

Abstract
Rev1 is a protein scaffold of the translesion synthesis (TLS) pathway, which employs low-fidelity DNA polymerases for replication of damaged DNA. The TLS pathway helps cancers tolerate DNA damage induced by genotoxic chemotherapy, and increases mutagenesis in tumors, accelerating the onset of chemoresistance. TLS inhibitors have emerged as potential adjuvant drugs to enhance efficacy of first-line chemotherapy, with the majority of reported inhibitors targeting protein-protein interactions (PPIs) of the Rev1 C-terminal domain (Rev1-CT). We previously identified phenazopyridine (PAP) as a scaffold to disrupt Rev1-CT PPIs with Rev1 interacting regions (RIRs) of TLS polymerases. To explore structure-activity relationships for this scaffold, here we developed a protocol for co-crystallization of compounds that target the RIR-binding site on Rev1-CT with a triple Rev1-CT/Rev7R124A/Rev3-RBM1 complex, and solved an X-ray crystal structure of Rev1-CT bound to the most potent PAP analogue. The structure revealed an unexpected binding pose of the compound and informed changes to the scaffold to improve its affinity for Rev1-CT. We synthesized 8 additional PAP derivatives, with modifications to the scaffold driven by the structure, and evaluated their binding to Rev1-CT by microscale thermophoresis (MST). Several second-generation PAP derivatives showed over an order of magnitude improved affinity for Rev1-CT, validating structure-based assumptions that went into the compound design.
Keywords: cancer chemotherapy, translesion synthesis inhibitors, structure activity relationships, crystallography, protein-protein interactions
Graphical Abstract
Translesion synthesis (TLS) inhibitors have emerged as adjuvant drugs to enhance efficacy of first-line cancer chemotherapy. The phenazopyridine (PAP) scaffold inhibits TLS by disrupting interactions of the Rev1-CT domain with the RIR motifs of TLS DNA polymerases. We solved a co-crystal structure of Rev1-CT in complex with the most potent of the earlier identified PAP analogues. Informed by this structure, we designed second-generation PAP derivatives with binding affinity for Rev1-CT improved by an order of magnitude.
Introduction
Genotoxic platinating and alkylating drugs are commonly used in first-line chemotherapy to treat numerous cancers.[1–5] For example, the platinating agent cisplatin originally licensed for clinical use in 1978 is now employed for treatment of 50% of cancer patients. Platinating and alkylating chemotherapies covalently bind to DNA bases and cause replication-blocking DNA lesions, triggering the DNA damage response that leads to cancer cell death. Often, tumors initially respond well to genotoxic therapy, but over time acquire resistance to drugs.[6–11] This resistance manifests through various mechanisms such as drug avoidance by overexpression of drug efflux pumps, upregulation of DNA damage repair pathways, or the use of DNA damage tolerance (DDT) pathways that enable replication of damaged DNA.
Translesion synthesis (TLS) is an error-prone DDT pathway implicated in resistance to platinating and alkylating agents.[9–13] In TLS, mutagenic DNA polymerases, Polη, Polι, Polκ, Rev1, and Polζ, are recruited to copy over DNA lesions that the replicative polymerases, Polδ and Polε, are unable to bypass.[14–17] TLS promotes drug resistance by different mechanisms. First, TLS allows replication over DNA adducts caused by genotoxic agents, thereby increasing cancer cell survival following chemotherapy. Thus, upregulation of the TLS pathway was reported in several drug resistant cancers. TLS polymerase Polζ is overexpressed in cisplatin resistant metastatic melanoma cells.[18] Resistance to platinum-based therapy in head and neck cancer correlated with high expression of Polη.[19] Second, error-prone TLS accelerates mutagenesis in cancer cells contributing to tumor heterogeneity and the onset of deleterious phenotypes such as metastasis and chemoresistance.[7–8] Therefore, TLS inhibitors that sensitize cancers to first-line chemotherapy and avert chemoresistance have emerged as a new class of potential anti-cancer drugs.[9–11]
