

Abstract
Glioblastoma (GBM) is the most devastating brain cancer, and cures remain elusive with currently available neurosurgical, pharmacological and radiation approaches. While retrovirus- and adenovirus-mediated suicide gene therapy using DNA encoding herpes simplex virus-thymidine kinase (HSV-tk) and prodrug ganciclovir has been suggested as a promising strategy, a non-viral approach for treatment in an orthotopic human primary brain tumor model has not previously been demonstrated. Delivery challenges include nanoparticle penetration through brain tumors, efficient cancer cell uptake, endosomal escape to the cytosol, and biodegradability. To meet these challenges, we synthesized poly(ethylene glycol)-modified poly(beta-amino ester) (PEG-PBAE) polymers to improve extracellular delivery and co-encapsulated plasmid DNA with end-modified poly(beta-amino ester) (ePBAE) polymers to improve intracellular delivery as well. We created and evaluated a library of PEG-PBAE/ePBAE NPs for effective gene therapy against two independent primary human stem-like brain tumor initiating cells, a putative target to prevent GBM recurrence. The optimally engineered PEG-PBAE/ePBAE NP formulation demonstrated 54% and 82% transfection efficacies in GBM1A and BTIC375 cells respectively, in comparison to 37% and 66% for optimized PBAE NPs without PEG. The leading PEG-PBAE NP formulation also maintained sub-250 nm particle size up to 5 h, while PBAE NPs without PEG showed aggregation over time to micron-sized complexes. The comparative advantage demonstrated in vitro successfully translated into improved in vivo diffusion, with a higher amount of PEG-PBAE NPs penetrating to a distance of 2 mm from the injection site. A significant increase in median survival from 53.5 days to 67 days by PEG-PBAE/pHSV-tk NP and systemic ganciclovir treatment compared to a control group in orthotopic murine model of human glioblastoma demonstrates the potential of PEG-PBAE-based NPs as an effective gene therapy platform for the treatment of human brain tumors.
Keywords: Glioblastoma, Suicide gene therapy, Gene delivery, Poly(beta-amino ester), Poly(ethylene glycol), Nanoparticles
1. Introduction
Glioblastoma (GBM) is the most common form of primary brain tumors in adults that accounts for more than 10,000 deaths in the United States despite tumor resection, chemotherapy, and radiation1, 2. GBM still has one of the highest fatality rates among cancers due to its high recurrence and treatment resistance3–5. It has been recently reported that the GBM tumor mass consists of both fully differentiated cancer cells and less differentiated brain tumor initiating cells (BTIC)6, 7. These stem-like BTICs that are insensitive to chemotherapy and radiation therapy, differentiate into cancer cells, and ultimately lead to tumor recurrence8. Novel therapies evaluated in an in vivo model that accurately presents this challenge posed by BTICs in human GBM tissue are needed to improve the poor prognosis of GBM patients.
Gene therapy using DNA, mRNA, and RNAi has introduced many novel approaches to treating diseases of genetic origin, including cancer. Specifically, tumor-suppressing proteins encoded by exogenous DNA and mRNA, such as TNF-related apoptosis-inducing ligand (TRAIL) and p53, can be expressed by tumor cells to prevent tumor growth9, 10. Suicide gene therapy is another DNA-based approach that delivers plasmid DNA to tumor cells to express proteins, such as herpes simplex virus thymidine kinase (pHSV-tk), which then activates small prodrug molecules in situ to induce apoptosis11. Because the prodrug molecule is non-toxic until activated, therapy is localized to the site of gene transfection and systemic side effects are minimized. Furthermore, suicide gene therapy has been shown to be more effective in chemotherapy-resistant tumors and synergize with radiotherapy12, 13.
One of the major components of a successful gene therapy is a safe and efficient gene delivery vector. A number of non-viral vectors, including lipids, polymers, and inorganic materials, have been explored as gene carriers in the preclinical stage based on their biocompatibility, cargo capacity, and tunability14, 15 and tested in clinical trials16. In particular, poly(beta-amino ester)s (PBAEs), a class of biodegradable cationic polymers, can form nanoparticles (NP) by electrostatic interaction with nucleic acids, including DNA1, 17, miRNA18, 19, and siRNA20, 21, and successfully deliver them to various types of cells with minimal toxicity22–24. A library of PBAEs with diverse physicochemical properties can be synthesized from a wide selection of monomers, which allows for a high-throughput screening process to identify the most optimal system against barriers to efficient gene transfection. For example, at the cellular level, PBAEs of specific structures have been correlated with cell-specific transfection1, 17, 20, 22, 24, 25 as well as steps leading up to successful transfection, including differential efficiency in cell uptake, endosomal escape, and genetic cargo release26–28. Furthermore, chemical modifications of PBAE polymers or resulting NPs, including conjugation of poly(ethylene glycol) (PEG) or surface modification with polyelectrolytes, have altered NP properties, such as particle stability and geometry, that has led to enhanced pharmacokinetics and specific tissue targeting22, 29, 30. Specifically, increased stability of NPs is important in tumor treatment as it can enable greater diffusion and transfection in larger areas of dense tumor tissue.
Our group has recently validated PBAE polymers as potent non-viral gene delivery vectors for suicide gene therapy using pHSV-tk and ganciclovir in a rat glioma model31. While this approach demonstrated promise, it also highlighted the need for an improved therapeutic nanobiotechnology that can promote tumor penetration and stability, efficiently deliver the DNA cargo to stem-like BTICs, and extend survival in an orthotopic model with human patient-derived GBM tumors. In this study, we utilize an orthotopic xenograft model in mice with primary human stem-like GBM cells from a patient to validate poly(ethylene glycol)-modified PBAE (PEG-PBAE) and ePBAE-based NPs as safe and efficient vectors to deliver DNA in vivo. The optimal candidate ePBAE polymer, identified through transfection screening of BTICs with a combinatorial nanomaterial library, was blended with PEG-PBAE to formulate NPs designed to penetrate the tumor core and also diffuse beyond the tumor margin to a greater area of brain tissue for widespread transfection. Delivery of exogenous pHSV-tk plasmid DNA and the prodrug ganciclovir was used to evaluate the utility of the enhanced gene delivery nanoparticles.
2. Methods
2.1. Materials
1,4-butanediol diacrylate (B4), 4-amino-1-butanol (S4), 5-amino-1-pentanol (S5), 1-(3-aminopropyl)-4-methylpiperazine (E7) (Alfa Aesar), pentane-1,3-diamine (E3) (TCI America), 2-(3-aminopropylamino)ethanol (E6) (Fluka), poly(ethylene glycol) methyl ether thiol (800 Da and 2000 Da), (Sigma-Aldrich), α-mercaptoethyl-ω-methoxy polyoxyethylene (5000 Da) (NOF America Corporation), and cell culture media components were purchased and used as received. HSV-tk gene cloned into the pcDNA3.1 vector (Life Technologies), and pEGFP-N1 (EGFP) and pDsRed (DsRed) DNA (Elim Biopharmaceuticals) were amplified by Aldevron. Ganciclovir (Invivogen), Label IT-Tracker Cy5 kit (Mirus Bio LLC), and CellTiter 96 AQueous One MTS assay (Promega) were obtained from commercial vendors and used per manufacturer’s instructions.
2.2. Poly(β-amino ester) (PBAE) synthesis
PBAE polymers were synthesized in a two-step reaction using commercially-available molecules. Briefly, a base polymer was first synthesized via Michael addition of a diacrylate monomer and a primary-amine containing side-chain monomer at 1:1:1 molar ratios by stirring at 90 °C for 24 h in DMSO (Fig. 1A-D). The molecular weight and chemical structure of the base polymer was analyzed by Bruker Avance III 500 MHz NMR spectrometer in CDCl3 after purification with diethyl ether and 24 h in vacuum for drying. In the second step, base polymers were end-capped with either a small molecule (ePBAE) or a PEG molecule (PEG-PBAE). End-capping with a small molecule followed Michael addition reaction by dissolving base polymer and 30-fold molar excess amount of end-capping molecule in THF and stirring at room temperature for 3 h. To end-cap base PBAE polymer with a PEG-thiol molecule, 1-(3-aminopropyl)-4-methylpiperazine (E7) was added to initiate amine-catalyzed, thiol-ene Michael addition reaction. The base polymer, methoxy PEG-thiol, and E7 were mixed at 1:2.5:0.2 molar ratios in DMSO and stirred at room temperature for 24 h at 1000 rpm. Final ePBAE and PEG-PBAE polymers were purified in diethyl ether by allowing polymer to precipitate without centrifugation, dried under vacuum for 24 h, and stored with desiccant at −20 °C as 100 mg/mL solution in DMSO.
Figure 1. Polymer synthesis and screening strategy.
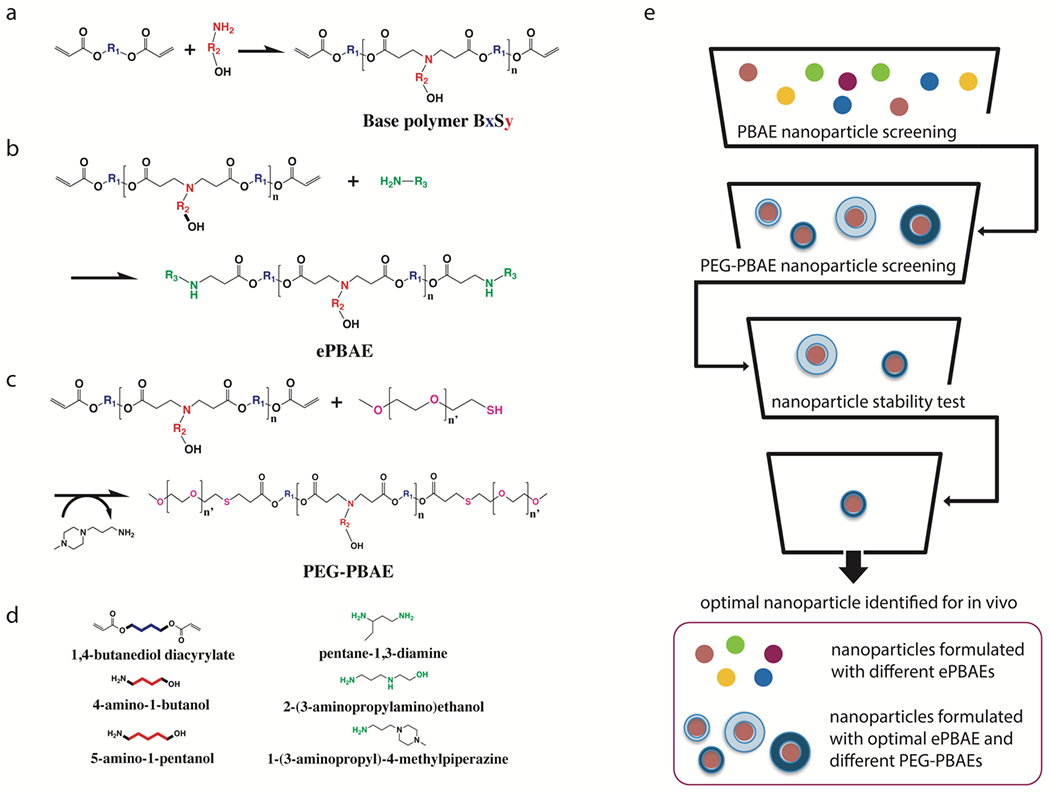
(a) Synthesis scheme of acrylate-terminated poly(β-amino ester)s (PBAE) base polymer, (b) conventionally end-capped ePBAEs, (c) poly(ethylene glycol)-co-poly(β-amino ester)s (PEG-PBAEs), and (d) monomers used in the synthesis of a library of PBAE-based polymers. (e) Workflow diagram of ePBAE and PEG-PBAE screening process for identification of the most optimal PEG-PBAE NP formulation.
