

Abstract
Background –
Glycemic control is a strong predictor of long-term cardiovascular risk in patients with diabetes mellitus, and poor glycemic control influences long-term risk of cardiovascular disease even decades after optimal medical management. This phenomenon, termed glycemic memory, has been proposed to occur due to stable programs of cardiac and endothelial cell gene expression. This transcriptional remodeling has been shown to occur in the vascular endothelium through a yet undefined mechanism of cellular reprogramming.
Methods:
In the current study, we quantified genome-wide DNA methylation of cultured human endothelial aortic cells (HAECs) via reduced-representation bisulfite sequencing (RRBS) following exposure to diabetic (250 mg/dL), pre-diabetic (125 mg/dL), or euglycemic (100mg/dL) glucose concentrations for 72 hrs (n = 2).
Results:
We discovered glucose-dependent methylation of genomic regions (DMRs) encompassing 2,199 genes, with a disproportionate number found among genes associated with angiogenesis and nitric oxide (NO) signaling-related pathways. Multi-omics analysis revealed differential methylation and gene expression of VEGF (↑5.6% DMR, ↑3.6-fold expression), and NOS3 (↓20.3% DMR, ↓1.6-fold expression), nodal regulators of angiogenesis and NO signaling, respectively.
Conclusion:
In the current exploratory study, we examine glucose-dependent and dose-responsive alterations in endothelial DNA methylation to examine a putative epigenetic mechanism underlying diabetic vasculopathy. Specifically, we uncover the disproportionate glucose-dependent methylation and gene expression of VEGF and NO signaling cascades, a physiologic imbalance known to cause endothelial dysfunction in diabetes. We therefore hypothesize that epigenetic mechanisms encode a glycemic memory within endothelial cells.
Keywords: Whole-genome DNA methylation, glycemic memory, epigenomics, computational biology, type 2 diabetes mellitus, diabetic cardiomyopathy
Introduction
Diabetes mellitus (DM) affects roughly 10% of the global population, and nearly two-thirds of these patients dying from cardiovascular (CVD)-related complications [1, 2]. Independent of comorbid illnesses, DM confers a four-fold increased risk of CVD mortality. Although DM is a multifactorial disease, the cardiovascular risk of DM correlates with degree of glycemic control [3–5], and growing body of evidence supports that transient hyperglycemia increases susceptibility for CVD long after the initial insult, a phenomenon termed “glycemic memory” [2, 6–8]. Although the precise molecular mechanisms are poorly understood, mounting interest has shifted to uncovering the epigenetic contribution of these metabolic memories [9–11].
Dysregulation of the vascular endothelium is a common underlying factor in the pathogenesis of DM-associated end-organ complications. Endothelial dysfunction (ED) refers to a physiologic state in which alterations to the systemic vasculature promote thrombosis and pro-atherosclerotic changes to increase likelihood of plaque rupture [12, 13]. Transient hyperglycemia is a known potent cytotoxic stressor capable of acutely damaging the vascular endothelium through several known processes, including formation of extracellular Advance Glycation End-products (AGEs) [14], profound metabolic perturbations [15], dysautonomia, immunologic disruption and downregulation of endothelial progenitor cell number via SIRT1 [16]. Our laboratory has previously shown that tight glycemic control mitigates the adverse effects of DM on circulating endothelial progenitor cells to restore their bioavailability [17]. Interestingly, the formation both of AGEs and glycoxidized low-density lipoprotein (LDL) downregulates endothelial nitric oxide synthase in human coronary cells perhaps contributing to ED [18]. However, the precise mechanism by which a glucose-dependent disruption of endothelial function is potentiated remains unknown.
The persistence of DM-associated CVD susceptibility has led to the search for epigenetic mechanisms of glucose-mediated endothelial dysfunction. DNA methylation is an epigenetic modification directly to DNA that is associated with alterations to genomic architecture and gene accessibility. Several studies have proposed endothelial DNA methylation as a molecular determinant of “glycemic memory,” particularly as the early molecular underpinnings of vascular damage [19, 20], leading also to diabetic end-organ complications including diabetic nephropathy and retinopathy [10, 21–27]. However, a genome-wide analysis of glucose-sensitive DNA methylation in the human endothelium has not yet been performed.
The current preliminary study therefore examines the impact of elevated glucose concentrations on genome-wide DNA methylation in human aortic endothelial cells (HAECs). Using reduced-representation bisulfite sequencing analysis of human aortic endothelial cells (HAECs) exposed to varying concentrations of glucose, we find that elevated glucose concentrations disproportionately affect genomic methylation at intronic regions of genes involved in angiogenesis, endothelin signaling, and PI3K/AKT-mediated insulin signaling. Specifically, we implicate differential methylation as a putative underlying mechanism of the physiologic uncoupling between VEGF and NO signaling pathways. As a result, this pilot study supports ongoing efforts to characterize the epigenetic modifications that may contribute to endothelial glycemic memory.
Methods
Ethics Statement.
Stable human aortic endothelial cells were obtained from Lonza© (CC-2535, Basel, Switzerland), which have been collected from patients who tested negative for mycoplasma, bacteria, yeast, and fungi. HIV-1, hepatitis B and hepatitis C. All human genome-wide DNA methylation data have been uploaded to the NCBI Gene Expression Omnibus database for public access and reanalysis (GSE163510). The protocol of the study was approved by the Investigation Review Board of IRCCS SDN, Naples, Italy.
Cell culture and glucose treatment.
Human aortic endothelial cells (HAECs) were obtained from Lonza, cultured in euglycemic (5.55mM) medium EGM-2MV bullet kit (Lonza) supplemented with 10% FBS and antibiotics and maintained in a 5% CO2 incubator at 37°C in a humidified atmosphere. When reaching 80–90% confluence, cells were detached with 0.5 mM EDTA/0.05% trypsin (Gibco) for 5 minutes at 37°C and then re-plated, as previously described [28, 29].
D-Glucose was purchased from Sigma-Aldrich (Milan, Italy). HAECs were seeded at a cell density of 5 × 103 cells/cm2. Cell number was determined using a Burker chamber, according to the manufacturer’s instructions and cell viability was evaluated using Trypan Blue vital dye. After cell attachment, the medium was changed to fresh culture medium containing 125 mg/dL and 250 mg/dL D-Glucose. After 48 hours, the D-glucose containing culture medium was refreshed for another 24 hours (total treatment 72 hours). Each experiment was conducted in duplicate following the third passage. Absence of mycoplasma contamination was confirmed by PCR with specific primers.
DNA extraction.
Genomic DNA was isolated from HEACs treated with 125 mg/dL and 250 mg/dL D-glucose using DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s instructions. DNA concentration and purity were determined using NanoDrop 2000 (Thermo Scientific). DNA integrity was verified using electrophoresis on 1% agarose gel.
Reduced-representation bisulfite sequencing (RRBS).