TLS of DNA adducts caused by platinating and alkylating agents is a two-step process whereby an ‘inserter’ TLS polymerase, Polη, Polκ, or Polι, inserts nucleotides across the site of DNA damage, and then the ‘extender’ polymerase Polζ, copies past the DNA lesion.[14–17,20] The ‘inserter’ and ‘extender’ polymerases are assembled into a multiprotein TLS complex with the aid of the ubiquitinated sliding clamp PCNA and the TLS polymerase Rev1, which play scaffolding roles in TLS by mediating protein-protein interactions (PPIs).[17,21–22] For example, the Rev1 C-terminal domain (Rev1-CT) binds Rev1-intracting regions (RIRs) of Polη, Polι, and Polκ,[23–25] and Polζ’s regulatory subunit Rev7[26] via two independent binding interfaces[27] (Figure 1A). Interestingly, the second interface also interacts with the RIR motif of Polζ’s accessory subunit, PolD3, and thereby is implicated in ‘inserter’ to ‘extender’ polymerase switching during TLS.[28]
Figure 1:

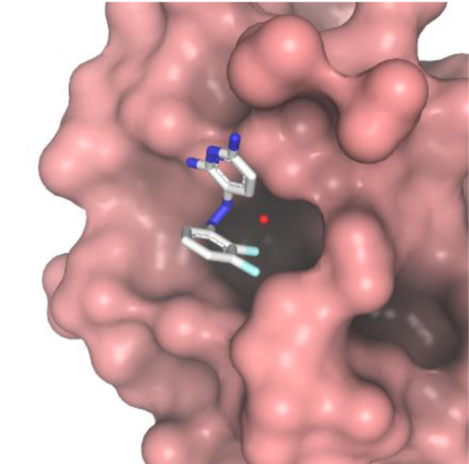
A) The FF residues of Polη-RIR bind a hydrophobic pocket on Rev1-CT. F532 forms T-shaped π-π interactions with W1175, while the backbone amides of F532 and F531 form charge-dipole interactions with D1186.[24] B) Surface representation of docked Rev1-CT complex with PAP analogue 1 (carbon is white, nitrogen is blue, and fluorine is cyan.[31]
One strategy to inhibit TLS for chemotherapeutic sensitivity is to disrupt essential PPIs that control assembly of the multiprotein TLS complex with small molecules.[9,29–33] Our attempts to inhibit TLS focused on blocking PPIs of Rev1-CT with RIR motifs from Polη, Polι, Polκ, and PolD3. The RIR motif, defined as ‘nFFhhhh’ (where ‘n’ is N-capping residue, ‘h’ is helix-forming residue), forms an α-helix upon binding, which inserts side-chains of the two consecutive phenylalanine residues in the binding pocket on Rev1-CT (Figure 1A).[23–25] The majority of contacts between Rev1-CT and RIR are mediated by the FF pair and mutation of these residues completely abrogates the PPI.[23–25] We have used biochemical high-throughput screening,[29–30] virtual screening,[32] and rational design approaches[31] to identify small molecules that disrupt Rev1-CT/RIR PPIs. The identified scaffolds demonstrated concentration-dependent displacement of FAM-Polκ-RIR peptide from its complex with Rev1-CT in fluorescence polarization and/or intensity (FP/FI) assays, as well as direct binding to the RIR-site on Rev1-CT in NMR titration experiments. Cellular assays revealed the compounds increase cancer cell sensitivity to cisplatin and reduce cisplatin-induced mutagenesis, making them candidates for further development as anti-cancer agents.
In our previous study, phenazopyridine (PAP) analogues with two aromatic rings that mimic FF residues of the RIR motif were rationally designed to bind and block the RIR-site on Rev1-CT.[31] The PAP and related azobenzene derivatives can adopt cis- and trans configurations and are susceptible to photoisomerization.[34] Equilibration of cis- and trans-isomers for azobenzenes in polar aqueous environment is a relatively fast process occurring on the time-scale of seconds or faster.[35] Therefore, for the purpose of our studies, PAP analogues dissolved in water may be considered as an equilibrium mixture of isomers, while their configuration in complex with Rev1-CT is primarily governed by protein-ligand interactions. Computational modeling predicted that the PAP scaffold binds to Rev1-CT in trans-configuration, in which the phenyl ring inserts in the hydrophobic pocket and forms π-π interactions with W1175 (Figure 1B).[31] The amino groups of the pyridine ring were modelled to interact with D1186 and E1174 via water bridges. NMR titration experiments validated the PAP compounds bind to the RIR interface of Rev1-CT. Furthermore, 2,3-fluorination of the PAP benzene ring, which is predicted to insert in the hydrophobic pocket on Rev1-CT, improved the FAM-Polκ-RIR displacement in the FI assay, providing initial structure activity relationships (SAR) for this scaffold.