2.3. PBAE nanoparticle formulation and characterization
Nanoparticles (NP) with a single type of ePBAE were formulated based on w/w weight ratios (mass ratios) between the PBAE polymer and DNA. Briefly, in order to make NP at 30 w/w ratio, polymer and DNA were diluted to 1.8 and 0.6 mg/mL, respectively, with 25 mM sodium acetate buffer (pH = 5). Equal volumes of polymer and DNA solutions was mixed together and incubated for 10 min for complexation.
NPs with a blend of ePBAE and PEG-PBAE polymers were made similarly at 30, 60, and 90 w/w ratios of total polymer to DNA. The total polymer used consisted of a mixture of ePBAE and PEG-PBAE at 1:2, 1:1, or 2:1 mass ratios. For example, in order to make NPs with ePBAE:PEG-PBAE 2:1 w/w and total polymer:DNA 90 w/w ratios, 50 μg of ePBAE and 25 μg of PEG-PBAE were diluted to 5.4 mg/mL with 25 mM sodium acetate buffer (pH = 5), and then mixed with diluted DNA solution at 0.06 mg/mL at equal volume. The effective mass ratios of ePBAE, PEG-PBAE, and DNA in all NPs formulated are listed in Table S1.
NP size, zeta potential, and stability over time were determined by dynamic light scattering (DLS) using Malvern Zetasizer Nano ZS (Malvern Instruments, detection angle 173°, 633 nm laser). NP was prepared at DNA concentration of 0.1 mg/mL and diluted into artificial cerebrospinal fluid (ACSF) for size measurement. To determine NP stability over time, NP was prepared and incubated at room temperature until size was measured at 2, 5, and 24 h time points. All reported measurements are intensity-weighted Z-average values that passed the quality control expert advice criteria. For zeta potential measurements, samples were prepared at DNA concentration of 0.03 mg/mL and diluted 2-fold into 25 mM sodium acetate buffer (pH = 5.0).
2.4. Cell culture
GBM1A and BTIC375 human primary brain tumor initiating cells, established and characterized by Vescovi32 and Goldman33 groups, respectively, were cultured at 37 °C and 5% CO2 in DMEM/ F-12 (1:1) (Cellgro 10-009-CV) with 1X B-27 supplement, 1% antibiotic-antimycotic (Invitrogen), 20 ng/mL basic fibroblast growth factor (bFGF), and 20 ng/mL epidermal growth factor (EGF)34, 35.
2.5. In vitro screening assays
2.5.1. Transfection
96-well tissue culture plates were incubated with laminin (5 μg/mL) for 2 h prior to cell seeding. GBM1A and BTIC375 cells were plated in laminin-coated 96-well plates at a density of 15,000 cells/well (100 μL/well) and incubated overnight before NP treatment. NPs were prepared as described above at 0.03 mg/mL DNA concentration, and 20 μL of NP solution was added to 100 μL of serum-containing medium in each well. Cells were incubated with NPs for 2 h, then washed twice with heparin-containing PBS and replenished with 100 μL fresh media.
2.5.2. Cell viability assay
Following NP treatment, cells were incubated for an additional 24 h at 37 °C until cell viability assay was performed. After the 24 h incubation, media in the wells was aspirated and 100 μL of fresh media with 20 μL of CellTiter 96 AqueousOne MTS reagent were added per well. Cells were then incubated with reagent at 37 °C, and absorbance was measured at 490 nm using a Synergy 2 plate reader (Biotek) after 2 h. Normalized absorbance signal to untreated control group is reported.
2.5.3. Flow cytometry
Following NP treatment, cells were incubated for 48 h until pEGFP transfection efficacy was determined by flow cytometry (Accuri C6 with HyperCyt high-throughput adaptor). To prepare for flow cytometry, cells were first detached from the wells using 30 μL of 0.05% trypsin and resuspended in 170 μL of 2% v/v FBS solution in PBS (FACS buffer) to neutralize trypsin. Propidium iodide (PI) (Invitrogen, Carlsbad, CA) was added to FACS buffer at 1:200 to gate out cells undergoing apoptosis. Cells were then transferred to a round-bottom 96-well plate, centrifuged for 5 min at 800 rpm at 4 °C, and resuspended in 30 μL. Using FlowJo 7.6.5 software, % of cells with EGFP expression was determined by first selecting PI- subpopulation in FSC-H vs. FL2, and then gating for EGFP+ in FL1 vs. FL2.
2.6. Delivery of pHSV-tk and ganciclovir
Cells were treated with NPs prepared using pHSV-tk for 2 h at 37°C, then incubated with 100 μL of fresh media for 24 h at 37°C. The media was then replaced with fresh media containing 0, 5, 10, 20, 50, 100, or 200 μg/mL of ganciclovir. Stock ganciclovir solution was prepared per manufacturer’s instructions. Media with ganciclovir was replaced every 2-3 days. Cell death was measured by staining cells with 1 μg/mL Hoechst 33342 dye, imaging the stained cells by fluorescence microscopy, and quantifying by image analysis.
2.7. Bystander effect
Dye transfer activity between GBM cells were determined using a gap junction permeable dye, Calcein AM (Invitrogen, C1430) following a previously established protocol36. Briefly, GBM1A cells were first labeled with 0.5 μM Calcein AM for 30 min. Then, Calcein AM-labelled GBM1A cells were co-cultured with mCherry protein-expressing GBM1A cells (GBM1A-mCherry) at 1:1 ratio for 24h. Following 24h, the transfer of Calcein AM dye from Calcein AM-labeled GBM1A cells to GBM1A-mCherry cells were observed under a confocal microscope (Zeiss LSM800). Dye transfer was estimated using flow cytometry (Attune NxT, Invitrogen). For flow cytometry, co-cultured cells were harvested and then re-suspended in 2% FACS buffer at 1×106 cells/ml. For live-dead cell discrimination, SYTOX Blue was added to the cell solutions at 1:1000 (Thermo Fischer Scientific, S34857). Then, flow cytometric data was collected on Attune NxT (Invitrogen) and analyzed using Attune NxT software. A minimum of 10,000 live cells events were recorded per sample.
2.8. Orthotopic glioblastoma xenograft formation
All in vivo studies were performed following animal protocols approved by Institutional Animal Care and Use Committee of Mayo Clinic, USA. Intracranial GBM tumor implantation was performed using previously established protocol23, 37, 38. Briefly, athymic nude male mice were anesthetized using isoflurane and then placed into a stereotactic frame. A midline incision was made to expose the cranium, and a burr hole was created through the cranium using an electric drill at coordinates: Lateral (L) 1.34 mm (right from the bregma), Antero-Posterior (AP) 1.5 mm from the bregma, Dorso-Ventral (DV) 3.5 mm. 5 × 105 EGFP-luciferase expressing GBM1A cells (GBM1A-EGFP-Luc) in 2 μL of PBS was injected at this coordinate using a 10 μl Hamilton syringe fitted with a needle. Cells were injected at 0.5 μL/min. Tumor growth was monitored by bioluminescence imagine (BLI) using IVIS Spectrum (PerkinElmer).
2.9. Intra-cranial cannula implantation and convection-enhanced delivery (CED) of nanoparticles
To enable multiple injections of NPs, a cannula was implanted after three weeks of GBM1A cell inoculation at the same coordinates where GBM cells were implanted using previously established procedure19, 39. A customized guide and internal cannula were used made by PlasticsOne (Roanoke, VA). The guide cannula was fitted with a mesh under the pedestal which helped to secure the cannula to the cranium and cut 3.5 mm from the mesh. The internal cannula was designed to fit inside the guide cannula with a 0.5 mm projection extending past the guide cannula (Fig. S1). Three weeks after GBM cell implantation, mice were anesthetized using isoflurane and placed on a stereotactic frame. A midline incision was made and the cranium was exposed. The cannula was implanted through the burr hole that was made during GBM cell inoculation. Surgical glue was used on the mesh so that the guide cannula remained secured to the cranium. The coordinates for guide cannula implantation were identical to the coordinates used for GBM inoculation.
For NP administrations, the internal cannula was connected to a Hamilton syringe by a sterile tube, and then the internal cannula was placed through the guide cannula into the mouse right striatum. Lyophilized NPs were re-suspended just before administration and loaded into the Hamilton syringe. 2.5 μL of NP solution was injected into the brain for 5 minutes at 0.5 μL/min.
2.10. In vivo nanoparticle diffusion in orthotopic glioblastoma xenograft
To compare and track the penetration between ePBAE and PEG-PBAE NPs in intracranial tumor-bearing mice, Cy5-labeled pDsRED was used to form the NPs. The cannula was implanted 3 weeks after GBM inoculation. One week after cannula implantation, 2.5 μL of Cy5-labeled NPs (n=3 mice per group) was infused slowly (0.5 μL/min). The mice were perfused 24 hours after NP infusion using 4% paraformaldehyde (PFA). Whole brain was collected and stored in 4% PFA for another day before placing the brains in 30% sucrose for one day at 4℃. Brain tissue was frozen in optimum cutting temperature (O.C.T.) medium followed by cryo-sectioning of 30 μm thin sections using Leica CM1950 cryostat.
