

Abstract
Small molecule colloidal aggregates adsorb and partially denature proteins, inhibiting them artifactually. Oddly, this inhibition is typically time-dependent. Two mechanisms might explain this: low concentrations of the colloid and enzyme might mean low encounter rates, or colloid-based protein denaturation might impose a kinetic barrier. These two mechanisms should have different concentration dependencies. Perplexingly, when enzyme concentration was increased, incubation times actually lengthened, inconsistent with both models and with classical chemical kinetics of solution species. We therefore considered molecular crowding, where colloids with lower protein surface density demand a shorter incubation time than more crowded colloids. To test this, we grew and shrank colloid surface area. As the surface area shrank, the incubation time lengthened, while as it increased, the converse was true. These observations support a crowding effect on protein binding to colloidal aggregates. Implications for drug delivery and for detecting aggregation-based inhibition will be discussed.
Graphical Abstract
INTRODUCTION
Many organic molecules, including drugs, investigational new drugs, clinical candidates,1–4 and especially early leads in drug discovery,5–8 aggregate into densely packed colloids at micromolar or even submicromolar concentrations in bio-chemical buffers.9 These aggregates sequester 105 to 106 protein molecules,10,11 leading to their partial denaturation12,13 and typically their inhibition. Such inhibition is among the dominant mechanisms for false-positive results in early drug discovery.14–17 Understanding its mechanism has been key for avoiding this phenomenon and, increasingly, for exploiting the unusual properties of the colloidal particles for useful applications, such as drug delivery.18–22
A curious feature of colloidal inhibition is that it typically increases on pre-incubation of the protein with the particle, up to some saturating point.12 Why this should happen has remained unclear. Slow onset inhibition is well known in ligand binding and classically may be attributed to two mechanisms: very low concentrations, which slow the formation of encounter complexes via diffusional barriers (unusual for most ligand-protein systems), or slow off-rates, reflecting either kinetic barriers or simply very tight binding.23,24 Both mechanisms are plausible for colloidal-based inhibition. While the concentration of the aggregating monomer may be in the μM to the high-nM range, the concentration of the colloidal particles themselves is mid-fM to low-pM.10 Mean-while, the proteins being inhibited can easily be in the low-nM range; these concentrations plausibly are low enough to constitute kinetic barriers to associations (Derivation S1). Conversely, proteins bind tightly to colloidal aggregates, with KD values in the pM range or better,10,11 and protein partial unfolding on colloidal surfaces could be a kinetic barrier to association and disassociation.
These two mechanisms imply different concentration dependencies. Low concentration, kinetic barriers to association should be sensitive to concentration changes in either the colloid or the protein. Off-rate barriers to disassociation, either because of high affinity, or because of kinetic barriers to protein folding, should be pseudo-zero order in enzyme concentration. Said another way, increasing the concentration of either the colloid or protein should reduce the incubation effect if this effect reflects association barriers, while for the mechanism reflecting kinetic off-rates, the incubation effect should be insensitive to enzyme concentration.
To distinguish between the two mechanisms, we investigated concentration effects on the kinetics of the inhibition buildup between model enzymes and colloidal aggregators. Unexpectedly, our results were inconsistent with both mechanisms. Instead of classical concentration-rate effects, we observed that increasing enzyme concentration slowed inhibition buildup, increasing the incubation time necessary to achieve a certain inhibition level. This seemed most consistent with a third, non-classical mechanism, molecular crowding on the colloidal surface, something we explored in depth in subsequent experiments. Since an incubation effect has been a harbinger of colloidal aggregation since it was first described,9 this mechanism has implications for the rapid detection of aggregation in early discovery and may influence how we exploit drug colloid formation in formulation and delivery.
RESULTS
To distinguish between the two initial mechanisms—low concentration encounter barriers or off-rate effects—we first investigated increasing the concentration of the colloids. The effect of incubation was measured at increasing concentrations of aggregating small molecules, each above their critical aggregation concentration (CAC). Within a certain range, the concentration of colloidal particles depends linearly on the amount of the monomer added over the CAC;25 as more monomers are added over the CAC, more colloidal particles are formed. For both the model enzymes AmpC β-lactamase and malate dehydrogenase (MDH),26 tested against the well-studied colloidal aggregators Sorafenib and fulvestrant, the incubation time necessary to reach a certain amount of inhibition fell monotonically as the concentration of the colloidal particles increased (Figure 1A–D). For instance, we incubated the colloidal aggregator Sorafenib for varying times with AmpC and then measured the rate of hydrolysis by initiating the reaction with the substrate (Figure 1A). Especially at lower concentrations of aggregating Sorafenib, as the time of incubation increases (x axis), so does inhibition. At a 5 μM Sorafenib monomer (perhaps mid-fM colloid), the reaction is 40% inhibited after a minute of pre-incubation, 80% inhibited after 2 min of pre-incubation, and is nearly fully inhibited after 5 min. As one increases the concentration of Sorafenib, the inhibition at every incubation time point increases. Thus, as one increases the concentration of the aggregating molecule, the incubation period decreases (the onset of inhibition gets faster). This may be also seen as plots of the time of incubation necessary to achieve 90% inhibition (T90, Figure 1E,F; we note the effect plateaus, reflecting limits to our ability to distinguish among the very fast T90s at high colloidal concentrations). This supports an encounter reaction that is first order in the concentration of the colloidal particles and is at least consistent with a low concentration encounter barrier model for the incubation effect.
Figure 1.