Sequencing experiments were performed at the Genomix4Life S.r.l. (Italy) with subsequent bioinformatics performed at the University of Alabama at Birmingham (USA), as described [30]. Briefly, 1 µg of genomic DNA were used for each library preparation. Each DNA sample was digested by MspI restriction enzyme. The digested products were purified with the GeneJet PCR Purification Kit (Thermo Fisher Scientific) and libraries were prepared by TruSeq Library Prep Kit (Illumina, USA). Fragments were bisulfite converted using the EZ DNA Methylation-Gold Kit (Zymo Research, USA). The converted DNA was amplified using PfuTurbo Cx Hotstart DNA Polymerase (Agilent Technologies, USA). The amplified fragments were purified by AMPure XP Beads and further quantified by the Agilent 4200 TapeStation (Agilent Technologies, USA). Each DNA library was analyzed by paired-end sequencing read (2 × 75 cycles) on Illumina Nextseq 500.
Bioinformatic analysis and data visualization.
A detailed description of the analysis performed, including coding scripts, is available as an online supplement and GitHub data repository: https://github.com/mepepin/Glycemic.Memory.HAECs. To assess RRBS quality, FastQC (0.11.9) was used both before and after adapter trimming (PHRED < 30) via TrimGalore (0.4.4). The bisulfite-reduced and sequenced reads were then aligned to the CT- and GA-converted human hg38 (GRCh38.p12) genome assembly via BWA-meth [31] to quantify relative alignment of methylated and unmethylated CpGs, respectively [32]. The alignment yielded a mapping efficiency of 98.4±0.2% (32.9±7.2 million) paired-end reads following quality trimming and alignment. Differential DNA methylation was computed using 500-base windows to exploit the regional CpG methylation analysis afforded by RRBS using the R package methylKit (1.16.0). Statistical significance of differential-methylation was assumed based on an over-correction adjusted Chi-squared test, as recommended by Wreczycka et al. [33]. To adjust P-values for multiple testing, a post hoc sliding linear model (SLIM) method was applied [34].
Gene-set enrichment and response element identification.
Computational enrichment analysis was performed using Enrichr [35] to identify biological pathways which are disproportionately affected by glucose-dependent differential methylation. The PANTHER pathway database [36] was parsed using genes containing differentially-methylated promoters (DMRs) at a statistical threshold of Q < 0.05. Pathways were then sorted by P-value significance per the Fisher’s exact test. Heatmap and hierarchical clustering generation was performed using pheatmap package (1.0.8) in R (4.0.3). Unless otherwise indicated, statistical significance of RRBS data was determined via unpaired two-tailed Bonferroni-adjusted P-value (Q) < 0.05.
Results
Dose-responsive regions of differential methylation in HAECs.
Quantification of differential methylation identified 2,605 genomic regions (DMRs) (Q < 0.05) found to be in association with 2,199 known genes that exhibit differential methylation (Table S1). Hierarchical clustering and heatmap visualization demonstrated a clustering of HAEC samples treated with glucose, with sub-grouping according to dosing (Fig. 1A).
Figure 1: Differential DNA methylation is glucose-responsive in human endothelial cells.
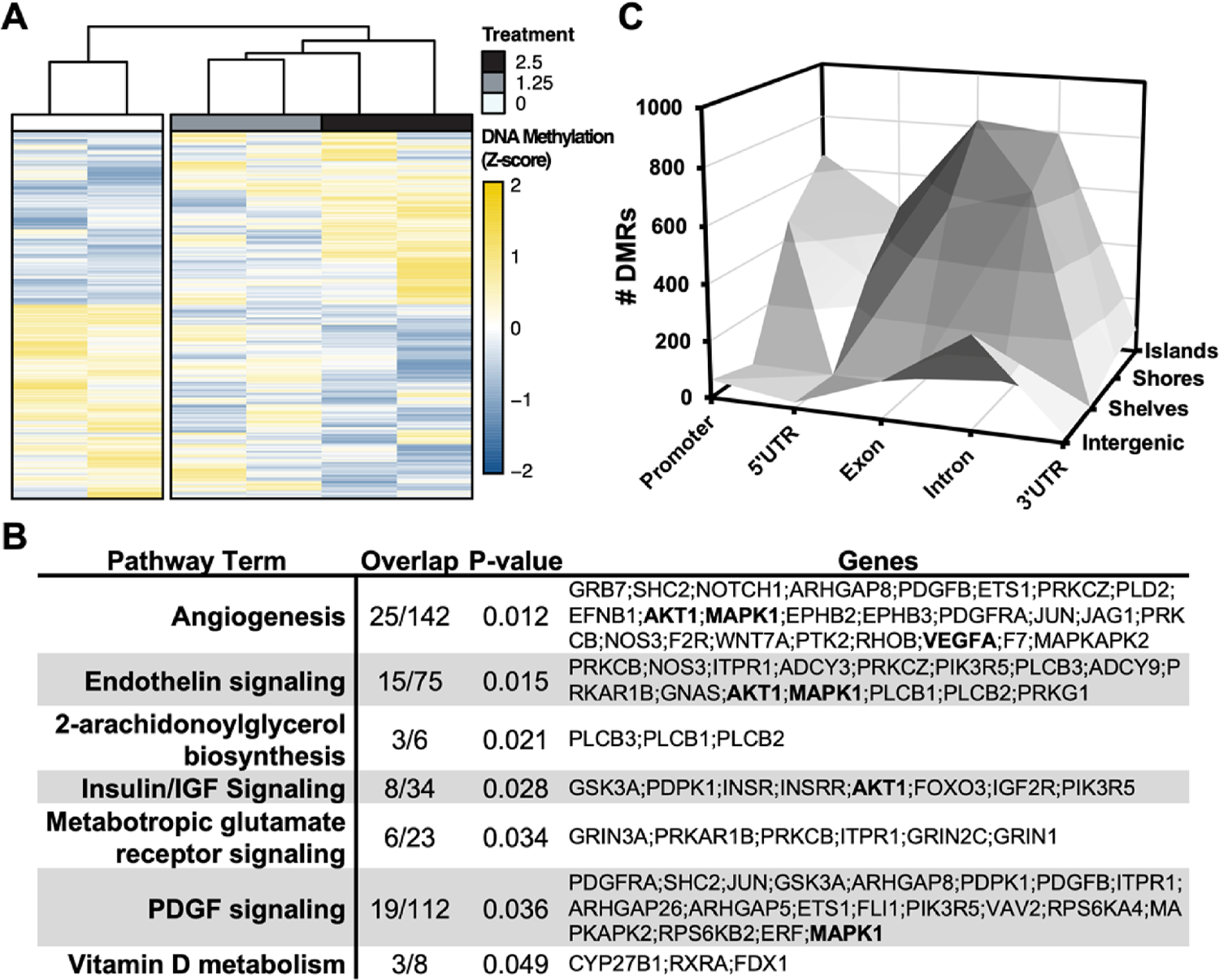
A. Hierarchical clustering via Wald.D2 Test and dendrogram constructed by Euclidean distance with heatmap visualization of differentially methylated regions (DMRs)*. B. 3-dimensional bar plot depicting the distribution of DMRs according to both genic annotation (promoter, intron, exon, and 5’ untranslated regions) and proximity to CpG Islands (CpG Island, Shore, and Shelf). *statistical significance of DMRs assumed at Q < 0.05
Owing to the site-specific nature of cytosine DNA methylation on transcriptional activity [37, 38], differentially-methylated regions (DMRs ,Q < 0.05) were mapped onto both genetic regions (promoter, 5’UTR, gene body, and 3’UTR) and according to proximity from CpG-rich genomic territories termed “CpG Islands” (Fig. 1B, Table S2). DMRs were found to disproportionately occur within intronic and exonic CpG islands, suggesting that the glucose-responsive CpG methylation dynamics predominantly occur in the gene body. Gene body methylation may directly correlate with endothelial gene expression in the setting of diabetes mellitus, as has been described elsewhere [39].
