notably higher levels of interleukin-6 (IL-6) production for BE-MLMs with HSCs compared to those without HSCs (Figure 2C). In contrast, similar production levels of tumor necrosis factor-α (TNF-α) were observed between the groups (Figure 2D). This indicates that HSCs in BE-MLMs may interact with KCs to induce their production of IL-6 rather than TNF-α. Interestingly, production levels of TGFβ1 in BE-MLMs with HSCs were significantly higher than compared to those without HSCs on days 14 and 21 (Figure 2E). The increase of TGFβ1 production may be due to the existence of HSCs interacting with other hepatic cells.[30] Matrix metalloproteinase-2 (MMP-2) is related to ECM remodeling, and it is often highly expressed in myofibroblasts during the liver fibrosis progression.[1] Both BE-MLMs with and without HSCs displayed increasing profiles in relative levels of MMP2 in the presence of FFAs for 14 days. However, lower (but not significantly, p > 0.05) production levels of MMP2 were observed on day 21 for BE-MLMs with HSCs compared to those without HSCs (Figure 2F).
An increase in the synthesis of α-smooth muscle actin (αSMA) implies the activation of HSCs and their differentiation to myofibroblasts. Collagens and other ECM proteins can be produced from the differentiated myofibroblasts, and they may play critical roles in the formation of scar during liver fibrosis. Fibrosis is accompanied through the extreme deposition of ECM proteins (e.g., collagen I and III and fibronectin). The alteration in ECM composition reduces fenestra and forms a basement membrane, making capillarization by hepatic sinusoidal endothelial cells difficult.[31] This process may hamper the regular transport of nutrients between sinusoidal blood and hepatocytes. In addition, the extension of liver fibrogenesis may lead to further advanced stages including liver cirrhosis and carcinoma.[32] To examine the activation of HSCs and their remodeling of ECM by depositing fibrosis-associated proteins, we assessed levels of fibrosis marker (i.e., collagen I, fibronectin, and αSMA) expression for all the groups of BE-MLMs (Figure 2G–I). It is expected that the FFAs supplementation accelerates advanced stages of NAFLD and the presence of HSCs is important for liver fibrosis progression. In line with the expectation, we observed that BE-MLMs with HSCs supplemented with FFAs displayed higher expression levels of collagen I, fibronectin, and αSMA than the other groups (especially without HSCs groups) on days 14 and 21. Together, the BE-MLMs with HSCs and FFAs supplementation can be efficiently used for the establishment of in vitro liver fibrosis model.
2.3. Comparison between Fibrotic BE-MLMs Induced by the Natural Progression of NAFLD and TGFβ1 Supplement
The natural progression of NAFLD is described by the continuous fat accumulation in hepatocytes that leads to inflammation by activated KCs and then fibrosis by activated HSCs.[3] Among this process, TGFβ1, as one of the profibrotic mediators, can lead to transdifferentiation of HSCs to ECM-secreting myofibroblasts, which are the dominant source of hepatic fibrosis.[33] In this regard, the supplement of TGFβ1 is intended to induce the hepatic fibrosis, which is supported by several studies that have shown TGFβ1-induced fibrosis models.[16,32] However, as we hypothesized, the fibrosis model induced by TGFβ1 may differ from our model that induces NAFLD through the natural progression, starting from the fat accumulation. To compare these models, we first cultured 200 cells (61% HepaRG + 13% HUVECs + 13% KCs + 13% HSCs) in each inverse pyramidal microwell for 4 days to generate BE-MLMs. After encapsulating the produced BE-MLMs in GelMA hydrogels, they were supplemented with FFAs or TGFβ1 for 21 days (control: neither FFAs nor TGFβ1 supplement). Then, we assessed cell viability as well as levels of lipid accumulation and ROS expression across the conditions on days 7, 14, and 21. Relative portions of dead cells showed no significant difference between TGFβ1 and FFAs groups (Figure 3A). However, as expected, FFAs supplementation markedly led to the significantly elevated accumulated amounts of lipid in BE-MLMs compared to the TGFβ1-incubated group (~2.4-fold, ~6.0-fold, and ~5.7-fold on day 7, 14, and 21, respectively) (Figure 3B). BE-MLMs accompanied with FFAs displayed higher expression levels of ROS compared to the TGFβ1 group (~1.3-fold, ~1.7-fold, and ~1.5-fold on day 7, 14, and 21, respectively), despite no significant variance shown between the groups (Figure 3C). To compare levels of fibrosis, BE-MLMs supplemented with TGFβ1 or FFAs were stained with fibrosis-specific markers (i.e., collagen I, fibronectin, and αSMA) on days 7, 14, and 21 (Figure 3D–F). The IF staining results exhibited that BE-MLMs supplemented with FFAs had significantly higher expression levels of the fibrosis markers, collagen I (~2.8-fold and ~3.3-fold on day 14 and 21, respectively), fibronectin (~2.9-fold and ~2.7-fold on day 14 and 21, respectively), and αSMA (~2.4-fold on day 14), relative to control. In addition, the FFAs-supplemented group showed higher expression levels of the fibrosis markers than the TGFβ1-treated group during the incubation period, although there was no significant difference. We also found that the trends for the expression levels of fibrosis markers for both FFAs- and TGFβ1-treated groups were likely saturated after 14 days in culture. No notable difference may be because our natural progression of NAFLD to induce fibrosis may take more time (several sequences including steatosis and inflammation to reach to hepatic fibrosis) relative to the TGFβ1 induction that directly activates stellate cells.
Figure 3. Comparison between fibrotic BE-MLMs induced by the natural progression of NAFLD and TGFβ1 supplement.
