

Abstract
Inflammation in arterial walls leads to coronary artery disease (CAD). We previously reported that a high omega-3 fatty index was associated with prevention of progression of coronary atherosclerosis, a disease of chronic inflammation in the arterial wall. However, the mechanism of such benefit is unclear. The two main omega-3 fatty acids, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are precursors of specialized pro-resolving lipid mediators (SPMs) - resolvins and maresins - which actively resolve chronic inflammation. To explore whether SPMs are associated with coronary plaque progression, levels of SPMs and proinflammatory mediators (leukotriene B4 [LTB4] and prostaglandins) were measured using liquid chromatography-tandem mass spectrometry in 31 statin-treated patients with stable CAD randomized to either EPA and DHA, 3.36 g daily, or no EPA/DHA (control). Coronary plaque volume was measured by coronary computed tomographic angiography at baseline and at 30-month follow-up. Higher plasma levels of EPA+DHA were associated with significantly increased levels of two SPMs - resolvin E1 and maresin 1- and 18-hydroxy-eicosapentaenoic acid (HEPE), the precursor of resolvin E1. Those with low plasma EPA+DHA levels had a low (18-HEPE+resolvin E1)/LTB4 ratio and significant plaque progression. Those with high plasma EPA+DHA levels had either low (18-HEPE+resolvin E1)/LTB4 ratios with significant plaque progression or high (18-HEPE+resolvin E1)/LTB4 ratios with significant plaque regression. These findings suggest that an imbalance between pro-resolving and proinflammatory lipid mediators is associated with plaque progression and potentially mediates the beneficial effects of EPA and DHA in CAD patients.
Clinical Trial Registration:
ClinicalTrials.gov Identifier: NCT01624727
Keywords: Metabololipidomics, Resolvins, Omega-3 fatty acids, Coronary Plaque Regression, Inflammation
1. INTRODUCTION
Atherosclerotic coronary artery disease (CAD) remains a major source of morbidity and mortality worldwide. A high level of residual risk of atherosclerotic plaque progression and cardiovascular disease (CVD) events remains despite achieving low-density-lipoprotein cholesterol (LDL-C) levels ≤ 70 mg/dL with statin treatment.1–3 Therefore, additional modalities to reduce residual risk are needed. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are very long chain omega-3 fatty acids. In the Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention trial (REDUCE-IT), those randomized to high-dose icosapent ethyl, a derivative of EPA, added to statin treatment had a significant 25% reduction in major adverse cardiovascular events compared to those on statin alone.4 These findings suggest that high-dose EPA added to statin therapy can reduce residual cardiovascular risk; however, the mechanisms underlying this significant benefit are not entirely clear.
Atherosclerosis is a disease of chronic inflammation in the arterial wall involving the innate immune system and characterized by monocyte and neutrophil infiltration and differentiation of monocytes to macrophages in the subendothelial space, leading to foam cell and fatty streak formation and advanced plaques.5–7 Inflammation is a protective host response designed to resist invasion and injury by enhancing antimicrobial activity and initiating healing to minimize tissue damage and restore normal function. In the initiation phase of inflammation, the omega-6 fatty acid, arachidonic acid, is converted via cyclooxygenases to proinflammatory, pro-thrombotic and vasoactive eicosanoids that include prostaglandin (PG) E2, PGF2α and thromboxane A2 and via 5-lipoxygenase to the proinflammatory leukotriene (LT) B4, a potent chemoattractant that recruits leukocytes into tissues to remove necrotic debris and apoptotic cells.8,9 As the inflammatory response proceeds, lipid mediator class switching occurs in which PGE2 activates biosynthesis of the specialized pro-resolving lipid mediators (SPMs) – lipoxins (LX), resolvins (Rv), protectins and maresins (MaR) - which are involved in pro-resolution and anti-inflammatory pathways that can actively terminate inflammation, including activation of endogenous clearance mechanisms.10–13 SPMs stimulate the resolution of acute inflammation by stopping further neutrophil recruitment to inflamed tissues and by stimulating non-phlogistic infiltration of monocytes that differentiate into reparative, anti-inflammatory, resolution macrophages.11 These resolution macrophages then phagocytize and clear apoptotic neutrophils and debris, steps that are key to resolution and prevention of chronic inflammation.11,14 Therefore, SPMs stimulate the resolution of acute inflammation and prevent chronic inflammation.11,14,15
We previously reported that patients with stable CAD on statin therapy randomized to high-dose EPA and DHA in the Slowing HEART diSease with lifestyle and omega-3 fatty acids (HEARTS) trial, had prevention of coronary plaque progression when an omega-3 fatty acid index ≥ 4% was achieved.16 EPA is the precursor of 18-HEPE and the E-series resolvins, and DHA is the precursor of the D-series resolvins and maresin 1 (MaR1).11,12 In a subset of the HEARTS trial, we reported that SPMs are deficient in CAD patients and supplementation with omega-3 ethyl ester restored levels of SPMs.17 To explore potential mechanisms for lack of plaque progression in the HEARTS trial, we measured levels of SPMs and proinflammatory mediators and correlated with coronary plaque progression and regression.
2. MATERIALS AND METHODS
Data availability statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
2.1. Study design
The HEARTS trial is a randomized, controlled clinical trial conducted at Beth Israel Deaconess Medical Center (BIDMC), Boston, MA. The protocol was approved by the BIDMC Institutional Review Board and conducted according to the Declaration of Helsinki. All subjects signed informed consent. The primary endpoint of the trial is the effect of high-dose omega-3 ethyl esters on progression of coronary arterial plaque at 30 months of follow-up and has been previously reported.16,18
2.2. Participants and study intervention
To explore the association between achieved levels of omega-3 fatty acids and pro-resolving and proinflammatory lipid mediators as well as coronary plaque progression, SPM levels were measured in 31 subjects with the highest and lowest plasma omega-3 fatty acid index at 30-month follow-up. Eligible subjects were aged 37 to 80 years and had stable CAD as defined previously.18 Subjects were randomized to either open-label omega-3 ethyl esters (Lovaza, GlaxoSmithKline, Research Triangle Park, NC) 4 capsules daily for a total daily dose of 1.86 g EPA and 1.5 g DHA or no omega-3 ethyl ester (control) for 30 months. Subjects were taking statin and aspirin therapy unless intolerant. Subjects were counselled not to take over-the-counter fish oil. At baseline and 6-month intervals, a detailed history and physical examination were performed. Blood samples were obtained after a 12-hour fast and measured at Quest Diagnostics (Cambridge, MA). High-sensitivity C-reactive protein (hs-CRP) was measured as previously described.16
2.3. Image acquisition, reconstruction and coronary plaque analysis
Coronary computed tomographic angiography (CCTA) imaging was performed at BIDMC at baseline and 30-month follow-up using a 320-row detector scanner (Aquilion ONE, Toshiba Medical Systems, Otawara, Japan) with prospective electrocardiogram gating as previously described.16,18–20 CCTA images underwent 3-dimensional reconstruction for coronary segment plaque volume analysis using semiautomated software (SUREPlaque, version 6.3.2, Vital Images, Minnetonka, MN, USA). Segments with prior revascularization or significant calcification causing calcium-bloom artifact were excluded. Branches or focal calcification served as fiducial markers to ensure measurement of the same segment at 30-month follow-up.