Here, we aimed to further explore SAR for the PAP analogues to optimize their Rev1-CT binding properties, which is a necessary step in their development as anti-cancer agents. While docking models of Rev1-CT with the compounds can provide useful insights into their potential binding poses, rigorous optimization of the analogues requires high-resolution experimental structures that reveal precise intermolecular contacts and binding-induced ligand configuration. Therefore, we developed a crystallization system for Rev1-CT with small molecule compounds that target its RIR-binding site, and solved an X-ray co-crystal structure of Rev1-CT bound to the most potent 2,3-diflurobenzene substituted PAP analogue 1.[31] The structure revealed an unexpected cis-configuration of 1 in complex with Rev1-CT, and guided design of the second-generation PAP derivatives with binding affinity for Rev1-CT improved by an order of magnitude.
Results and Discussion
Crystallization system for Rev1-CT with small molecules
Previously, we determined solution NMR structures of human Rev1-CT alone and in complexes with Polη and PolD3 RIR motifs.[24] NMR structure of the mouse Rev1-CT/Polκ-RIR complex was also reported.[25] These structures revealed the mechanism of Rev1-CT/RIR recognition whereby the RIR motif folds into an α-helix that binds to an interface formed by the N-terminal β-hairpin (βHP) and the first two helices of the Rev1-CT four-helix bundle (Figure 1A). The side-chains of FF residues in the first turn of the RIR α-helix bind to a pre-formed pocket in the N-terminal part of Rev1-CT, while their backbone amides interact with negatively charged D1186 (Figure 1A).[24] Our 15N NMR relaxation dispersion study revealed that both apo and RIR-bound Rev1-CT exhibit μs-ms conformational exchange at temperatures above 15 °C. [24] This conformational plasticity is likely the reason why crystallization protocols for apo Rev1-CT or its binary complexes with RIR motifs have never been reported.
Rev1-CT has a second interface, which mediates the PPI with Polζ’s accessory subunit Rev7, comprising of the loop between α-helices H2 and H3 and the C-terminal tail following the helix H4 (Figure 1A).[26–27] In turn, Rev7 – a HORMA domain protein[36] – interacts with the two Rev7-binding motifs (RBMs) of Polζ’s catalytic subunit Rev3 through the mechanism whereby Rev7’s “safety-belt” loop closes on the top of Rev3-RBM, creating a binding interface for Rev1-CT.[37–38] X-ray crystal structure was reported for a triple Rev1-CT/Rev7R124A/Rev3-RBM1 complex, in which the RIR-binding site in the Rev1-CT N-terminus is open and accessible for interactions.[26] Furthermore, a crystal structure was solved for a quadruple Rev1-CT/Polκ-RIR/Rev7R124A/Rev3-RBM1 complex,[39] suggesting that, when bound to Rev7, Rev1-CT can be co-crystallized with ligands that target its RIR-binding site.
Here, we decided to pursue a strategy in which small molecules that bind to the RIR-site are co-crystallized with Rev1-CT in the context of the triple Rev1-CT/Rev7R124A/Rev3-RBM1 complex.[26] We successfully reproduced the crystallization of this complex in the P3121 space group and determined its crystal structure at 2.2 Å resolution, thereby establishing a system for crystal soaking or co-crystallization of ligands with Rev1-CT (Table 1; Figure 2A; PDB 6WS0). Our structure is in agreement with the published structure of the triple complex (PDB 3VU7)[26] with all-atom RMSD of 0.45 Å for Rev7/Rev3-RBM1 and 0.35 Å for Rev1-CT.
Table 1:
Data collection and refinement statistics for apo and inhibitor 1 bound hetero-trimeric Rev1-CT/Rev7R124A/Rev3-RBM1 complex structures.
| Apo | Inhibitor bound | ||
|---|---|---|---|
| Data Collection | Wavelength (A) | 0.98 | 0.92 |
| Space group | P3121 | P212121 | |
| Cell Size (Å) | a 70 | a 50 | |
| Cell Angle | α 90° | α 90° | |
| Resolution | 29.0 – 2.2 | 29.5 – 2.4 | |
| Observed reflections | 133024 | 74886 | |
| Unique reflections | 14749 | 11712 | |
| Rmeas | 0.107 | 0.073 | |
| Completeness (%) | 99.4 | 99.5 | |
| I/sigma(I) | 12.9 | 13.7 | |
| Refinement Statistics | Refined reflections | 14716 | 11673 |
| Free reflections | 771 | 620 | |
| R | 0.20 | 0.20 | |
| Rfree | 0.23 | 0.24 | |
| RMSD bond length | 0.010 | 0.014 | |
| RMSD bond angle | 1.8 | 1.9 |
Figure 2:

Co-crystallization of the Rev1-CT/Rev7R124A/Rev3-RBM1 complex bound to 1. A) Crystal structures of the apo (light colors) and inhibitor bound (dark colors) triple complexes. B) Close up views of intermolecular interactions between 1 and Rev1-CT. The water molecule is shown as red sphere.