To understand the ontological networks most affected by glucose-dependent alterations in DNA methylation, gene-set enrichment analysis (GSEA) was performed on DMRs using the PANTHER database [40]. Several canonical diabetes-associated pathways were found to be enriched, among which “angiogenesis” (18% enriched, P = 0.01), “endothelin signaling” (20% enriched, P = 0.02), and “Insulin/IGF signaling” (24% enriched, P = 0.03) were present among the five most-enriched pathways (Fig. 2, Table S3). Inspection of the genes containing DMRs revealed a subset present in multiple pathways. Specifically, 11.4% hypermethylation of the Protein kinase B (AKT1) proximal promoter occurred in the pre-diabetic range of glucose exposure, but this DMR was conversely hypomethylated 7.5% in the diabetic range (Table S1). Vascular endothelial growth factor A (VEGFA) was found to contain promoter hypermethylation in both pre-diabetic (3.7%, Q < 0.05) and diabetic (5.6%, Q < 0.001) treatments. By contrast, endothelial NOS (NOS3) possessed hypomethylation (intron-exon, 20.3% hypomethylated, Q = 0.002) only at the diabetic-range of glucose exposure.
Figure 2: Hierarchical Pathway Enrichment Clustering.
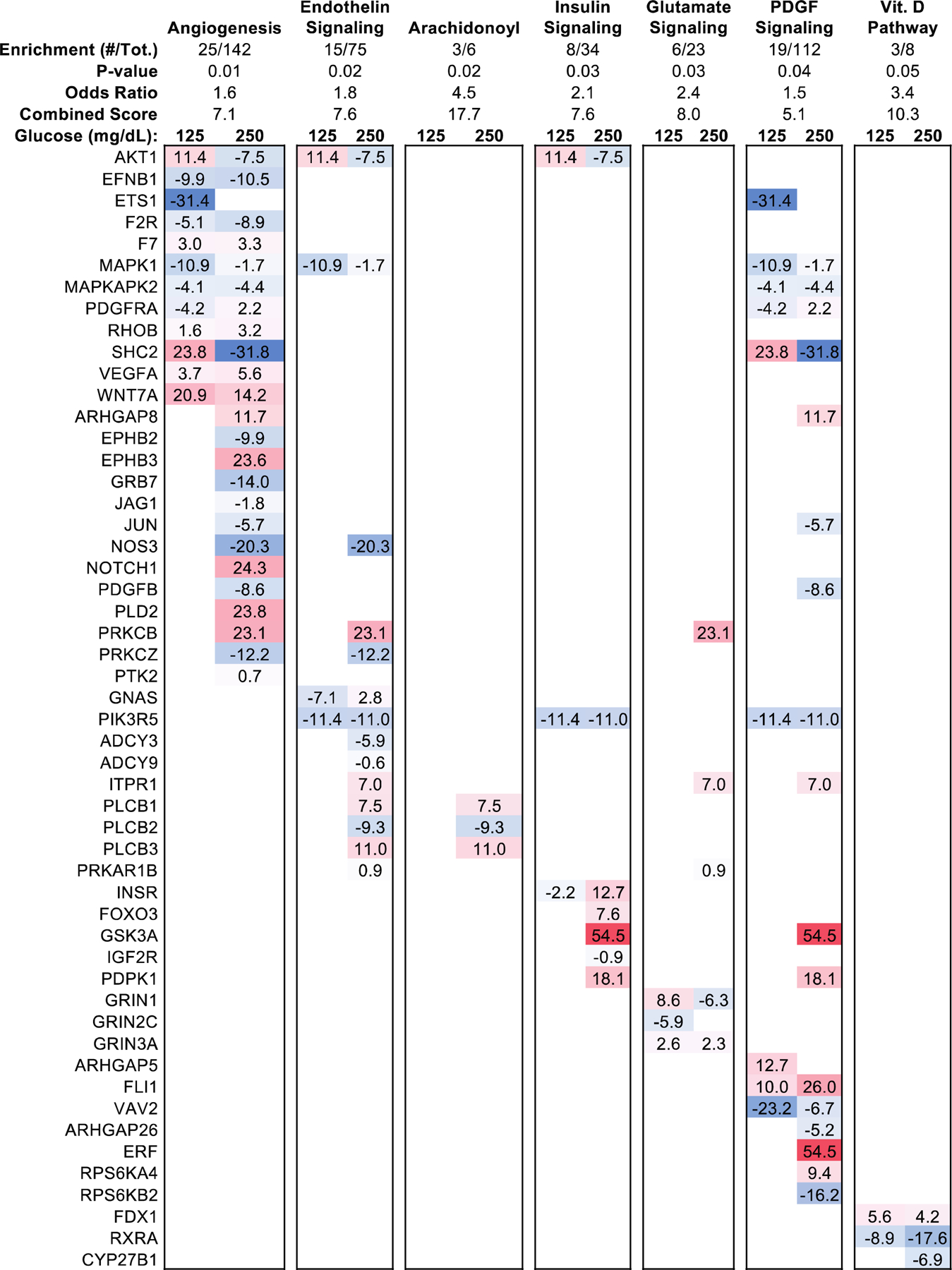
Hypergeometric gene set enrichment analysis of glucose-responsive DMRs with statistical significance of DMRs assumed at Q < 0.05. Pathways were sorted by Fisher’s exact test statistic of gene set enrichment, with top 5 most-enriched pathways shown. The genes containing DMRs within the top 5 most-enriched pathways were reported based on the percent methylation difference from glucose treatment (125mg/dL, 250mg/dL).
Among genes associated with the “Angiogenesis” pathway, the top most-enriched among dose-responsive DMRs associated with several genes found to be robustly hypermethylated only in diabetic-range glucose concentrations and otherwise unchanged in the pre-diabetic range of glucose exposure (Fig. 2): Rho GTPase activating protein 8 (ARHGAP8; intronic, 11.7% hypermethylated, Q = 0.04), notch receptor 1 (NOTCH1, inter-genic, 24.3% hypermethylated, Q = 2.3 × 10−5), receptor-tyrosine kinase HEK2 (EPHB3; intronic, 23.6% hypermethylated, Q = 3.4 × 10−13), Phosphatidylcholine-Hydrolyzing Phospholipase D2 (PLD2; exon-intron, 23.8% hypermethylated), and Protein kinase C beta (PRKCB; inter-genic, 23% hypermethylated, Q < 0.05). In contrast, hypomethylation was found to occur within NOS3 (intron-exon, 20.3% hypomethylated, Q = 0.002), PRKCZ (intron, 14%, 1.0 × 10−5), JUN (exon, −18.8%, Q = 8.4 × 10−5), PDGFB (intron, −11.5%, Q = 0.002), EPHB2 (intron, −9,9%, Q = 0.03), and GRB7 (exon, −14.0%, Q = 0.005).