Plaque analysis was performed independently by two readers blinded to treatment allocation. Hounsfield unit (HU) densities were used to define plaques as fatty (−100 to 49 HU) and fibrous (50 to 150 HU).18 Noncalcified plaque is the sum of fatty and fibrous plaque. The intra-observer and inter-observer agreement indexes were 0.99 and 0.98, respectively, showing excellent correlation between readings.18 Plaque volume was indexed to length of vessel examined and expressed in mm3/mm. Progression of coronary plaque was defined as a percent change greater than 0 and regression of coronary plaque was defined as a percent change < 0 in fibrous and noncalcified plaque.
2.4. Measurement of fatty acid levels
EPA, DHA, arachidonic acid and total fatty acids were measured as previously described.16 The omega-3 fatty acid index was calculated as the percentage of EPA and DHA of total fatty acid level.
2.5. Targeted metabololipidomics
To measure levels of SPMs and the proinflammatory mediators - prostaglandins and leukotrienes - that are derived from EPA, DHA and arachidonic acid functional-metabolomes, we carried out targeted metabololipidomics using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) approach as previously described.21–23 Serum samples stored at −80 °C were used. Samples for LC-MS-MS based metabololipidomics were solid-phase (C-18)-extracted and injected into a Qtrap 6500 mass spectrometer (SCIEX, Framingham, MA, USA) equipped with a Shimadzu HPLC system (Shimadzu Corp, Kyoto, Japan). A Poroshell 120 EC-C18 column (100x 4.6mm x 2.7 μm; Agilent technologies, Santa Clara, CA, USA) was used for separation. To identify and quantify lipid mediators (LM), multiple reaction monitoring (MRM) was used with signature ion fragments (m/z) for each molecule. Identification was conducted using published criteria and matching of at least six diagnostic ions and retention times of standard compounds.21–23 The complete stereochemistry of each of the SPMs, i.e. RvD1 and RvE1, were previously determined.21 The coefficients of variation for measurements were LTB4: 6.2%, MaR1: 6.5%, RvE1: 11.2%, PGD2: 13.1% and PGE2: 10%.23 Reproducibility between labs and a methodological validation have been reported.24
2.6. Statistical Analysis
Categorical variables were expressed as counts and percentages. Normality tests were conducted using the Shapiro-Wilk test. Continuous variables were reported as the mean and standard deviation (SD) for normally distributed variables or median and interquartile range [IQR] for non-normally distributed variables. Plaque volumes and percent change were not normally distributed and therefore, were reported as median [IQR]. Continuous variables were compared using unpaired Student’s t-tests for normally distributed variables or the Mann-Whitney-U test for non-normally distributed variables. Categorical variables were compared with either Chi-square or Fisher’s exact tests. Correlations were determined using Pearson correlation coefficient for normally distributed variables or Spearman’s rank correlation coefficient for non-normally distributed variables. A 2-sided P value ≤ 0.05 was considered statistically significant. Data analyses were performed using SPSS 20.0 for Windows (IBM Corp. Armonk, NY, USA).
3. RESULTS
3.1. Study patients
Baseline characteristics stratified by the omega-3 fatty acid index: high (median 8.4% [IQR 8.1 to 9.6]) and low (median 2.4% [IQR 2.3 to 2.7]) are shown in Table 1. Those with a high omega-3 fatty acid index were more likely to be women and to have lower white blood cell and neutrophil counts compared to those with a low omega-3 fatty acid index. Both baseline low-density lipoprotein cholesterol (LDL-C) levels (mean ± SD, 2.07 ± 0.60 vs. 2.26 ± 0.89 mmol/L; P = 0.51) and high-sensitivity C-reactive protein levels (median [IQR], 21.9 [1.9, 39.1] vs. 7.6 [3.8, 24.8] nmol/L; P = 0.98) were well-controlled and not significantly different in the low omega-3 fatty acid index and high omega-3 fatty acid index groups, respectively. Other baseline characteristics were not significantly different.
Table 1.
Baseline characteristics stratified by low and high omega-3 fatty acid index groups
| Low Omega-3 Fatty Acid Index (n = 15) | High Omega-3 Fatty Acid Index (n = 16) | P value | |
|---|---|---|---|
| Omega-3 fatty acid index, median (IQR) | 2.4% [2.3,2.7] | 8.4% [8.1, 9.6] | <0.001 |
| Characteristic, n (%) | |||
| Age, mean (SD) | 63.1 (5.4) | 62.8 (8.2) | 0.90 |
| Female sex | 1 (6.7) | 6 (37.5) | 0.04 |
| History of myocardial infarction | 6 (40.0) | 7 (43.8) | 0.83 |
| History of PCI | 8 (53.3) | 8 (50.0) | 0.85 |
| History of CABG | 3 (20.0) | 4 (25.0) | 0.74 |
| Hypertension | 12 (80.0) | 8 (50.0) | 0.08 |
| Diabetes | 0 (0.0) | 0 (0.0) | NA |
| Anthropometrics and Blood Pressure, mean (SD) | |||
| Weight, kg | 89.3 (13.6) | 92.7 (14.3) | 0.51 |
| Body-mass index, kg/m2 | 30.5 (2.7) | 31.6 (3.3) | 0.37 |
| Waist circumference, cm | 106.4 (7.7) | 108.7 (11.5) | 0.51 |
| Blood pressure, mm Hg | |||
| Systolic | 125.6 (16.1) | 115.3 (14.3) | 0.07 |
| Diastolic | 74.2 (9.9) | 72.7 (11.8) | 0.71 |
| Biochemical Profile, mean (SD), except hs-CRP which is median [interquartile range] | |||
| Glycated hemoglobin, % | 5.8 (0.3) | 5.8 (0.4) | 0.67 |
| Glucose, mmol/L mg/dl | 5.22 (0.49) | 5.14 (0.61) | 0.69 |
| 94.0 (8.9) | 92.6 (11.0) | ||
| hs-CRP, nmol/L mg/L | 21.9 [1.9, 39.1] | 7.6 [3.8, 24.8] | 0.98 |
| 2.3 [0.2,4.1] | 0.8 [0.4,2.6] | ||
| Lipid profile, mean (SD) except triglyceride which is median [interquartile range] | |||
| Total cholesterol, mmol/L mg/dL | 4.00 (0.80) | 4.29 (1.06) | 0.41 |
| 154.5 (30.7) | 165.6 (40.8) | ||
| Triglyceride, mmol/L mg/dL | 1.41 [0.80,2.12] | 1.39 [0.98,2.06] | 0.74 |
| 125.0 [71.0,188.0] | 123.0 [87.0,182.0] | ||
| HDL-C, mmol/L mg/dL | 1.20 (0.32) | 1.32 (0.32) | 0.30 |
| 46.3 (12.3) | 51.1 (12.2) | ||
| LDL-C, mmol/L mg/dL | 80.1 (23.2) | 87.3 (34.5) | 0.51 |
| 2.07 (0.60) | 2.26 (0.89) | ||
| Complete blood count, mean (SD) | |||
| White blood cells, 109 cells/L | 7.5 (2.4) | 6.1 (1.2) | 0.047 |
| Monocytes, cells/μL | 569.5 (180.5) | 485.6 (142.6) | 0.16 |
| Neutrophils, cells/μL | 5188.0 (1969.9) | 3728.6 (1032.5) | 0.014 |
| Lymphocytes, cells/μL | 1550.0 (724.0) | 1726.7 (332.3) | 0.39 |
| Platelets, cells/μL | 187.0 (65.3) | 210.9 (34.5) | 0.21 |
| Concomitant medication, n (%) | |||
| Statin | 15 (100.0) | 15 (93.8) | 0.33 |
| Aspirin | 14 (93.3) | 15 (93.8) | 0.96 |
| ACE-I | 8 (53.3) | 8 (50.0) | 0.85 |
| ARB | 2 (13.3) | 1 (6.3) | 0.51 |
| Hydrochlorothiazide | 2 (13.3) | 1 (6.3) | 0.51 |
| Furosemide | 1 (6.7) | 1 (6.3) | 0.96 |
| Calcium-channel blocker | 3 (20.0) | 3 (18.8) | 0.93 |
| Beta-blocker | 10 (66.7) | 9 (56.3) | 0.55 |
ACE-I, angiotensin converting enzyme inhibitors; ARB, angiotensin receptor blocker; CABG, coronary artery bypass graft surgery; HDL-C, high-density lipoprotein cholesterol; hs-CRP, high-sensitivity C-reactive protein; LDL-C, low- density lipoprotein cholesterol; PCI, percutaneous coronary intervention.