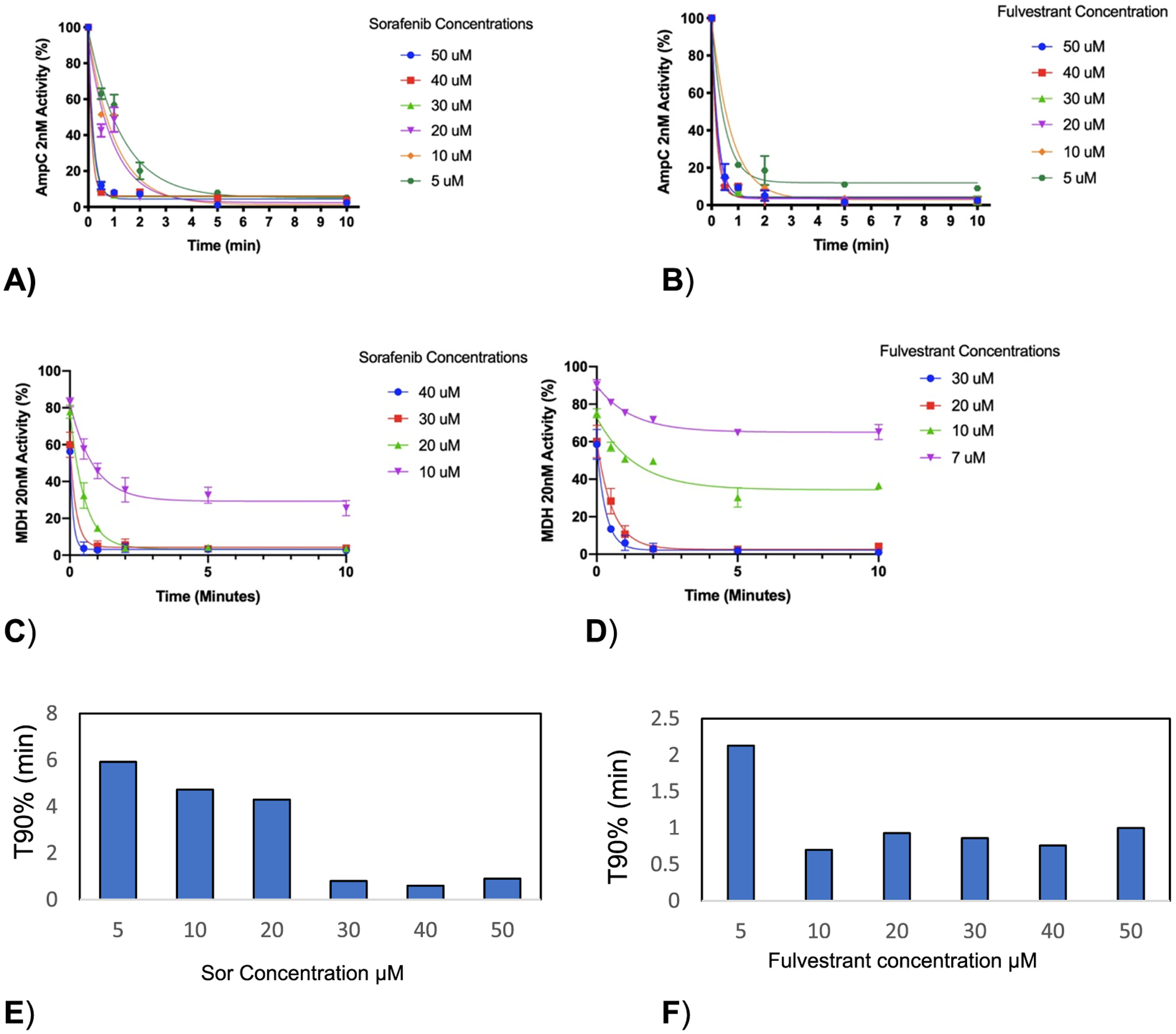
(A, B) The onset of inhibition of AmpC ß-lactamase at 2 nM is expedited by increasing Sorafenib and fulvestrant concentrations. (C, D) Inhibition of malate dehydrogenase at 20 nM is expedited to be the same. (E, F) Incubation time required to reach 10% of activity (90% inhibition) (T90) decreases with increasing colloidal concentration.
If the effect of increasing colloidal concentration readily fits at least one of our hypotheses, the results were unexpected when we turned to increasing enzyme concentration. In contrast to what classic rate kinetics would predict, the onset of inhibition—the length of incubation time necessary to reach a certain level of inhibition—increased with enzyme concentration, for both AmpC and MDH, in the presence of constant Sorafenib or constant fulvestrant colloidal aggregates (Figure 2). For instance, a 30 s pre-incubation of 2 nM AmpC with a 10 μM Sorafenib monomer (likely a mid-fM Sorafenib colloid) resulted in 67% inhibition, and a 5 min pre-incubation sufficed to essentially fully inhibit the enzyme. Doing the same at the 10 nM enzyme, however, led to only 40% inhibition after 30 s of pre-incubation, and even after 5 min, at least 20% of enzyme activity remained (Figure 2A). Quantitatively, T90 values monotonically increased with enzyme concentrations for both AmpC and MDH and for both colloidal aggregators (Figure 2D and Figure 2E, respectively). In short, as enzyme concentration increased, the onset of inhibition took longer. This is inconsistent with both classical models for the time dependence of colloidal inhibition.
Figure 2.
