

Abstract
Genetically engineered mouse (GEM) models are commonly used in biomedical research. Generating GEMs involve complex set of experimental procedures requiring sophisticated equipment and highly skilled technical staff. Because of these reasons, most research institutes set up centralized core facilities where custom GEMs are created for research groups. Researchers, on the other hand, when they begin thinking about generating GEMs for their research, several questions arise in their minds. For example, what type of model(s) would be best useful for my research, how do I design them, what are the latest technologies and tools available for developing my model(s), and finally how to breed GEMs in my research. As there are several considerations and options in mouse designs, and as it is an expensive and time-consuming endeavor, careful planning upfront can ensure the highest chance of success. In this article, we provide brief answers to several frequently asked questions that arise when researchers begin thinking about generating mouse model(s) for their work.
Keywords: CRISPR, transgenic mouse, genetic engineering, knockout mouse, conditional knockout mouse, knock-in mouse
Introduction
Developing mouse models to probe biological questions—whether related to neuroscience, cancer, physiology, or pharmacology—is quite an undertaking for any researcher. There are many reasons for this. First, it is time-consuming: it takes two or more years to generate publishable results to address the hypothesis for which a novel mouse model is generated. Second, it is expensive to generate and breed the model to produce enough animal cohorts for the intended study. Third, a variety of technologies and tools to create mouse models are available. Investigators may not be familiar with the range of possible options. Fourth, and the most important one, is that the numerous design strategies used to create genetically engineered mouse models are quite confusing to researchers not familiar with the recent technological developments. This is where many researchers begin to lose their way as questions multiply.
In this article, we provide answers to some frequently asked questions that normally come to mind when researchers begin a new mouse model generation project. Note that this is not a comprehensive review on the subject; rather, this article provides short answers to frequently asked questions. Wherever possible, we provide references for further reading.
The set of questions range from basic to advanced, keeping in mind a broad readership. The questions are in three areas: Ⅰ) general questions about the types of mouse models; Ⅱ) questions about model design considerations; Ⅲ) questions specific to mouse models generated using CRISPR-based methods.
General questions about the types of mouse models
I am interested in studying the function of my gene of interest (GOI) using genetically engineered mice. I do not know if I should overexpress or delete (knockout) or replace (knock-in) the gene in mice. Where do I begin to learn about this? This is one of the most fundamental and the most common questions that comes to any researcher who begins to think about using the mouse as a model organism. Also, answers to questions like whether to overexpress or knockout my gene require several considerations, particularly the biological function of the gene, which will be touched on later in this article. To begin, we guide the reader through the concept of overexpressing (transgenic) and deleting (knockout) a gene.
What is a transgenic mouse? The term transgenic mouse is often used loosely referring to any type of genetically engineered mouse (including knockout mouse, for example). However, the term transgenic mouse refers to a genetically engineered mouse containing an exogenous DNA cassette, and the cassette is typically introduced into the mouse genome via pronuclear microinjection of one-cell stage zygotes[1–2].
Once the transgene (overexpression DNA cassette of interest) is injected, does it get inserted in the genome at a specific site? DNA cassettes usually do not get inserted at a specific site in the genome. For example, Brinster et al (1989) microinjected 10 602 zygotes with the intention of inserting a transgene in a specific locus[3]. A total of 506 transgenic mice were produced, but only one mouse had inserted the transgene in the targeted locus. Transgenes insert at random locations, sometimes at multiple sites, often as a multi-copy concatemer, and sometimes the cassettes are fragmented and/or mix with genomic segments in an unpredictable way[4–7]. The first generation of transgenic mice (F0/G0) generated through pronuclear injection (PNI) of DNA will have a unique transgene insertion site, often called as founder mouse lines. Each independent founder line is used to establish germline transmitted mice by breeding one generation (F1) transgenic mice. Mice from each line, in the F1 generation, are screened for desirable expression of the cassette before expanding the line(s) for the research studies. It is important to remember a few additional points about transgenic mice. Transgenic DNA cassettes can integrate at the coding genes (sometimes disrupting their expression) and/or regulatory genes affecting their function leading to a phenotype, which can be mistaken for the phenotype of the transgene[7–8]. Randomly integrated transgenes can be susceptible to 'position effect variegation' that often causes impaired transgene expression[9]. Some transgenes can produce a too high level of expression (especially if they are multi-copy insertions), and/or can undergo transgene silencing, a phenomenon where the expression becomes silenced in the mouse colony after a few generations. Transgene silencing is thought to occur due to the epigenetic changes of the transgene[10]. For these reasons, it is prudent to establish and analyze 2–4 independent lines of transgenic mice for each project.
What is the main reason researchers consider generating a transgenic mouse? To overexpress a gene (or DNA cassette) of interest. Transgenic mouse generation via PNI is a well-established method. It is a popular method because of its relatively low cost and quickness, compared to the targeted knock-in strategies that require lengthy and complex steps of gene-targeting in mouse embryonic stem (ES) cells (see below knock-in mouse). The PNI methods were first developed in the 1960s and 1970s[2,11–12] and the first transgenic mouse containing exogenously introduced DNA was created in 1980[13]. Many tens of thousands of successful transgenic mice have been generated since then that have tremendously helped advance biomedical and therapeutic research.