P values comparing proportions were calculated with either Chi-square test or Fisher’s exact.
P values comparing continuous variables were calculated using unpaired Student’s t-test or Mann-Whitney U test.
At 30-month follow-up (Table 2), the high omega-3 fatty acid index group had a significant reduction in triglyceride level compared to the low omega-3 fatty acid index group (median percent change, −18.3% versus +14.9%, respectively, between group P = 0.025) and a significant increase in lymphocyte count (median percent change, 6.3% versus −18.3% respectively, between group P = 0.008). Otherwise, there were no significant differences.
Table 2.
Characteristics and percent change at 30-month follow-up stratified by omega-3 fatty acid index group
| Low Omega-3 Fatty Acid Index (n = 15) 2.4% [2.3, 2.7] |
High Omega-3 Fatty Acid Index (n = 16) 8.4% [8.1, 9.6] |
||||
|---|---|---|---|---|---|
| 30-Month Absolute Value (IQR or SD) | Median % Change [IQR] at 30 months compared to Baseline | 30-Month Absolute Value (IQR or SD) | Median % Change [IQR] at 30 months compared to Baseline | Between group P value † | |
| BMI, kg/m2 | 31.2 (2.6) | 2.7 [−1.2, 5.4] | 31.0 (3.6) | −3.26 [−4.4, 4.0] | 0.097 |
| Blood Pressure, mm Hg, mean (SD) | |||||
| Systolic BP | 121.1 (14.2) | −5.8 [−11.9, 6.9] | 116.3 (10.1) | 3.3 [−7.1, 9.4] | 0.26 |
| Diastolic BP | 74.6 (7.7) | −3.5 [−10.6, 17.8] | 71.0 (8.7) | 0.0 [−4.5, 3.6] | 0.88 |
| Biochemical Profile, mean (SD) except hs-CRP which is median [interquartile range] | |||||
| Glucose, mmol/L mg/dL | 5.82 (1.98) | 1.0 [−4.3, 7.1] | 4.90 (0.49) | −1.58 [−4.1, 4.1] | 0.24 |
| 104.8 (35.7) | 88.3 (8.9) | ||||
| hs-CRP, nmol/L mg/L | 18.1 [6.7, 36.2] | 26.5 [−57.3, 56.5] | 8.6 [1.9, 30.5] | −48.39 [−66.0, −4.6] | 0.14 |
| 1.9 [0.7, 3.8] | 0.9 [0.2, 3.2] | ||||
| Lipid profile, mean (SD) except triglyceride which is median [interquartile range] | |||||
| Total cholesterol, mmol/L mg/dL | 4.13 (0.93) | 0.0 [−10.2, 18.4] | 4.21 (1.27) | −10.6 [−15.0, 6.1] | 0.28 |
| 159.3 (35.9) | 162.7 (49.2) | ||||
| Triglyceride, mmol/L mg/dL | 1.48 [1.10,2.59] | 14.9 [−14.4, 25.1] | 0.95 [0.76,1.47] | −18.3 [−31.0, −7.8] | 0.025 |
| 131.0 [97.0, 229.0] | 84.5 [67.5, 130.0] | ||||
| HDL-C, mmol/L mg/dL | 1.10 (0.26) | −5.3 [−10.6, 4.4] | 1.30 (0.27) | −5.7 [−17.2, 4.5] | 0.92 |
| 42.4 (9.9) | 50.3 (10.6) | ||||
| LDL-C, mmol/L mg/dL | 2.19 (0.90) | 16.3 [−18.0, 32.2] | 2.32 (1.01) | −8.8 [−23.0, 12.2] | 0.50 |
| 84.6 (34.7) | 89.4 (39.1) | ||||
| Complete blood count, mean (SD) | |||||
| WBC, 109 cells/L | 6.8 (2.7) | −6.9 [−21.4, 4.8] | 6.5 (1.7) | −0.62 [−6.0, 7.1] | 0.20 |
| Monocytes, cells/μL | 527.5 (154.3) | −6.29 [−24.3, 19.0] | 477.8 (157.2) | −0.8 [−10.4, 17.4] | 0.44 |
| Neutrophils, cells/μL | 4841.5 (2633.7) | −4.71 [−24.7, 8.3] | 3837.8 (1481.5) | −5.6 [−20.7, 7.6] | 0.70 |
| Lymphocytes, cells/μL | 1252.9 (655.0) | −18.33 [−32.4, −8.2] | 1954.9 (722.7) | 6.3 [−16.4, 29.4] | 0.008 |
P Value comparing % change between groups (low omega-3 fatty acid index compared to high omega-3 fatty acid index) at 30-month follow-up with Mann-Whitney U test.
BMI, body mass index; BP, blood pressure; HDL-C, high-density lipoprotein cholesterol; hs-CRP, high-sensitivity C-reactive protein; LDL-C, low-density lipoprotein cholesterol; WBC, white blood cell count.
3.2. High plasma omega-3 fatty acid index is associated with higher SPM levels in subjects with CAD
Representative MRM chromatograms of selected ion pairs for 18-HEPE, RvE1, MaR1 and 17-HDHA along with representative LC-MS/MS spectra and diagnostic ions employed for their identification are shown in Figure 1A. Table 3 reports that, compared to those with a low omega-3 fatty acid index, those with a high omega-3 fatty acid index had significantly higher levels of the pro-resolving and anti-inflammatory mediators, RvE1 (P = 0.015), 5-fold higher levels of the intermediary precursor of RvE1, 18-HEPE (P = 0.0016) and 2-fold higher levels of MaR1 (P = 0.003). As shown in Figure 1B (data shown in Supplemental Table 1), EPA levels were significantly directly correlated with their downstream intermediary, 18-HEPE, and its product, RvE1, and DHA levels were significantly directly correlated with its downstream intermediary, 17-HDHA, and downstream product, MaR1. Both EPA and DHA were significantly inversely correlated with PGD2.
Figure 1. Pro-resolving lipid mediator measurement and correlation with EPA, DHA and omega-3 fatty acid index.

(A) Representative Multiple Reaction Monitoring Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Chromatograms of Selected Ion Pairs for Lipid Mediators. 18-HEPE, resolvin E1, maresin 1 and 17-HDHA are reported along with representative MS/MS spectra and diagnostic ions employed for their identification; representative of n = 31. M/Z represents molecular mass divided by charge state.
(B) Heat map representing the Pearson correlation coefficients between the precursors, eicosapentaenoic acid, docosahexaenoic acid and omega-3 fatty acid index, and their downstream lipid mediator products at 30-month follow-up. Numerical data are in Supplemental Table 1.
17-HDHA, 17-hydroxy-docosahexaenoic acid; 18-HEPE, 18-hydroxy-eicosapentaenoic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; MaR1, maresin 1; PGD2, prostaglandin D2; RvE1, resolvin E1.