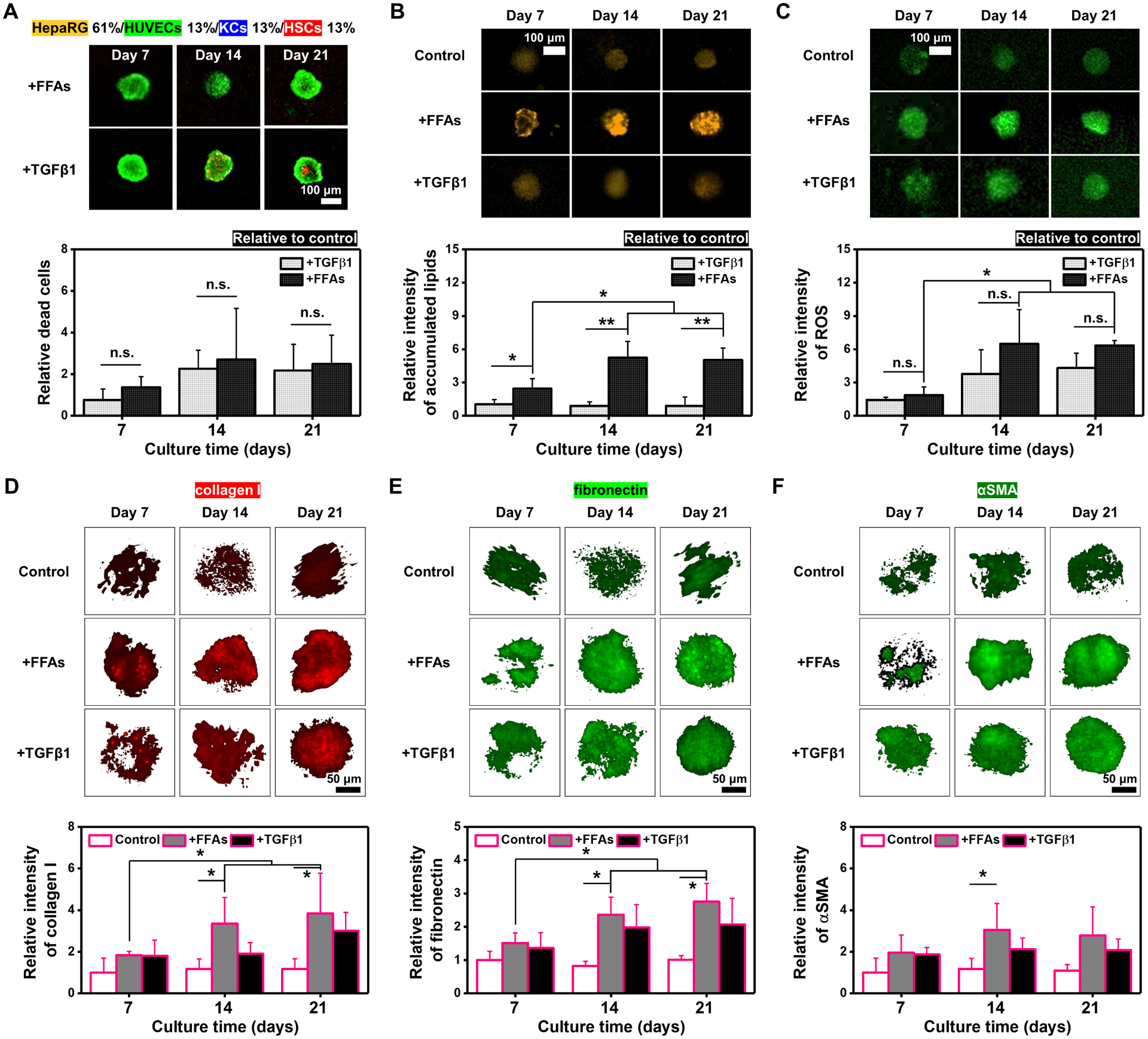
A) Representative live (green)/dead (red) images of BE-MLMs supplemented with FFAs or TGFβ1 in the presence of HSCs on days 7, 14, and 21. Quantification of dead cells on days 7, 14, and 21 relative to control without the supplement of FFAs and TGFβ1 (n = 4). Representative confocal images and quantification of B) accumulated lipids and C) produced ROS in BE-MLMs supplemented with FFAs or TGFβ1 for 21 days (n = 4). Representative confocal images and relative intensity of D) collagen I, E) fibronectin, and F) αSMA in BE-MLMs supplemented with FFAs or TGFβ1 on days 7, 14, and 21 (n = 4). Error bars indicate standard deviation. ANOVA, *P < 0.05, **P < 0.005, between specified groups.
2.4. Drug Screening and Drug Transporters/Enzymes Identification Tests in the Microfluidics-Assisted Liver-on-a-Chip System
Fibrogenesis can be triggered by a combination of multiple factors ranging from ROS-induced oxidative stress to various signaling pathways (i.e., TGFβ, connective tissue growth factor (CTGF), and platelet-derived growth factor (PDGF)).[32] Although not statistically different, we hypothesize the slightly higher expression levels of ROS and fibrotic markers may contribute to differences in drug screening outcomes. To further explore how our liver fibrosis model with the natural progression of NAFLD differs from TGFβ1-treated fibrosis models in terms of drug-dose response and application to drug transport and metabolism studies, we employed a perfusable and gradient-generating microfluidic device that permits perfusion culture. The microfluidic device was designed for generating drug concentration gradients (in eight channels from 0 to 1 mM) while maintaining the same velocity and shear stress applied to BE-MLMs in GelMA hydrogels (Figure S6). We employed pirfenidone, one of the anti-fibrotic agents, to evaluate the reversibility of liver fibrosis (induced by FFAs or TGFβ1 treatment) to a healthy state. First, HepaRG-based BE-MLMs in GelMA hydrogels were cultured for 14 days with FFAs or TGFβ1 to induce fibrosis and then transferred to the gradient-generating microfluidic devices for treatment with different concentrations of pirfenidone for 7 days. We decided to induce hepatic fibrosis for 14 days since the trends for the expression levels of fibrosis markers (fibronectin and αSMA) were likely saturated after 14 days in culture (Figure 3D–F). In addition, the pre-cultured BE-MLMs in microfluidic devices were not supplemented with FFAs or TGFβ1 to examine whether the BE-MLMs without sustained fibrosis-induction could switch back to a healthy state (reversible) or remain in a disease state (irreversible). First, we observed that no drug application (concentration: 0 mM) presented significant differences in expression levels of fibronectin and αSMA in FFAs- (~3.7-fold for fibronectin & ~2.6-fold for αSMA) and TGFβ1-treated (~2.9-fold for fibronectin & ~2.6-fold for αSMA) groups compared to control (precultured without treatment of FFAs and TGFβ1 for 14 days) (Figure 4A and B). This result indicates that, unlike the steatosis state being reversed to the healthy state without drug treatment within a week,[20] the fibrosis state of BE-MLMs induced by the supplementation of FFAs or TGFβ1 may be irreversible.[2] However, both FFAs- and TGFβ1-supplemented BE-MLMs showed decreased expression levels of fibrosis markers with increasing concentrations of pirfenidone up to 1 mM (approximately 0.12, 0.26, 0.42, 0.58, 0.73, 0.88, and 1.00 mM). Fibrosis induced by FFAs or TGFβ1 could be reversed entirely after treatment with the drug at the maximum concentration (1 mM) for 7 days, although possible cytotoxicity as a confounding factor was not ruled out. Note that the HepaRG-based BE-MLMs with TGFβ1-induced fibrotic state showed entirely reversed expression levels of αSMA at low concentrations (~0.42 mM) of pirfenidone, which led to significantly lower expression levels of αSMA compared to those with FFAs-induced fibrotic state. This outcome demonstrates that fibrosis induced by FFAs or TGFβ1 may show different dose-dependent recovery of αSMA expression levels. This means that lower drug concentrations may be required for TGFβ1-induced fibrosis state to switch back to the healthy state compared to FFAs-induced fibrosis state.