The slices were imaged by confocal microscope (Zeiss LSM800) at10X magnification and images were analyzed by ImageJ software. The tumor area was determined by the presence of EGFP (GBM1A EGFP-Luc) positive regions and the nanoparticle distribution area was measured by the Cy5 positive (red fluorescence) regions. Tumor penetration by the NPs was represented as percent of tumor volume with Cy5 signal. The volume of the tumor/NP penetration was calculated using the following formula (1) developed based on Cavalieri’s principle40. Estimated volume Est(V) of a tumor sectioned in a number of parallel sections spaced by a constant distance (t):
Est(V) = t* (A1 + A2 + A3 +……+ An), where An is the area of the respective tissue section
To determine the extent of brain penetration of the NPs, brain tissue slices were imaged at 10X magnification using confocal microscopy (Zeiss LSM800). The images were divided into rectangles at 500 μm intervals beginning at the site of catheter implantation (Fig. S2). The fluorescence intensity of the Cy5-labeled NPs (red fluorescence) was quantified using ImageJ in each interval from the catheter implantation site. The fluorescence intensity at each interval was normalized to the fluorescence intensity at the catheter implantation site representing NPs that did not penetrate the brain from the injection site. This was expressed as the percentage of NPs at each distance from the injection site relative to the amount of NPs found at the injection site.
2.11. Survival study
Intracranial tumor was established using 5 × 105 EGFP-luciferase expressing GBM1A cells. Three weeks after tumor inoculation, the cannula was implanted. Mice were randomized into two groups: a) PEG-PBAE/pDsRED NPs (n=4) and b) PEG-PBAE/pHSV-tk NPs (n=5). One week after cannula implantation, mice were started on the treatment infusion schedule with 2.5 μL of NPs via cannula, two times a week. Intraperitoneal administration of ganciclovir (GCV, 50 mg/kg) was started next day onwards after NP infusion once every day and continued until mice reached humane endpoint. Mice were observed every day and were perfused when mice reached humane endpoint. Survival analysis was performed using the Kaplan-Meier estimator method and statistical significance was estimated by the log rank test.
2.12. Statistics
All statistical analysis was performed with GraphPad Prism 5 software package. One-way ANOVA with post-hoc Dunnett test was used to test statistical significance of multiple conditions against the control group (p < 0.05) and one-way ANOVA with post-hoc Bonferroni test was used to test statistical significance between multiple groups (p < 0.05). The survival study was analyzed by Kaplan-Meier analysis and compared between groups using the long-rank (Mantel-Cox) test and p value lower than 0.05 was considered as statically significant.
3. Results
3.1. Synthesis and characterization of ePBAE and PEG-PBAE polymers
Two acrylate-terminated base polymers, poly(1,4-butanediol diacrylate-co-4-amino-1-butanol) (B4S4) and poly(1,4-butanediol diacrylate-co-5-amino-1-pentanol) (B4S5), were first synthesized (Fig. 1a-d). The molecular weights of the two base polymers were analyzed by 1H NMR to be 6,500 and 6,100 Da, respectively. Five ePBAEs were then synthesized using the two base polymers and three small molecules E3, E6, and E7. An example of ePBAE is “457”, which consists of base polymer B4S5 end-capped with E7. Two PEG-PBAEs, 44-PEG0.8k and 44-PEG2k, were synthesized using B4S4 base polymer and two PEG molecules of 800 and 2,000 Da, respectively. 1H NMR spectrum showed the disappearance of peaks for protons on acrylate groups in base polymer, indicating the reaction between B4S4 base polymer and PEG-thiol molecule went to completion (Fig. S3).
3.2. Screening of ePBAEs based on nanoparticle’s toxicity and transfection efficacy
Given a number of NP formulation parameters in which to design the PBAE-based NPs, including five ePBAE structures, two PEG-PBAE structures, various ePBAE:PEG-PBAE weight ratios, and various total polymer:DNA weight ratios, we needed a strategic workflow to screen the library of NPs (Fig. 1e). First, NPs formulated with each of the five ePBAEs without PEG-PBAE were screened based on in vitro cell viability and transfection efficacy. ePBAE NPs with B4S4 as the base polymer generally showed less toxicity on GBM1A cells, where 446 and 447 ePBAE NPs showed 100% viability at all conditions but only lower ePBAE:DNA weight ratios for 456 and 457 showed above 90% viability (Fig. 2a). Also, the use of ePBAE NPs with E3 resulted in significantly decreased viability compared to untreated control, showing polymer dose-dependent toxicity. In BTIC375 cells, a similar trend was observed, as 453 ePBAE NPs showed the strongest toxicity, while B4S4-based ePBAE NPs had more weight ratio conditions with greater than 80% cell viability than B4S5-based ePBAE NPs (Fig. 2b).
Figure 2. ePBAE screening based on cell viability and transfection efficacy.
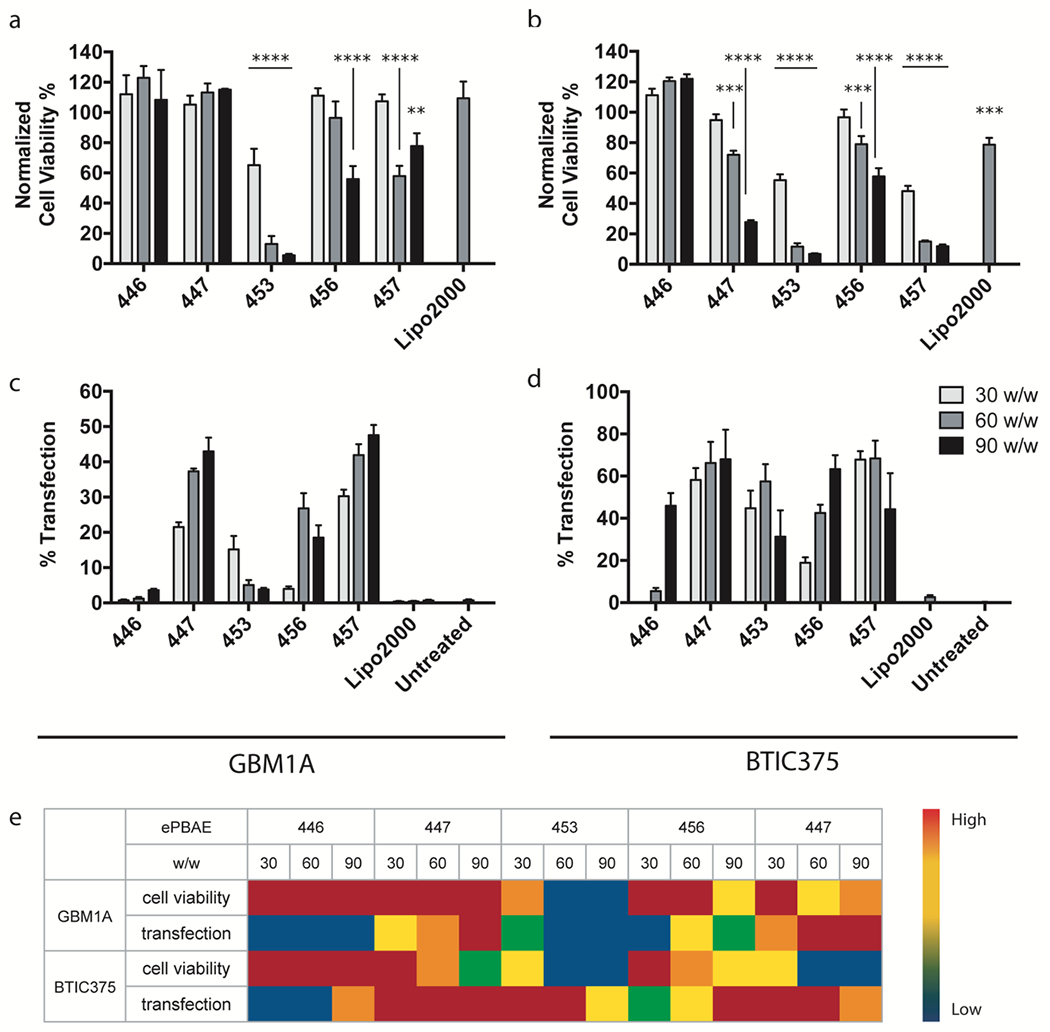
Cell viability of (a) GBM1A and (b) BTIC375 cells from incubation with 15 different ePBAE NP formulations normalized to untreated control determined by MTS assay (n = 4, mean ± s.d., *: p < 0.05 compared to untreated control). pEGFP transfection efficacy of 15 ePBAE NP formulations in (c) GBM1A and (d) BTIC375 cells determined by flow cytometry (n = 4, mean ± s.d.). (e) 5-scale heat map of cell viability and transfection efficacy from 15 ePBAE NP formulations in both cell types.
For transfection, only 447, 456, and 457 ePBAE NPs resulted in greater than 20% transfection efficacy in GBM1A cells. As shown in Figure 2c, 457 ePBAE 90 w/w NP showed the highest transfection efficacy of 47% followed by 447 ePBAE 30 w/w NP at 43%. Both showed increasing transfection efficacy with greater polymer:DNA weight ratios; however, high efficacy with 457 ePBAE 60 w/w and 90 w/w NPs should be considered in conjunction with its high toxicity level. 447 ePBAE NPs again showed the highest transfection with all weight ratio conditions in BTIC375 cells, yielding more than 60% EGFP-positive cells (Fig. 2d).
Interestingly, 453 ePBAE NPs were able to transfect 62% of BTIC375 cells, although the utility of this formulation is limited by its relatively high toxicity. Intriguingly, the small differences to polymer structure (4 carbons in the hydroxyl side chain of the B4S4 polymers compared to the 5 carbons in the hydroxyl side chain of the B4S5 polymers) as well as variance to just the terminal end-group of the linear polymers, which makes up only a small amount of the polymer by mass, both made dramatic differences to the efficacy and toxicity of using these polymers as vectors for transfection of human primary glioblastoma cells.
In order to identify the optimal ePBAE polymer for DNA delivery to both human primary brain tumor initiating cell types, % normalized cell viability and % transfection efficacy values were considered together (as categorized and depicted in a heat map for visualization in Figure 2e) ePBAE 447 and 60 w/w NP were selected and used in the following experiments as the lead ePBAE polymer and PBAE NP formulation, respectively.