X-ray crystal structure of Rev1-CT bound to PAP
Recently, we used the rational design approach to identify the PAP scaffold as Rev1-CT/RIR inhibitor, which mimics the FF residue pair found in all RIR motifs responsible for the majority of binding free energy (Figure 1B).[31] To further explore the SAR for this scaffold and optimize its binding affinity for Rev1-CT, the structural characterization of this binding interaction was required. Therefore, our objective was to obtain an X-ray co-crystal structure of Rev1-CT bound to the most potent PAP analogue 1 identified in our previous study (Figure 1B).[31]
To generate a crystal structure of the Rev1-CT/1 complex, we initially tried crystal soaking, which either dissolved the crystals or adulterated the diffraction, or resulted in structures of the Rev1-CT/Rev7R124A/Rev3-RBM1 complex with no inhibitor bound. Therefore, co-crystallization was attempted. By mixing 1 with the triple complex at an 8:1 ratio, co-crystals were obtained in the dark that belonged to the P212121 space group and diffracted at 2.4 Å (Table 1). Despite the change in space group, the structure of the triple complex bound to 1 is in agreement with the structure of the apo-complex (Figure 2A, Figure S1; PDB 6WS5) with all-atom RMSD of 0.44 Å for Rev7/Rev3-RBM1 and 0.25 Å for Rev1-CT.
As expected, PAP analogue 1 binds and blocks the RIR-site in the N-terminal part of Rev1-CT, with the compound binding resulting in minimal structural changes in the domain (Figure 2A). However, the observed binding mechanism differs from that predicted by our computational modelling,[31] which suggested 1 would bind Rev1-CT in trans-configuration with the phenyl ring entering the binding pocket on Rev1-CT and forming stacking π-π interactions with W1175 (Figure 1B). This is not observed in the crystal structure (Figure 2B). Instead, binding to Rev1-CT favors cis-configuration of 1, which is further stabilized by intramolecular π-π interactions between the PAP phenyl and pyridine rings (3.5 Å distance). The phenyl ring of the PAP scaffold does not enter the hydrophobic pocket, nor does it participate in π-π interactions with W1175 (Figure 2B). Instead, fluorines of the 2,3-diflurobenzene ring of 1 form Van der Waals contacts with the methyl group of A1160 flanking the binding pocket. The pocket itself is filled with a water molecule, which loosely interacts with the side-chain of D1186. The pyridine ring of 1 forms T-shaped π-π interactions with W1175 (6.8 and 8.2 Å distances between the pyridine and the two indole rings that are longer than optimal, but are within the range expected for this type of interactions at protein-ligand interfaces[40]). The two amino groups attached to the pyridine ring form weak hydrogen bonds with the side-chains of M1183 and D1186.
Overall, the structure of Rev1-CT bound to the PAP compound 1 (Figure 2) revealed an unexpected binding mechanism whereby the inhibitor obstructs the hydrophobic pocket on Rev1-CT that accommodates the conserved FF residues of the RIR motif rather than penetrating and binding inside the pocket. While our previous modeling suggested the PAP phenyl ring can insert in the hydrophobic pocket if the compound is bound in trans-configuration,[31] the obtained crystal structure has shown that 1 in complex with Rev1-CT (i) is locked in cis-configuration by intramolecular π-π interactions between its phenyl and pyridine rings, and/or (ii) is unable to adopt trans-configuration presumably due to suboptimal interactions of its phenyl moiety with residues that form the hydrophobic pocket on Rev1-CT.
Structure-based design of improved PAP derivatives
Informed by the binding mechanism observed in the crystal structure of the Rev1-CT/1 complex, we aimed to investigate SAR for the PAP scaffold to improve its Rev1-CT binding affinity. Our first structure-based assumption was that the intramolecular π-π interaction between the phenyl and pyridine rings locks the PAP scaffold in cis-configuration in complex with the Rev1-CT domain and prevents its phenyl ring entering the binding pocket on Rev1-CT. Therefore, we explored ring substitutions of the PAP scaffold that may favor its trans-configuration when bound to Rev1-CT by reducing intramolecular π-π stacking. Previous studies of the effect of heteroatoms on aromatic π-π interactions showed that the benzene-pyridine pair binds stronger than a benzene dimer in parallel-displaced configuration similar to that observed for 1 in complex with Rev1-CT.[41] This led us to investigate the effect a pyridine to phenyl substitution in the PAP scaffold would have on its binding affinity to Rev1-CT. Our second assumption was that modification of the PAP phenyl with bulky hydrophobic groups could improve its fit in the binding pocket and favor trans-configuration of the PAP scaffold when bound to Rev1-CT. Therefore, we also studied the effects of phenyl methylation and phenyl-naphthalene substitutions on Rev1-CT binding affinities of PAP derivatives. Following this strategy, a total of 8 second-generation PAP analogues with changes to both aromatic ring systems were designed and synthesized through standard diazotization coupling procedures (Scheme 1; Table 2).[31]
Scheme 1.