Figure 4. Drug screening and drug transporters/enzymes identification tests in the microfluidics-assisted liver-on-a-chip system.
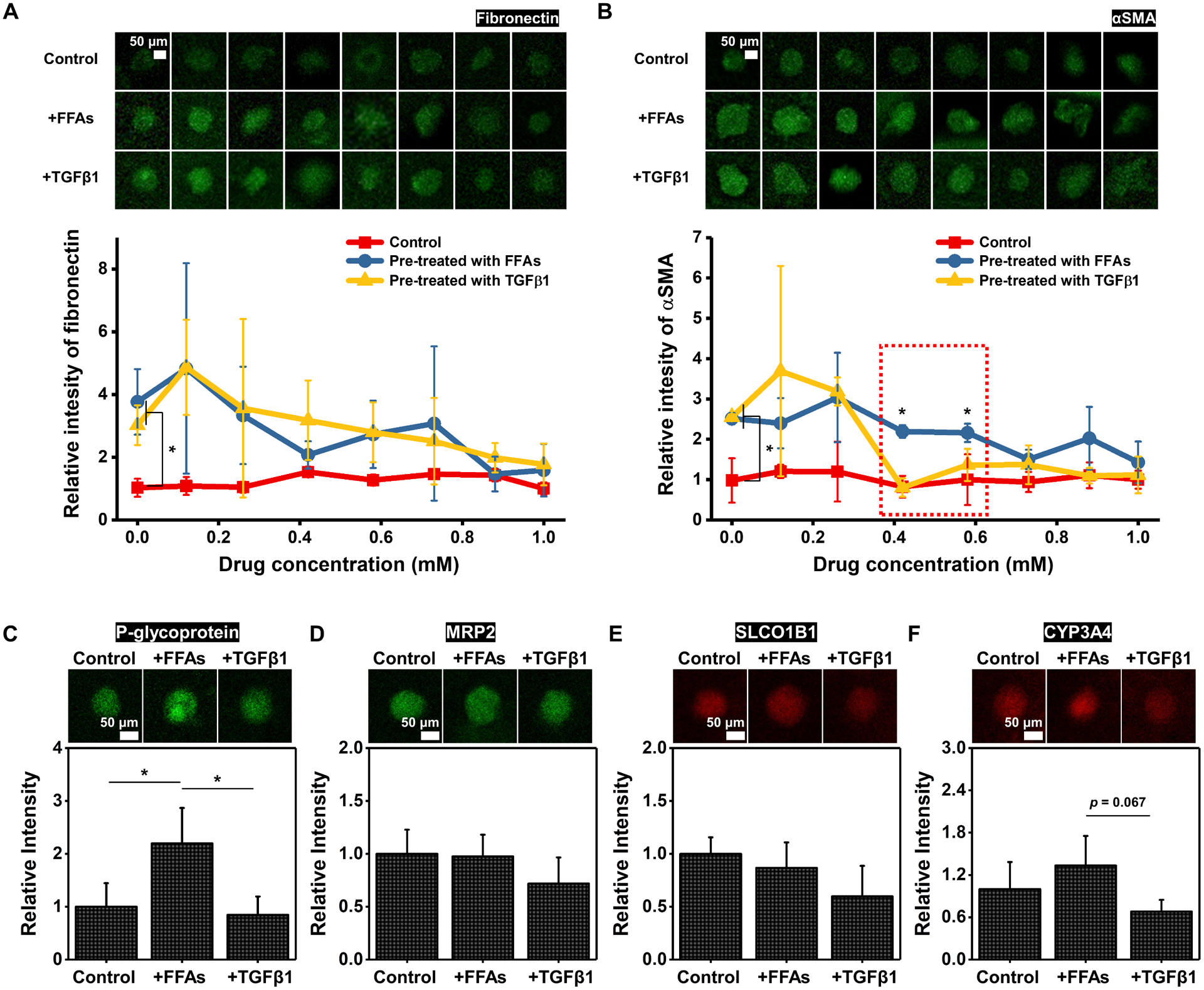
Representative confocal images and relative intensity of A) fibronectin and B) αSMA expression levels for control and groups pre-treated with FFAs or TGFβ1 after culturing them with different concentrations of pirfenidone in microfluidic devices (n = 3). The relative ratio of the control group is set as 1, and those of other groups are presented. The relative intensity of C) P-glycoprotein, D) MRP2, E) SLCO1B1, and F) CYP3A4 in control, FFAs-treated, and TGFβ1-treated groups on day 21 (n = 3). The relative intensity of the control group is set as 1, and relative ratios of those in FFAs-treated and TGFβ1-treated groups are calculated. Error bars indicate standard deviation. ANOVA, *P < 0.05, between specified groups.
To precisely and efficiently apply our proposed NAFLD-on-a-chip system for drug discovery and development, studying drug transporters and enzymes is required. Multidrug resistance-associated protein 2 (MRP2) and P-glycoprotein were selected as drug efflux transporters, and solute carrier organic anion transporter family member 1B1 (SLCO1B1) and CYP3A4 were employed as an organic anion transporter and drug-metabolizing enzyme, respectively (Figure 4C–F). BE-MLMs with the FFAs supplement exhibited notably higher expression level of P-glycoprotein compared to control and TGFβ1-supplemented group, while similar MRP2 and SLCO1B1 levels were presented in all the groups. It implies that P-glycoprotein is more likely to function as part of major drug transporters in fibrotic HepaRG-based BE-MLMs supplemented with FFAs compared to those induced by TGFβ1.