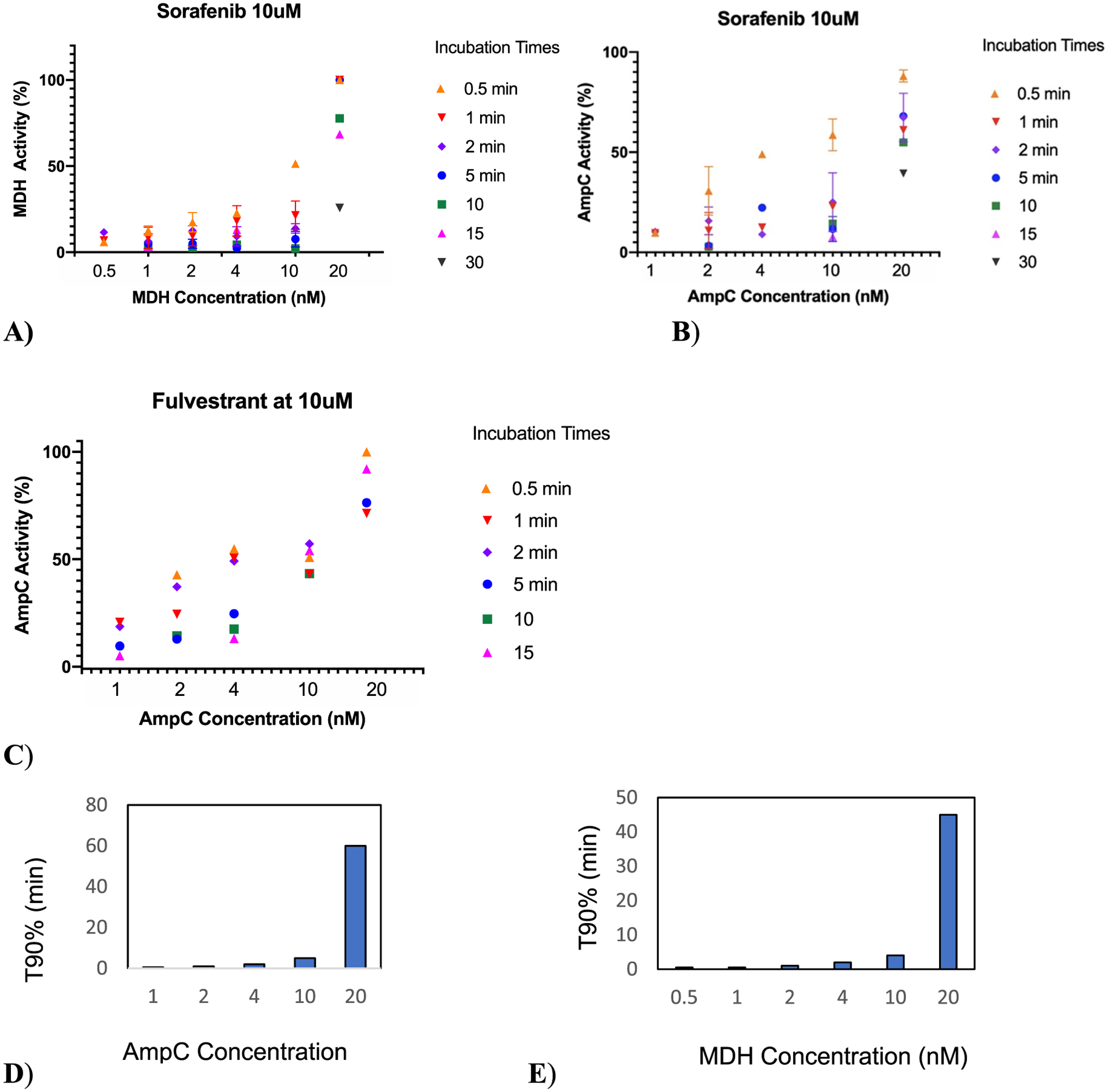
Change of the inhibition onset with incubation time as a function of (A) MDH concentration at 10 μM Sorafenib. (B) AmpC concentrations at 10 μM Sorafenib and (C) AmpC concentration at 10 μM fulvestrant. (D) T90 for varying concentration AmpC inhibition by Sorafenib and (E) T90 for varying concentration MDH inhibition by Sorafenib.
This observation led us to investigate a protein crowding mechanism on the colloidal surface, where inhibition appears to occur.12,26 In this non-classical model, as protein binds to the particle surface, the rate at which the next protein molecule can bind is reduced by crowding. We therefore investigated how varying the colloidal surface area changed the incubation time—the onset of inhibition—using four different strategies. First, we changed the buffer ionic strength, with lower ionic strengths leading to smaller colloids with less available surface areas per particle (more crowding) and higher ionic strengths leading to larger colloids with more surface areas per particle9 (less crowding). Second, we pre-incubated the colloid with another, functionally inert protein to see how it would affect the inhibition onset of the monitored enzyme; this increases protein crowding on the colloidal surface. Third, we co-formulated the colloids with Congo Red, which substantially decreases colloid size, again increasing crowding. Fourth, we investigated the rates of the inhibition onset on an enzyme much larger than AmpC and MDH, β-galactosidase—the larger size of this enzyme should itself increase crowding leading to a slower inhibition onset (Figure 3).
Figure 3.
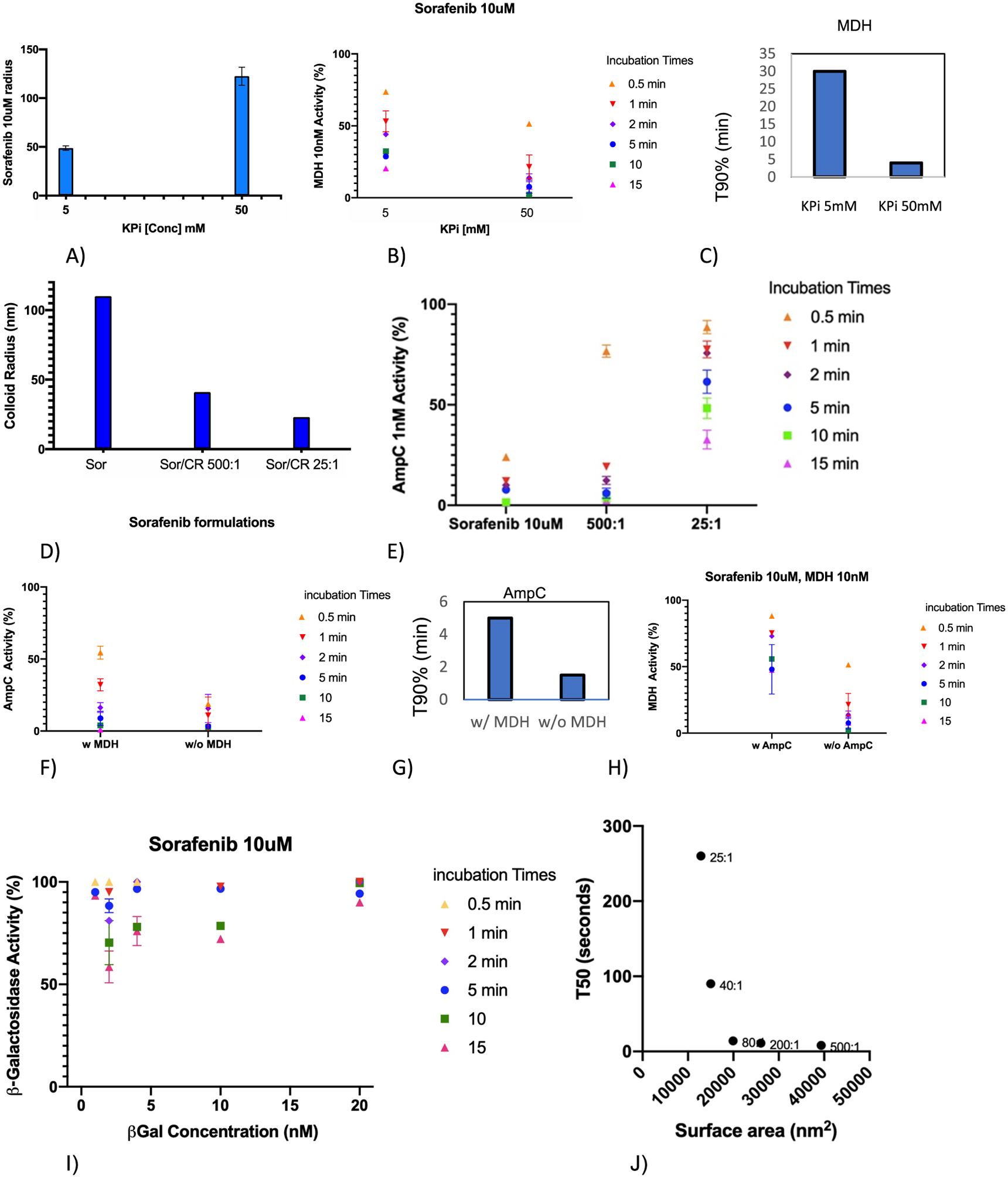
Decreasing the available surface area of colloidal particles increases the incubation time, slowing the attainment of full inhibition. (A) Sorafenib colloid radii (nm) vary with ionic strength. (B) The smaller radii colloids slow the buildup of MDH inhibition. (C) T90% of MDH inhibition at varying buffer strengths. (D) Co-formulation of Sorafenib with Congo Red reduces the radii of the colloidal particles. (E) As the particle size diminishes in the co-formulated colloids, the time of incubation necessary to inhibit increases (the onset of inhibition slows). (F) Pre-incubating Sorafenib colloids with MDH increases the incubation time necessary to inhibit AmpC (the onset of inhibition slows). (G) T90% of AmpC with and without MDH. (H) Correspondingly, pre-incubating Sorafenib colloids with AmpC increases the incubation time necessary to inhibit MDH (the onset of inhibition slows). (I) Compared to the rate of the inhibition onset with smaller enzymes, that of the unusually large enzyme β-galactosidase is far slower (compared to Figure 2A,B). (J) Relationship of the colloidal particle size with the incubation time necessary to reach 50% inhibition (T50). Colloid radii were varied by changing the co-formulation ratio of Sorafenib and Congo Red; these ratios are indicated.