Is it possible to insert the transgene at a specific locus in the genome (instead of at a random location)? Inserting the transgene at a specific locus is possible, which was typically done (prior to the advent of CRISPR technology) via ES cell-based gene-targeting approaches. The latter approaches are similar to the steps followed for developing a knockout mouse (see next). However, using the traditional PNI approach, targeting a transgene to a specific locus is almost impossible because of very low efficiency[1,3]. A method called PNI-based targeted transgenesis (PITT), developed in 2010, and its improved version (called i-PITT), and also TARGATT, developed in 2011, allows this by first creating a mouse containing landing pad elements on a chromosome (for example modified loxP sites and/or attP sites), and the zygotes from such a mouse are then used to insert the plasmid donors (containing modified loxP sites and/or attB sites) with Cre recombinase and/or PhiC31 integrase enzymes[14–16]. PITT, i-PITT, and TARGATT are elegant approaches, but require the production and maintenance of seed mouse strains containing the landing pad sequences. Targeted transgenic mice (also called knock-in mice; see below), can also be created readily using direct PNI of CRISPR reagents. Strategies using either long single-stranded DNA or linear- or circular- double-stranded DNAs have been employed as donors in the repair process of double-stranded DNA breaks created by the CRISPR-Cas system.
What is a knockout mouse? A genetically engineered mouse in which a specific gene is disrupted or deleted is called a knockout mouse. By studying what went wrong in a specific knockout mouse can help decipher the function of the deleted gene. Developing a knockout mouse via PNI is now routinely performed using the CRISPR-Cas system (see below). However, the PNI approach to generate knockout mice was not practical until the development of targeted nucleases (ZFNs, TALENs, and CRISPR-Cas systems; see below). Previous methods relied on homologous recombination in ES cells, where the targeted gene segment in the mouse genome was replaced by a drug selection cassette. Correctly targeted ES cell clones are identified through genotyping the targeted gene. Then gene-targeted ES cell clones are introduced into 3.5 day-old mouse embryos (called blastocysts) to generate ES-cell mouse chimeras formed with contributions from the host embryo (blastocyst) and the targeted ES cells. The chimeras are bred to wild-type mice and the offspring are screened for inheritance of the targeted mutation in the ES cells[17].
What is the most common method to create knockout mice in this day and age? Knockout mice can be created very efficiently and rapidly by simply injecting pronuclei with guide RNA(s), along with Cas9 protein, to create double-stranded breaks at a desired site. Both guide RNA and Cas9 protein are commercially available from numerous vendors. Prior to the CRISPR era, creating knockout mice was a tedious and time-consuming endeavor, because those methods relied on the use of ES cell-based approaches. Even though most of the simple models such as knockout and point mutation knock-in models (see below) can be generated via direct injection of CRISPR reagents into zygotes, ES cell-based approaches remain the most reliable strategy for large knock-ins such as multi-kb constructs.
What is a conditional knockout mouse? Conditional knockout (cKO) mouse systems rely on a classical genetic recombination tool called Cre-loxP, derived from bacteriophage P1[18]. Cre is an enzyme that recombines loxP sites and deletes the DNA segment between the two loxP sites. A loxP site is 34 base pair-long sequence containing 13 bases of a palindromic repeat, an 8 base pair spacer, followed by the 13 base palindrome. cKO mice are developed by inserting two loxP sites so that they surround a protein coding portion of a gene (called exons). Insertion of loxP sites in introns (non-coding parts of the gene) typically do not affect the expression of the target gene, and thus the cKO mouse does not behave like a knockout mouse until it is bred to a mouse that expresses Cre recombinase gene under a ubiquitous, inducible, or tissue-specific promoter. The schematics of Cre-loxP recombination system are shown in Fig. 1 . Mice containing two loxP sites are also referred to as floxed mice because they contain genetic elements flanked by loxP sites. The International Knockout Mouse Consortium to produce knockout and conditional alleles for every gene in the mouse genome is based on the use of Cre-loxP technology[19]. cKO mouse models are very versatile because they can be used to produce global knockout mice for study, in addition to cell-specific gene knockouts with Cre-expressing mice, or by the localized delivery of Cre with a viral vector.
Figure 1.

Schematics of the Cre-loxP recombination system.
How do the traditional methods of generating knockout and cKO mice compare? Both methods were very tedious because they relied on ES cell-based homologous recombination approaches. The first step, generating the targeting DNA constructs, was particularly complex. This involved stitching together long, genomic DNA segments in a precise way into a bacterial plasmid vector, and the constructs also needed to contain additional elements such as positive selection (e.g. neomycin resistance gene), and negative selection (thymidine kinase or diphtheria toxin A chain) cassettes. Generating cKO constructs was more tedious compared to complete knockout constructs. One reason for this was the necessity to stitch at least three separate segments of the genomic DNA to engineer two loxP sites around a critical exon (to generate cKO constructs), whereas the complete knockout DNA constructs just needed to join only two separate pieces of the genomic DNA into a bacterial plasmid vector.