2.5. Modeling of Hepatic Fibrosis Using PHH-Based BE-MLMs
The use of hepatocytes derived from primary or induced pluripotent stem cells (iPSCs) in combination with hepatic non-parenchymal stem cells is more representative of in vivo cell population and enables more physiologically and pathologically relevant fibrosis models of human NAFLD. In particular, the process of generating organoid-like liver structure with PHHs is easier than with iPSCs, which requires multiple steps to differentiate into hepatocyte-like cells. Based on the BE-MLM platform developed using HepaRG cells, we further modeled hepatic fibrosis using PHH-based BE-MLMs. First, 61% PHHs + 13% HUVECs + 13% KCs + 13% HSCs were mixed in inverse pyramidal microwells and cultured for 4 days to generate PHH-based BE-MLMs. Then, the BE-MLMs were encapsulated in GelMA hydrogels to establish a more physiologically and pathologically relevant fibrosis model of the human NAFLD. We assessed the number of dead cells on days 7, 14, and 21. The average values of dead cell percentages in PHH-based BE-MLMs supplemented with FFAs were slightly higher than the control and TGFβ1-supplemented groups (Figure S7A). For lipid accumulation in the presence of FFAs or TGFβ1, the FFAs-supplemented group exhibited significantly higher levels of accumulated lipids relative to control and TGFβ1-treated group on days 7, 14, and 21 (Figure S7B). ROS expression levels in the FFAs-supplemented group were significantly higher than the control (Figure S7C). Moreover, the FFAs-supplemented group displayed higher levels of ROS expression (~1.1-fold, ~1.5-fold, and ~1.3-fold on day 7, 14, and 21, respectively) compared to TGFβ1-supplemented group. These trends in the dead cell fraction, lipid accumulation, and ROS production correspond to the outcomes from HepaRG-based BE-MLMs. To compare levels of fibrosis, PHH-based BE-MLMs supplemented with TGFβ1 or FFAs were stained with fibrosis-specific markers (i.e., collagen I, fibronectin, and αSMA) on days 7, 14, and 21 (Figure 5A–C). The IF staining results showed that BE-MLMs supplemented with FFAs had significantly higher expression levels of the fibrosis markers, collagen I (~2.3-fold on day 14), fibronectin (~1.6-fold and ~2.2-fold on day 14 and 21, respectively), and αSMA (~1.9-fold on day 14), relative to control. Moreover, in line with the results from HepaRG-based BE-MLMs, the FFAs-supplemented group showed higher expression levels of the fibrosis markers, collagen I (~1.2-fold on days 7, 14, and 21), fibronectin (~1.1-fold and ~1.2-fold on day 14 and 21, respectively), and αSMA (~1.0-fold, ~1.2-fold, ~1.1-fold on day 7, 14, and 21, respectively), than the TGFβ1-treated group during the incubation period.
Figure 5. Modeling of hepatic fibrosis using PHH-based BE-MLMs.
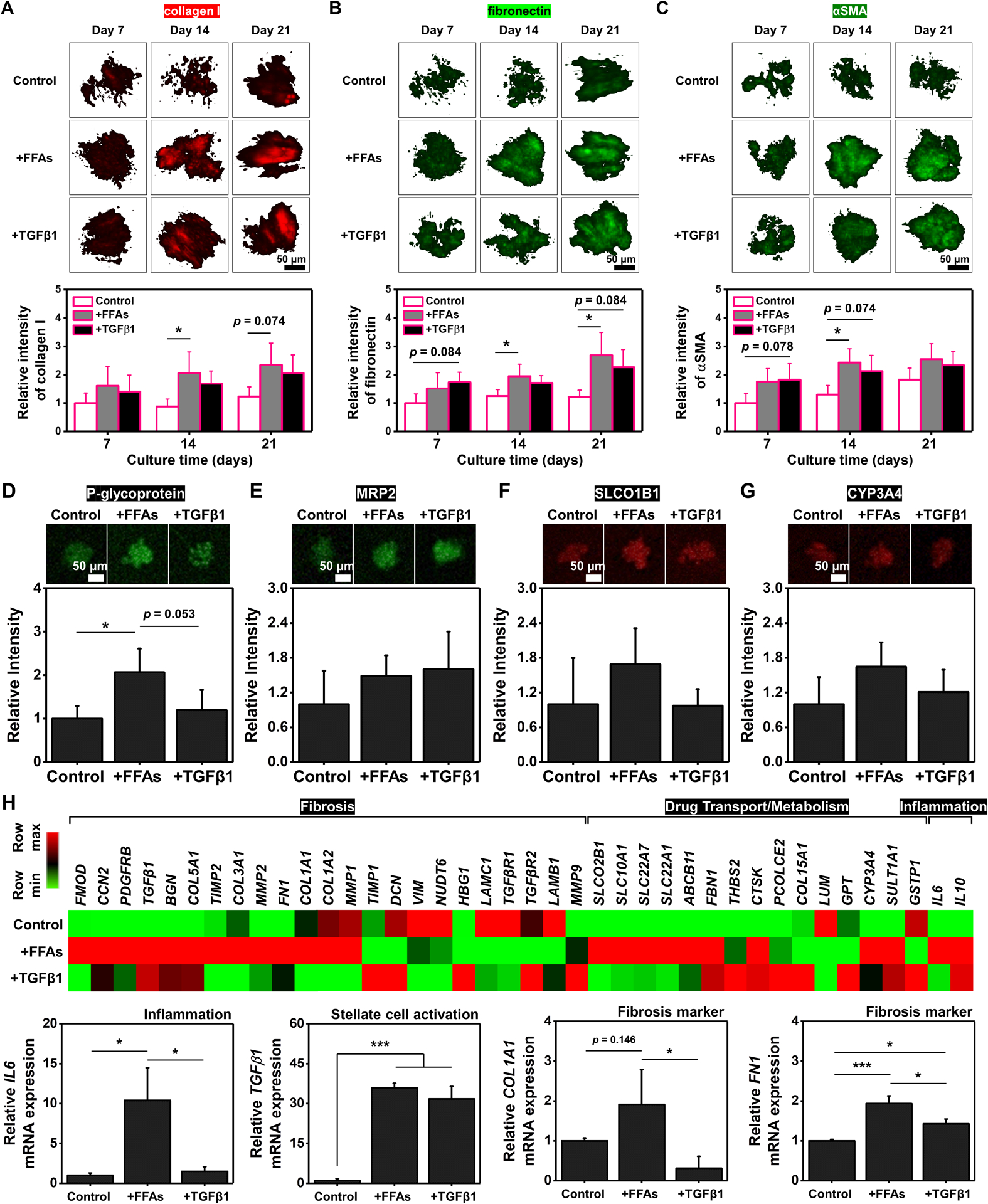
Representative confocal images and relative intensity of A) collagen I, B) fibronectin, and C) αSMA in BE-MLMs supplemented with FFAs or TGFβ1 on days 7, 14, and 21 (n = 4). The relative intensity of D) P-glycoprotein, E) MRP2, F) SLCO1B1, and G) CYP3A4 in control, FFAs-treated, and TGFβ1-treated groups on day 21 (n = 3). H) Expression heat map of transcripts linked to fibrosis, drug transport/metabolism, and inflammation for control, FFAs-treated, and TGFβ1-treated groups (n = 3). Outcomes from gene expression analysis of IL6, TGFβ1, COL1A1, and FN1 of BE-MLMs supplemented with FFAs or TGFβ1 on day 21 (n = 3). The relative intensity of the control group is set as 1, and relative ratios of those in FFAs-treated and TGFβ1-treated groups are calculated. Error bars indicate standard deviation. ANOVA, *P < 0.05, between specified groups.