In the first experiment, we measured inhibition at 5 mM KPi versus 50 mM KPi. At the lower ionic strength, Sorafenib colloids shrank to 48 from 121 nm radii at 50 mM KPi (Figure 3A and Figure S1 and Table S1), as expected.9 At this reduced size, inhibition increased more slowly, demanding longer incubation times (Figure 3B), and T90 values went from 5 min at the larger radii to >15 min at the smaller radii. Because ionic strength can affect other aspects of the kinetics of the inhibition onset, we also investigated the effects of reducing colloid size by co-formulating the Sorafenib colloids with the dye Congo Red.21 At a ratio of 500:1 Sorafenib to dye, the particle size shrank from a radius of 110 to 41 nm, and at a ratio of 25:1, the particles further shrank to a radius of 23 nm (Figure 3C and Figure S1 and Table S1). As particles shrunk, the onset of inhibition lengthened, and the time necessary to incubate to reach a certain level of inhibition increased, also consistent with the crowding hypothesis. This observation is striking because the concentration of these smaller co-formulated colloids scales as the cube of the ratio of the radii such that going from 110 nM to 23 will increase colloidal concentration by over 100-fold. All things being equal, the pseudo-first order reaction rate should thus increase by over 100-fold (Derivation S1), and the t1/2 should decrease by the same factor. Instead, the time over which the reaction occurred was substantially longer (the kinetics slowed). To quantify how the particle size affected the incubation time necessary to inhibit, we plotted the T50 of the reaction (the lengthening of the incubation time) against the particle size, where the latter was varied by changing the ratio of Sorafenib to co-formulated Congo Red. The T50 values scaled monotonically with the inverse of particle surface area (Figure 3J). The apparent exponential shape of the curve may reflect the severity of crowding as the particles shrink, and potentially saturation effects.
In a third set of experiments, we investigated the effect on the inhibition onset of pre-loading colloidal particles with an effectively inert protein before exposing them to the enzyme being monitored. If crowding affects the rate of the inhibition onset, then we would expect pre-loading the inert protein to reduce the onset of inhibition of the monitored enzyme without affecting the total level of inhibition ultimately achieved. We pre-loaded Sorafenib colloids with 2 nM MDH for 5 min and investigated how that affected the rate of the inhibition onset of AmpC by those colloids versus how they inhibited AmpC in the absence of MDH pre-loading. When pre-loaded with MDH, AmpC inhibition grew substantially slower than when the colloids had not been pre-loaded with MDH (Figure 3E). For instance, a 30 s pre-incubation of AmpC with Sorafenib colloids without MDH pre-loading led to 80% inhibition, and by a 5 min pre-incubation, enzyme activity was barely measurable. Conversely, the same 30 s pre-incubation after pre-loading the colloids with MDH led to only 45% inhibition, and even after a 5 min incubation with AmpC, the enzyme retained a still measurable activity. Correspondingly, T90 values lengthened from 1 min in the absence of MDH pre-loading to 5 min with MDH pre-loading. The same effect on MDH activity, when AmpC was used as the pre-loaded enzyme, was also observed (Figure 3F,G). These results, too, support the surface crowding hypothesis.
Turning to the enzyme size as a variable, the 0.5 megadalton (520 kDa) ß-galactosidase was used as an unusually large enzyme, compared to 35 kD MDH and the 40 kD AmpC, to induce more crowding on the colloidal surface per mol of the enzyme absorbed. At every concentration of β-galactosidase, the onset of inhibition was slower than MDH or AmpC with Sorafenib colloids (Figure 3I vs Figure 2A and Figure 2B), also consistent with the crowding hypothesis.