Synthesis of PAP analogues:
i. 1 M HCl (compounds 1–6) or ii. CHCl3, ~5 drops conc. HCl (compounds 7–9), 1.25 eq NaNO2 dissolved in H2O, on ice 20 min
iii. 2,6-diaminopyridine or m-phenylenediamine dissolved in H2O, pH adjusted with sodium acetate, room temperature for 1 hour.
Table 2:
Aromatic ring modifications of the PAP scaffold and MST-derived binding affinities between Rev1-CT and PAP analogues.
| Compound | R1 | R2 | Kd (μM)[a] |
|---|---|---|---|
| 1 | 2,3-difluorobenzene | N | 21 ± 1 μM |
| 2 | 2,3-difluorobenzene | C | 18 ± 4 μM |
| 3 | 2,3-dimethylbenzene | N | 32 ± 20 μM |
| 4 | 2,3-dimethylbenzene | C | 2.9 ± 0.4 μM |
| 5 | 3,4-dimethylbenzene | N | 17± 1 μM |
| 6 | 3,4-dimethylbenzene | C | 3.9 ± 0.2 μM |
| 7 | 3,4-naphthalene | N | 1.4 ± 0.1 μM |
| 8 | 3,4-naphthalene | C | 1.5 ± 0.2 μM |
| 9 | 2,3-naphthalene | N | 1.4 ± 0.1 μM |
Kd values and errors were calculated from three repeat experiments. Binding affinities that improved over 5-fold relative to 1 are boldened.
Binding studies of Rev1-CT with PAP derivatives
To probe binding between Rev1-CT and synthesized analogues (1–9) we utilized the microscale thermophoresis (MST) technique, which allows measurement of a dissociation constant (Kd) for a complex of fluorescently-labeled protein with the unlabeled ligand from the temperature-induced changes in protein fluorescence obtained at different concentrations of added ligand.[42] For each of the analogues (1–9), a series of 16 dilutions (500 uM to 10 nM concentrations) and 20 nM fluorescently labelled Rev1-CT were incubated and loaded into standard capillaries. The MST traces were collected and analyzed to extract binding affinities using the NanoTemper Monolith MO Affinity analysis software (Figure 3, Figure S2, Table 2).
Figure 3:

MST binding data for compounds 1 (A) and 9 (B), both graphs are a representative curve of 3 separate experiments.
The MST-derived disassociation constants (Kd) revealed two groups of PAP analogues with binding affinities for Rev1-CT that differ by an order of magnitude (Table 2). The weaker binding compounds 1–3 and 5 have Kd values for Rev1-CT of 17–32 μM. The stronger binders 4 and 6–9 have Kd values for Rev1-CT of 1.4–3.9 μM, which is comparable to the affinity of the tightest RIR-mediated PPI between Rev1 and Polκ (1.7 μM obtained by SPR[28]). 2,3-fluorobenzene substituted PAP analogues 1 and 2 have similar weaker binding affinities for Rev1-CT (21 and 18 μM), which indicates the pyridine to benzene substitution in 2 that weaken intramolecular π-π interaction[41] is likely insufficient to flip the PAP scaffold in complex with Rev1-CT from cis- to trans-configuration that would ensure the phenyl ring enters the binding pocket on Rev1-CT. The most interesting group of compounds include 2,3- and 3,4-dimethylbenzene substituted PAP analogues 3–6, in which the phenyl ring is modified with the two hydrophobic methyl groups. These substitutions are expected to strengthen interaction of the PAP phenyl ring with the hydrophobic pocket on Rev1-CT. The dimethylbenzene substituted compounds 3 and 5 displayed weaker binding affinities for Rev1-CT (32 and 17 μM), indicating these substitutions alone may be insufficient to favor trans-configuration of the PAP scaffold. In contrast, a combination of the phenyl methylation and pyridine to phenyl substitution in 4 and 6 resulted in an order of magnitude improvement of their affinity for Rev1-CT (2.9 and 3.9 μM) consistent with these compounds binding to Rev1-CT in trans-configuration. The PAP phenyl ring substitution with the bulky naphthalene moiety is expected to further strengthen interaction with the hydrophobic pocket on Rev1-CT. Both pyridine and pyridine to phenyl substituted analogues 7–9 exhibited Kd of 1.4–1.5 μM, suggesting they likely bind to Rev1-CT in trans-configuration irrespective of pyridine to phenyl substitution.