3.3. Screening of PEG-PBAEs based on nanoparticle toxicity and transfection efficacy
Next, we investigated the effect of introducing PEG-PBAE polymer into ePBAE 447-based NPs on viability and transfection efficacy. Two PEG-PBAE polymers, 44-PEG0.8k and 44-PEG2k, were used to evaluate different lengths (molecular weights) of PEG molecules in formulated PEG-PBAE/ePBAE NPs. Each of the two PEG-PBAE polymers was blended in with ePBAE 447 at mass ratios of 2:1, 1:1, and 1:2 to form NPs with plasmid DNA at total polymer to DNA mass ratios of 30, 60, and 90. A total of 18 PEG-PBAE-based NP formulations were similarly evaluated for the cell toxicity and transfection efficacy. In GBM1A, all formulations tested showed cell viability above 95% (Fig. 3a), which was hypothesized as ePBAE 447 exhibited low cytotoxicity and blending PEG-PBAE into the nanoparticle effectively reduces the amount of positive charge and hydrophobic ePBAE 447 polymer in the formulations further. In BTIC375 cells, which are more sensitive, only 30 and 60 w/w ratios were evaluated because the high 447 ePBAE amount at 90 w/w ratio exhibited significant toxicity (Fig. 3b). While formulations that differ only by the type of PEG-PBAE polymer showed similar cell viability, there was 447 ePBAE dose-dependent toxicity for a given total polymer:DNA w/w ratio. Formulations with lower total polymer:DNA w/w ratio and an equal or greater proportion of PEG-PBAE polymer to 447 ePBAE polymer showed minimal cytotoxicity. Specifically, both PEG-PBAE polymers mixed with ePBAE 447 at 1:1 30 w/w, 1:2 30 w/w, and 1:2 60 w/w resulted in greater than 80% cell viability (Fig. 3).
Figure 3. PEG-PBAE screening based on cell viability and transfection efficacy.
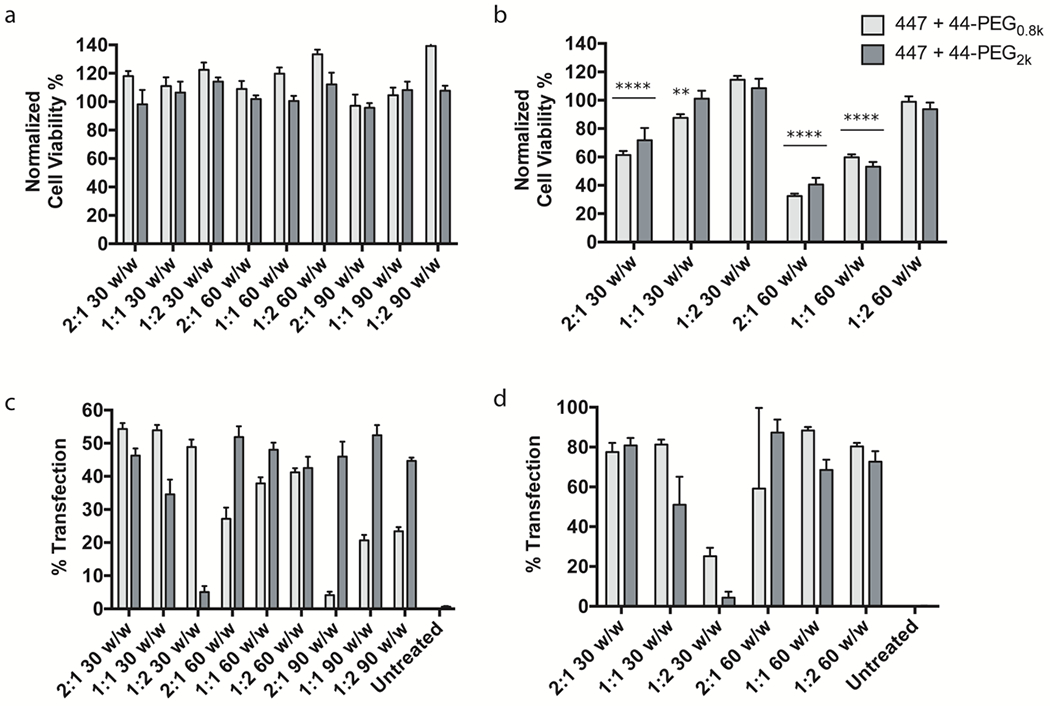
447 ePBAE was combined with 2 different PEG-PBAEs to generate 18 different PEG-PBAE NP formulations. Cell viability of (a) GBM1A and (b) BTIC375 cells from incubation with 18 and 12 PEG-PBAE NP formulations, respectively, normalized to untreated control determined by MTS assay (n = 4, mean ± s.d., *: p < 0.05 compared to untreated control). pEGFP transfection efficacy of 18 PEG-PBAE NP formulations in (c) GBM1A and (d) BTIC375 cells determined by flow cytometry (n = 4, mean ± s.d.).
PEG-PBAE-based NPs were able to transfect GBM1A cells at comparable levels as 447 ePBAE NPs despite the presence of PEG, which is known to often inhibit efficient cellular uptake of diverse NPs due to charge neutralization and steric hindrance. When 447 ePBAE was mixed with 44-PEG0.8k, transfection efficacy was greatest with the lowest w/w ratios of 30 w/w, while blending with 44-PEG2k showed greater transfection efficacy at 60 and 90 w/w (Fig. 3c and 3d). This can potentially be explained by the greater absolute amount of ePBAE necessary in the presence of longer PEG chain, as end-capping molecules in ePBAEs, such as E7, were demonstrated to be a critical parameter leading to enhanced transfection. 447 ePBAE + 44-PEG0.8k 2:1 30 w/w, 447 ePBAE + 44-PEG0.8k 1:1 30 w/w, and 447 ePBAE + 44-PEG2k 2:1 60 w/w resulted in approximately 53% transfection efficacy.
Interestingly, two PEG-PBAE-based formulations with PEG0.8k had the greatest fold increase in the geometric mean GFP intensity, which is a measure of how much GFP is expressed on average per cell due to successful transfection (Fig. S4). This is an important criterion because the expression level of exogenous gene could directly correlate to the functional output. In applications such as suicide gene therapy, enhanced expression of HSV-tk increases the probability of ganciclovir phosphorylation and cell killing efficacy. For BTIC375, among the formulations with greater than 80% cell viability, three of them, 447 + 44-PEG0.8k 1:1 30 w/w, 447 + 44-PEG0.8k 1:2 60 w/w, and 447 + 44-PEG2k 1:2 60 w/w, showed comparable transfection efficacies to 447 ePBAE NPs of approximately 80% (Fig. 3d/S4b). Based on low toxicity and high transfection in both GBM1A and BTIC375 cells, 447 + 44-PEG0.8k 1:1 30 w/w as well as 447 + 44-PEG2k 1:2 60 w/w formulations were selected for further stability screening.
3.4. Characterization of nanoparticles
As the final step to identify the optimal PEG-PBAE-based NP formulation, the particle size and stability over time were investigated via dynamic light scattering. Hydrodynamic diameter of 447 ePBAE 60 w/w NP and two selected PEG-PBAE NP formulations from transfection screening were measured in artificial cerebrospinal fluid to mimic the physiological environment in the brain tissue. Figure 4a shows that 447 + 44-PEG0.8k 1:1 30 w/w NP was the smallest NP with 141 nm hydrodynamic diameter followed by 447 + 44-PEG2k 1:2 60 w/w NP at 139 nm, which were both significantly smaller than 447 60 w/w at 160 nm in size. When incubated in artificial cerebrospinal fluid over a 24 h period, 447 60 w/w and 447 + 44-PEG2k 1:2 60 w/w increased their size to 347 and 338 nm respectively by 2 h, and to 1.6 and 1.3 μm by 5 h (Fig. 4b). However, 447 + 44-PEG0.8k 1:1 30 w/w NP maintained its size to sub 250 nm up to 5 h. Although all three formulations reached micron-sized aggregates over 24 h, only 447 + 44-PEG0.8k 1:1 30 w/w NP showed extended period of stability between 2 and 5 h. This would allow smaller 447 + 44-PEG0.8k 1:1 30 w/w NPs to penetrate farther through the brain tumor tissue by convection and diffusion following injection in the first 5 h. Transmission electron microscope images of 447 60 w/w and 447 + 44-PEG0.8k 1:1 30 w/w NPs show spherical complexes that ranged from approximately 50 – 120 nm in size (Fig. 4c). As hypothesized due to shielding, PEG surface coating on 447 + 44-PEG0.8k 1:1 30 w/w formulation resulted in near neutral zeta potential, in contrast to highly positive surface charge of 447 60 w/w NP due to positively charged 447 ePBAE polymer (Fig. 4d). 447 60 w/w (PBAE NP) and 447 + 44-PEG0.8k 1:1 30 w/w (PEG-PBAE NP) formulations were identified as the optimal NPs based on the in vitro evaluation of cytotoxicity, transfection efficacy, and stability.
Figure 4. PBAE and PEG-PBAE NP characterization.