If decreasing colloid size, increasing protein crowding, slows the inhibition onset and increases incubation times, then we might expect that increasing colloid size and decreasing protein crowding should speed the inhibition onset, decreasing the incubation time necessary to reach a certain level of inhibition. Accordingly, we investigated two small molecules that naturally form larger colloids than Sorafenib (100 nm average radius) and fulvestrant (82 nm average radius): nicardipine (300 nm average radius) and clofazimine (570 nm average radius). In studies with MDH, inhibition kinetics by both of the larger colloids was bell-shaped, speeding up as enzyme concentration increased until reaching a minimum at around the 4 nM enzyme, after which the inhibition onset began to slow (Figure 4A,B). Unlike with the smaller colloidal particles, here in the first part of the curve, inhibition kinetics actually do increase with enzyme concentration, consistent with simple diffusion-governed inhibition in this domain. As higher concentrations of the enzyme are reached, for these larger particles, crowding-governed behavior seems again to take hold. Intriguingly, the larger β-galactosidase, even on the larger nicardipine colloids, retained the non-classical crowding mechanism throughout as the inhibition onset grew longer as the enzyme concentration increased (Figure 4C).
Figure 4.
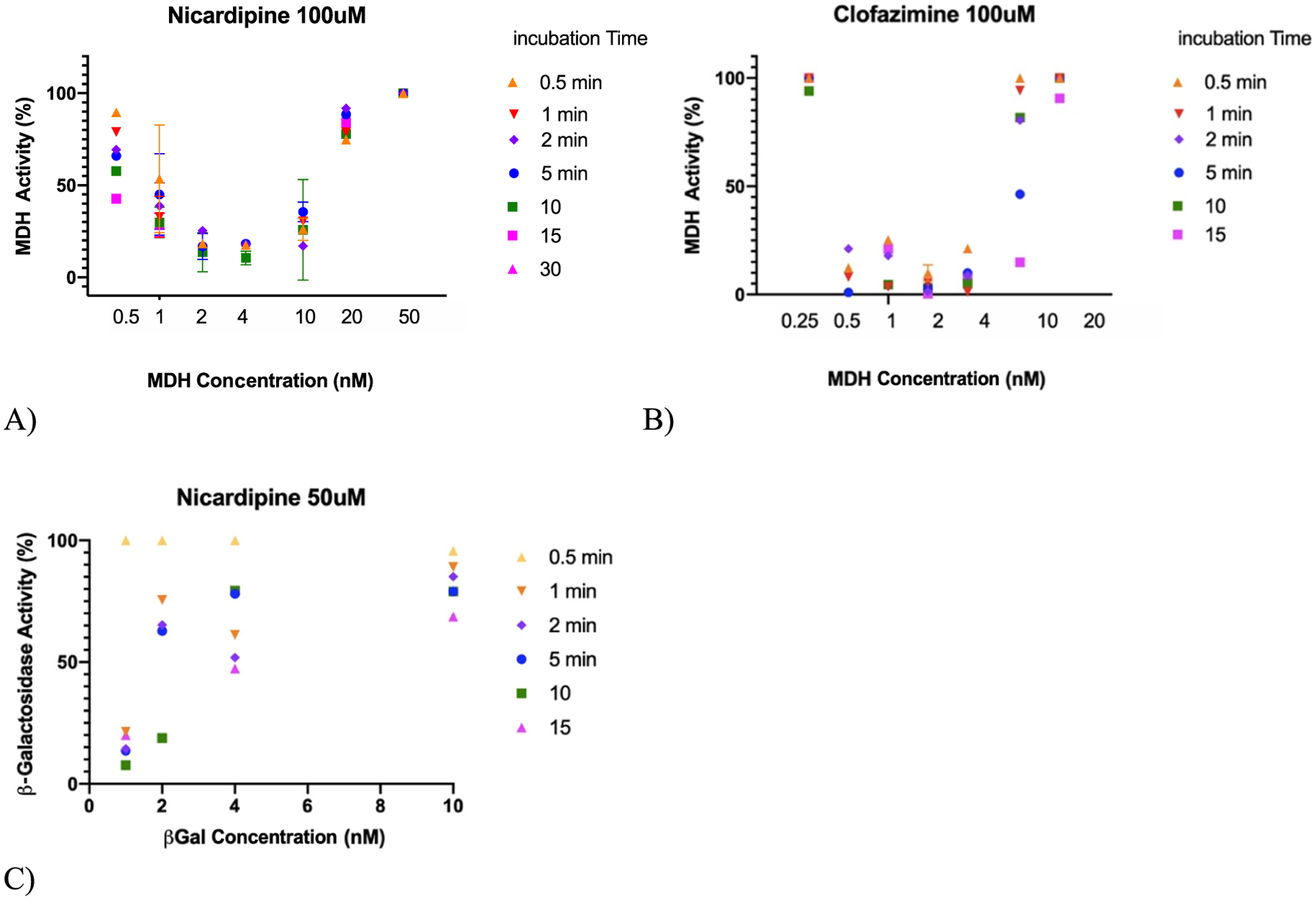
Large colloids relieve the crowding effect, returning to classical kinetic concentration dependence in a low enzyme concentration domain. At higher enzyme concentration, crowding returns, leading to bell-shaped curves with MDH. (A) Nicardipine colloids, ~300 nm radius, inhibiting MDH. (B) Clofazimine, 570 nm radius size, inhibiting MDH. (C) With the larger enzyme, β-galactosidase, crowding once again dominates even with nicardipine.
DISCUSSION AND CONCLUSIONS
The slow onset of colloid-based inhibition—the incubation effect that has long been considered a characteristic of the phenomenon—reflects protein crowding on the colloidal surface. To our surprise, our results invalidated the two other hypotheses that we had favored to explain this effect: simple kinetic barriers deriving from femtomolar concentrations of the colloidal particles, or the kinetic barrier of enzyme unfolding. Although increasing colloidal concentration sped the enzyme inhibition onset (Figure 1), consistent with the kinetics of association hypothesis (Derivation S1), neither model explains the slowing of the inhibition onset, the lengthening of incubation necessary to reach a given level of inhibition, as enzyme concentration is increased (Figure 2). This observation led us to investigate a model of surface crowding, where as the colloidal surface becomes increasingly occupied with protein; the rate of new protein binding to that surface slows.