What is a knock-in mouse? A knock-in mouse is created by changing or replacing DNA sequences in genes of the mouse genome. Knock-in mice can carry simple mutations containing just one or few amino acid differences from the wild-type gene, one of a few exons may be changed, or they can have the entire gene replaced with a modified or a new gene from a different species. The mouse created by insertion of an extra gene into a specific locus without making any deletion of original mouse sequence is also called knock-in mouse. Before the CRISPR era, developing any type of knock-in model was quite time-consuming, because the steps required ES cell targeting, and creating small changes (such as one or a few amino acid change) was relatively quite difficult. In contrast, the process of CRISPR-based PNI approaches can very easily create such models. More importantly, targeted integration sites will not have insertions of other genetic elements (for example a positive selection marker) close to the targeted site, which was one of the major constraints using ES cell-based traditional approaches. Nevertheless, as mentioned above, ES cell-based approaches remain as methods of choice for large knock-ins because there are not many reliable CRISPR-based approaches for creating very large knock-ins (such as >10 kb).
I am working on a novel gene; should I go for a knockout mouse (to delete the gene), a transgenic mouse (to overexpress it), or knock-in (to modify/replace it)? This is a very hard question to answer, but it largely depends on the central focus of the research problem. Although multiple different mutant mouse models can be generated for a given gene, collectively, they can serve as complementary tools to answer a research question (or to understand the function of that gene more extensively). Practically, many researchers may not be able to create all kinds of models for the gene in question because it requires a lot of resources and time. The guiding principle to choose the ideal mouse model is to make some intelligent guesses based on what is known about the gene or the gene pathway from other studies involving that gene. Here we mention a couple of obvious examples. If the gene was identified in a high-throughput screen as being upregulated or downregulated, the first mouse model considered for creation would be transgenic or knockout, respectively. If the GOI was found in a human genome-wide association study as containing a pathogenic mutation, then creating a knock-in mouse for that mutation would serve as the best model (instead of a transgenic or knockout).
I am planning to create a knockout model; should I delete it completely (whole body knockout), or create a cKO model using the Cre-loxP system (tissue-specific knockout)? Creating complete knockout mice is faster and cheaper compared to cKO models. If the gene knockout is lethal, the model will not be of much use. In a survey of 489 knockout mouse models, it was observed that 29% were embryonic lethal[20]. Thus, it is preferable to create a cKO model, instead of a complete knockout model, in case the global knockout model of the GOI is embryonic lethal. The cKO models, unlike complete knockout models, are invariably viable (the insertion of loxP sites rarely affect unknown regulatory elements in the introns[21]), and can be used for breeding with a ubiquitous Cre driver, such as CMV-Cre or Actin-Cre, to generate whole body KO mice, and many different tissue-specific Cre mice, to study various biological questions.
I am creating a cKO model; at this stage, do I need to think about what Cre models are available for breeding my cKO model to? It is very critical that researchers learn about the available Cre driver lines for their work. Oftentimes sufficient options of Cre driver lines may not be available. A convenient resource to identify interesting Cre mouse lines is the CrePortal database at the Mouse Genome Database[22]. It is also the case that the available Cre drivers (in the tissue- or cell-type of interest) may not be thoroughly validated for the sensitivity and specificity. Sometimes good Cre driver lines are not commercially available, and consideration needs to be given to creating a Cre driver mouse line for use with the floxed gene.
What is a Cre reporter mouse? A Cre-reporter mouse contains a reporter (like GFP or RFP or LacZ) driven by a ubiquitous promoter, but the reporter is kept in an inactive state by placing a loxP-Stop-loxP (LSL) cassette between the promoter and the reporter. The reporter is not expressed because of the multiple polyadenylation signals between the promoter and the reporter gene. When the reporter mouse is bred to a Cre driver line, the stop cassette between the promoter and the reporter gene gets deleted by Cre-mediated recombination, activating expression and leading to the expression of the reporter protein. The reporter gene will be expressed only in those cells or tissues where the Cre gene is expressed. This property of reporter expression serves as an indicator of Cre recombination in the target tissues. If you breed your newly created cKO model with a Cre driver and a reporter line, you can ensure specificity and sensitivity of Cre expression by relying on the reporter expression.
Should I also consider strain differences between my cKO model and Cre-expressing models that I would be breeding together? It is ideal to maintain strain uniformity when multiple mutant mice are bred together. The available Cre driver lines may not have been originally generated in the same strain as you plan to generate your cKO model. The majority of the available Cre driver mice, developed either by random transgenesis or targeted transgenesis, were in the non-standard or mixed strain backgrounds although several of them are backcrossed to pure genetic backgrounds such as C57BL6/J. If you intend to develop and maintain your cKO model in particular strain background, you will need to ensure that the Cre line(s) that you would like to breed them to are in the same genetic background. If they are not, it may need backcrossing up to 10 generations, or expedite the process through speed congenic breeding[23].