To study the drug-dose response and drug transport and metabolism in FFAs- and TGFβ1-supplemented groups, PHH-based BE-MLMs in GelMA hydrogels were pre-cultured for 14 days with FFAs or TGFβ1. Then, the BE-MLMs were transferred to the perfusable and gradient-generating microfluidic device described in Figure 4A to treat them with different concentrations of pirfenidone for 7 days. In line with the outcomes from HepaRG-based BE-MLMs, (1) no drug application caused significant differences in expression levels of fibronectin and αSMA groups compared to control, (2) both FFAs- and TGFβ1-supplemented groups showed decreased levels of fibrosis marker expression with increasing concentrations of pirfenidone, and (3) TGFβ1-supplemented group showed generally lower expression levels of αSMA compared to FFAs-supplemented group (Figure S8). Expression levels of P-glycoprotein, MRP2, SLCO1B1, and CYP3A4 in PHH-based BE-MLMs were also quantitatively analyzed (Figure 5D–G). As observed in Figure 4D, the FFAs treatment led to elevated levels of the P-glycoprotein expression compared to control (~2.1-fold) and TGFβ1-supplemented group (~1.7-fold) (Figure 5D). Moreover, PHH-based BE-MLMs supplemented with FFAs exhibited higher expression levels of SLCO1B1 and CYP3A4 compared to control and TGFβ1-supplemented group (Figure 5F and G).
We observed that the FFAs-treated group showed no significantly different expression levels (despite even higher levels) of ROS (Figure 3C) and fibrosis markers (Figure 3D–F) compared to the TGFβ1-treated group. To better understand the differential fibrotic state driven by the supplement of FFAs or TGFβ1, we analyzed RNA transcripts linked to fibrosis (ranging from FMOD to MMP9), drug transport/metabolism (ranging from SLCO2B1 to GSTP1), and inflammation (IL6 and IL10) using quantitative polymerase chain reaction (qPCR) (Figure 5H and S9). We found an increase in expression levels of the genes in PHH-based BE-MLMs supplemented with FFAs compared to expression levels in control or those supplemented with TGFβ1, especially from FMOD to MMP1 for fibrosis-related genes, from SLCO2B1 to FBN1 for drug transport/metabolism-related genes, as well as IL6 and IL10 for inflammation-related genes (arranged in descending order of fold change for FFAs-supplemented group over control from left to right). Among those genes, we observed the FFAs-supplemented group exhibited higher expression levels of IL6 (corresponding to the results of HepaRG-based BE-MLMs in Figure 2C), TGFβ1 (corresponding to the results of HepaRG-based BE-MLMs in Figure 2E), COL1A1 (corresponding to the results of HepaRG-based BE-MLMs in Figure 3D and PHH-based BE-MLMs in Figure 5A), and FN1 (corresponding to the results of HepaRG-based BE-MLMs in Figure 3E and PHH-based BE-MLMs in Figure 5B). Overall, our results from PHH-based BE-MLMs are consistent with the outcomes from HepaRG-based BE-MLMs and demonstrate that the natural progression of NAFLD promotes a fibrotic state that differs from artificial one induced by the supplement of TGFβ1. Therefore, the FFAs-induced BE-MLM model of fibrosis is more representative of native NAFLD disease pathogenesis and may be better suited for studying disease progression and evaluating drug candidates.
3. Conclusion
We have developed a NAFLD-on-a-chip platform with PHH-based BE-MLMs consisting of four major liver cell types (PHHs, stellate cells, Kupffer cells, and HUVECs) to model hepatic fibrosis driven by NAFLD. By using the platform, we explored how a fibrotic state induced by the natural progression of NAFLD differs from the TGFβ1-supplemented artificial fibrosis model. First, we demonstrated that HSCs in BE-MLMs are crucial for inducing higher levels of lipid accumulation, ROS production, inflammation, and fibrosis marker expression compared to those in the absence of HSCs. Next, by applying the developed platform from HepaRG-based BE-MLMs to PHH-based BE-MLMs, we showed that our fibrosis model generated with the pathological progression of NAFLD could achieve better fibrotic phenotypes with increased levels of ROS production and fibrosis marker expression. Furthermore, by employing microfluidic devices that create a precise gradient of an anti-fibrotic agent, we showed increased efficiency levels of drug transport and metabolism for fibrotic BE-MLMs induced by FFAs compared to those supplemented with TGFβ1. However, we employed HUVECs, which have morphologic and functional differences from the liver sinusoidal endothelial cell, for proof-of-concept studies to compare the natural progression of NAFLD with TGFβ-induced fibrosis models. Further studies will be conducted by employing all four hepatic cell types including liver sinusoidal endothelial cells from the same donor to realize personalized medicine. Nevertheless, we believe that the suggested platform pathologically recapitulates the advanced stages of NAFLD and will be broadly applicable to understand the mechanisms underlying fibrosis progression in NAFLD as well as facilitate drug screening and development for patients with different stages of NAFLD.
4. Experimental Section
Cell Culture:
Primary HUVECs (PCS-100-010) were purchased from American Type Culture Collection (ATCC). HUVECs were cultured in 1% (w/v) gelatin (from porcine skin, G18890, Sigma-Aldrich)-coated flasks. Gelatin was solubilized in deionized water by incubating for around 5‒10 min at 80 °C. Filtered gelatin solution was added to the culture flask, incubated in a humid incubator (5% CO2 and 37 °C) for 1‒2 h, and it was eliminated. HUVECs were grown in Endothelial Cell Basal Medium 2, including Endothelial Cell Growth Medium Kit (22111, Promo Cell), vascular endothelial growth factor (22011, Promo Cell), and pen/strep (1%, v/v).[15] HepaRG cells (CVCL 9720) and primary KCs (HUKCCS) were acquired from Thermo Fisher Scientific (Waltham, MA, USA). HepaRG cells were supplemented with William’s E medium (A1217601, containing HepaRG addition (HPRG620), Thermo Fisher Scientific).[25] Primary HKCs were thawed by using the mixed medium, including 91.4% of advanced DMEM (12-491-015, Thermo Fisher Scientific), 5% FBS, and 3.6% thawing/plating cocktail A (CM3000, Thermo Fisher Scientific).[25] HKCs were seeded, incubated for 6 h, and cultured with fresh thawing medium for 24 h. Then, HKCs were incubated with maintenance medium (91% advanced DMEM, 5% FBS, and 4% maintenance cocktail B (CM4000, Thermo Fisher Scientific)) for 2‒3 days. Those cells were employed for the subsequent studies. PHHs were purchased from ZenBio Inc. PHHs were serially cultured with hepatocyte thawing medium (MHT), hepatocyte plating medium (MHP), and hepatocyte maintenance medium (MHM), which were provided by ZenBio Inc. HSCs were supplied by Lonza. HSCs were cultured with Stellate Cell Growth Medium (MCST250, Lonza). For co-culturing cells, each kind of medium was blended according to the cell portion. All types of cells were cultured at 37 °C with 5% CO2 supply.