Because regional alterations in genomic methylation correspond with alterations to genomic architecture and gene accessibility, we inspected genome-wide distribution of DMRs to identify genes and genomic regions likely influenced by differential methylation (Fig. 3). From this circular genome plot, we observed several hyperdynamic regions of differential DNA methylation in response to diabetic-range, but not pre-diabetic levels, of glucose exposure. Specifically, chromosome 19 possessed highest density of DMRs and was associated with differential methylation of 246 DMRs, 22 of which exceeded 25% difference in methylation at diabetic-range glucose exposure. Among them, robust alterations in DNA methylation of INSR (27.6% hypermethylated, intergenic, Q = 0.004) and GSK3A (54.3% hypermethylated, promoter, Q = 3.5 × 10−18) was noted.
Figure 3: Epigenetic topography of glucose-responsive DMRs.
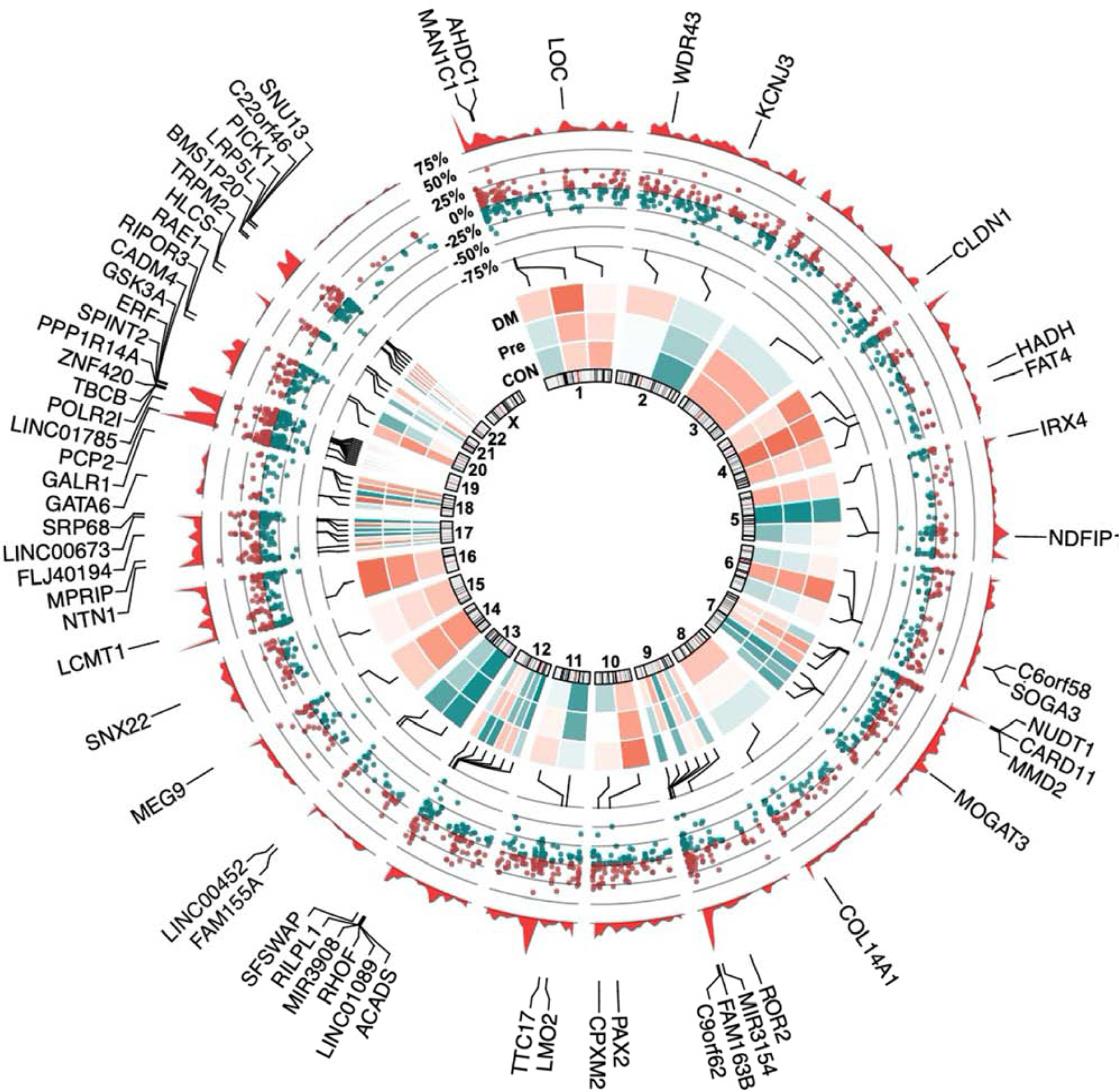
Circular genome plot illustrating the distribution of regional methylation, with methylation density plotted as the outermost data track (red). Genes possessing DMRs > 25% methylation under diabetic-range (250mg/dL) glucose were labeled, with the middle track representing percent methylation of increased (red) and decreased (green) DMRs (Q < 0.05, |Percent Methylation| > 25%). The innermost track depicts a normalized heatmap of average percent methylation for control, pre-diabetic, and diabetic-range glucose concentration (n = 2).
DNA methylation has been shown in other contexts to regulate transcriptional activity by interfering with regulatory binding to genomic elements [41]. Therefore, we performed a motif enrichment analysis to identify the regulatory elements that may be disproportionately affected by alterations in DNA methylation across the proximal promoter, intronic and exonic coding regions (Fig. 4A). Consequently, we identified SRY-box transcription factor 6 (SOX6), RAR-related orphan receptor alpha (RORA), and (Scratch Family Transcriptional Repressor 1 (SCRT1) as the most enriched transcriptional regulators among the DMRs within proximal promoter, intronic and exonic regions, respectively.
Fig. 4.

Locus-specific motif enrichment and pathway analysis of differential DNA methylation. De novo motif enrichment and gene set enrichment analyses were performed using DMRs* based on location relative to known gene annotations, specifically examining the gene promoter (green), introns (yellow), and exons (grey). Locus-specific gene set enrichment of DMRs annotated to reside within A. Promoter, B. Intron, and C. Exon regions. *Q < 0.05, |Percent Methylation| > 25%.
GO-term enrichment of genes containing glucose-responsive DMRs identified several known and novel endothelial pathways within HAECs affected by the diabetic milieu. Among the pathways most affected by promoter methylation, “Endothelial NOS activation” was most significant (P = 2.1 × 10−4), followed by “ERBB2 signaling” (P = 2.1 × 10−4) and “VEGF Receptor 1 Signaling” (P = 3.3 × 10−4) (Fig. 4B). By contrast, intronic and exonic DMRs concordantly enriched “RhoA Activity” (PExon = 2.4 × 10−4, PIntron = 4.5 × 10−6) and “Rho GTPase cycle” (PExon = 4.0 × 10−5, PIntron = 4.0 × 10−4) pathways (Fig. 4C, D). Exonic DMRs additionally enriched “Oxidative stress signaling via NRF2” (P = 0.006), a pathway implicated in the pathogenesis of endothelial inflammation and integrity [42]. Together, these observations suggest that differential DNA methylation occurs in a regional manner to impact both established pathological processes.