The crowding model made testable predictions: shrinking a colloid should increase crowding, slowing the inhibition onset, as should pre-loading the colloid with a second inert protein. Similarly, increasing the size of the enzyme, without changing the colloid size, should slow the inhibition onset, while larger colloidal particles should speed the inhibition onset, decreasing incubation times. Each of these was supported by the experiment. When colloids were shrunk by either decreasing ionic strength (Figure 3A,B) or by co-formulating the colloids with Congo Red (Figure 3C,D), the inhibition onset slowed dramatically. Similarly, pre-loading the colloids with a second inert protein had the same effect, lengthening incubation times necessary to reach a given level of inhibition (Figure 3E,F). Moving to the much larger β-galactosidase slowed the inhibition buildup compared to smaller AmpC and MDH, with the same colloidal particles (Figure 3I). Finally, relieving crowding with larger colloidal particles, as formed by nicardipine and clofazimine, led to faster inhibition onsets and, more compelling still, a return to a domain where increasing enzyme concentration increased the rate of the inhibition onset, leading to greater levels of inhibition in faster times, as would be classically expected. This held over a certain concentration domain, after which, presumably as crowding increased too far on the colloidal surfaces, the rate of the inhibition onset began again to slow, leading to bell-shaped curves with enzyme concentration (Figure 4). Integrating previous studies that investigated the stoichiometry and concentration of colloidal particles,10,27 the partial denaturation of proteins on colloidal surfaces12,13 with the current study, the model that emerges is one where the kinetics of protein binding to colloidal aggregates is at least influenced by, and in many domains dominated by, crowding on the surface of the particle (Figure 5).
Figure 5.
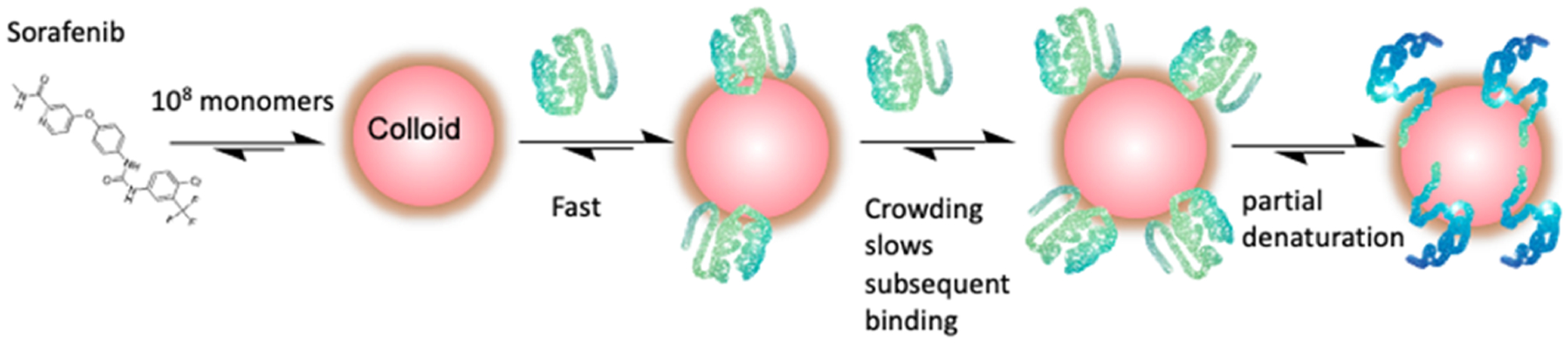
Combined crowding and partial denaturation model for protein adsorption on the surface of colloidal aggregates. Typically, between 106 and 108, monomers aggregate to form colloidal particles, whose concentrations are often in the mid-femtomolar to low picomolar range.10 This work suggests that the rate of protein adsorption is, in many domains, dominated by protein crowding at the surface. Ultimately, proteins are partially and reversibly denatured at the protein surface;12,13 how the rate of that unfolding compares with the rate of protein adsorption remains unclear.
It is interesting to model at what point protein saturation of the colloidal surface begins to slow the inhibition onset, lengthening the incubation time necessary to see a given level of inhibition. To do so, we must know not only the concentration of the enzyme and the surface area of the particles, which are readily known, but also the concentration of the colloidal particles themselves, which are difficult to measure. Fortunately, such measurements have been made for nicardipine colloids,10 which we also study here. It was found that, for every 1 μM nicardipine added over a critical aggregation concentration of 32 μM, 2000 colloidal particles were formed.10 From this, it could be calculated that each nicardipine colloidal particle adsorbed about 1.3 × 104 molecules of AmpC after a 5 min incubation. If each AmpC molecule was adsorbed along its longest axis (leading to the greatest coverage), then about 16% of the colloidal surface would be consumed. The MDH enzyme studied here with nicardipine is a dimer of monomers, each of which is only slightly smaller than AmpC; with the same assumptions, one nicardipine colloid should absorb about 0.6 × 104 MDH dimers. Thus, 100 μM nicardipine should lead to 8.9 × 107 colloidal particles in 1 mL, adsorbing 1 nM MDH, consuming between 6 and 15% surface area, depending on how the enzyme is oriented on the particle surface. Intriguingly, from 0.5 to 4 nM MDH, there is no evidence of crowding (Figure 4A). As MDH concentration increases further, however, we re-enter the crowding domain, with the rate of the inhibition onset slowing as the enzyme concentration increases (Figure 4A). By 10 nM MDH, the enzyme concentration might be within 50% of the total loading capacity of the particle after a 5 min incubation (50% surface area coverage), and by 20–50 nM enzyme concentration, the particles might be fully saturated. This is borne out by the course of the inhibition buildup, which by 50 nM enzyme is barely observed, presumably because the capacity to adsorb more enzymes, even for long incubations, has been exceeded. Obviously, these are rough calculations as the amount of space on the colloid consumed by each MDH molecule is uncertain. Taken at face value, they suggest that the time dependence of the inhibition onset may occur as the amount of the enzyme to be adsorbed begins to approach about 50% of the physical limits of the colloidal particle.