Preparation and Culture of BE-MLMs:
BE-MLMs were prepared with microwell-embodied culture plates (AggreWell TM400, 24-well, Stem Cell Technologies). The new plate was rinsed with an anti-adherence solution (07010, Stem Cell Technologies) according to the manufacturer’s guideline. The cell suspension was placed into the microwell-embodied plate, and BE-MLMs were spontaneously formed with seeded cells by self-aggregation at 200 cells per BE-MLMs density. To trace each cell type in the BE-MLMs, all kinds of cells were labeled with different cell tracker dyes following the manufacturer’s instructions: CMRA (CellTracker Orange, C7001, Invitrogen) for HepaRG cells, CMFDA (CellTracker Green, C2925, Invitrogen) for HUVECs, CMAC (CellTracker Blue, C2110, Invitrogen) for KCs, and CM-Dil (CellTracker Deep Red, C34565, Invitrogen) for HSCs. BE-MLMs were prepared with different cell seeding ratios: HepaRG (100%), HepaRG/HUVECs (80%/20%), HepaRG/HUVECs/KCs (70%/15%/15%), and HepaRG/HUVECs/KCs/HSCs (61%/13%/13%/13%) as well as PHHs/HUVECs/KCs/HSCs (61%/13%/13%/13%). BE-MLMs were formed by using AggreWell for 4 days, and cell culture media were exchanged every other day. Circularity and aspect ratio were calculated with diameter measurement by fluorescence microscope every day. Labeled dyes from each cell were quantitatively determined using a confocal laser scanning microscope (LSM880, Zeiss). BE-MLMs composed of unlabeled (dye-free) cells were then encapsulated in GelMA hydrogels for further studies.
Preparation of GelMA Encapsulating BE-MLMs:
GelMA was prepared based on our previous report.[34] For making photo-cross-linked GelMA hydrogel, 10% (w/v) of GelMA and 0.5% (w/v) of 2-hydroxy-40-(2-hydroxyethoxy)-2-methylpropiophenone (410896, Sigma-Aldrich) as a photoinitiator (PI) were added in the corresponding cell culture media and dissolved at 70 °C for 3 min. On day 4, suspended BE-MLMs with mild pipetting were mixed with the GelMA dispersion with PI, and then the mixture was crosslinked under an ultraviolet (UV)-exposure at 20 mW/cm2 for 3 min.
Fibrosis Induction Using FFAs and TGFβ1 Supplements:
FFAs composed of OA (O1383, Sigma-Aldrich) and PA (P0500, Sigma-Aldrich) were applied to the BE-MLMs to assess the liver fibrosis induction efficiency.[25] A combination of PA (0.33 mM) and OA (0.66 mM) in methanol was prepared, and it was frozen at −80 °C before its use. PA/OA solution was diluted with corresponding cell culture media at 1000 times, and 1% (w/v) bovine serum albumin (BSA; A3983, Sigma-Aldrich) was also added for facilitating their uptake into the hepatocytes. Cell culture media, including FFAs, were exchanged every other day. In the case of TGFβ1-treated groups, cell culture media, including 10 ng/mL TGFβ1 (Recombinant Human TGF-β1, 100-21C, Peprotech, Inc.), were replaced every other day during the BE-MLM culture period.
Biochemical Assay:
Viability of cells in BE-MLMs was measured with LIVE/DEAD kit (L3224, Thermo Fisher Scientific) composed of calcein AM (for staining live cells, green) and ethidium homodimer-1 (for staining dead cells, red). BE-MLMs-incorporated GelMA hydrogels were rinsed with phosphate-buffered saline (PBS), and they were placed in an incubator with dye mixture made of calcein AM and ethidium homodimer-1 (0.5 μL/mL and 2 μL/mL, respectively) for 20 min. Then, GelMA hydrogels were washed with PBS, and cellular images of green and red fluorescence signals were acquired by imaging using a confocal microscope. Percentages of dead cell populations were determined in each incubation time point.
ELISA Assay:
Secreted albumin, urea, IL-6, TNF-α, TGFβ1, and MMP2 levels in the culture media were analyzed. Culture media were collected from BE-MLMs across the conditions. Secreted albumin (EHALB, Thermo Fisher Scientific), urea (MAK006, Sigma-Aldrich), IL-6 (KMC0061, Thermo Fisher Scientific), TNF-α (BMS607-3, Thermo Fisher Scientific), TGFβ1 (ab100647, Abcam), and MMP2 (ab100606, Abcam) were quantified according to the manufacturer’s guideline.
Evaluation of Intracellular Lipid Accumulation and ROS Production:
GelMA hydrogels containing BE-MLMs were collected, and the lipid accumulation degrees in the BE-MLMs were analyzed by imaging using a confocal microscope. Intracellular triglyceride deposition was assessed with the use of the AdipoRed assay (PT-7009, Lonza) according to the manufacturer’s guideline. In brief, GelMA hydrogels were washed with PBS and incubated with AdipoRed dye (3%, v/v) in PBS for 10‒15 min. ROS expression levels of BE-MLMs in GelMA hydrogels were evaluated. Carboxy-H2DCFDA (general oxidative stress indicator; C400, Thermo Fisher Scientific) dissolved in PBS (10 μM) was prepared by diluting its stock solution in dimethyl sulfoxide (10 mM). BE-MLMs-incorporated GelMA specimens were rinsed with PBS, and they were placed with carboxy-H2DCFDA solution for around 30 min at 37°C, followed by replacing the solution with PBS containing 10% FBS. Levels of accumulated lipid or cellular ROS was determined by imaging using a confocal microscope.