To determine the putative functional consequences of these early glucose-responsive epigenetic modifications, we analyzed a publicly-available microarray dataset generated by Hazra et al. of endothelial precursor (CD34+) cells obtained from diabetic and non-diabetic human subjects (n = 5) (GSE43950). This resource revealed increased expression of AKT1 (↑2.5-fold, P = 0.005), MAPK1 (↑2.6-fold, P = 0.001), and VEGFA (↑3.6-fold, P = 0.001) in samples from diabetic subjects relative to those of non-diabetics; conversely, we found decreased expression of both NOS3 (↓1.6-fold, P = 0.02) and CAV1 (↓1.2-fold, P = 0.01) (Fig. 5). Additionally, we found several epigenetic regulators to be increased in diabetic samples: DNMT3A (↑1.4X, P = 0.04), HDAC4 (↑3.5X, P = 0.0005), TET2 (↑4.3X, P =0.001), and GADD45B (↑4.5X, P = 0.0005).
Figure 5: Differential expression of VEGF signaling and NO signaling intermediate genes.
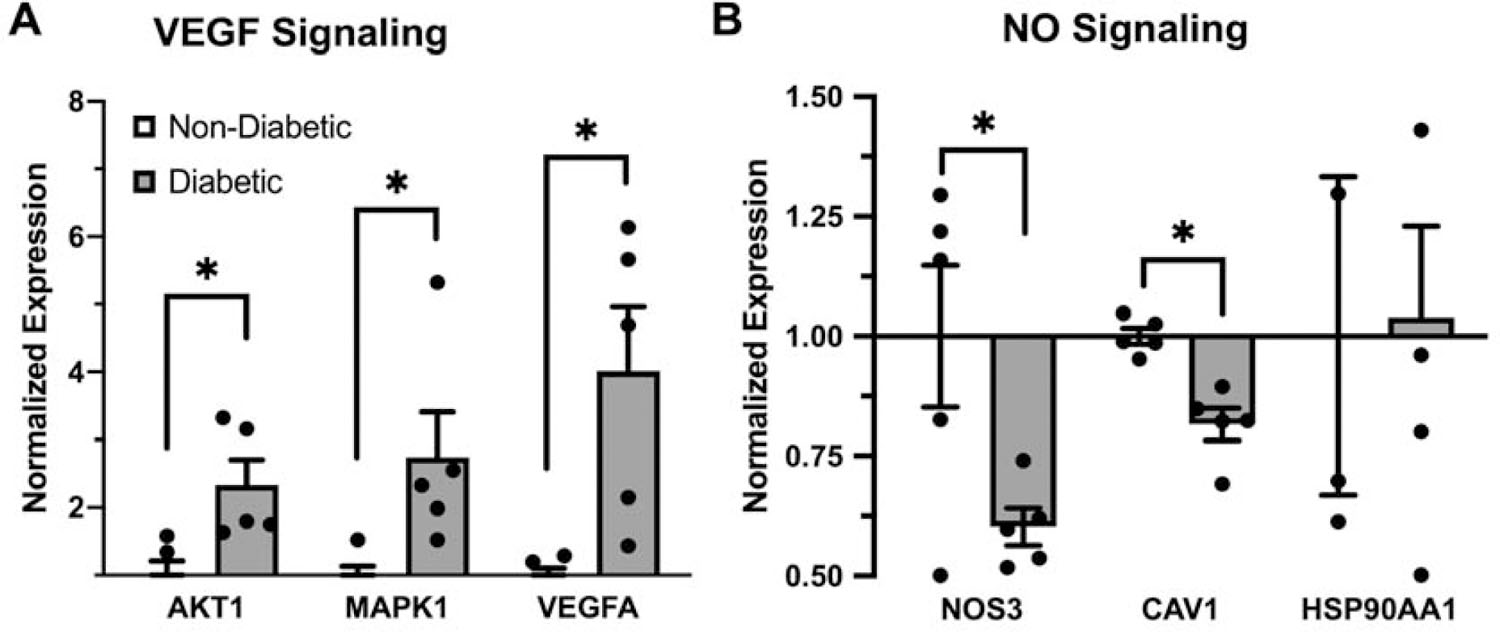
Bar graph of normalized gene expression via array-based transcriptomic analysis of diabetic and non-diabetic subjects (n =5) endothelium of key intermediate genes involved in A. VEGF signaling and B. NO signaling cascades. Bars shown as mean ± standard error of the mean, with statistical significance assumed via student’s t-test.
Discussion
As a potent driver of vascular dysfunction, diabetes mellitus is a leading cause of cardiovascular disease worldwide [43], though the precise mechanisms by which glycemic stress potentiates endothelial dysfunction remains poorly-understood. DM is a multifactorial disease involving both genetic and environmental factors, but the cardiovascular risk due to DM is most strongly associated with the degree of glycemic control [3–5]. Furthermore, evidence exists to suggest that even transient episodes of hyperglycemia can increase susceptibility for CVD long after the initial insult, a phenomenon termed “glycemic memory” [2, 6–8]. In the current study, we explore the role of epigenetic alterations, specifically DNA methylation dynamics, as a program activated in response to short-term glucose challenges.
Among the molecular pathways most associated with glucose-dependent differential DNA methylation, we found “Angiogenesis” as the most statistically enriched. Neoangiogenesis has long been associated with diabetic microvascular complications, with its disruption found to occur within the vascular networks of retinal [44], renal [45], and cardiac [46] tissues. Additionally, hyperglycemia has been demonstrated to cause an imbalance, or “uncoupling,” between the endothelial vascular endothelial growth factor (VEGF) and nitric oxide signaling cascades, wherein VEGF activation occurs alongside endothelial NOS suppression [47]. Although the molecular signals responsible for this uncoupling process remain poorly understood, genetic models have shown that genetic depletion of NOS3 causes a severe vascular phenotype in diabetic mice [48]. We observe gene body hypermethylation associated increase in expression of VEGFA with concomitant gene body hypomethylation and reduction in expression of NOS3. Although future mechanistic work is needed, we hypothesize that glucose-dependent epigenomic reprogramming of the vascular endothelium causes discordant regulation of VEGF and NO signaling.
Although the current study provides no molecular mechanism through which glucose reprograms the endothelial epigenome, we hypothesize that the transcriptional activation of growth arrest and DNA damage inducible protein beta (GADD45β) may contribute to this process. GADD45β is itself a molecular chaperone, or coactivator, required for the recruitment of DNA demethylases such as the ten-eleven translocases (TETs) in the removal of methyl groups via 5-hydroxymethylation in a DNA repair-like mechanism [49–51]. This mechanism of active demethylation is established in the context of memory formation in the brain [51, 52], but has not been explored in the context of diabetes. Additionally, GADD45β is required for the liver’s oxidative stress-mediated induction of apoptosis via TGFβ signaling [53]. As a stress sensor, GADD45β responds to both oxidative and osmotic stressors, serving a central role in initiating intrinsic apoptosis [53–58]. We hypothesize that endothelial GADD45β induction in diabetes leads to pathological reprogramming of endothelial DNA methylation and, consequently, diabetic vasculopathy.