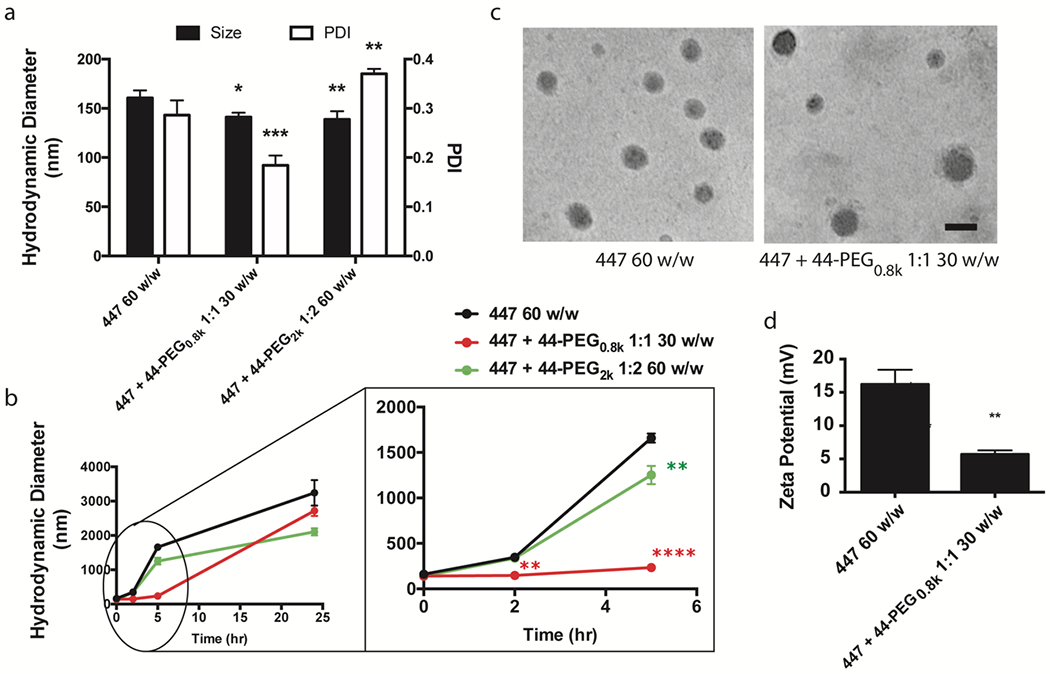
(a) Hydrodynamic diameter, polydispersity index (PDI), and (b) 24-h size stability of 447 60 w/w, 447 + 44-PEG0.8k 1:1 30 w/w, and 447 + 44-PEG2k 1:2 60 w/w formulations in artificial cerebrospinal fluid measured by dynamic light scattering (n = 3, mean ± s.d., *: p < 0.05 compared to 447 60 w/w NP). (c) Representative transmission electron microscopy images (scale bar = 100 nm) and (d) zeta potential of 447 60 w/w and 447 + 44-PEG0.8k 1:1 30 w/w formulations (n = 3, mean ± s.d., *: p < 0.05 compared to 447 60 w/w NP).
3.5. Therapeutic activity of ganciclovir in GBM1A transfected with PBAE and PEG-PBAE NPs carrying pHSV-tk
We next examined the functional efficacy of suicide gene therapy on GBM1A cells in vitro using ganciclovir as the prodrug and PBAE and PEG-PBAE NPs delivering pHSV-tk. HSV-tk, when expressed by transfected cells, will phosphorylate ganciclovir (GCV) into its active form, which then induces cell death by disrupting DNA replication41, 42. As a control, PBAE and PEG-PBAE NPs with reporter pEGFP DNA did not cause cell death when incubated with a range of GCV concentrations (Fig. 5a), demonstrating that GCV becomes toxic only in the presence of HSV-tk. Figure 5b/d shows PBAE NP carrying pHSV-tk kills GBM1A cells more efficiently after incubation with increasing concentration of GCV. Cell death occurred starting on day 3, leading to approximately 60% cell killing on day 6 with 100 μg/mL GCV. Cells treated with PEG-PBAE NP began to undergo apoptosis faster by day 2 and reached maximum cell death of 80% by day 3 with 100 μg/mL GCV (Fig. 5c/d). Also, cell death from PEG-PBAE NP treatment had greater GCV dose-dependence on day 3 than that from PBAE NP, showing that NP transfection efficacy is responsible for the level of GCV activation required to reach its active form. The observed difference in cell killing efficiency between PEG-PBAE and PBAE NPs correlates closely with their respective EGFP reporter transfection efficacies of 54% and 37%. Further, incubation with the lowest GCV concentration of 20 μg/mL GCV in cells treated with PEG-PBAE/pHSV-tk NP did not show significant difference in magnitude of cell killing at day 6 in comparison to 100 μg/mL GCV condition in cells treated with PBAE/pHSV-tk NP, again demonstrating the importance of NP transfection efficiency in the overall cell killing effect of suicide gene therapy.
Figure 5. GBM1A cell death mediated by in vitro transfection with HSV-tk and ganciclovir treatment.
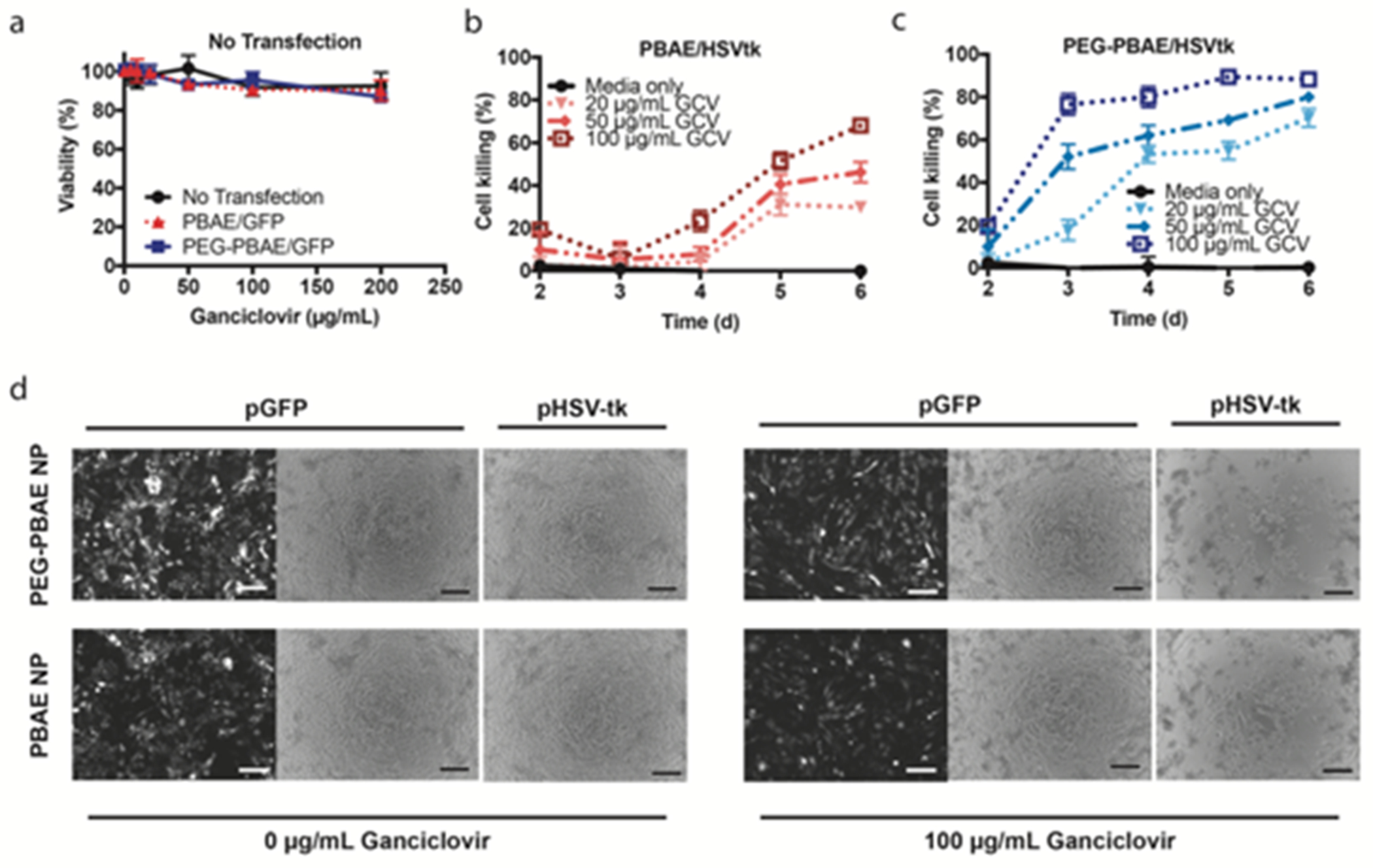
Viability of GBM1A cells measured by MTS assay (a) in the absence of HSV-tk transfection after exposure to ganciclovir or (b-c) after HSV-tk transfection using PBAE or PEG-PBAE NPs and incubation with increasing concentrations of ganciclovir (n = 4, mean ± s.d., *: p < 0.05, statistical significance given in Table S2). Cell viability is calculated by normalizing cell counts to those of GFP-transfected cells exposed to the same concentrations of ganciclovir. (d) Representative bright-field microscopy images (scale bar = 200 μm) of GBM1A cells six days after transfection.
3.6. In vivo tissue penetration of nanoparticles to the tumor core and in the tumor penumbra
A major roadblock of gene therapy using non-viral methods to treat brain tumors is suboptimal tumor and tissue penetration. To compare the tumor penetration, distribution, and retention in the brain between PBAE and PEG-PBAE NPs, both NPs were formed with Cy5-labeled pDsRed DNA to observe the diffusion area via confocal imaging. Intracranial GBM tumor-bearing mice were slowly injected over 5 minutes with NPs using a form of convection enhanced delivery (CED). Twenty-four hours after NP injection, mice were perfused and brain slices were imaged. Both PBAE and PEG-PBAE NPs (Fig. 6) exhibited uniform distribution from the injection site; however, PEG-PBAE NPs exhibited 28.5% tumor volume penetration which was significantly higher (p<0.001) compared to PBAE NPs which penetrated only 14.3% of the tumor volume. Next, we analyzed the extent of NP penetration beyond the tumor core. A higher distribution of PEG-PBAE NPs (Fig. 6b) versus PBAE NPs (Fig. 6a) was observed in the tumor penumbra. Further, we compared the penetration of NPs by examining the distance travelled by the nanoparticles from the injection site, using fluorescence intensity. With 100% NP presence at the injection point, the percentage of NPs reaching progressively farther distance from the injection site was calculated. While PEG-PBAE and PBAE NPs showed similar level of penetration to 1000 μm from the injection site, only PEG-PBAE NPs had particle presence of 29% at a 2000 μm distance (Fig. 6d) compared to 0% for PBAE NPs. Thus, the current investigation suggests that the PEG-PBAE NPs, which are validated in vitro to be smaller, more neutrally charged, and more stable under physiological conditions of the brain than the non-PEGylated NPs, also penetrate deeper both within the tumor core as well as in the tumor penumbra. Future studies would be beneficial to further support this finding.
Figure 6. NP distribution in the brain after CED-assisted administration.