Do I need to always use a reporter mouse with my cKO:Cre double mutant mouse? It is generally advised to breed mice with the floxed gene, and the Cre recombinase gene with a Cre-reporter line[16–17,24]. This will ensure the sensitivity and specificity of Cre recombination in the desired target cells. While the use of reporters helps ensure Cre recombination in the target tissues, it should be noted that reporters may not reliably reflect the faithful Cre recombination at your cKO locus[25–27]. Because establishing breeder stock mice that carry multiple mutations (floxed gene+Cre driver+Cre-reporter gene) takes a very long time (typically requiring more than a year to achieve and to produce breeder stocks), it is prudent to make plans for procuring the Cre and a Cre-reporter model prior to beginning a cKO model generation project. It would be best to obtain Cre mice (preferably homozygous if that is an option) and homozygous Cre-reporters from another project, from a collaborator, or a repository. Breeding of the cKO model to such mouse colonies (homozygous for both Cre expression and Cre reporter genes) will expedite the breeding process.
Questions about model design considerations
Questions related to transgenic mice designs
I am creating a transgenic model; what promoter should I use for expressing my GOI? There are multiple options. Choosing a suitable promoter largely depends on the overall research goal. Here we provide a couple of examples of situations and the guiding principles in choosing a suitable promoter. If a researcher wants the GOI to be expressed constitutively and ubiquitously (i.e., in all tissues) at very high levels, one of the most preferred promoters would be a CAG (CMV enhancer, chicken beta-actin promoter and rabbit beta-globin splice acceptor site) promoter[28]. Often researchers also consider expressing their GOI under a promoter of another gene that they have previously worked with, which may result in a model that is suitable for expressing GOI in a specific cell type of choice. The model may then be used for studying a well-defined question, but often the model may be less useable for answering broader questions.
How can I express my GOI conditionally or make it inducible? Yes, the promoters like CAG (or any such promoter for that matter) can be designed to make them conditional (instead of constitutive) by placing a classical LSL element in between the promoter and the GOI[19]. Tetracycline response element driver promoter system is routinely used for expressing GOI in an inducible manner. It should be remembered that in conditional and inducible systems, the transgenic mouse will need to be bred with another mouse line prior to using it for the research studies. For example, the newly created conditional transgenic mouse will need to be bred with a Cre driver to remove the stop cassette upstream of the GOI, or with a tetracycline trans-activator (tTA) or reverse tTA (rtTA) mouse, to induce/repress GOI expression by administration of doxycycline[29]. The schematics of Tet-Off and Tet-On systems are shown in Fig. 2 .
Figure 2.
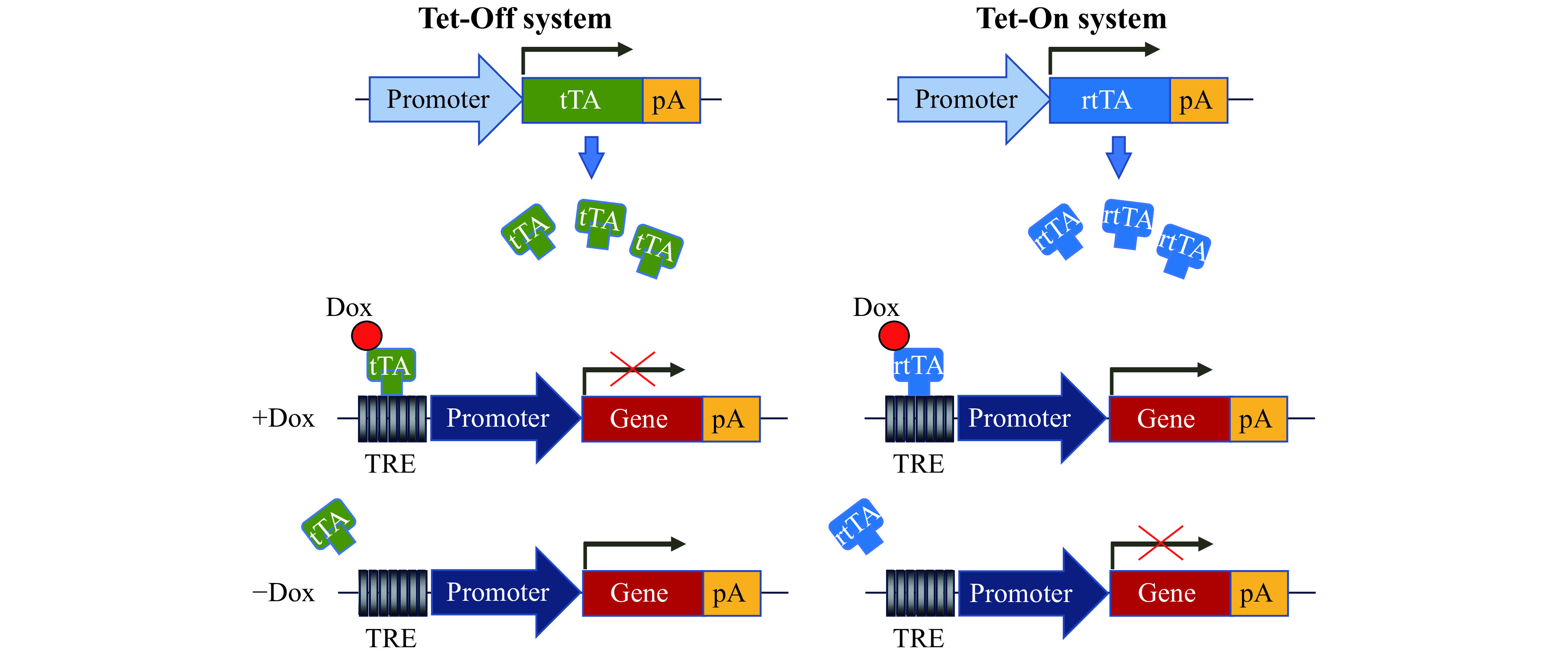
Schematics of Tet-Off and Tet-On systems.