Other caveats also bear airing. Most importantly, none of the perturbations we make to the colloid size are perfect experiments—they all perturb more than one aspect of the system. For instance, changing the ionic strength changes gross aspects of the buffer, in addition to colloid size, while co-formulating Sorafenib with Congo Red likely changes the surface properties of the colloids (though the size change should dominate). Also, there are differences in behavior from protein to protein, something likely driven by the different properties of the proteins themselves,28 including their stability toward denaturation,13 and we do not pretend that crowding explains every aspect of protein-colloid association.
Notwithstanding these provisos, the main conclusion of this study should be clear: the onset of protein inhibition by colloidal aggregates typically does not follow classical kinetics of association but is instead usually dominated by surface crowding. This has several pragmatic implications. An incubation effect has long been considered a hallmark of colloidal aggregation and is used to recognize it in early discovery.11 What this study teaches is that this incubation effect is itself sensitive to enzyme concentrations. At higher enzyme concentrations, the typical 5 min incubation times advocated in the past may be too short; substantial inhibition may only build up over longer times. A better criterion might be observing the impact of enzyme concentrations on incubation times; only for a colloidal mechanism would one expect incubation times to increase with enzyme concentration. Correspondingly, simply incubating proteins with the colloids longer should increase the protein load harbored by the colloids, something useful for vehicles designed to deliver drugs and their targeting proteins, for instance, as colloid-antibody or colloid-transferin conjugates.29,20 Thus, we expect that this effort to understand the fundamental kinetic bases for colloid-protein association will have pragmatic implications, as has been true of previous mechanistic studies of colloidal aggregation12,13,30–32
EXPERIMENTAL SECTION
Dynamic Light Scattering.
Colloid radii were measured by dynamic light scattering (DLS) using a DynaPro Plate Reader II (Wyatt Technologies), with a 60 mW laser at a 830 nm wavelength and a detector angle of 158°; the beam size of the instrument was increased by the manufacturer to better enable detection of the larger colloidal species. Samples were measured in 384 well plates with 30 μL of loading and 10 acquisitions per sample. Compounds were dissolved in DMSO in 100× concentration and were further diluted by adding filtered 50 mM KPi (pH 7.0) to obtain a final 1% DMSO concentration. For the co-formulations, a 25:1 and 500:1 ratio of DMSO-dissolved Sorafenib and Congo Red were first mixed, and KPi was then added to this mixture to obtain a final volume of 1 mL, a final Sorafenib concentration at 10 μM, CR at 400 and 20 nM, and final 1.5% (v/v) DMSO.
Enzyme Inhibition.
Enzyme inhibition assays were performed at room temperature on an HP8453a spectrophotometer in kinetic mode using UV–vis Chemstation software (Agilent Technologies) in methacrylate cuvettes (Fisher Scientific, 14955128) with a final volume of 1 mL for both control and test reactions. Varying concentrations of the enzyme were mixed with varying concentrations of the small molecule in 50 mM KPi pH 7 buffer and were incubated at intervals between 30 s and 15 min. The enzyme-colloid aqueous solution was mixed by repeated pipetting of 150 μL before incubation. The substrate was added at the end of the incubation followed by mixing. The reactions were monitored for 150 s alongside a rate control reaction that lacked the colloidal aggregator, and initial rates were divided by the initial rate of the negative control to obtain % activity of the enzyme. The activity versus time plots were generated using GraphPad Prism.
For the AmpC ß-lactamase assay, CENTA (Millipore Sigma, 219475) at 70 μM was used as the chromogenic substrate and the change in absorbance was monitored at 405 nm. For malate dehydrogenase (MDH) (from Porcine Heart, 901643, Sigma-Millipore), the reaction was initiated by 200 μM nicotinamide adenine dinucleotide (54839, Sigma Aldrich) and 200 μM oxaloacetic acid (324427, Sigma Aldrich) and the rate was monitored at 340 nm. For ß-galactosidase (from Escherichia coli overproducer, 10105031001, Sigma Aldrich), the reaction was initiated by 300 μM ortho-nitrophenyl-β-galactoside and reaction was monitored at 320 nm. For assays where an inert enzyme was used, Sorafenib was first incubated with the inert enzyme for 5 min at room temperature, the second enzyme was then added, and the mixture was further incubated at room temperature for varying times.
For the 25:1 Sorafenib:Congo Red co-formulation, a DMSO stock was prepared with concentrations of 1 mM Sorafenib and 40 μM CR (Sigma C6277-25G). For the 500:1 formulation, a DMSO stock was prepared with concentrations of 1 mM Sorafenib and 2 μM CR. Sorafenib was added first followed by Congo Red and buffer. The solution was mixed well, and the enzyme was mixed with Sorafenib for various incubation times.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by U.S. NIH grants R35GM122481 (to B.K.S.) and GM118303 (to M.P.J.).
ABBREVIATIONS USED
- DLS
dynamic light scattering
- DMSO
dimethyl sulfoxide
- β-Gal
β-galactosidase
- KPi
potassium phosphate pH 7
- MDH
malate dehydrogenase
- Sor/CR
Sorafenib/Congo Red
- T90
time at which 90% of enzyme inhibition is observed
- UV–vis
ultraviolet–visible
- v/v
volume/volume
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c02253.