Immunofluorescence:
Expression levels of CD31, collagen I, fibronectin, αSMA, P-glycoprotein, CYP3A4, MRP2, and SLCO1B1 in BE-MLMs incubated with or without FFAs or TGFβ1 were quantitatively analyzed with IF staining. After fixing cells with 4% paraformaldehyde (Sigma-Aldrich) for 20 min, Triton X-100 (T8787, Sigma-Aldrich) (0.1%) was added to each GelMA hydrogels including BE-MLMs, and they were incubated for 30 min. After eliminating it, blocking was performed with BSA (1%, w/v) in PBS for 15 min. Following removing BSA solution, primary antibody (Ab) in BSA (1%, w/v) solution was added to each GelMA hydrogels containing BE-MLMs, and they were incubated for overnight. Anti-CD31 Ab (rabbit, ab28364, Abcam), collagen I Ab (rabbit, NB600-408, Novus Biologicals, LLC), fibronectin Ab (mouse, NBP2-22113, Novus Biologicals), αSMA Ab (mouse, NBP2-33006, Novus Biologicals), anti-P-glycoprotein Ab (mouse, ab3366, Abcam), anti-CYP3A4 Ab (rabbit, ab135813, Abcam), anti-MRP2 Ab (mouse, ab3373, Abcam), or anti-SLCO1B1 Ab (rabbit, SAB4300781, Sigma-Aldrich) were used for IF staining in this study. BE-MLMs after overnight incubation with primary Ab were washed twice with PBS. Samples were incubated with 2% goat serum (GS) in BSA (1%, w/v) solution for 15 min. Then, labeling goat 594-anti-rabbit or goat 488-anti-mouse (ab150080 and ab150113, respectively, Abcam) was performed in 2% GS in BSA (1%, w/v) solution for 25 min in CO2 incubator at 37°C. Samples were then rinsed thrice with PBS and stored in the fridge (4°C) for imaging stained cells using a confocal microscope.
Preparation of Gradient-Generating Microfluidics Devices:
PDMS-based microfluidic devices were fabricated as previously described.[35] Briefly, the design schematic was first drawn on CorelDRAW following a previously published design for microfluidic gradient mixing.[35] In this design, the channel width is 500 μm, and the size of the culture chamber is 5 mm × 5 mm. This design, along with a 50 mm × 50 mm poly(methyl methacrylate) (PMMA) base, was laser cut using an ILS Laser Platform (Universal Laser Systems) into a PMMA sheet (1.5 mm-thickness) (McMaster Carr). The resulting PMMA design was fixed to the PMMA base using Weldon #4 Acrylic Cement and allowed to solidify for 24 h. PDMS was prepared in a 10:1 (elastomer to curing agent) ratio following Sylguard 184 Kit instructions (Fisher Scientific), poured onto the laser-cut mold inside a square petri dish (Fisher Scientific), and desiccated for 45 min to remove bubbles. The dish was then transferred to an 80°C oven for 2 h to cure. Following, the cured PDMS was carefully peeled from the PMMA mold and cut into appropriate dimensions. The resulting PDMS gradient mixer device that enables 8 distinct concentrations from 2 inlets has channels with a fixed cross-sectional area of 0.75 mm2 (0.5 mm diameter × 1.5 mm height), 5 layers of mixing channels, and 8 culture chambers.
Simulation Study:
In order to perform a numerical study on the gradient generation in the proposed device, numerical simulation was achieved with commercial finite element method solver (Comsol Multiphysics version 5.3, Comsol, Inc.). After generating a proper unstructured mesh for the fluid flow domain, the mesh independency of the solution was performed. By applying proper boundary circumstances at the inlets and outlets as constant velocity at inlets (corresponding to our used flow rate: 100 μL/h at both inlets) and atmospheric pressure at all outlets, the central calculations for liquid flow including momentum equations (Navier-Stokes equations) and continuity calculation (mass preservation) were solved numerically to find the flow field information including velocity field and pressure distribution inside the channel. For numerical simulation of convection-diffusion calculation for determining the concentration gradient, by using appropriate border settings for the inlets as 0 mM for one of the inlets and 1 mM for another inlet as pirfenidone (P2116, Sigma-Aldrich) concentration, equation for concentration distribution inside the channel was resolved. The no-slip condition was considered for fluid flow study, and water was considered as a working fluid and its properties, including 1000 kg/m3 for density and 0.001 Pa∙s for viscosity for the walls of the microchannel. The no-mass flux border condition was used for the walls of the microchannel, and constant diffusivity for water (1.67×10−9 m2/s) was used for mass transport study.
Drug Study:
Pharmacological efficacies of pirfenidone in HepaRG-based or PHH-based BE-MLMs were assessed in the microfluidic device system. BE-MLMs pre-cultured in GelMA hydrogels for 14 days were cultured in the microfluidic device with different concentrations of pirfenidone (0–1 mM) for 7 days. On day 21, BE-MLMs in GelMA hydrogels were collected from the devices, and the pirfenidone concentration dependent-expression levels of fibronectin and αSMA in BE-MLMs were assessed by IF staining.
qPCR assay:
The in vitro samples (n = 3 per each group) were collected and stored in the −80°C deep freezer until further evaluation. According to the manufacturer’s guidelines, total RNA was obtained with a RNeasy Plus Mini Kit (Qiagen Inc., Hilden, Germany). RNA quality test was completed on all samples using a NanoDrop One Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific) to ensure the value range of 260/280 nm within 1.7‒2.1. Total RNA (1 μg) was obtained by cDNA synthesis using a QuantiTect Reverse Transcription Kit (Qiagen). The primers are listed in supplementary data (Table S1). PowerUp Sybr Green Master Mix (Thermo Fisher Scientific) was employed, and the real-time PCR amplification was performed for 45 cycles. Melting curve analysis was conducted to verify the amplification of specific qPCR products. To calculate the expression fold change, the ΔΔCt method was used. Reference genes (glyceraldehyde 3-phosphate dehydrogenase (GAPDH), beta actin (β actin), and 18s ribosomal RNA (18s rRNA)) were employed for the normalization, and the results were expressed as fold changes of the threshold cycle (Ct) (using 2−ΔΔCt method).
Data analysis:
ImageJ was applied to analyze and quantify the fluorescence intensities. Average intensity in the sections of interest in the cell was quantified in the data of lipid staining and ROS assay data.
Statistical analysis:
All data were obtained from at least three repeats. Error bars show the standard deviation. One-way analysis of variance (ANOVA, Tukey HSD as a post-hoc test) was done for statistical analyses to evaluate multiple assessments. A p-value below than 0.05 was regarded as statistically important.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge funding from the National Institutes of Health (1UG3TR003148-01 and 1R01GM126571-01). In addition, this paper was sponsored by the Office of the Secretary of Defense and was accomplished under Agreement Number W911NF-17-3-003. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Office of the Secretary of Defense or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein. This research was funded in part by the Advanced Regenerative Manufacturing Institute, Inc. (“ARMI”) through the above-referenced agreement. The views and conclusions contained in this document are those of the authors and should not be interpreted as the views of ARMI. H-J.C. acknowledges the support of a research grant of Kangwon National University in 2019.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Hyun-Jong Cho, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; College of Pharmacy, Kangwon National University, Chuncheon, Gangwon 24341, Republic of Korea.