* P<0.05, **P<0.01, ***P<0.001, Pearson correlation.
TABLE 3.
Levels of lipid mediators according to omega-3 fatty acid index group
| Low Omega-3 Fatty Acid Index 2.4% [2.3, 2.7] (n = 15) | High Omega-3 Fatty Acid Index 8.4% [8.1, 9.6] (n = 16) | P value | |
|---|---|---|---|
| Docosahexaenoic acid metabolome, pg/ml, median [interquartile range] | |||
| Resolvin D1 | 10.3 [0.0,17.4] | 4.5 [1.0,10.3] | 0.45 |
| Resolvin D2 | −† | − | NA |
| Resolvin D3 | 0.8 [0.0,2.2] | 1.0 [0.0,1.7] | 0.47 |
| Resolvin D4 | − | − | NA |
| Resolvin D5 | − | − | NA |
| 17-HDHA, 17-hydroxy-docosahexaenoic acid ‡ | 152.0 [118.1,298.5] | 314.0 [204.6,329.7] | 0.10 |
| Protectin D1 | − | − | NA |
| 14-HDHA (14-hydroxy-docosahexaenoic acid) | 41.4 [22.4,289.6] | 87.4 [54.7,233.2] | 0.17 |
| Maresin 1 | 24.1 [0.0,41.3] | 47.0 [35.5,97.3] | 0.003 |
| Eicosapentaenoic acid metabolomes, pg/ml, median [interquartile range] | |||
| Resolvin E1 | 0.0 [0.0,0.0] | 1.1 [0.0,3.1] | 0.015 |
| 18-HEPE (18-hydroxy-eicosapentaenoic acid) § | 67.5 [42.1,79.0] | 341.2 [239.7,451.9] | 0.0016 |
| Arachidonic acid metabolomes, pg/ml, median [interquartile range] | |||
| Lipoxin A4 | − | − | NA |
| Lipoxin B4 | − | − | NA |
| Leukotriene B4 | 32.5 [19.2,76.9] | 21.1 [14.7,61.6] | 0.35 |
| Prostaglandin D2 | 4.9 [3.6,14.8] | 3.4 [1.2,3.6] | 0.19 |
| Prostaglandin E2 | 1.7 [0.0,27.6] | 1.2 [0.0,7.2] | 0.44 |
− Below limit of detection. Limit of detection ~ 0.1 pg/ml.
17-hydroxy-docosahexaenoic acid (17-HDHA) is the downstream product of docosahexaenoic acid and intermediary precursor of resolvin D1 to resolvin D5 and protectin D1.
18-hydroxy-eicosapentaenoic acid (18-HEPE) is the downstream product of eicosapentaenoic acid and intermediary precursor of resolvin E1.
P values were calculated using the Mann-Whitney U test.
NA, not applicable.
3.3. SPM levels correlate with change in coronary plaque volume in CAD patients
Coronary plaque volumes were measured using CCTA. A representative image of a coronary artery segment with plaque components is shown in Figure 2A. All omega-3 fatty acid levels and ratios were significantly inversely correlated with change of all plaque subtypes with EPA having the strongest inverse correlation with plaque volume (Figure 2B with data in Supplemental Table 2). The levels of 18-HEPE, RvE1 and MaR1 were significantly inversely correlated with change in total plaque, suggesting that pro-resolution capacity leads to coronary plaque regression. The proinflammatory mediator, PGD2, and the proinflammatory precursor, arachidonic acid, were significantly directly correlated with progression in total plaque, a finding consistent with their known proinflammatory effects. As expected, higher plasma omega-3 fatty acid levels were inversely associated with triglyceride levels. However, the change in triglyceride was not correlated with change in any of the plaque types (r = 0.238; P = 0.21 for total plaque) (data not in table).
Figure 2. Correlation between fatty acid levels, pro-resolving and proinflammatory lipid mediators and change in coronary plaque volume.
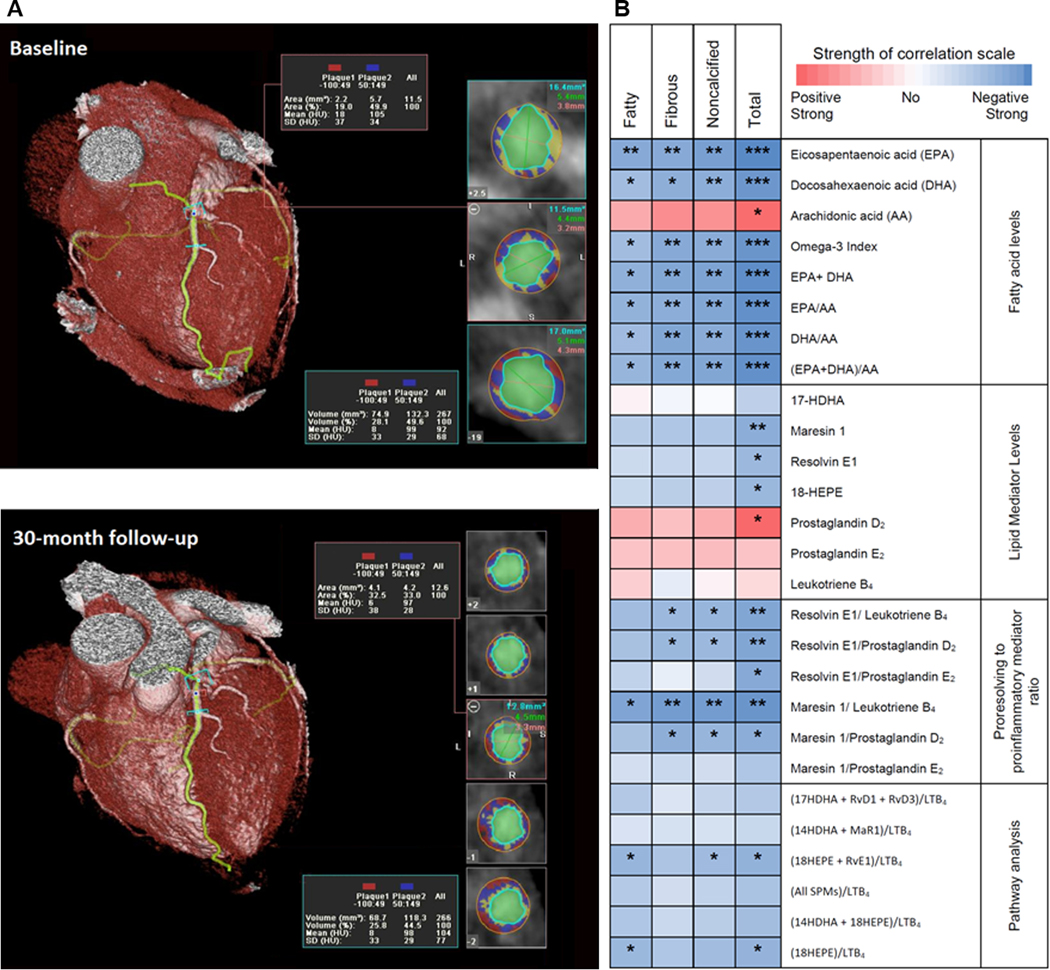
(A) Representative image of a coronary artery segment. The components of coronary plaque in a coronary computed tomographic angiogram were defined by Hounsfield Densities. Fatty plaque was between −100 and 49 HU (shown in red); fibrous plaque: 50 to 150 HU (shown in blue); calcified > 150 (shown in yellow). The vessel lumen is shown in green.