I am creating a transgenic mouse to overexpress a human gene. Should I fuse the gene with a reporter (for example, GFP)? It is always prudent to think about a reliable method for differentiating and detecting your GOI before creating the transgenic model. One of the most important considerations is whether you have reliable and specific antibodies against your GOI that easily differentiate it from the endogenous copy of the protein. Ideally, if your GOI is a human protein, and you have two different antibodies that differentiate mouse and human proteins of the GOI, you are well prepared to detect transgene expression. In the event that is not the case (which is true for many GOIs), then fusing a reporter (like GFP), or a small immunoaffinity tag (like FLAG, Myc, HA), can serve as a valuable tool. It should be very important to make sure that fusing the tag does not affect protein function or localization. If any previous studies used a fusion strategy for your GOI, either in cell culture systems or in other model organisms, they would serve as ready reference for deciding on fusions. Another important consideration is to decide whether you want to add the fusion tag to N or C terminus of your protein. Again, this should be decided based on the knowledge of the protein structure (if available), and if other studies employed fusion strategies with the GOI. It may be helpful to place a fusion protein linker between the GOI and the reporter protein[30].
I would like to express two genes driven by a single promoter; are there any tools that will allow this? Sometimes, researchers would like to express a second gene such as a reporter or a drug-selection cassette along with their GOI under the same promoter. This is achieved by using one of the two commonly used genetic tools called internal ribosome entry site (IRES) and 2A peptidase. The concepts of IRES and 2A peptidases are shown in Fig. 3A . IRES allows expression of the second protein without fusing it with your GOI protein. The architecture of the expression cassette constitutes your GOI followed by an IRES and the second gene. The mRNA from such a cassette is translated as two separate polypeptides by the cellular ribosomal translation machinery. The IRES tool has been used in a large number of overexpression constructs. One drawback of the IRES system is that the gene placed downstream of the IRES may not be expressed at the same level as the gene placed upstream of it. Lower expression of the reporter (placed distal to IRES) often underestimates the expression of GOI, therefore, inferring the expression of GOI solely based on the expression of the reporter will not be accurate. The 2A peptide tool overcomes the problem of unequal expression of the two proteins. 2A is a small peptide (~20 amino acids long) derived from picornaviruses that is self-cleaving at the C terminus[31]. 2A proteins from different picornaviruses have been used: F2A is derived from foot and mouth disease virus; E2A is derived from equine rhinitis A virus; P2A is derived from porcine teschovirus-1; and T2A is derived from thosea asigna virus. Of these, P2A seems to have highest efficiency, followed by T2A[32]. One of the important considerations when using 2A peptide fusions is that, after cleavage, amino acids from the 2A peptide will be added to the end of GOI and a proline residue will be added to the beginning of the reporter protein (or the beginning of the downstream protein when two GOIs are expressed). This is an important consideration, especially in situations where the researcher does not want to take chances with adding extra amino acids, which may modify the structure and function of the protein. Again, knowledge about the protein from other structure function studies and fusion experiments conducted in cell culture should be considered when deciding which end of the GOI to be fused with the 2A peptide. The reporter fusion, via 2A or IRES, allows detection of cells expressing GOI, but subcellular localization of the GOI and the reporter may not be the same. In other words, GOI can be localized in cell membrane or the cytoplasm, but the reporter may be localized in the nucleus. The schematics of IRES and 2A tools are shown in Fig. 3 .
Figure 3.