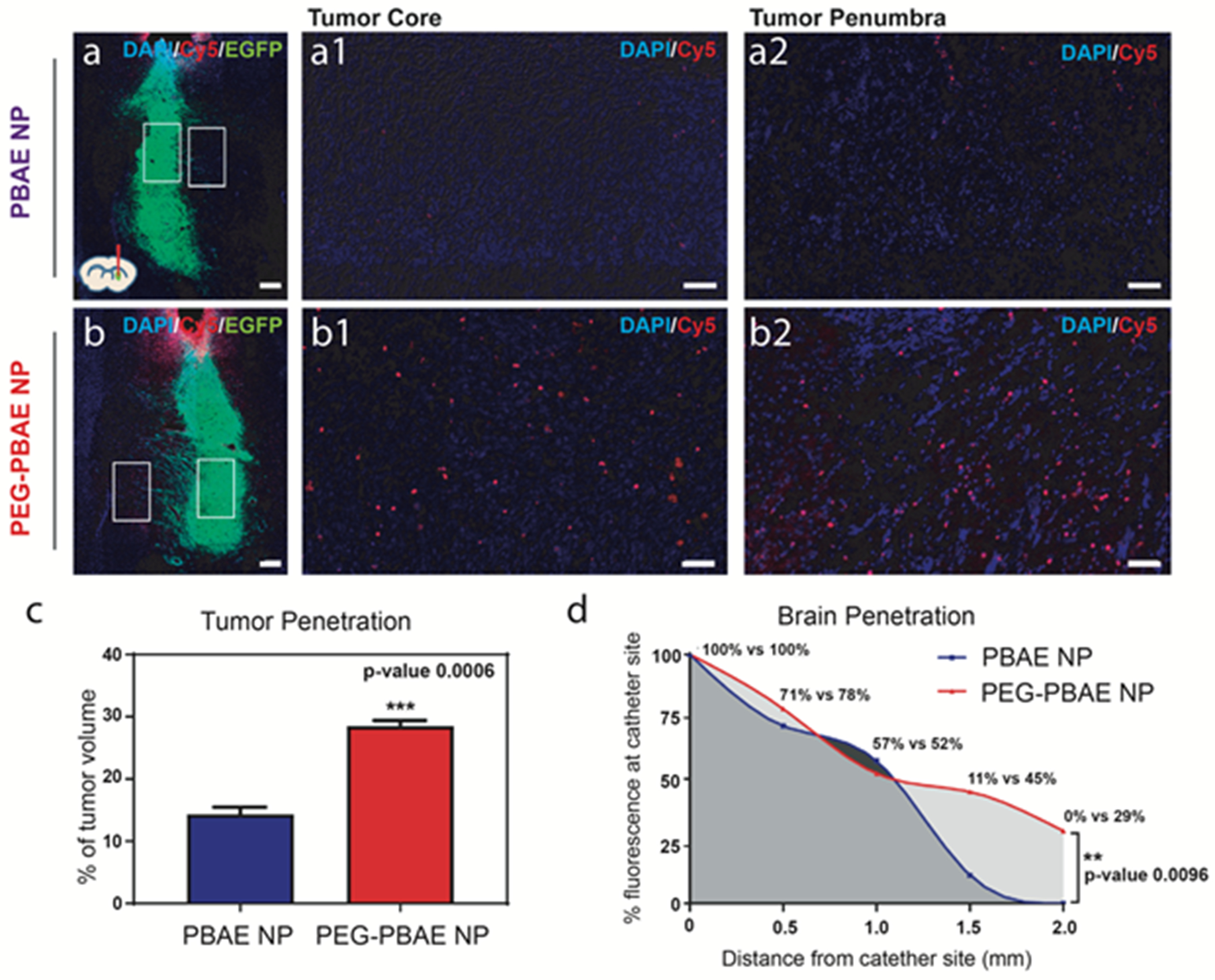
Confocal microscopy images of representative frozen sections from the brain injected with (a) PBAE (447 60 w/w) or (b) PEG-PBAE (447 + 44-PEG0.8k 1:1 30 w/w) NPs (scale bar = 200 μm, DAPI: nuclei, Cy5: NP, EGFP: GBM1A cells). Higher magnification images of (a1/b1) tumor core regions and (a2/b2) tumor penumbra regions indicated by red and orange boxes respectively in (a) and (b) (scale bar = 50 μm). (c) Quantification of tumor penetration by % tumor volume, analyzed from the confocal images with ImageJ and Cavalieri’s principle (n = 3, mean ± s.d., *: p < 0.05). (d) Quantification of brain penetration, analyzed as mean % Cy5 fluorescence intensity in subdivided rectangular ROI in confocal images at 500μm intervals beginning at the site of catheter implantation (n = 2, *: p < 0.05).
3.7. Prolonged survival of human glioblastoma-bearing mice
Our therapeutic strategy of PEG-PBAE/pHSV-tk NP + GCV system is based on the transfection of pHSV-tk via CED followed by systemic administration of GCV pro-drug. To achieve the robust effect of this therapy, it is important that transfected cells are capable of transferring active phosphorylated GCV drug to nearby non-transfected cells through gap junctions and induce cell death. This secondary effect is called the “bystander effect” (Fig. S5).
A dye transfer study was performed to show that GBM1A cells are susceptible to the bystander effect. First, we labeled the GBM1A wildtype (GBM1A WT) cells with calcein-AM (green) and then co-cultured with GBM1A-mCherry (red) cells. Calcein-AM is cell impermeant dye, and it can transfer to nearby cells only if these cells are in direct contact through gap junctions43. Following co-culture, we found (Fig. 7a) the presence of calcein-AM in GBM1A-mCherry cells (white arrow), indicating the transfer of calcein-AM from GBM1A WT to GBM1A-mCherry cells. Further, flow cytometry-based quantitative evaluation showed that after co-culture, all GBM1A-mCherry cells became positive for calcein-AM which indicates that calcein-AM transferred from GBM1A-WT to GBM1A-mCherry cells. Collectively this dye transfer study validates the presence of active gap junctions in GBM 1A cells.
Figure 7. Efficacy of suicide gene therapy in glioblastoma using PEG-PBAE/pHSV-tk NP and ganciclovir.
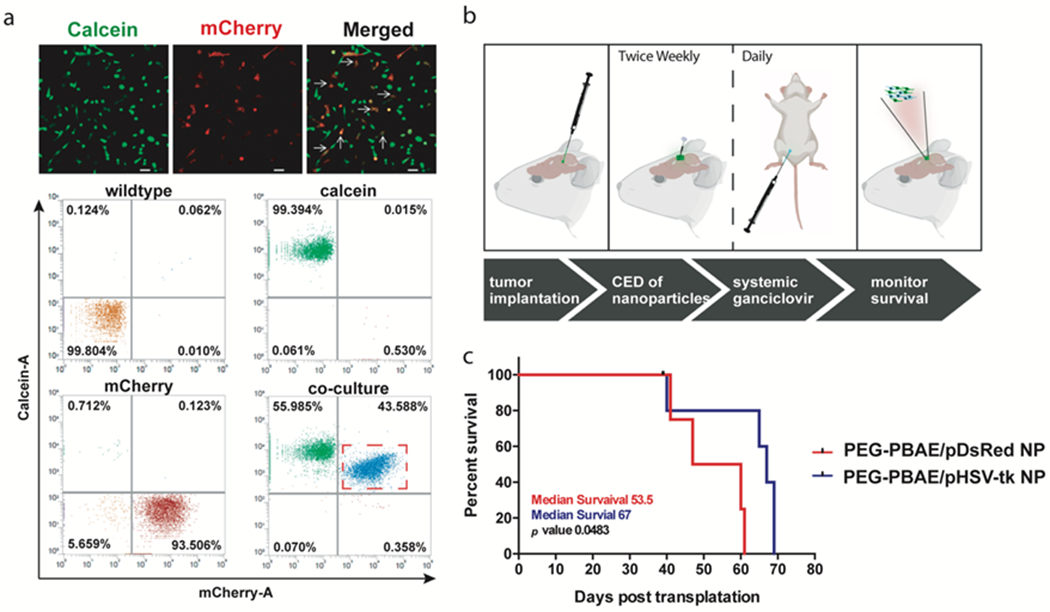
(A) Confocal images and flow cytometry scatter plots demonstrating the efficient transfer of calcein-AM dye from pre-incubated GBM1A WT cells into co-cultured GBM1A-mCherry cells through active gap junctions (scale bar = 50 μm). (B) Experimental schedule for survival study in GBM1A-bearing orthotopic glioblastoma model in mice, and (C) Kaplan-Meier survival plot between mice treated with intratumoral injection of either PEG-PBAE/pHSV-tk NP or PEG-PBAE/pDsRed NPs in conjunction with systemic ganciclovir (n ≥ 4 per group).
After demonstrating the presence of active gap junctions in GBM1A, we chose a GBM1A-based tumor model to investigate the efficacy of the PEG-PBAE/pHSV-tk NP + GCV system.
Figure 7b shows the timeline of the survival study on human glioblastoma-bearing mouse, which involved intracranial delivery of PEG-PBAE NPs carrying either pHSV-tk or pDsRed via CED, followed by intraperitoneal injection of GCV. Results showed that PEG-PBAE/pHSV-tk NP treatment significantly (p<0.05) prolonged the survival of mice compared to mice treated with PEG-PBAE/pDsRed NPs (Fig. 7c). A 25% increase in the median survival was demonstrated after treatment with PEG-PBAE/pHSV-tk NP (median survival 67 days) versus the control group (median survival 53.5 days). The survival study successfully corroborates the results from the in vitro performance as well as in vivo tumor penetration of PEG-PBAE NP, and validates PEG-PBAE/pHSV-tk NP as one of the potential non-viral gene therapy systems against glioblastoma.
4. Discussion
Shortcomings of previous non-viral gene delivery vectors suggest the need for improving the biomaterial design. For example, limited diffusion of nanoparticles from the injected site into the peripheral region of the tumor tissue prevents efficacious tumor killing. Convection-enhanced delivery (CED) offers an exciting alternative for local delivery in the brain. However, the positive surface charge of cationic polymer-based gene-carrying nanoparticles can still lead to adsorption of proteins, destabilization, or aggregation post-administration, which hinders their wide distribution into the brain tumor tissue. Wide distribution of nanoparticles is necessary for the successful treatment of brain cancer—although the brain tumor bulk is often removed through surgery, residual infiltrating BTICs persist and ultimately lead to tumor recurrence19. Furthermore, existing therapies cannot reach these invading cells resulting in treatment failure. Modification of the surface of nanoparticles by conjugating neutral, water-soluble PEG molecules is a widely employed strategy to improve transport in physiological environments44. We and other groups have reported that PEGylation not only reduces surface charge and thereby enhances potential safety of the vector22, 45, but also limits protein adsorption through steric hindrance, which allows for improved stability22, 30, pharmacokinetics46, and diffusivity into the brain parenchyma47. These are important factors to enhance the distribution of the nanoparticles and enable a widespread effect against brain cancer.