(B) Heat map representing Spearman correlation coefficients measuring the correlation between the absolute levels and ratios of fatty acid precursors and lipid mediators and their ratios with percent change in coronary plaque volume. Numerical data are shown in Supplemental Table 2.
17-HDHA, 17-hydroxy-docosahexaenoic acid; 18-HEPE,18-hydroxy-eicosapentaenoic acid;
AA, arachidonic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid.
* P<0.05, ** P<0.01, *** P<0.001, Spearman correlation.
To evaluate the balance between pro-resolution to proinflammatory lipid mediators, we calculated ratios of SPMs to the proinflammatory mediators, LTB4 and PGD2 (Figure 2B with data in Supplemental Table 2). Higher ratios of RvE1, MaR1 or 18-HEPE + RvE1 to the proinflammatory mediator, LTB4, were all significantly inversely associated with noncalcified and total plaque volume change over 30-months. In addition to a significant inverse correlation with noncalcified and total plaque volume change, the ratios of 18-HEPE + RvE1 to LTB4 or MaR1 to LTB4 were also significantly inversely correlated with change in fatty plaque, the type most likely to rupture and cause acute coronary syndrome. The MaR1/LTB4 ratio had the strongest inverse correlation with changes in plaque subtypes and was similar to that of EPA alone for total plaque (r =−0.61, P = 0.001).
3.4. When omega-3 fatty acid index is high, a high pro-resolution to proinflammatory ratio is associated with coronary plaque regression
Subjects with a low omega-3 fatty acid index had significant progression of all plaque types (Figure 3 with data in Supplemental Table 3). Subjects with a high omega-3 fatty acid index fell into two groups: Eleven subjects had significant regression of fatty, fibrous and noncalcified plaque volume whereas five had significant progression of fatty, fibrous, noncalcified and total plaque volume. Table 4 reports that those with a low omega-3 fatty acid index had plaque progression and a low ratio of (18-HEPE + RvE1)/LTB4 (median, 1.5 [IQR, 0.5 to 2.7]) compared to those with a high omega-3 fatty acid index who had plaque regression and a significantly higher ratio of (18-HEPE + RvE1)/LTB4 (median, 10.8 [IQR, 2.7, 20.8]) (between group P = 0.009). Next, we examined whether an imbalance of pro-resolution to proinflammatory lipid mediators determines plaque progression or regression among those with a high plasma omega-3 fatty acid index. The (18-HEPE + RvE1)/LTB4 ratio for the high omega-3 fatty acid index regressor group was significantly higher compared to the high omega-3 fatty acid index progressor group (median, 10.8 [IQR, 2.7, 20.8] vs 1.1 [IQR, 0.4, 2.1], respectively, between group P = 0.028) (Table 4). The (18-HEPE + RvE1)/LTB4 ratio was not significantly different for the low omega-3 fatty acid index progressors and high omega-3 fatty acid index progressors (P = 0.33). Neither RvE1 nor MaR1 alone distinguished between the high-index progressors and high-index regressors (Table 4). In contrast, the ratios of 18-HEPE or 18-HEPE+ RvE1 to the proinflammatory mediator, LTB4, were the only ratios to distinguish between the high-index progressors and high-index regressors. The MaR1/LTB4 ratio distinguished the low-index progressors from the high index regressors (P = 0.009) but not the high-index regressors and high-index progressors (P = 0.063). As seen in Supplemental Table 4, no difference was observed in baseline characteristics stratified by the 3 groups based on omega-3 fatty acid index and regression or progression of coronary plaque.
Figure 3. Percent change in coronary plaque at 30-month follow-up stratified by low and high omega-3 fatty acid index and regression or progression of plaque.

* P value compares within group change at 30 months compared to baseline and is 0.001 for all plaque subtypes. † P value compares within group change at 30 months compared to baseline and is 0.043 for all plaque subtypes.
Black bar indicates median value of percent change in plaque volume.
Within group P values were calculated using Wilcoxon signed-rank test and between group P values with Mann-Whitney U test.
FA, fatty acid.
TABLE 4.
Lipid mediator levels and specialized pro-resolving to proinflammatory lipid mediator ratios at 30-month follow-up stratified by regression or progression of fibrous and noncalcified plaque
| Low Omega-3 Fatty Acid Index 2.4% [2.3, 2.7] | High Omega-3 Fatty Acid Index 8.4% [8.1, 9.6] | ||||
|---|---|---|---|---|---|
| Plaque Progression (n = 15) | Plaque Regression (n = 11) | Plaque Progression (n = 5) | P value† | P value‡ | |
| Docosahexaenoic acid metabolomes, pg/ml, median [interquartile range] | |||||
| 17-hydroxy-docosahexaenoic acid (17-HDHA) § | 152.0 [118.1, 298.5] | 252.5 [189.1, 337.5] | 344.3 [321.8, 745.7] | 0.058 | 0.027 |
| Maresin 1 | 24.1 [0.0, 41.3] | 47.0 [27.6, 115.0] | 51.2 [46.3, 124.4] | 0.004 | 0.46 |
| Eicosapentaenoic acid metabolomes, pg/ml, median [interquartile range] | |||||
| Resolvin E1 | 0.0 [0.0, 0.0] | 1.1 [0.0, 4.7] | 3.7 [0.0, 7.7] | 0.009 | 0.525 |
| 18-hydroxy-eicosapentaenoic acid (18-HEPE) ¶ | 67.5 [42.1, 79.0] | 351.7 [238.9, 471.7] | 341.2 [247.1, 500.1] | 0.001 | 0.69 |
| Arachidonic acid metabolomes, pg/ml, median [interquartile range] | |||||
| Leukotriene B4 | 32.5 [19.2, 76.9] | 19.1 [17.1, 97.9] | 201.5 [0.0, 293.0] | 0.34 | 0.69 |
| Prostaglandin D2 | 4.9 [3.6, 14.8] | 1.9 [0.9, 3.7] | 1.4 [1.3, 6.9] | 0.004 | 1.00 |
| Prostaglandin E2 | 1.7 [0.0, 27.6] | 0.0 [0.0, 8.3] | 1.7 [1.6, 3.1] | 0.43 | 0.38 |
| Specialized pro-resolving to proinflammatory mediator ratios | |||||
| Resolvin E1/ Leukotriene B4 | 0.0 [0.0, 0.0] | 0.1 [0.0, 0.1] | 0.0 [0.0, 0.0] | 0.005 | 0.39 |
| Resolvin E1/Prostaglandin D2 | 0.0 [0.0, 0.0] | 0.6 [0.0, 1.9] | 0.3 [0.0, 1.1] | 0.005 | 0.52 |
| Resolvin E1/Prostaglandin E2 | 0.0 [0.0, 0.0] | 0.1 [0.0, 1.2] | 0.7 [0.1, 3.6] | 0.012 | 0.46 |
| Maresin 1/Leukotriene B4 | 0.1 [0.0, 0.7] | 2.3 [0.5, 2.7] | 0.2 [0.1, 0.4] | 0.009 | 0.063 |
| Maresin 1/Prostaglandin D2 | 1.4 [0.0, 5.0] | 19.9 [11.7, 41.4] | 22.0 [9.2, 34.3] | 0.002 | 0.64 |
| Maresin 1/Prostaglandin E2 | 1.4 [0.0, 16.2] | 12.9 [3.7, 13.0] | 29.5 [15.8, 39.4] | 0.26 | 0.62 |
| 18-HEPE/Leukotriene B4 | 1.5[0.5, 2.7] | 10.8 [2.6, 20.8] | 1.1 [0.4, 2.1] | 0.011 | 0.028 |
| Pathway Analysis | |||||
| (17-HDHA+RvD1+RvD3)/LTB4 | 4.1 [1.6, 6.3] | 10.1 [2.3, 18.4] | 1.9 [1.4, 8.5] | 0.082 | 0.24 |
| (14-HDHA+MaR1)/LTB4 | 1.4 [0.9, 4.6] | 6.2 [2.2, 15.5] | 0.6 [0.5, 0.8] | 0.17 | 0.063 |
| (18-HEPE+RvE1)/LTB4 | 1.5 [0.5, 2.7] | 10.8 [2.7, 20.8] | 1.1 [0.4, 2.1] | 0.009 | 0.028 |
P value for high-index regressors compared to low-index progressors with Mann-Whitney U test.