Furthermore, our analysis identified robust differential methylation within the coding region of genes following treatment with diabetic-range glucose. Gene body methylation has been proposed to be “epiphenomenal,” or the spurious result of genomic accessibility to epigenetic machinery [37]. Nevertheless, gene body methylation has been widely shown to correspond with a positive effect on associated genes’ expression through an as-yet undefined mechanism [59]. The precise physiologic role(s) of epigenetics in the pathogenesis glycemic remains poorly understood, continued efforts exist to elucidate both the metabolic signals and functional consequences of glycemic reprogramming [11, 60].
Although we provide novel insights, the current study is limited in several ways. Furthermore, our limited sample size (n = 2) limits our ability to make broader inferences regarding the DNA methylation events detected. Although our in vitro use of human aortic endothelial cells permitted us to examine genome-wide DNA methylation at different glucose concentrations, our experimental conditions are unlikely to reflect the heterogeneity and clinical relevance of in vivo studies. Lastly, the current experimental design does not provide an understanding of how long these alterations exist, particularly once euglycemic conditions are restored. We therefore recommend follow-up experiments to address these mechanistic and experimental limitations.
Conclusions
In the current study, we provide novel evidence that hyperglycemia triggers dose-responsive changes in DNA methylation dynamics affecting key physiological processes involved in maintaining endothelial function. As a preliminary study of these events, we propose future experiments which test the hypothesis that de novo alterations in endothelial DNA methylation cause a glucose-dependent physiologic uncoupling of VEGF and NO signaling to cause endothelial dysfunction. As such, the current study advances our current insights into a putative mechanism whereby glycemic memory is encoded within cells to potentiate short-term cytotoxic stress.
Supplementary Material
Figure 6: Working Model.
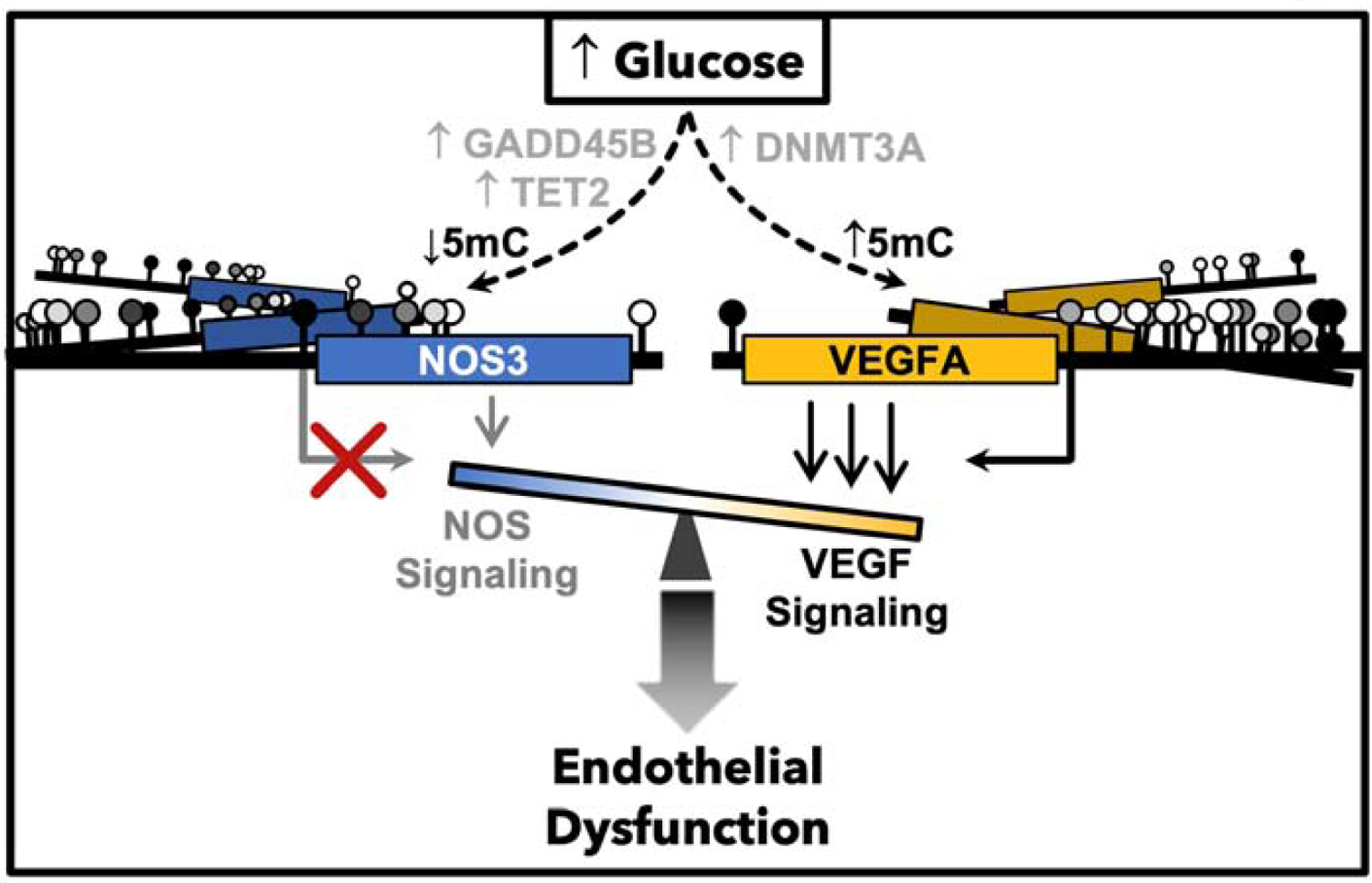
Hypothesized role of DNA methylation as a glucose responsive mediator of endothelial glycemic memory, encoding the pathophysiologic imbalance of VEGF and NO signaling.
Funding
Financial support for this work to C.N. was provided by progetto PRIN 2017 (2017F8ZB89), funded by Italian Ministry of Research. Financial support for this work provided to A.R.W. by the NIH NHLBI R01 HL133011. Training support was provided to M.E.P. by an NIH F30 HL137240 and postdoctoral research fellowship through the Alexander Humboldt Foundation. G.B. is a PhD Student of Translational Medicine supported by an Educational Grant from the University of Campania “Luigi Vanvitelli”, Naples, IT.
Abbreviations:
- HAECs
human aortic endothelial cells
- DMR
differentially methylated genomic region
- DM
type 2 diabetes mellitus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors have no conflicts of interest to disclose.
Data availability
All data generated or analyzed during this study are included in this article and are found on the NCBI Gene Expression Omnibus (GEO) repository (GSE163510), as well as at the following repository: https://github.com/mepepin/Glycemic.Memory.HAECs.