While PEGylation of the cationic polymer polyethylenimine (PEI) as a non-viral transfection agent has been investigated in the literature for approximately 20 years and found to improve transfection to tumors and reduce PEI’s potential systemic toxicity48, PEI is highly charged and non-degradable, and consequently its cytotoxicity concerns are hard to mitigate49. An alternative approach is the use of biodegradable cationic polymers, such as PBAEs28, 50, which exhibit hydrolytic degradation and relative safety27. Previous studies have demonstrated the safety of PBAE NPs to treat brain cancer based on histological and hematological analyses when administered either intracranially or intravenously19, 31, 37. In addition, studies have shown that PEGylation can reduce the in vivo toxicity score of cationic polymeric NPs, such as those formed with PEI, when they are injected into the brain45. The current investigation demonstrates that PEG-PBAE NPs have even lower cytotoxicity in vitro than unPEGylated PBAE NPs.
Another property of non-viral vectors that is important for successful functional outcome is intracellular delivery and cell specificity. PBAE polymers have been observed to have a correlation between end-capping molecule structure and cellular functional activity, including cell-specific uptake and transfection. For example, ePBAE 457 and 536 were demonstrated to exhibit specific plasmid DNA uptake and transfection of small cell lung cancer cells and hepatocellular carcinoma cells, respectively, over their corresponding healthy cells and these differences were driven by end-capping molecule22, 24. Also, bioreducible PBAE with the (E6) end-group was shown to efficiently deliver siRNA to GBM cells over neural progenitor cells20. Lastly, our group previously reported selective in vivo transfection of patient-derived GBM cells over healthy brain using ePBAE 4471, 17, 25, 51. This is in concert with our finding that polymer ePBAE 447 has the highest transfection efficacy amongst ePBAEs evaluated against the GBM1A and BTIC375 patient cells.
Here, we successfully synthesized a PEG-PBAE copolymer and generated PEGylated NPs with reduced surface charge and improved stability in aCSF. Enhanced transport shown in vivo is a significant advantage of PEG-PBAE NPs as it translates to a larger potential area of transfection. In order to combine cancer cell specificity and high transfection efficiency with nanoparticle stability and improved transport, we sought to evaluate mixtures of ePBAE and PEG-PBAE polymers with pDNA to create PEG-PBAE NP formulations. Multiple stages of the characterization process enabled identification of the most optimal PEG chain length and the mass ratio between ePBAE and PEG-PBAE polymers for the resulting NP.
We have recently evaluated the potential of PEG-PBAE-based NPs in small cell lung cancer cells in vitro, and found them to be effective22. In the realm of brain cancer, PBAE NPs carrying HSV-tk have recently been evaluated in rat glioma and found to lead to increased animal survival31. An alternative synthesis approach to formulate PEG-PBAE NPs that focused on PBAE 446 and a larger 5,000 Da PEG end-capping group, found improvements to NP penetration and efficacy with PEGylation in rat glioma models as well52. However, none of these previous studies investigated a model with human glioma generally, or patient-derived glioblastoma, in particular, making it unclear if these findings could extend to potential clinical therapeutic use.
To investigate the potential of our non-viral gene delivery vector for the treatment of GBM, the pathophysiology of the disease was carefully considered in designing the experiment. Given the intratumoral heterogeneity of human GBM and its frequent recurrence, the efficacy of novel treatments is clinically more impactful if evaluated on stem-like cells that are harvested from patients. In this study, we used primary brain tumor initiating cells from patient tumor tissue. The primary GBM cells used in this study were evaluated and shown to be capable of intercellular transport of molecules through active gap junctions. This maximizes the potential of suicide gene therapy using HSV-tk via the bystander effect, as successful gene transfection in a subset of the cancer cell population in the tumor tissue could lead to an amplified cancer killing effect. Further, it has been shown that in immunocompetent models, HSV-tk treatment has been demonstrated to lead to strong anti-tumor cellular immune responses with CD4+ and CD8+ T cell infiltration, a process not able to be captured in our human orthotopic model, but one that may significantly amplify its potency in a clinical setting53. Therefore, the cancer cell killing demonstrated by PEG-PBAE/ePBAE 447/pHSV-tk NPs treating human glioblastoma, in addition to killing BTICs and leading to a bystander effect, may also be able to generate a similar immunologic response in the presence of T cells, and thus has the potential for an even stronger effect in the clinic.
5. Conclusion
The aim of this work was to modify and enhance an existing gene delivery vector, PBAE polymer, by chemical modification with PEG in order to generate nanoparticles with improved efficacy, stability, and brain tumor penetration. We successfully synthesized ePBAE and PEG-PBAE polymers, and identified the optimal blend of these two components using a multiparametric approach and screening process based on cell viability, transfection efficacy, and particle stability. PEG-PBAE NPs showed enhanced penetration through patient-derived brain tumors and brain parenchyma from the site of injection compared to PBAE NPs in an orthotopic xenograft model of human glioblastoma in mice. PEG-PBAE NPs were utilized for suicide gene therapy by encapsulating pHSV-tk and treating with ganciclovir and demonstrated increased median survival in vivo. These non-viral vectors are promising for the treatment of human brain cancer.
Supplementary Material
Acknowledgements
The authors thank Drs. J. Laterra and A. Vescovi for kindly providing GBM1A cells. A.Q.H. was supported by the Mayo Clinic Professorship and a Clinician Investigator award. J.J.G. was supported by the Bloomberg–Kimmel Institute for Cancer Immunotherapy. The authors thank the NIH for support (R01CA228133, R01EB016721, R01CA195503, R01CA200399, R01CA183827). J.K. received fellowship support from Samsung Scholarship. AGB received fellowship support from the Howard Hughes Medical Institute.
Footnotes
Declaration of Interest
Authors declare no conflict of interest.
References
- 1.Guerrero-Cazares H; Tzeng SY; Young NP; Abutaleb AO; Quinones-Hinojosa A; Green JJ, Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano 2014, 8 (5), 5141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R; Mason WP; van den Bent MJ; Weller M; Fisher B; Taphoorn MJ; Belanger K; Brandes AA; Marosi C; Bogdahn U; Curschmann J; Janzer RC; Ludwin SK; Gorlia T; Allgeier A; Lacombe D; Cairncross JG; Eisenhauer E; Mirimanoff RO; European Organisation for, R.; Treatment of Cancer Brain, T.; Radiotherapy, G.; National Cancer Institute of Canada Clinical Trials, G., Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005, 352 (10), 987–96. [DOI] [PubMed] [Google Scholar]
- 3.Brade AM; Tannock IF, Scheduling of radiation and chemotherapy for limited-stage small-cell lung cancer: repopulation as a cause of treatment failure? J Clin Oncol 2006, 24 (7), 1020–2. [DOI] [PubMed] [Google Scholar]
- 4.Lara-Velazquez M; Al-Kharboosh R; Jeanneret S; Vazquez-Ramos C; Mahato D; Tavanaiepour D; Rahmathulla G; Quinones-Hinojosa A, Advances in Brain Tumor Surgery for Glioblastoma in Adults. Brain Sci 2017, 7 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegel R; Naishadham D; Jemal A, Cancer statistics, 2012. CA Cancer J Clin 2012, 62 (1), 10–29. [DOI] [PubMed] [Google Scholar]
- 6.Chesler DA; Berger MS; Quinones-Hinojosa A, The potential origin of glioblastoma initiating cells. Front Biosci (Schol Ed) 2012, 4, 190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinones-Hinojosa A; Chaichana K, The human subventricular zone: a source of new cells and a potential source of brain tumors. Exp Neurol 2007, 205 (2), 313–24. [DOI] [PubMed] [Google Scholar]
- 8.Jackson M; Hassiotou F; Nowak A, Glioblastoma stem-like cells: at the root of tumor recurrence and a therapeutic target. Carcinogenesis 2015, 36 (2), 177–85. [DOI] [PubMed] [Google Scholar]
- 9.Lang FF; Bruner JM; Fuller GN; Aldape K; Prados MD; Chang S; Berger MS; McDermott MW; Kunwar SM; Junck LR; Chandler W; Zwiebel JA; Kaplan RS; Yung WK, Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol 2003, 21 (13), 2508–18. [DOI] [PubMed] [Google Scholar]
- 10.Wang S; El-Deiry WS, TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22 (53), 8628–33. [DOI] [PubMed] [Google Scholar]
- 11.Moolten FL, Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986, 46 (10), 5276–81. [PubMed] [Google Scholar]
- 12.Michaelsen SR; Christensen CL; Sehested M; Cramer F; Poulsen TT; Patterson AV; Poulsen HS, Single agent- and combination treatment with two targeted suicide gene therapy systems is effective in chemoresistant small cell lung cancer cells. J Gene Med 2012, 14 (7), 445–58. [DOI] [PubMed] [Google Scholar]
- 13.Sun X; Xing L; Deng X; Hsiao HT; Manami A; Koutcher JA; Clifton Ling C; Li GC, Hypoxia targeted bifunctional suicide gene expression enhances radiotherapy in vitro and in vivo. Radiother Oncol 2012, 105 (1), 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hollon T, Researchers and regulators reflect on first gene therapy death. Am J Ophthalmol 2000, 129 (5), 701. [DOI] [PubMed] [Google Scholar]
- 15.Pack DW; Hoffman AS; Pun S; Stayton PS, Design and development of polymers for gene delivery. Nat Rev Drug Discov 2005, 4 (7), 581–93. [DOI] [PubMed] [Google Scholar]
- 16.Ginn SL; Amaya AK; Alexander IE; Edelstein M; Abedi MR, Gene therapy clinical trials worldwide to 2017: An update. J Gene Med 2018, 20 (5), e3015. [DOI] [PubMed] [Google Scholar]