P value for high omega-3 fatty acid index regressors compared to high omega-3 fatty acid index progressors with Mann-Whitney U test.
17-hydroxy-docosahexaenoic acid is the downstream product of docosahexaenoic acid and intermediary precursor of resolvin D1 to resolvin D5 and protectin D1.
18-hydroxy-eicosapentaenoic acid (18-HEPE) is the downstream product of eicosapentaenoic acid and intermediary precursor of resolvin E1.
4. DISCUSSION
In the current study, we focused on the relationship between progression and regression of coronary artery plaque and production of bioactive lipid mediators from EPA and DHA. High-dose EPA and DHA supplementation, leading to a high plasma level of the omega-3 fatty acid index, significantly increased levels of their down-stream pro-resolving SPM products, RvE1 and MaR1, and the precursor of RvE1, 18-HEPE, in patients with CAD compared to those with a low omega-3 fatty acid index. In addition, levels of EPA were most strongly correlated with its downstream products, 18-HEPE and RvE1, and DHA was most strongly correlated with its downstream products, 17-HDHA and MaR1. The plasma omega-3 fatty acid index was strongly correlated with levels of 17-HDHA, 18-HEPE, RvE1 and MaR1. These findings are consistent with their known precursor/product relationships, supporting the contention that increasing plasma omega-3 fatty acids by dietary provision of EPA and DHA influences downstream production of their SPMs in humans over a 30-month period. Both EPA and DHA were inversely correlated with PGD2, a finding suggesting that EPA and DHA also lower proinflammatory mediators in CAD patients.
Subjects with a low plasma omega-3 fatty acid index had low SPM levels leading to a low ratio of the SPM pathway derived from EPA, 18-HEPE + RvE1, to the proinflammatory mediator, LTB4; these subjects had significant progression of coronary plaque. In contrast, all subjects who attained a high plasma omega-3 fatty acid index had high levels of the downstream products of EPA and DHA, the SPMs, as would be expected, but they fell into two groups when plaque change was examined. The majority had low levels of the proinflammatory mediators leading to a high (18-HEPE + RvE1)/ LTB4 ratio; these subjects had significant plaque regression. However, several subjects in the high omega-3 fatty acid index group had high levels of proinflammatory mediators, leading to a low ratio of SPM/LTB4 - either (18-HEPE + RvE1)/ LTB4 or MaR1/LTB4 or RvE1/LTB4; these subjects had significant progression of plaque. The (18-HEPE + RvE1)/ LTB4 ratio was the only SPM ratio to distinguish the high omega-3 fatty acid index regressors from the high omega-3 fatty acid index progressors. Consequently, the (18-HEPE + RvE1)/ LTB4 ratio could potentially be used as a blood diagnostic marker to predict progression or regression of coronary plaque. SPM levels are high in the high omega-3 fatty acid index group who had plaque progression; therefore, the reason for the low SPM/LTB4 ratio is a persistently high level of the proinflammatory mediator, LTB4. The reason for a high level of proinflammatory mediator is unknown but deserves further study since it may account for the residual CVD risk seen in some subjects on statin therapy. Whether even higher intakes of EPA and DHA would be necessary to lower LTB4 would require further study.
The higher ratio of SPM to proinflammatory mediators with EPA and DHA supplementation in the high omega-3 fatty acid index group who had plaque regression suggests a shift in the balance toward pro-resolving over proinflammatory mediators such that progression of coronary plaque is prevented and in fact, at highest levels, regression is seen. These findings highlight the importance of the pro-resolution to proinflammatory balance which may mediate the effect of an elevated plasma omega-3 fatty acid index on the change in coronary plaque volume. The current findings show that the 18-HEPE and RvE1 pathway may be an important determinant of response to elevated omega-3 fatty acid levels when proinflammatory mediators are taken into account. The value of measuring both SPMs and proinflammatory mediators offers an explanation as to mechanisms and affords a better measure of the likelihood of progression or regression of coronary plaque than do EPA and DHA levels. The benefit in the current study occurred in the setting of well-controlled LDL-C levels on statin therapy and low hs-CRP levels,16 findings which support a benefit for a favorable ratio of SPM/LTB4 on coronary plaque progression independent of LDL-C reduction and hs-CRP. Moreover, the SPM/LTB4 ratio may be a measure of residual inflammation in the setting of low levels of hs-CRP.
The current results are supported by data from isolated human cells and animal studies which support a role of the downstream products of EPA and DHA, the 18-HEPE + RvE1 pathway and MaR1, in resolving inflammation and reducing atherosclerosis. Laguna-Fernandez et al.25 demonstrated that RvE1 signaling via its receptor, ERV1/ChemR23, exerts protection in atherosclerosis by decreasing uptake of oxidized LDL by macrophages and enhances macrophage phagocytosis, thereby, offering protection against lesion development and necrotic core formation. In RvE1 receptor knockout mice, there was an increase in pro-atherogenic macrophages, increased oxidized LDL and reduced phagocytosis, findings which resulted in increased atherosclerotic plaque size and plaque necrotic core formation.25 Both RvE1 and its precursor, 18-HEPE, are anti-inflammatory and stop polymorphonuclear migration31 and promote polymorphonuclear apoptosis and phagocytic removal of apoptotic cells, thereby, stimulating efferocytosis and resolution of inflammation.27,28 RvE1 downregulates leukocyte adhesive molecules (i.e., CD11/ CD18) and ADP-dependent platelet activation,29,30 blocks IL-12 production,31 reduces tumor necrosis factor alpha (TNF-α), IL-1β and IL-626 and inhibits migration of vascular smooth muscle cells, thereby, providing potential mechanisms for inhibition of plaque progression. RvE1 and RvE2 each potently stimulate IL-10 and macrophage phagocytosis.26,32 In apoE*3 Leiden mice, both low- and high-dose RvE1 supplementation reduced the size of atherosclerotic lesions by 35% (P<0.05) and attenuated the formation of severe lesions in the absence of change in cholesterol levels and provided additional benefit to atorvastatin treatment.33 When administered topically to the periodontium at the onset of high-fat, high-cholesterol diet feeding in rabbits, RvE1 decreased aortic plaque formation, a finding suggesting that higher levels of RvE1 early on may be important for delaying atherogenesis.34 RvE1 through its receptor, ChemR23, has also been shown to reduce intimal hyperplasia35,36 and diminish vascular calcification by lowering the expression of bone morphogenetic protein 2 in vascular smooth muscle cells.37
In cultured human saphenous vein endothelial cells and human vascular smooth muscle cells, MaR1 decreased TNF-α induced monocyte adhesion and production of reactive oxygen species through up-regulation of cyclic adenosine monophosphate and down-regulation of NF-ĸB in a time-dependent manner.38, reviewed in 39 When apoE −/− mice were fed a high fat diet, levels of resolving lipid mediators, MaR1 and RvD2, decreased as atherosclerosis progressed. LTB4 and PGE2 directly correlated with plaque vulnerability whereas RvD2 and MaR1 were associated with plaque stability.40 Supplementation with MaR1 and RvD2 prevented progression of atherosclerosis. Compromised clearance mechanisms at the plaque level may be a major contributing factor to plaque build-up and instability, suggesting a local imbalance between proinflammatory events and counter-acting resolution mechanisms of the immune system, a finding suggesting that resolution may be dysfunctional.10 In support of this concept, SPMs were higher in stable versus vulnerable plaque regions from human carotid arteries.41 In line with animal studies, the current findings in humans with CAD show that low SPM levels and a low ratio between pro-resolving and proinflammatory lipid mediators may serve as a biomarker of non-resolving inflammation underlying atherosclerosis. Furthermore, these findings suggest that impaired resolution of inflammation manifested as an imbalance of pro-resolving and proinflammatory lipid mediators may contribute to the progression of coronary atherosclerosis in CAD patients with well-controlled LDL-C levels on statin therapy
We previously reported significantly lower rates of intercurrent infections seen in those taking high-dose EPA and DHA compared to controls.18 EPA and DHA through resolvins have been shown to enhance bacterial killing42 which could account for the lower rates of infections observed in those receiving EPA and DHA in the HEARTS trial.18 A prior trial of canakinumab, a monoclonal antibody against IL-1β, showed benefit with lowering inflammation on cardiovascular events.43 However, a significant increase in death from infections was noted due to immunosuppression from canakinumab. In contrast, pro-resolving mediators have the advantage of resolving inflammation without compromising host-defense.11 Our data suggest that high-dose supplementation with EPA and DHA can modify the inflammatory response via increased production of SPMs that act as resolving mediators that modulate, rather than block, a pathway, and consequently results in decreased production of proinflammatory mediators, thus decreasing the atherosclerotic inflammatory process by promoting the resolution phase and terminating inflammation without causing immunosuppression.