References
- [1].Napoli C, Benincasa G, Schiano C, Salvatore M, Differential Epigenetic Factors in the Prediction of Cardiovascular Risk in Diabetic Patients, Eur Heart J Cardiovasc Pharmacother (2019). [DOI] [PMC free article] [PubMed]
- [2].Ceriello A, Monnier L, Owens D, Glycaemic variability in diabetes: clinical and therapeutic implications, Lancet Diabetes Endocrinol 7 (2019) 221–230. [DOI] [PubMed] [Google Scholar]
- [3].Kannel WB, Hjortland M, Castelli WP, Role of diabetes in congestive heart failure: the Framingham study, Am J Cardiol 34 (1974) 29–34. [DOI] [PubMed] [Google Scholar]
- [4].Malchoff CD, Diagnosis and classification of diabetes mellitus, Conn Med 55 (1991) 625–629. [PubMed] [Google Scholar]
- [5].Liguori A, Abete P, Hayden JM, Cacciatore F, Rengo F, Ambrosio G, Bonaduce D, Condorelli M, Reaven PD, Napoli C, Effect of glycaemic control and age on low-density lipoprotein susceptibility to oxidation in diabetes mellitus type 1, Eur Heart J 22 (2001) 2075–2084. [DOI] [PubMed] [Google Scholar]
- [6].Diabetes C, Complications Trial I /Epidemiology of Diabetes, G. Complications Study Research, Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up, Diabetes Care 39 (2016) 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Skyler JS, Bergenstal R, Bonow RO, Buse J, Deedwania P, Gale EA, Howard BV, Kirkman MS, Kosiborod M, Reaven P, Sherwin RS, American Diabetes A, American F College of Cardiology, A. American Heart, Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA diabetes trials: a position statement of the American Diabetes Association and a scientific statement of the American College of Cardiology Foundation and the American Heart Association, Circulation 119 (2009) 351–357. [DOI] [PubMed] [Google Scholar]
- [8].Schernthaner G, Diabetes and Cardiovascular Disease: Is intensive glucose control beneficial or deadly? Lessons from ACCORD, ADVANCE, VADT, UKPDS, PROactive, and NICE-SUGAR, Wien Med Wochenschr 160 (2010) 8–19. [DOI] [PubMed] [Google Scholar]
- [9].Coco C, Sgarra L, Potenza MA, Nacci C, Pasculli B, Barbano R, Parrella P, Montagnani M, Can Epigenetics of Endothelial Dysfunction Represent the Key to Precision Medicine in Type 2 Diabetes Mellitus?, Int J Mol Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sommese L, Zullo A, Mancini FP, Fabbricini R, Soricelli A, Napoli C, Clinical relevance of epigenetics in the onset and management of type 2 diabetes mellitus, Epigenetics 12 (2017) 401–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pepin ME, Wende AR, Epigenetics in the development of diabetic cardiomyopathy, Epigenomics 11 (2019) 469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Funk SD, Yurdagul A Jr., Orr AW, Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes, Int J Vasc Med 2012 (2012) 569654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Corban MT, Lerman LO, Lerman A, Endothelial Dysfunction, Arterioscler Thromb Vasc Biol 39 (2019) 1272–1274. [DOI] [PubMed] [Google Scholar]
- [14].Chilelli NC, Burlina S, Lapolla A, AGEs, rather than hyperglycemia, are responsible for microvascular complications in diabetes: a “glycoxidation-centric” point of view, Nutr Metab Cardiovasc Dis 23 (2013) 913–919. [DOI] [PubMed] [Google Scholar]
- [15].Silva C, Sampaio-Pinto V, Andrade S, Rodrigues I, Costa R, Guerreiro S, Carvalho E, Pinto-do OP, Nascimento DS, Soares R, Establishing a Link between Endothelial Cell Metabolism and Vascular Behaviour in a Type 1 Diabetes Mouse Model, Cell Physiol Biochem 52 (2019) 503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Balestrieri ML, Rienzo M, Felice F, Rossiello R, Grimaldi V, Milone L, Casamassimi A, Servillo L, Farzati B, Giovane A, Napoli C, High glucose downregulates endothelial progenitor cell number via SIRT1, Biochim Biophys Acta 1784 (2008) 936–945. [DOI] [PubMed] [Google Scholar]
- [17].De Pascale MR, Bruzzese G, Crimi E, Grimaldi V, Liguori A, Brongo S, Barbieri M, Picascia A, Schiano C, Sommese L, Ferrara N, Paolisso G, Napoli C, Severe Type 2 Diabetes Induces Reversible Modifications of Endothelial Progenitor Cells Which are Ameliorate by Glycemic Control, Int J Stem Cells 9 (2016) 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Napoli C, Lerman LO, de Nigris F, Loscalzo J, Ignarro LJ, Glycoxidized low-density lipoprotein downregulates endothelial nitricoxide synthase in human coronary cells, J Am Coll Cardiol 40 (2002) 1515–1522. [DOI] [PubMed] [Google Scholar]
- [19].Schiano C, Benincasa G, Franzese M, Della Mura N, Pane K, Salvatore M, Napoli C, Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases, Pharmacol Ther (2020) 107514. [DOI] [PubMed]
- [20].Napoli C, Infante T, Casamassimi A, Maternal-foetal epigenetic interactions in the beginning of cardiovascular damage, Cardiovasc Res 92 (2011) 367–374. [DOI] [PubMed] [Google Scholar]
- [21].Perrone L, Matrone C, Singh LP, Epigenetic modifications and potential new treatment targets in diabetic retinopathy, J Ophthalmol 2014 (2014) 789120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gautier JF, Porcher R, Abi Khalil C, Bellili-Munoz N, Fetita LS, Travert F, Choukem SP, Riveline JP, Hadjadj S, Larger E, Boudou P, Blondeau B, Roussel R, Ferre P, Ravussin E, Rouzet F, Marre M, Kidney Dysfunction in Adult Offspring Exposed In Utero to Type 1 Diabetes Is Associated with Alterations in Genome-Wide DNA Methylation, PLoS One 10 (2015) e0134654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alam F, Islam MA, Gan SH, Mohamed M, Sasongko TH, DNA Methylation: An Epigenetic Insight into Type 2 Diabetes Mellitus, Curr Pharm Des (2016). [DOI] [PubMed]
- [24].Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA, Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus, BMC Med Genomics 3 (2010) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Simmons RA, Programming of DNA methylation in type 2 diabetes, Diabetologia 56 (2013) 947–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, Poulsen P, Ribel-Madsen R, Pedersen NL, Almgren P, Fadista J, Ronn T, Klarlund Pedersen B, Scheele C, Vaag A, Ling C, Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes, Diabetes 63 (2014) 2962–2976. [DOI] [PubMed] [Google Scholar]
- [27].Maghbooli Z, Larijani B, Emamgholipour S, Amini M, Keshtkar A, Pasalar P, Aberrant DNA methylation patterns in diabetic nephropathy, J Diabetes Metab Disord 13 (2014) 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Nigris F, Mancini FP, Schiano C, Infante T, Zullo A, Minucci PB, Al-Omran M, Giordano A, Napoli C, Osteosarcoma cells induce endothelial cell proliferation during neo-angiogenesis, J Cell Physiol 228 (2013) 846–852. [DOI] [PubMed] [Google Scholar]
- [29].de Nigris F, Cacciatore F, Mancini FP, Vitale DF, Mansueto G, D’Armiento FP, Schiano C, Soricelli A, Napoli C, Epigenetic Hallmarks of Fetal Early Atherosclerotic Lesions in Humans, JAMA Cardiol 3 (2018) 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pepin ME, Infante T, Benincasa G, Schiano C, Miceli M, Ceccarelli S, Megiorni F, Anastasiadou E, Della Valle G, Fatone G, Faenza M, Docimo L, Nicoletti GF, Marchese C, Wende AR, Napoli C, Differential DNA Methylation Encodes Proliferation and Senescence Programs in Human Adipose-Derived Mesenchymal Stem Cells, Front Genet 11 (2020) 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brent S. Pedersen KE, De Subhajyoti, Yang Ivana V., Schwartz David A., Fast and accurate alignment of long bisulfite-seq reads, arXiv (2014).