- 17.Tzeng SY; Guerrero-Cazares H; Martinez EE; Sunshine JC; Quinones-Hinojosa A; Green JJ, Non-viral gene delivery nanoparticles based on poly(beta-amino esters) for treatment of glioblastoma. Biomaterials 2011, 32 (23), 5402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez-Bertoni H; Kotchetkov IS; Mihelson N; Lal B; Rui Y; Ames H; Lugo-Fagundo M; Guerrero-Cazares H; Quinones-Hinojosa A; Green JJ; Laterra J, A Sox2/miR-486–5p axis regulates survival of GBM cells by inhibiting tumor suppressor networks. Cancer Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Bertoni H; Kozielski KL; Rui Y; Lal B; Vaughan H; Wilson DR; Mihelson N; Eberhart CG; Laterra J; Green JJ, Bioreducible Polymeric Nanoparticles Containing Multiplexed Cancer Stem Cell Regulating miRNAs Inhibit Glioblastoma Growth and Prolong Survival. Nano Lett 2018, 18 (7), 4086–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozielski KL; Ruiz-Valls A; Tzeng SY; Guerrero-Cazares H; Rui Y; Li Y; Vaughan HJ; Gionet-Gonzales M; Vantucci C; Kim J; Schiapparelli P; Al-Kharboosh R; Quinones-Hinojosa A; Green JJ, Cancer-selective nanoparticles for combinatorial siRNA delivery to primary human GBM in vitro and in vivo. Biomaterials 2019, 209, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozielski KL; Tzeng SY; De Mendoza BA; Green JJ, Bioreducible cationic polymer-based nanoparticles for efficient and environmentally triggered cytoplasmic siRNA delivery to primary human brain cancer cells. ACS Nano 2014, 8 (4), 3232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J; Kang Y; Tzeng SY; Green JJ, Synthesis and application of poly(ethylene glycol)-co-poly(beta-amino ester) copolymers for small cell lung cancer gene therapy. Acta Biomater 2016, 41, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mangraviti A; Tzeng SY; Gullotti D; Kozielski KL; Kim JE; Seng M; Abbadi S; Schiapparelli P; Sarabia-Estrada R; Vescovi A; Brem H; Olivi A; Tyler B; Green JJ; Quinones-Hinojosa A, Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials 2016, 100, 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamboni CG; Kozielski KL; Vaughan HJ; Nakata MM; Kim J; Higgins LJ; Pomper MG; Green JJ, Polymeric nanoparticles as cancer-specific DNA delivery vectors to human hepatocellular carcinoma. J Control Release 2017, 263, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzeng SY; Wilson DR; Hansen SK; Quinones-Hinojosa A; Green JJ, Polymeric nanoparticle-based delivery of TRAIL DNA for cancer-specific killing. Bioeng Transl Med 2016, 1 (2), 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J; Sunshine JC; Green JJ, Differential polymer structure tunes mechanism of cellular uptake and transfection routes of poly(beta-amino ester) polyplexes in human breast cancer cells. Bioconjug Chem 2014, 25 (1), 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunshine JC; Peng DY; Green JJ, Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm 2012, 9 (11), 3375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop CJ; Ketola TM; Tzeng SY; Sunshine JC; Urtti A; Lemmetyinen H; Vuorimaa-Laukkanen E; Yliperttula M; Green JJ, The effect and role of carbon atoms in poly(beta-amino ester)s for DNA binding and gene delivery. J Am Chem Soc 2013, 135 (18), 6951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris TJ; Green JJ; Fung PW; Langer R; Anderson DG; Bhatia SN, Tissue-specific gene delivery via nanoparticle coating. Biomaterials 2010, 31 (5), 998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J; Shamul JG; Shah SR; Shin A; Lee BJ; Quinones-Hinojosa A; Green JJ, Verteporfin-Loaded Poly(ethylene glycol)-Poly(beta-amino ester)-Poly(ethylene glycol) Triblock Micelles for Cancer Therapy. Biomacromolecules 2018, 19 (8), 3361–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mangraviti A; Tzeng SY; Kozielski KL; Wang Y; Jin Y; Gullotti D; Pedone M; Buaron N; Liu A; Wilson DR; Hansen SK; Rodriguez FJ; Gao GD; DiMeco F; Brem H; Olivi A; Tyler B; Green JJ, Polymeric nanoparticles for nonviral gene therapy extend brain tumor survival in vivo. ACS Nano 2015, 9 (2), 1236–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galli R; Binda E; Orfanelli U; Cipelletti B; Gritti A; De Vitis S; Fiocco R; Foroni C; Dimeco F; Vescovi A, Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004, 64 (19), 7011–21. [DOI] [PubMed] [Google Scholar]
- 33.Auvergne R; Wu C; Connell A; Au S; Cornwell A; Osipovitch M; Benraiss A; Dangelmajer S; Guerrero-Cazares H; Quinones-Hinojosa A; Goldman SA, PAR1 inhibition suppresses the self-renewal and growth of A2B5-defined glioma progenitor cells and their derived gliomas in vivo. Oncogene 2016, 35 (29), 3817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiapparelli P; Zhang P; Lara-Velazquez M; Guerrero-Cazares H; Lin R; Su H; Chakroun RW; Tusa M; Quinones-Hinojosa A; Cui H, Self-assembling and self-formulating prodrug hydrogelator extends survival in a glioblastoma resection and recurrence model. J Control Release 2020, 319, 311–321. [DOI] [PubMed] [Google Scholar]
- 35.Shah SR; Kim J; Schiapparelli P; Vazquez-Ramos CA; Martinez-Gutierrez JC; Ruiz-Valls A; Inman K; Shamul JG; Green JJ; Quinones-Hinojosa A, Verteporfin-Loaded Polymeric Microparticles for Intratumoral Treatment of Brain Cancer. Mol Pharm 2019, 16 (4), 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suhaeri M; Noh MH; Moon JH; Kim IG; Oh SJ; Ha SS; Lee JH; Park K, Novel skin patch combining human fibroblast-derived matrix and ciprofloxacin for infected wound healing. Theranostics 2018, 8 (18), 5025–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karlsson J; Rui Y; Kozielski KL; Placone AL; Choi O; Tzeng SY; Kim J; Keyes JJ; Bogorad MI; Gabrielson K; Guerrero-Cazares H; Quinones-Hinojosa A; Searson PC; Green JJ, Engineered nanoparticles for systemic siRNA delivery to malignant brain tumours. Nanoscale 2019, 11 (42), 20045–20057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q; Wijesekera O; Salas SJ; Wang JY; Zhu M; Aprhys C; Chaichana KL; Chesler DA; Zhang H; Smith CL; Guerrero-Cazares H; Levchenko A; Quinones-Hinojosa A, Mesenchymal stem cells from human fat engineered to secrete BMP4 are nononcogenic, suppress brain cancer, and prolong survival. Clin Cancer Res 2014, 20 (9), 2375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez-Perez O; Guerrero-Cazares H; Quinones-Hinojosa A, Targeting of deep brain structures with microinjections for delivery of drugs, viral vectors, or cell transplants. J Vis Exp 2010, (46). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gundersen HJ; Jensen EB, The efficiency of systematic sampling in stereology and its prediction. J Microsc 1987, 147 (Pt 3), 229–63. [DOI] [PubMed] [Google Scholar]
- 41.Rubsam LZ; Boucher PD; Murphy PJ; KuKuruga M; Shewach DS, Cytotoxicity and accumulation of ganciclovir triphosphate in bystander cells cocultured with herpes simplex virus type 1 thymidine kinase-expressing human glioblastoma cells. Cancer Res 1999, 59 (3), 669–75. [PubMed] [Google Scholar]
- 42.Tomicic MT; Thust R; Kaina B, Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene 2002, 21 (14), 2141–53. [DOI] [PubMed] [Google Scholar]
- 43.Ryu S; Yoo J; Jang Y; Han J; Yu SJ; Park J; Jung SY; Ahn KH; Im SG; Char K; Kim BS, Nanothin Coculture Membranes with Tunable Pore Architecture and Thermoresponsive Functionality for Transfer-Printable Stem Cell-Derived Cardiac Sheets. ACS Nano 2015, 9 (10), 10186–202. [DOI] [PubMed] [Google Scholar]
- 44.Suk JS; Xu Q; Kim N; Hanes J; Ensign LM, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016, 99 (Pt A), 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mastorakos P; Zhang C; Berry S; Oh Y; Lee S; Eberhart CG; Woodworth GF; Suk JS; Hanes J, Highly PEGylated DNA Nanoparticles Provide Uniform and Widespread Gene Transfer in the Brain. Adv Healthc Mater 2015, 4 (7), 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shamul JG; Shah SR; Kim J; Schiapparelli P; Vazquez-Ramos CA; Lee BJ; Patel KK; Shin A; Quinones-Hinojosa A; Green JJ, Verteporfin-Loaded Anisotropic Poly(Beta-Amino Ester)-Based Micelles Demonstrate Brain Cancer-Selective Cytotoxicity and Enhanced Pharmacokinetics. Int J Nanomedicine 2019, 14, 10047–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nance EA; Woodworth GF; Sailor KA; Shih TY; Xu Q; Swaminathan G; Xiang D; Eberhart C; Hanes J, A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med 2012, 4 (149), 149ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogris M; Brunner S; Schuller S; Kircheis R; Wagner E, PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther 1999, 6 (4), 595–605. [DOI] [PubMed] [Google Scholar]
- 49.Moghimi SM; Symonds P; Murray JC; Hunter AC; Debska G; Szewczyk A, A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther 2005, 11 (6), 990–5. [DOI] [PubMed] [Google Scholar]
- 50.Tzeng SY; Green JJ, Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for siRNA and DNA delivery to human brain cancer. Adv Healthc Mater 2013, 2 (3), 468–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tzeng SY; Higgins LJ; Pomper MG; Green JJ, Student award winner in the Ph.D. category for the 2013 society for biomaterials annual meeting and exposition, april 10–13, 2013, Boston, Massachusetts : biomaterial-mediated cancer-specific DNA delivery to liver cell cultures using synthetic poly(beta-amino ester)s. J Biomed Mater Res A 2013, 101 (7), 1837–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mastorakos P; Zhang C; Song E; Kim YE; Park HW; Berry S; Choi WK; Hanes J; Suk JS, Biodegradable brain-penetrating DNA nanocomplexes and their use to treat malignant brain tumors. J Control Release 2017, 262, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuriyama S; Kikukawa M; Masui K; Okuda H; Nakatani T; Akahane T; Mitoro A; Tominaga K; Tsujinoue H; Yoshiji H; Okamoto S; Fukui H; Ikenaka K, Cancer gene therapy with HSV-tk/GCV system depends on T-cell-mediated immune responses and causes apoptotic death of tumor cells in vivo. Int J Cancer 1999, 83 (3), 374–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.