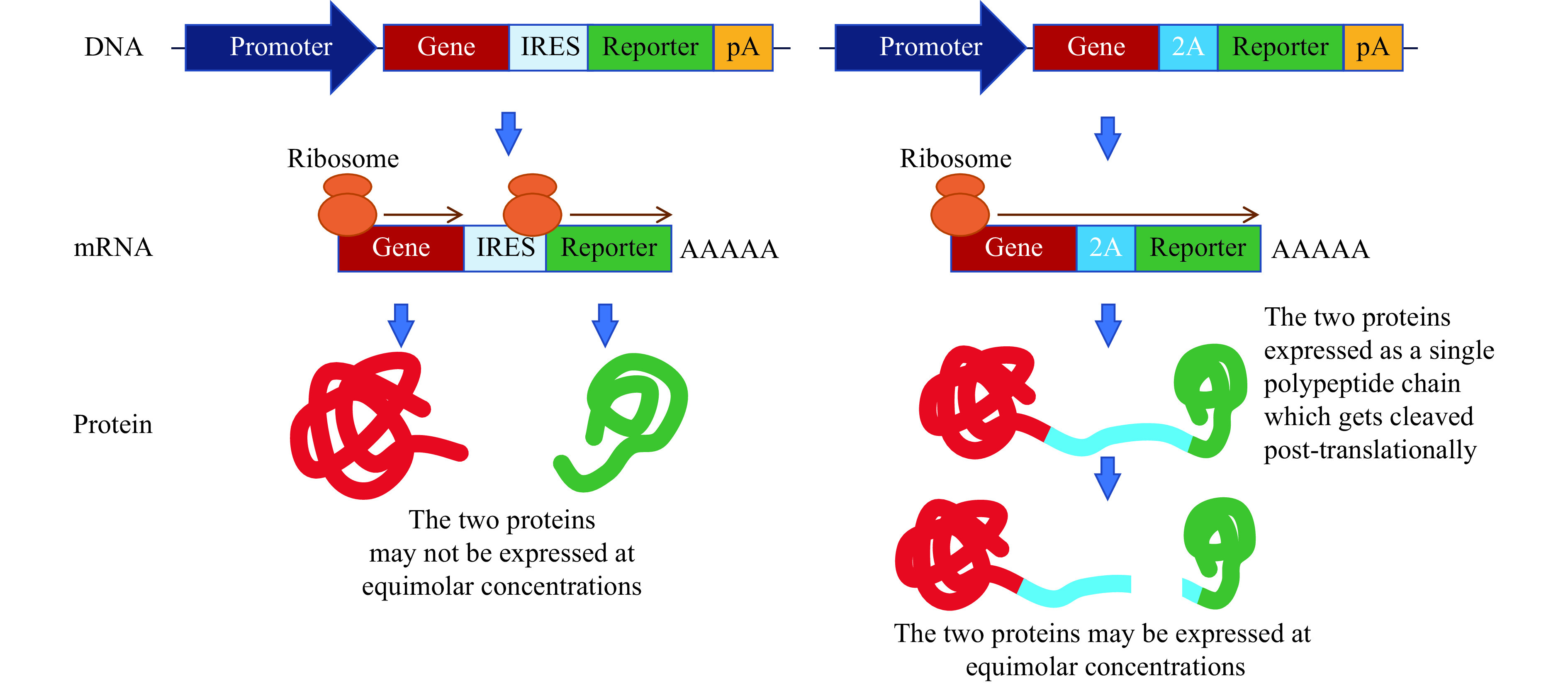
Schematics of IRES and 2A tools.
IRES: internal ribosome entry site.
Questions related to knockout and cKO mice designs
Can you briefly explain what are the components of CRISPR system used for generating knockout or cKO mice? Generating a simple knockout mouse using the CRISPR system requires two components: guide RNA/s and the Cas9 protein to cleave and disrupt a GOI. In addition to these components, generating cKO model requires a third component: a donor DNA to insert the LoxP sites at the genomic cleavage sites. More details and specifics about these components are answered in the next few questions. The schematics of CRISPR-Cas components are shown in Fig. 4 .
Figure 4.
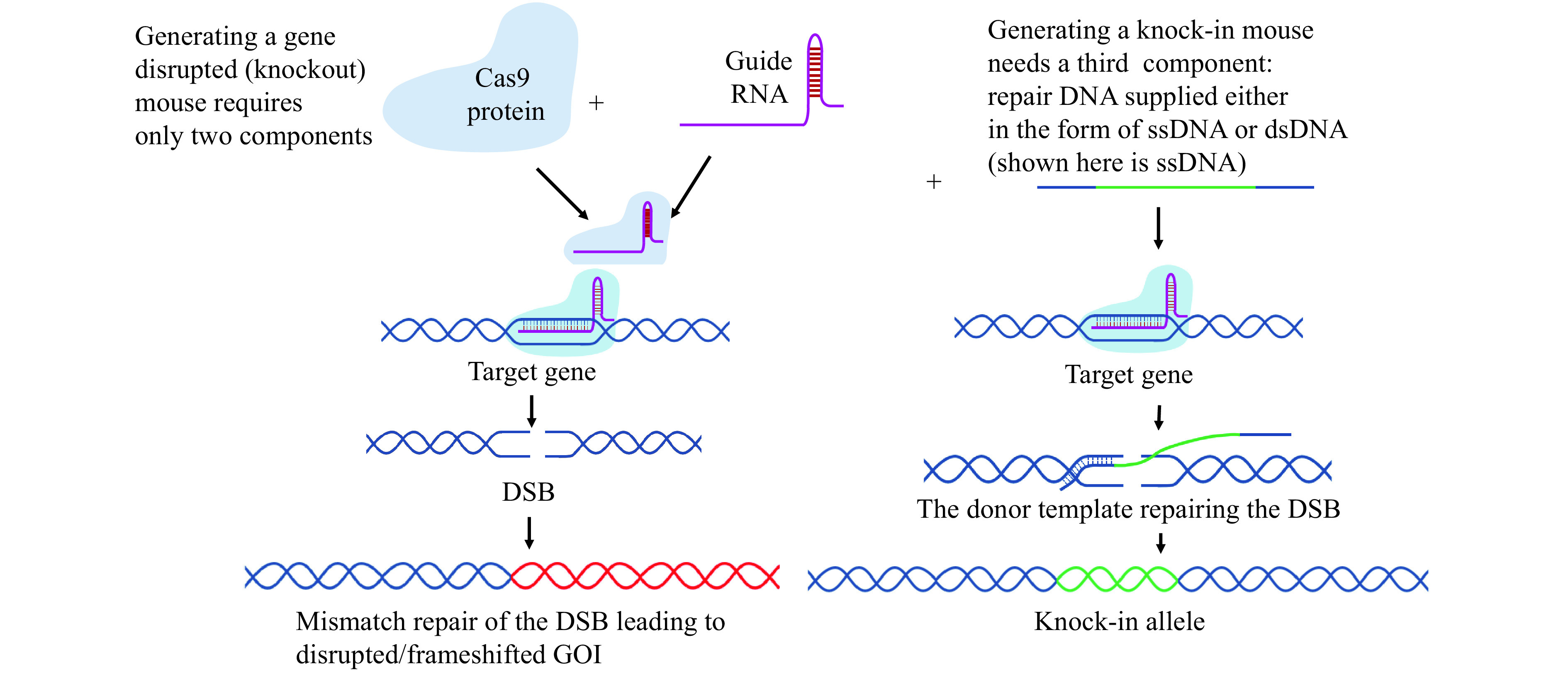
CRISPR-Cas genome editing components.