- [32].Sun X, Han Y, Zhou L, Chen E, Lu B, Liu Y, Pan X, Cowley AW Jr., Liang M, Wu Q, Lu Y, Liu P, A comprehensive evaluation of alignment software for reduced representation bisulfite sequencing data, Bioinformatics 34 (2018) 2715–2723. [DOI] [PubMed] [Google Scholar]
- [33].Wreczycka K, Gosdschan A, Yusuf D, Gruning B, Assenov Y, Akalin A, Strategies for analyzing bisulfite sequencing data, J Biotechnol 261 (2017) 105–115. [DOI] [PubMed] [Google Scholar]
- [34].Wang HQ, Tuominen LK, Tsai CJ, SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures, Bioinformatics 27 (2011) 225–231. [DOI] [PubMed] [Google Scholar]
- [35].Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool, BMC Bioinformatics 14 (2013) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA, Doremieux O, PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification, Nucleic Acids Res 31 (2003) 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK, On the presence and role of human gene-body DNA methylation, Oncotarget 3 (2012) 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bird AP, CpG-rich islands and the function of DNA methylation, Nature 321 (1986) 209–213. [DOI] [PubMed] [Google Scholar]
- [39].Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM, Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells, Nat Biotechnol 27 (2009) 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD, PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements, Nucleic Acids Res 45 (2017) D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].D’Anna F, Van Dyck L, Xiong J, Zhao H, Berrens RV, Qian J, Bieniasz-Krzywiec P, Chandra V, Schoonjans L, Matthews J, De Smedt J, Minnoye L, Amorim R, Khorasanizadeh S, Yu Q, Zhao L, De Borre M, Savvides SN, Simon MC, Carmeliet P, Reik W, Rastinejad F, Mazzone M, Thienpont B, Lambrechts D, DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance, Genome Biol 21 (2020) 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C, Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression, Am J Physiol Heart Circ Physiol 290 (2006) H1862–1870. [DOI] [PubMed] [Google Scholar]
- [43].Tabish SA, Is Diabetes Becoming the Biggest Epidemic of the Twenty-first Century?, Int J Health Sci (Qassim) 1 (2007) V–VIII. [PMC free article] [PubMed] [Google Scholar]
- [44].Stitt AW, McGoldrick C, Rice-McCaldin A, McCance DR, Glenn JV, Hsu DK, Liu FT, Thorpe SR, Gardiner TA, Impaired retinal angiogenesis in diabetes: role of advanced glycation end products and galectin-3, Diabetes 54 (2005) 785–794. [DOI] [PubMed] [Google Scholar]
- [45].Nakagawa T, Kosugi T, Haneda M, Rivard CJ, Long DA, Abnormal angiogenesis in diabetic nephropathy, Diabetes 58 (2009) 1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, Marra C, Nasti R, Marfella R, Cozzolino D, Indolfi C, Cotrufo M, Torella R, Salvatore T, Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease, J Am Coll Cardiol 46 (2005) 827–834. [DOI] [PubMed] [Google Scholar]
- [47].Nakagawa T, Uncoupling of VEGF with NO as a mechanism for diabetic nephropathy, Diabetes Res Clin Pract 82 Suppl 1 (2008) S67–69. [DOI] [PubMed] [Google Scholar]
- [48].Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson MA, Johnson RJ, Croker B, Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy, J Am Soc Nephrol 18 (2007) 539–550. [DOI] [PubMed] [Google Scholar]
- [49].Sultan FA, Wang J, Tront J, Liebermann DA, Sweatt JD, Genetic deletion of Gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity, J Neurosci 32 (2012) 17059–17066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Leach PT, Poplawski SG, Kenney JW, Hoffman B, Liebermann DA, Abel T, Gould TJ, Gadd45b knockout mice exhibit selective deficits in hippocampus-dependent long-term memory, Learn Mem 19 (2012) 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gavin DP, Kusumo H, Sharma RP, Guizzetti M, Guidotti A, Pandey SC, Gadd45b and N-methyl-D-aspartate induced DNA demethylation in postmitotic neurons, Epigenomics 7 (2015) 567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gavin DP, Sharma RP, Chase KA, Matrisciano F, Dong E, Guidotti A, Growth arrest and DNA-damage-inducible, beta (GADD45b)-mediated DNA demethylation in major psychosis, Neuropsychopharmacology 37 (2012) 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr., Liebermann DA, Bottinger EP, Roberts AB, Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation, J Biol Chem 278 (2003) 43001–43007. [DOI] [PubMed] [Google Scholar]
- [54].Zumbrun SD, Hoffman B, Liebermann DA, Distinct mechanisms are utilized to induce stress sensor gadd45b by different stress stimuli, J Cell Biochem 108 (2009) 1220–1231. [DOI] [PubMed] [Google Scholar]
- [55].Kim JH, Qu A, Reddy JK, Gao B, Gonzalez FJ, Hepatic oxidative stress activates the Gadd45b gene by way of degradation of the transcriptional repressor STAT3, Hepatology 59 (2014) 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen Z, Wan X, Hou Q, Shi S, Wang L, Chen P, Zhu X, Zeng C, Qin W, Zhou W, Liu Z, GADD45B mediates podocyte injury in zebrafish by activating the ROS-GADD45B-p38 pathway, Cell Death Dis 7 (2016) e2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cho HJ, Park SM, Hwang EM, Baek KE, Kim IK, Nam IK, Im MJ, Park SH, Bae S, Park JY, Yoo J, Gadd45b mediates Fas-induced apoptosis by enhancing the interaction between p38 and retinoblastoma tumor suppressor, J Biol Chem 285 (2010) 25500–25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].He G, Xu W, Tong L, Li S, Su S, Tan X, Li C, Gadd45b prevents autophagy and apoptosis against rat cerebral neuron oxygen-glucose deprivation/reperfusion injury, Apoptosis 21 (2016) 390–403. [DOI] [PubMed] [Google Scholar]
- [59].Arechederra M, Daian F, Yim A, Bazai SK, Richelme S, Dono R, Saurin AJ, Habermann BH, Maina F, Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer, Nat Commun 9 (2018) 3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Marfella R, Amarelli C, Cacciatore F, Balestrieri ML, Mansueto G, D’Onofrio N, Esposito S, Mattucci I, Salerno G, De Feo M, D’Amico M, Golino P, Maiello C, Paolisso G, Napoli C, Lipid Accumulation in Hearts Transplanted From Nondiabetic Donors to Diabetic Recipients, J Am Coll Cardiol 75 (2020) 1249–1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article and are found on the NCBI Gene Expression Omnibus (GEO) repository (GSE163510), as well as at the following repository: https://github.com/mepepin/Glycemic.Memory.HAECs.