Han-Jun Kim, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
KangJu Lee, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Soufian Lasli, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA.
Aly Ung, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA.
Tyler Hoffman, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA.
Rohollah Nasiri, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Praveen Bandaru, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Samad Ahadian, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Mehmet R. Dokmeci, Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA; Department of Radiological Sciences, David Geffen School of Medicine, University of California-Los Angeles, Los Angeles, CA 90095, USA.
Junmin Lee, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA.
Ali Khademhosseini, Department of Bioengineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Center for Minimally Invasive Therapeutics (C-MIT), University of California-Los Angeles, Los Angeles, CA 90095, USA; Terasaki Institute for Biomedical Innovation, Los Angeles, CA 90064, USA; Department of Radiological Sciences, David Geffen School of Medicine, University of California-Los Angeles, Los Angeles, CA 90095, USA; Department of Chemical and Biomolecular Engineering, Henry Samueli School of Engineering and Applied Sciences, University of California-Los Angeles, Los Angeles, CA 90095, USA; Jonsson Comprehensive Cancer Centre, University of California, Los Angeles, CA 90095, USA.
References
- [1].Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R, Nat. Rev. Rheumatol 2019, 15, 705. [DOI] [PubMed] [Google Scholar]
- [2].Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA, Rinella ME, Nat. Rev. Dis. Primers 2015, 1, 15080. [DOI] [PubMed] [Google Scholar]
- [3].Tsuchida T, Friedman SL, Nat. Rev. Gastroenterol. Hepatol 2017, 14, 397. [DOI] [PubMed] [Google Scholar]
- [4].Fabregat I, Caballero-Diaz D, Front. Oncol 2018, 8, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, Grønbæk H, Nat. Rev. Gastroenterol. Hepatol 2019, 16, 145. [DOI] [PubMed] [Google Scholar]
- [6].Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ, Nat. Med 2018, 24, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shetty A, Syn WK, Health and economic burden of non-alcoholic fatty liver disease in the United States and its impact on veterans. Fed. Pract 2019, 36, 14. [PMC free article] [PubMed] [Google Scholar]
- [8].Van Herck MA, Vonghia L, Francque SM, Nutrients 2017, 9, 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lau JKC, Zhang X, Yu J, Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J. Pathol 2017, 241, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chavez-Tapia NC, Rosso N, Tiribelli C, Curr. Med. Chem 2011, 18, 1079. [DOI] [PubMed] [Google Scholar]
- [11].Kanuri G, Bergheim I, Int. J Mol. Sci 2013, 14, 11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Boeckmans J, Natale A, Buyl K, Rogiers V, Kock JD, Vanhaecke T, Rodrigues RM, Pharmacol. Res 2018, 134, 257. [DOI] [PubMed] [Google Scholar]
- [13].Müller FA, Sturla SJ, Curr. Opin. Toxicol 2019, 16, 9. [Google Scholar]
- [14].Gerets HHJ, Tilmant K, Gerin B, Chanteux H, Depelchin BO, Dhalluin S, Atienzar FA, Cell Biol. Toxicol 2012, 28, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Berger B, Donzelli M, Maseneni S, Boess F, Roth A, Krähenbühl S, Haschke M, Front. Pharmacol 2016, 7, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Norona LM, Nguyen DG, Gerber DA, Presnell SC, Mosedale M, Watkins PB, Plos One. 2019, 14, e0208958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Feaver RE, Cole BK, Lawson MJ, Hoang SA, Marukian S, Blackman BR, Figler RA, Sanyal AJ, Wamhoff BR, Dash A, JCI Insight 2016, 1, e90954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cole BK, Feaver RE, Wamhoff BR, Dash A, Expert Opin. Drug Discov 2018, 13, 193. [DOI] [PubMed] [Google Scholar]
- [19].Fang Y, Eglen RM, Slas Discov. 2017, 22, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lasli S, Kim HJ, Lee K, Suurmond CAE, Goudie M, Bandaru P, Sun W, Zhang S, Zhang N, Ahadian S, Dokmeci MR, Lee J, Khademhosseini A, Adv. Biosystems 2019, 3, 1900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hosseini V, Maroufi NF, Saghati S, Asadi N, Darabi M, Ahmad SNS, Hosseinkhani H, Rahbarghazi R, J. Transl. Med 2019, 17, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, Rautou P-E, J Hepatol. 2017, 66, 212. [DOI] [PubMed] [Google Scholar]
- [23].Sitia G, Iannacone M, Aiolfi R, Isogawa M, van Rooijen N, Scozzesi C, Bianchi ME, von Andrian UH, Chisari FV, Guidotti LG, PLoS Pathog. 2011, 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hoffmann C, Djerir NEH, Danckaert A, Fernandes J, Roux P, Charrueau C, Lachagès A-M, Charlotte F, Brocheriou I, Clément K, Aron-Wisnewsky J, Foufelle F, Ratziu V, Hainque B, Bonnefont-Rousselot, Bigey P, Escriou V, Sci Rep. 2020, 10, 3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Suurmond CE, Lasli S, van den Dolder FW, Ung A, Kim HJ, Bandaru P, Lee K, Cho HJ, Ahadian S, Ashammakhi N, Dokmeci MR, Lee J, Khademhosseini A, Adv. Healthc. Mater 2019, 8, 1901379. [DOI] [PubMed] [Google Scholar]
- [26].Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Langan LM, Dodd NJF, Owen SF, Purcell WM, Jackson SK, Jha AN, PLoS ONE, 2016, 11, e0149492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deng J, Wei W, Chen Z, Lin B, Zhao W, Luo Y, Zhang X, Micromachines 2019, 10, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beckwitt CH, Clark AM, Wheeler S, Taylor DL, Stolz DB, Griffith L, Wells A, Exp. Cell. Res 2018, 363, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Seki E, Brenner DA, J. Hepatobiliary Pancreat. Sci 2015, 22, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].DeLeve LD, Hepatology. 2015, 62, 326.26108875 [Google Scholar]
- [32].Dewidar B, Meyer C, Dooley S, Meindl-Beinker N. Cells 2019, 8, 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fabregat I, Caballero-Díaz D, Front Oncol. 2018, 8, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bandaru P, Cefaloni G, Vajhadin F, Lee K, Kim H-J, Cho H-J, Hartel MC, Zhang S, Sun W, Goudie MJ, Ahadian S, Dokmeci MR, Lee J, Khademhosseini A, Small 2020, 16, e2001837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Burdick JA, Khademhosseini A, Langer R, Langmuir 2004, 20, 5153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.