DSB: double-stranded break; GOI: gene of interest.
Should I use one guide or more than one guide RNA to delete my GOI? Targeting one of the upstream exons of the GOI, using just one guide RNA, can create a frameshift mutation. However, in some cases this approach may not be sufficient because the truncated gene may still be expressed from distal exons, and it is also known that exon skipping can occur[33] and/or the cells can overcome the loss of protein expression by upregulating the expression of proteins with a similar function[34]. For example, an internal promoter was discovered in the Occludin gene after an upstream exon was removed by Cre[35]. Therefore, it is prudent to delete the gene by cleaving it at two places, preferably to include most of the coding exons in the deleted region.
Which exon(s) should I choose for conditionally targeting my GOI? Typically, one or more of the most upstream exons, avoiding the first coding exon, are targeted, for a number of reasons. Although some groups have successfully floxed promoter, and the first exon of the gene to achieve definite deletion, the first exon is generally avoided because it is hard to know the upstream boundary of the promoter, and placing a loxP upstream of that boundary may not be possible without knowing the location of the promoter and/or enhancer elements. In other words, floxing the promoter and the first exon of the gene can be an option to generate a true null allele without concern of undesired effects by alternative splicing (see below), provided a clear understanding of the promoter sequence of your GOI (particularly its length including any additional regulatory sequences) is known. It should also be noted that regulatory elements may reside within the introns, and inserting loxPs in those locations can affect the normal expression of the gene (i.e., in the cKO mouse before it is bred to a Cre mouse). The principle is to remove an exon, so as to cause a frameshift that will introduce a premature termination codon to occur. This will trigger nonsense-mediated mRNA decay, and failure to make a protein[36]. Another commonly used strategy is to flox the exon(s) that code for important functional domains of the protein such as enzymatic catalytic regions.
Should I flox many exons of my GOI, or is floxing just one, or a few exons, enough? Just deleting one of the smaller exons can achieve the purpose (if those exon/s happen to be one of the critical exons), but phenomena like exon-skipping can also occur with cKO strategies, and thus it is important to ensure that the deleted exon(s) lead to frameshifting if the distal exons are spliced with the undeleted upstream exons. Another consideration is alternative splicing and expression of different transcripts can occur from the same gene. If the intention is to delete all isoforms, target one of the exons closer to the 5′ end of the gene, which is present in all isoforms.
What other considerations I need to keep in mind in deciding which exon/s to flox? Extra caution must be paid, especially when deleting many (or all the) of your GOI, to avoid deleting functional elements, such as miRNAs and lncRNAs that may be present within the intronic regions. The CRISPR tool has been used recently to demonstrate the detection of a previously intractable protein in the mouse[37]. Sometimes just a subtle change in the genomic sequence can abrogate the function of the non-coding RNA sequences[38]. As more and more long noncoding genes have been identified in the genome, with many close to or overlapping mRNA genes, it is essential that the reader pay particular attention to the presence of such noncoding genes when designing a CRISPR targeting approach[39–41]. Critical information about your GOI, such as its exon-intron structure, different splice variants, 5′-UTR, 3′-UTR regions, its neighboring genes, basic information about regulatory regions like promoters, enhancers, overlapping noncoding genes or regulatory regions in opposite strand downstream promoters (with H3K4me3 data), can be found from genome browsers like Ensembl and UCSC genome browsers. The presence of conserved non-coding DNA sequences with potential to affect gene expression can be identified with software tools such as Vistapoint[42].
How far apart should I place the two loxPs? As noted above, just floxing one or a few exons is enough as long as they are critical for gene function. If those exons are within a 1 to 2 kb sequence, and if the flanking introns are sufficiently long (say 300 bases or longer), loxPs can be placed in those introns to flank the critical exons. If the bulk of the protein-coding sequence falls within this region, it can be even better. Placing the two loxPs far away from each other (say >5 kb apart) can pose two problems: creating such models can be difficult by using one targeting construct (discussed below), and the Cre recombination between the two loxPs may not occur efficiently. In the case of single exon genes, it may be possible to introduce an artificial intron in the protein coding sequence to obtain a conditional allele[43–44].