Overall, the structural information about Rev1-CT/PAP interaction obtained by X-ray crystallography and MST binding data may indicate that affinity of the Rev1-CT/PAP interaction depends on whether the PAP compounds bind Rev1-CT in cis- or trans-configurations, which in turn is determined by a balance between intramolecular aromatic π-π interactions in the PAP scaffold (that favor cis-configuration) and interaction of the PAP phenyl moiety (or its substitute) with the hydrophobic pocket in the N-terminal part of Rev1-CT (that favors trans-configuration). Fine-tuning of these contributions led us to design second-generation PAP derivatives 7–9 with binding affinity for Rev1-CT one order of magnitude stronger than that of the original compound 1.[31]
Conclusions
TLS inhibition has recently emerged as a strategy to increase chemotherapeutic sensitivity and avert resistance associated with first-line platinating and alkylating drugs.[9–11] Rev1’s scaffolding function is essential for TLS[17,21–22] and its inhibition provides a promising avenue for maintaining genotoxic chemotherapy sensitivity in cancer cells.[9,29–33] The RIR interface of Rev1-CT is an important binding site for the ‘inserter’ TLS polymerases Polι, Polκ, Polη, and the ‘extender’ polymerase Polζ (via its accessory subunit, PolD3).[23–25,28] Disruption of the Rev1-CT PPIs with RIR motifs is expected to impair assembly of the multi-protein TLS complex and polymerase switching, limiting the TLS pathway’s ability to bypass DNA damage induced by chemotherapeutics and introduce further mutations to cancer cells. Previously, we identified several small-molecule scaffolds that inhibit essential Rev1-CT/RIR PPIs.[29–32] To guide structure-based design of the improved analogues, here we developed a co-crystallization protocol for small molecules with the RIR-site on Rev1-CT in the context of the triple Rev1-CT/Rev7R124A/Rev3-RBM1 complex. This protocol was employed to solve the X-ray crystal structure of Rev1-CT with the most potent initial PAP scaffold compound 1.[31] The structure revealed an unexpected binding mechanism and provided critical SAR that led us to design next generation PAP analogues 6–9 with Rev1-CT binding affinity improved 15-fold. These optimized lead compounds will be evaluated for their anti-TLS an anti-cancer activity in our future cellular and in vivo studies.
Experimental Section
Protein expression and purification
E. coli BL21 cells were transformed with a pETDuet-1 co-expression plasmid encoding Rev7R124A and a Rev3-RBM1 peptide (residues 1847–1898), [37] or a Pet28b+ plasmid encoding human Rev1-CT (residues 1158–1251).[24] Cells were grown at 37 °C, and protein expression was induced at OD of 0.8–1.0 with 0.1 mM isopropyl 1-thio-b-D-galctopyranoside (IPTG) at 20 °C overnight. Cells were lysed in 50 mM Na phosphate, 300 mM NaCl, and 10 mM imidazole. Protein was purified by Co2+ affinity chromatography followed by size-exclusion chromatography on a Superdex 75 column (GE LifeSciences) in 20 mM Tris, 100 mM NaCl, 2 mM DTT, 1 mM EDTA, pH 7.4. Rev1-CT His-tag was removed by incubation with TEV protease at room temperature. Following cleavage, His-tag was separated from protein by Co2+ affinity chromatography. Rev1-CT was further purified by size-exclusion chromatography a second time. To assemble the triple Rev1-CT/Rev7R124A/Rev3-RBM1 complex, the purified Rev7R124A/Rev3-RBM1 complex was mixed with Rev1-CT at a 1:1.1 ratio, and the complex was co-eluted on a Superdex 75 column in 5 mM HEPES, 100 mM NaCl, 2 mM TCEP, pH 7.4.