Equation of encounter kinetics explaining minute-range incubation effects for colloids in the fM concentration range, DLS curves showing colloids of different radii, and radii of colloids under different conditions (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c02253
REFERENCES
- (1).Seidler J; McGovern SL; Doman TN; Shoichet BK Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem 2003, 46, 4477–4486. [DOI] [PubMed] [Google Scholar]
- (2).Frenkel YV; Clark AD; Das K; Wang Y-H; Lewi PJ; Janssen PAJ; Arnold E Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J. Med. Chem 2005, 48, 1974–1983. [DOI] [PubMed] [Google Scholar]
- (3).Doak AK; Wille H; Prusiner SB; Shoichet BK Colloid formation by drugs in simulated intestinal fluid. J. Med. Chem 2010, 53, 4259–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Blevitt JM; Hack MD; Herman KL; Jackson PF; Krawczuk PJ; Lebsack AD; Liu AX; Mirzadegan T; Nelen MI; Patrick AN; Steinbacher S; Milla ME; Lumb KJ Structural basis of small-molecule aggregate induced inhibition of a protein-protein interaction. J. Med. Chem 2017, 60, 3511–3517. [DOI] [PubMed] [Google Scholar]
- (5).Feng BY; Simeonov A; Jadhav A; Babaoglu K; Inglese J; Shoichet BK; Austin CP A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem 2007, 50, 2385–2390. [DOI] [PubMed] [Google Scholar]
- (6).LaPlante SR; Aubry N; Bolger G; Bonneau P; Carson R; Coulombe R; Sturino C; Beaulieu PL Monitoring drug self-aggregation and potential for promiscuity in off-target in vitro pharmacology screens by a practical NMR strategy. J. Med. Chem 2013, 56, 7073–7083. [DOI] [PubMed] [Google Scholar]
- (7).Yang Z-Y; He J-H; Lu A-P; Hou T-J; Cao D-S Frequent hitters: nuisance artifacts in high-throughput screening. Drug Discovery Today 2020, 25, 657–667. [DOI] [PubMed] [Google Scholar]
- (8).Allen SJ; Dower CM; Liu AX; Lumb KJ Detection of small-molecule aggregation with high-throughput microplate bio-physical methods. Curr. Protoc. Chem. Biol 2020, 12, e78. [DOI] [PubMed] [Google Scholar]
- (9).McGovern SL; Caselli E; Grigorieff N; Shoichet BK A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem 2002, 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- (10).Coan KED; Shoichet BK Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc 2008, 130, 9606–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shoichet BK Screening in a spirit haunted world. Drug Discovery Today 2006, 11, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Coan KED; Maltby DA; Burlingame AL; Shoichet BK Promiscuous aggregate-based inhibitors promote enzyme unfolding. J. Med. Chem 2009, 52, 2067–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Torosyan H; Shoichet BK Protein stability effects in aggregate-based enzyme inhibition. J. Med. Chem 2019, 62, 9593–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Feng BY; Shelat A; Doman TN; Guy RK; Shoichet BK High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol 2005, 1, 146–148. [DOI] [PubMed] [Google Scholar]
- (15).Babaoglu K; Simeonov A; Irwin JJ; Nelson ME; Feng B; Thomas CJ; Cancian L; Costi MP; Maltby DA; Jadhav A; Inglese J; Austin CP; Shoichet BK Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J. Med. Chem 2008, 51, 2502–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jadhav A; Ferreira RS; Klumpp C; Mott BT; Austin CP; Inglese J; Thomas CJ; Maloney DJ; Shoichet BK; Simeonov A Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J. Med. Chem 2010, 53, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Aldrich C; Bertozzi C; Georg GI; Kiessling L; Lindsley C; Liotta D; Merz KM Jr.; Schepartz A; Wang S The ecstasy and agony of assay interference compounds. J. Med. Chem 2017, 60, 2165–2168. [DOI] [PubMed] [Google Scholar]
- (18).Shamay Y; Shah J; Isik M; Mizrachi A; Leibold J; Tschaharganeh DF; Roxbury D; Budhathoki-Uprety J; Nawaly K; Sugarman JL; Baut E; Neiman MR; Dacek M; Ganesh KS; Johnson DC; Sridharan R; Chu KL; Rajasekhar VK; Lowe SW; Chodera JD; Heller DA Quantitative self-assembly prediction yields targeted nanomedicines. Nat. Mater 2018, 17, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang J; Zhou J; He H; Wu D; Du X; Xu B Cell-compatible nanoprobes for imaging intracellular phosphatase activities. ChemBioChem 2019, 20, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Donders EN; Ganesh AN; Torosyan H; Lak P; Shoichet BK; Shoichet MS Triggered release enhances the cytotoxicity of stable colloidal drug aggregates. ACS Chem. Biol 2019, 14, 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McLaughlin CK; Duan D; Ganesh AN; Torosyan H; Shoichet BK; Shoichet MS Stable colloidal drug aggregates catch and release active enzymes. ACS Chem. Biol 2016, 11, 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ganesh AN; Aman A; Logie J; Barthel BL; Cogan P; Al-Awar R; Koch TH; Shoichet BK; Shoichet MS Colloidal drug aggregate stability in high serum conditions and pharmacokinetic consequence. ACS Chem. Biol 2019, 14, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Schloss JV Significance of slow-binding enzyme inhibition and its relationship to reaction-intermediate analogs. Acc. Chem. Res 1988, 21, 348–353. [Google Scholar]
- (24).Morrison JF; Walsh CT The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol 1988, 61, 201–301. [DOI] [PubMed] [Google Scholar]
- (25).Coan KED; Shoichet BK Stability and equilibria of promiscuous aggregates in high protein milieus. Mol. BioSyst 2007, 3, 208–213. [DOI] [PubMed] [Google Scholar]
- (26).Duan D; Doak AK; Nedyalkova L; Shoichet BK Colloidal aggregation and the in vitro activity of traditional Chinese medicines. ACS Chem. Biol 2015, 10, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shoichet BK Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem 2006, 49, 7274–7277. [DOI] [PubMed] [Google Scholar]
- (28).Duan D; Torosyan H; Elnatan D; McLaughlin CK; Logie J; Shoichet MS; Agard DA; Shoichet BK Internal structure and preferential protein binding of colloidal aggregates. ACS Chem. Biol 2017, 12, 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ganesh AN; Logie J; McLaughlin CK; Barthel BL; Koch TH; Shoichet BK; Shoichet MS Leveraging colloidal aggregation for drug-rich nanoparticle formulations. Mol. Pharmaceutics 2017, 14, 1852–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Raina SA; Alonzo DE; Zhang GGZ; Gao Y; Taylor LS Using environment-sensitive fluorescent probes to characterize liquid-liquid phase separation in supersaturated solutions of poorly water soluble compounds. Pharm. Res 2015, 32, 3660–3673. [DOI] [PubMed] [Google Scholar]
- (31).Ilevbare GA; Liu H; Pereira J; Edgar KJ; Taylor LS Influence of additives on the properties of nanodroplets formed in highly supersaturated aqueous solutions of ritonavir. Mol. Pharmaceutics 2013, 10, 3392–3403. [DOI] [PubMed] [Google Scholar]
- (32).McGovern SL; Helfand BT; Feng B; Shoichet BK A specific mechanism of nonspecific inhibition. J. Med. Chem 2003, 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.