In conclusion, the current results suggest that stimulation of the resolution pathway of inflammation by the downstream products of EPA and DHA, the SPMs, may prevent plaque progression in human subjects with CAD with well-controlled LDL-C levels on statin therapy. The results also suggest that a balance of downstream pro-resolving and proinflammatory lipid mediators must be maintained in order to attenuate and reduce plaque growth. As such, a low ratio of SPM to proinflammatory mediator, specifically (18-HEPE+RvE1)/LTB4, is a potential novel risk factor associated with coronary plaque progression and could potentially be used as a blood biomarker to predict progression. Thus, increased SPM production with high-dose omega-3 fatty acid supplementation may provide additional benefit and modify risk for CAD in patients with well-controlled LDL-C levels on statin treatment and may account for lower residual risk of CVD events in trials of omega-3 fatty acid supplementation in statin-treated patients. Examining new strategies such as dietary supplementation of high-dose EPA and DHA to increase SPMs to target the inflammatory response is a new therapeutic approach to treating CVD in addition to aspirin, statins and anti-platelet agents. Thus, new strategies to increase SPMs or the SPM/LTB4 ratio to promote resolution of inflammation may offer unique opportunities to combat chronic inflammation associated with CVD.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health P50HL083813 (Dr. Welty), Harvard Clinical and Translational Science Center Award NIH UL1 TR001102 (Dr. Welty) and Forsyth Pilot Grant FPILOT47 (Drs. Schulte and Hardt). The Forsyth Center for Salivary Diagnostics was funded by the Massachusetts Life Science Center.
Data presented at the American Heart Association Annual Scientific Sessions, November 11, 2018, in Chicago, Illinois.
Nonstandard abbreviations:
- 14-HDHA
14-hydroxy-4Z,7Z,10Z,12E,16Z,19Z-docosahexaenoic acid
- 17-HDHA
17-hydroxy-4Z, 7Z, 10Z, 13Z, 15E, 19Z-docosahexaenoic acid
- 18-HEPE
18-hydroxy-5Z, 8Z, 11Z, 14Z, 16E-eicosapentaenoic acid
- 5S,15S-diHETE
5S,15S-dihydroxy-eicosatetraenoic acid
- AA
arachidonic acid
- ACE
angiotensin-converting enzyme
- CAD
coronary artery disease
- CVD
cardiovascular disease
- CCTA
coronary computed tomographic angiography
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- GISSI
Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto
- HbA1c
hemoglobin A1c
- HDL-C
high-density lipoprotein cholesterol
- LC-MS-MS
liquid chromatography-tandem mass spectrometry
- LDL-C
low-density lipoprotein cholesterol
- LM
lipid mediators
- LT
leukotrienes
- LTB4
leukotriene B4 (5S,12R-dihydroxy-eicosa-6Z, 8E,10E,14Z-tetraenoic acid)
- LX
AA-derived lipoxin
- LXA4
lipoxin A4 (5S,6R,15S-trihydroxy-eicosa- 7E,9E,11Z,13E-tetraenoic acid)
- LXB4
lipoxin B4 (5S,14R,15S-trihydroxy-eicosa-6E,8Z,10E,12E-tetraenoic acid)
- MaR
maresins
- MaR1
maresin 1 (7R,14S-dihydroxy-docosa- 4Z,8E,10E,12Z,16Z,19Z-hexaenoic acid)
- PD
protectins
- PD1
protectin D1 (10R,17S-dihydroxy-docosa- 4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid)
- PG
prostaglandins
- PGD2
prostaglandin D2 (11-oxo-9, 15S-dihydroxy-prosta-5Z, 13E-dien-1-oic acid)
- PGE2
prostaglandin E2 (9-oxo-11,15S-dihydroxy-prosta-5Z,13E-dien-1-oic acid)
- PGF2α
prostaglandin F2α (9,11,15S-trihydroxy-prosta-5Z,13E-dienoic acid)
- RvD
DHA-series resolvins
- RvD1
resolvin D1 (7S,8R,17S-trihydroxy-docosa-4Z,9E,11E,13Z,15E,19Z-hexaenoic acid)
- RvD2
resolvin D2 (7S,16R,17S-trihydroxydocosa-4Z,8E,10Z,12E,14E,19Z-hexaenoic acid)
- RvD3
resolvin D3 (4S,11R,17S-trihydroxydocosa-5Z,7E,9E,13Z,15E,19Z-hexaenoic acid)
- RvD5
resolvin D5 (7S,17S-dihydroxy-docosa- 4Z,8E,10Z,13Z,15E,19Z-hexaenoic acid)
- RvE
EPA-series resolvins
- RvE1
resolvin E1 (5S,12R,18R-trihydroxy-eicosa-6Z,8E,10E,14Z,16E-pentaenoic acid)
- SPM(s)
specialized pro-resolving lipid mediator(s)
- Tx
thromboxane
- TxB2
thromboxane B2 (9,11,15S-trihydroxythromba-5Z,13E-dien-1-oic acid)
Footnotes
CONFLICT OF INTEREST
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
REFERENCES
- 1.Sampson UK, Fazio S, Linton MF. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atheroscler Rep. 2012;14:1–10. doi: 10.1007/s11883-011-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bayturan O, Kapadia S, Nicholls SJ, Tuzcu EM, Shao M, Uno K, Shreevatsa A, Lavoie AJ, Wolski K, Schoenhagen P, Nissen SE. Clinical predictors of plaque progression despite very low levels of low-density lipoprotein cholesterol. J Am Coll Cardiol. 2010;55:2736–2742. doi: 10.1016/j.jacc.2010.01.050. [DOI] [PubMed] [Google Scholar]
- 3.Nicholls SJ, Hsu A, Wolski K, Hu B, Bayturan O, Lavoie A, Uno K, Tuzcu EM, Nissen SE. Intravascular ultrasound-derived measures of coronary atherosclerotic plaque burden and clinical outcome. J Am Coll Cardiol. 2010;55:2399–2407. doi: 10.1016/j.jacc.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr, Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM; REDUCE-IT Investigators. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med. 2019. January 3;380:11–22. doi: 10.1056/NEJMoa1812792. Epub 2018 Nov 10. [DOI] [PubMed] [Google Scholar]
- 5.Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114:1867–1879. doi: 10.1161/CIRCRESAHA.114.302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12: 204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 7.Bäck M, Yurdagul A, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samuelsson B. Role of basic science in the development of new medicines: examples from the eicosanoid field. J Biol Chem. 2012;28:10070–10080. doi: 10.1074/jbc.X112.351437. Epub 2012 Feb 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haeggström JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. Epub 2011 Sep 22. [DOI] [PubMed] [Google Scholar]