Protein crystallization and X-ray diffraction data analysis
Crystals of the triple complex were obtained by mixing 230 mM of protein with a crystallization buffer of 162 mM triammonium citrate and 18% w/v PEG 3350 at pH of 7, as was done previously.[26] Protein was crystallized at 20 °C using the hanging drop vapor diffusion method. For co-crystallization, PAP inhibitors were mixed with protein at an 8:1 molar ratio. To avoid photoisomerization, co-crystallization was performed in the dark. Data were collected at the AMX beamline at the Brookhaven National laboratory. Data sets were processed by DIALS software and reduced by Aimless; molecular replacement was done by MolRep with PDB 3VU7 as a reference structure, and the structure was refined by iterative model building with Refmac and Coot (as part of the CCP4 software package).[43] Atomic coordinates of the triple Rev1-CT/Rev7R124A/Rev3-RBM1 and quadruple Rev1-CT/Rev7R124A/Rev3-RBM1/1 complexes were deposited to Protein Data Bank (PDB) with the IDs 6WS0 and 6WS5.
Synthesis of PAP derivatives
PAP derivatives (1–9) were synthesized through standard diazotization coupling procedures (Scheme 1; Table 2).[31] The monocyclic anilines were dissolved in 1 M HCl (step i in Scheme 1; compounds 1–6) and the bicyclic anilines were dissolved in chloroform (step ii in Scheme 1; compounds 7–9); then ~5 drops of 12 M HCl was added to the solution. 1.25 equivalents of sodium nitrate dissolved in water was added dropwise to the aniline solution and this mixture was stirred for 20 minutes on ice. This crude reaction was added to a mixture of either 2,6-diaminopyridine or m-phenylenediamine dissolved in water and brought to pH of 5 with sodium acetate, this mixture was stirred at room temperature for 1 hour (Scheme 1). To work up the reaction, cold water was added to the reaction mixture and the precipitate was filtered and collected. If no solid crashes out, the product can be extracted in chloroform or ethyl acetate and water. Identities of the resulting compounds were confirmed by 1H and 13C NMR (Figure S3).
Microscale Thermophoresis
The MST experiments were performed on a NanoTemper Monolith instrument using the NanoTemper RED-NHS kit, which fluorescently labels protein lysine residues. Rev1-CT dissolved in the MST buffer (20 mM HEPES, 100 mM NaCl, 2 mM TCEP, pH 8.2) was labelled and purified according to the labelling kit protocol. The labelled protein concentration and degree of labelling were calculated from A205, A650, and A280 absorbance using equations in the labelling kit manual. All experiments were performed in the Expert Mode of MO.Control software. Stock concentrations of the synthesized compounds (1–9) were prepared in 100% DMSO (10 mM to 200 nM). The dilution set contained 19 μL of 20 nM labelled Rev1-CT (diluted in MST buffer with 0.005% Tween 20) and 1 μL of the compound stocks (final concentration 500 μM to 10 nM). The dilution set was incubated and then 10 μL loaded into standard capillaries. MO Affinity Analysis software was used to analyze the MST traces to calculate the binding affinities. Note that cis-trans isomerization of the PAP derivatives in aqueous solution is expected to be fast on the time-scale of our binding measurements,[35] which evaluated the equilibrium mixture of cis- and trans-isomers.
Supplementary Material
Acknowledgements
This work was supported by NIH R01CA233959 and CT Innovations PITCH2 grants to M.K.H and D.M.K.
References
- [1].Siddik ZH, Oncogene 2002, 22, 7265–7279. [DOI] [PubMed] [Google Scholar]
- [2].Kelland L, Nat. Rev. Cancer 2007, 7, 573–584. [DOI] [PubMed] [Google Scholar]
- [3].Wang D, Lippard SJ, Nat. Rev. Drug Discov 2005, 4, 307. [DOI] [PubMed] [Google Scholar]
- [4].Dasari S, Tchounwou PB, Eur. J. Pharmacol 2014, 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lajous H, Lelievre B, Vauleon E, Lecomte P, Garcion E, Trends. Pharmacol. Sci 2019, 40, 342–357. [DOI] [PubMed] [Google Scholar]
- [6].Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G, Oncogene 2012, 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- [7].Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, Cancer Res 2005, 65, 11704–11711. [DOI] [PubMed] [Google Scholar]
- [8].Xie K, Doles J, Hemann MT, Walker GC, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20792–20797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Korzhnev DM, Hadden MK, J. Med. Chem 2016, 59, 9321–9336. [DOI] [PubMed] [Google Scholar]