- 10.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10: 36–46. doi: 10.1038/nri2675. Epub 2009 Dec 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014; 510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 14.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. doi: 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128(7):2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al Faddagh A, Elajami TK, Saleh M, Mohebali D, Bistrian B, Welty FK. An omega-3 fatty acid plasma index ≥4% prevents progression of coronary artery plaque in patients with coronary artery disease on statin treatment. Atherosclerosis. 2019. June;285:153–162. doi: 10.1016/j.atherosclerosis.2019.04.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elajami TK, Colas RA, Dalli J, Chiang N, Serhan CN, Welty FK. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016. April 27;30:2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alfaddagh A, Elajami TK, Ashfaque H, Saleh M, Bistrian BR, Welty FK. Effect of Eicosapentaenoic and Docosahexaenoic Acids Added to Statin Therapy on Coronary Artery Plaque in Patients with Coronary Artery Disease: A Randomized Clinical Trial. J Am Heart Assoc. 2017; 6: e006981. 10.1161/JAHA.117.006981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khosa F, Khan AN, Nasir K, Bedayat A, Malik Z, Jon AF, Cheema AR, Clouse ME, Welty FK. Comparison of coronary plaque subtypes in male and female patients using 320-row MDCTA. Atherosclerosis. 2013;226:428–432. doi: 10.1016/j.atherosclerosis.2012.11.033. Epub 2012 Dec 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauser TH, Salastekar N, Schaefer EJ, Desai T, Goldfine HL, Fowler KM, Weber GM, Welty F, Clouse M, Shoelson SE, Goldfine AB. Targeting Inflammation Using Salsalate in Cardiovascular Disease (TINSAL-CVD) Study Team. Effect of Targeting Inflammation With Salsalate: The TINSAL-CVD Randomized Clinical Trial on Progression of Coronary Plaque in Overweight and Obese Patients Using Statins. JAMA Cardiol. 2016;1:413–423. doi: 10.1001/jamacardio.2016.0605. [DOI] [PubMed] [Google Scholar]
- 21.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol. 2014;307:C39–54. doi: 10.1152/ajpcell.00024.2014. Epub 2014 Apr 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang R, Chiang N, Oh SF, Serhan CN. Metabolomics-lipidomics of eicosanoids and docosanoids generated by phagocytes. Curr Protoc Immunol. 2011;Chapter 14: Unit 14.26. doi: 10.1002/0471142735.im1426s95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parashar K, Schulte F, Hardt M, Baker O.J. Sex-mediated elevation of the specialized pro-resolving lipid mediator levels in a Sjögren’s syndrome mouse model. FASEB J. 2020;34:7733–7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norris PC, Skulas-Ray AC, Riley I, Richter CK, Kris-Etherton PM, Jensen GL, Serhan CN, Maddipati KR. Identification of specialized pro-resolving mediator clusters from healthy adults after intravenous low-dose endotoxin and omega-3 supplementation: a methodological validation. Sci Rep. 2018. December 21;8(1):18050. doi: 10.1038/s41598-018-36679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laguna-Fernandez A, Checa A, Carracedo M, Artiach G, Petri MH, Baumgartner R, Forteza MJ, Jiang X, Andonova T, Walker ME, Dalli J, Arnardottir H, Gisterå A, Thul S, Wheelock CE, Paulsson-Berne G, Ketelhuth DFJ, Hansson GK, Bäck M. ERV1/ChemR23 Signaling Protects Against Atherosclerosis by Modifying Oxidized Low-Density Lipoprotein Uptake and Phagocytosis in Macrophages. Circulation. 2018;138:1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Invest. 2011;121:569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192:1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109:14983–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A, Cheng G, von Andrian UH, Serhan CN. Resolvin E1, an EPA-derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood. 2008;112:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fredman G, Serhan CN. Specialized proresolving mediator targets for RvE1 and RvD1 in peripheral blood and mechanisms of resolution. Biochem J. 2011;437:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN. Stereochemical assignment, anti-inflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005;201:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh SF, Dona M, Fredman G, Krishnamoorthy S, Irimia D, Serhan CN. Resolvin E2 formation and impact in inflammation resolution. J Immunol. 2012;188:4527–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salic K, Morrison MC, Verschuren L, Wielinga PY, Wu L, Kleemann R, Gjorstrup P, Kooistra T. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis. 2016;250:158–165. doi: 10.1016/j.atherosclerosis.2016.05.001. Epub 2016 May 2. [DOI] [PubMed] [Google Scholar]
- 34.Hasturk H, Abdallah R, Kantarci A, Nguyen D, Giordano N, Hamilton J, Van Dyke TE. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35:1123–1133. doi: 10.1161/ATVBAHA.115.305324. Epub 2015 Mar 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu G, Gong Y, Zhang R, Piao L, Li X, Liu Q, Yan S, Shen Y, Guo S, Zhu M, Yin H, Funk CD, Zhang J, Yu Y. Resolvin E1 attenuates injury-induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J. 2018;32:5413–5425. [DOI] [PubMed] [Google Scholar]
- 36.Artiach G, Carracedo M, Clària J, Laguna-Fernandez A, Bäck M. Opposing Effects on Vascular Smooth Muscle Cell Proliferation and Macrophage-induced Inflammation Reveal a Protective Role for the Proresolving Lipid Mediator Receptor ChemR23 in Intimal Hyperplasia. Front Pharmacol. 2018; November 20;9:1327. doi: 10.3389/fphar.2018.01327. eCollection 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carracedo M, Artiach G, Witasp A, Clària J, Carlström M, Laguna-Fernandez A, Stenvinkel P, Bäck M. The G-protein coupled receptor ChemR23 determines smooth muscle cell phenotypic switching to enhance high phosphate-induced vascular calcification. Cardiovasc Res. 2019;115:1557–1566. [DOI] [PubMed] [Google Scholar]
- 38.Chatterjee A, Sharma A, Chen M, Toy R, Mottola G, Conte MS. The pro-resolving lipid mediator maresin 1 (MaR1) attenuates inflammatory signaling pathways in vascular smooth muscle and endothelial cells. PLoS One. 2014;9:e113480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conte MS, Desai TA, Wu B, Schaller M, Werlin E. Pro-resolving lipid mediators in vascular disease. J Clin Invest. 2018. August 31;128:3727–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Döring Y, Drechsler M, Weber C, Zimmer R, Cenac N, Soehnlein O. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. 2016;119:1030–1038. [DOI] [PubMed] [Google Scholar]
- 41.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and proinflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016; September 23;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012. April 25;484(7395):524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131.doi: 10.1056/NEJMoa1707914. Epub 2017 Aug 27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.