- [10].Yamanaka K, Chatterjee N, Hemann MT, Walker GC, PLOS Genetics 2017, 13, e1006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zafar MK, Eoff RL, Chem. Res. Toxicol 2017, 30, 1942–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lange SS, Takata K-I, Wood RD, Nat. Rev. Cancer 2011, 11, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Knobel PA, Marti TM, Cancer Cell Int. 2011, 11, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sale JE, Lehmann AR, Woodgate R, Nat. Rev. Mol. Cell Biol 2012, 13, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vaisman A, Woodgate R, Crit. Rev. Biochem. Mol. Biol 2017, 52, 274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang W, Gao Y, Annu. Rev. Biochem 2018, 87, 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rizzo AA, Korzhnev DM, The Enzymes 2019, 45, 139–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Song L, McNeil EM, Ritchie AM, Astell KR, Gourley C, Melton DW, BMC cancer 2017, 17, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou W, Chen YW, Liu X, Chu P, Loria S, Wang Y, Yen Y, Chou KM, PloS one 2013, 8, e83978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Livneh Z, Shachar S, Cell Cycle 2010, 9, 729–735. [DOI] [PubMed] [Google Scholar]
- [21].Nelson JR, Gibbs PE, Nowicka AM, Hinkle DC, Lawrence CW, Mol. Microbiol 2000, 37, 549–554. [DOI] [PubMed] [Google Scholar]
- [22].Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I, Science 2005, 310, 1821–1824. [DOI] [PubMed] [Google Scholar]
- [23].Ohashi E, Hanafusa T, Kamei K, Song I, Tomida J, Hashimoto H, Vaziri C, Ohmori H, Genes to Cells 2009, 14, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pozhidaeva A, Pustovalova Y, D’Souza S, Bezsonova I, Walker GC, Korzhnev DM, Biochemistry 2012, 51, 5506–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wojtaszek J, Liu J, D’Souza S, Wang S, Xue Y, Walker GC, Zhou P, J. Biol. Chem 2012, 287, 26400–26408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kikuchi S, Hara K, Shimizu T, Sato M, Hashimoto H, J. Biol. Chem 2012, 287, 33847–33852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pustovalova Y, Bezsonova I, Korzhnev DM, FEBS Lett. 2012, 586, 3051–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pustovalova Y, Magalhães MT, D’Souza S, Rizzo AA, Korza G, Walker GC, Korzhnev DM, Biochemistry 2016, 55, 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sail V, Rizzo AA, Chatterjee N, Dash RC, Ozen Z, Walker GC, Korzhnev DM, Hadden MK, ACS Chem. Biol 2017, 12, 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ozen Z, Dash RC, McCarthy KR, Chow SA, Rizzo AA, Korzhnev DM, Hadden MK, Bioorg. Med. Chem 2018, 26, 4301–4309. [DOI] [PubMed] [Google Scholar]
- [31].Dash RC, Ozen Z, Rizzo AA, Lim S, Korzhnev DM, Hadden MK, J. Chem. Inf. Model 2018, 58, 2266–2277. [DOI] [PubMed] [Google Scholar]
- [32].Dash RC, Ozen Z, McCarthy KR, Chatterjee N, Harris CA, Rizzo AA, Walker GC, Korzhnev DM, Hadden MK, ChemMedChem 2019, 14, 1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, Hemann MT, Hong J, Walker GC, Zhou P, Cell 2019, 178, 152–159.e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Willner I, Rubin S, Angew. Chem. Int. Ed. Engl 1996, 35, 367–385. [Google Scholar]
- [35].Dunn NJ, Humphries WH, Offenbacher AR, King TL, Gray JA, J. Phys. Chem. A 2009, 113, 13144–13151. [DOI] [PubMed] [Google Scholar]
- [36].Aravind L, Koonin EV, Trends Biochem. Sci 1998, 23, 284–286. [DOI] [PubMed] [Google Scholar]
- [37].Hara K, Hashimoto H, Murakumo Y, Kobayashi S, Kogame T, Unzai S, Akashi S, Takeda S, Shimizu T, Sato M, J. Biol. Chem 2010, 285, 12299–12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rizzo AA, Vassel F-M, Chatterjee N, D’Souza S, Li Y, Hao B, Hemann MT, Walker GC, Korzhnev DM, Proc. Natl. Acad. Sci. U. S. A 2018, 115, E8191–E8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xie W, Yang X, Xu M, Jiang T, Protein Cell 2012, 3, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brylinski M, Chem. Biol. Drug. Des 2018, 91, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hohenstein EG, Sherrill CD, J. Phys. Chem. A 2009, 113, 878–886. [DOI] [PubMed] [Google Scholar]
- [42].Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S, Nat. Commun 2010, 1, 100. [DOI] [PubMed] [Google Scholar]
- [43].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, Acta Cryst. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.