Where should I place the loxP within intron? Intron sequences often contain some important sequences for splicing (e.g., splicing donor and acceptor, and branch site), and regulatory sequences for gene expression (e.g. enhancer or miRNA). loxP sites should not interrupt these important sequences. It is best to avoid placing loxP sites close to splice donor and acceptor sites (LoxP sites need to be more than 50 bases away from intron-exon boundary). In addition, you may need to check evolutionarily conserved sequences (e.g. between human and mouse), where functionally important sequences are possibly located, using genome browsers, such as UCSC.
Questions related to knock-in mouse designs
I plan to create a point mutation knock-in model to mimic a human disease mutation in mice. What are the basic considerations one needs to be aware of in developing such a model? Creating a point mutation knock-in is perhaps one of the easier mouse model design projects (creating a complete knockout mouse being the easiest). The process involves PNI (or electroporation) of a guide RNA that cleaves close to the mutation site, a donor oligonucleotide containing the new mutation, and the Cas9 protein. The first consideration is to choose a guide that cleaves as close to the mutation site as possible. The chance of success in generating the mutant is highest if there is a CRISPR guide target very close to the mutation site of interest, preferably within 1, 2 or 3 bases. The farther the guide cleavage site, the lower the efficiency of obtaining the desired mutant. If there is no good guide available close to the site, two flanking guides can be chosen to excise out a fragment followed by insertion of the desired sequence using a longer donor oligonucleotide[45]. The second consideration is to insert a silent mutation in the guide binding region of the donor, or the PAM region, to prevent re-cleavage. The third consideration is to engineer a restriction endonuclease (RE) site in the new mutation, which will serve as a valuable tool for genotyping the model using the restriction fragment length polymorphism approach. If the intended mutation site does not create a new RE site (or leads to loss of a pre-existing RE site), additional codons within the guide cleavage site can be considered to introduce silent mutations that may help create (or abolish) an RE site. When choosing silent coding changes to codons, it is helpful to refer to the table of mouse codon usage and to choose an alternative codon that has similar usage frequency as the codon that is being changed[46].
Can I achieve creating multiple point mutations in my GOI? It is possible to incorporate multiple point mutations in a gene, but there are some considerations. If the mutations are very close to each other, say within 1 to 2 kb, then they can all be changed using one long donor sequence. Achieving multiple mutations, using a pair of short oligonucleotide donors and guide RNAs for each of them, may work at much reduced efficiencies, because cleaving of the genomic DNA at multiple sites can lead to complex recombination events on the chromosome. If more than one mutation is incorporated in one experiment, it is essential to make sure that the mutations are inserted on the same copy of the chromosome (i.e., in-cis fashion).
Questions specific to mouse models generated using CRISPR-based methods
What are the commonly used CRISPR-based strategies for developing mouse models? CRISPR technology has greatly simplified the mouse model generation process. Specifically, most of the commonly used mouse models (simple knockouts, point mutation knock-ins, cKOs, reporter/Cre knock-ins) can be created quite efficiently and rapidly (within 2 to 3 months) using the CRISPR-based methods. Complete knockout mice can be created by simply injecting one or two guide RNAs and Cas9 protein. Point mutation knock-in mice are created by including a short single-stranded oligodeoxynucleotide (ssODN) as the donor template in the injection mix (in addition to a guide RNA and the Cas9 protein). cKO and reporter/Cre knock-in models can be generated efficiently by using long ssDNA donors (up to about 2 kb long). The method is known as Easi-CRISPR (Efficient additions with ssDNA inserts CRISPR)[47–48]. Creating large knock-ins (approximately 5 to 10 kb long) is generally achieved using plasmid DNAs as donors[49–52].
Can I create my model in the strain background of my choice using the CRISPR-based approaches? Yes, this is readily possible. The CRISPR approach can be used for generating genetically engineered mouse models for almost any strain of mouse, whereas the previous methods that relied on ES cell-based approaches could only be used for few strains.
I have heard that CRISPR reagents can be delivered into zygotes via electroporation rather than microinjection. Can you introduce me to those approaches? Microinjection was the primary method (perhaps the sole method) employed for delivering targeting constructs into mouse zygotes until CRISPR tools became available. Electroporation, as an alternative to microinjection of mouse zygotes, was first investigated in 2015, demonstrating that CRISPR gene targeting reagents can be introduced into the zygotes, and many zygotes can be processed simultaneously[53]. Since then, this approach has become quite popular at many laboratories, due to several obvious advantages: electroporation does not require sophisticated equipment or highly-skilled personnel; a large number of zygotes can be processed in each batch (unlike injecting one zygote at a time in the microinjection process); and, electroporation is much gentler than microinjection, as shown by higher zygote survival and birth rates from electroporation compared to microinjection. The electroporation approach has been used very efficiently to create complete knockout and point mutation knock-in models. One disadvantage of electroporation is that it is hard to introduce larger DNA constructs to create KI models. Knocking in donors about 200 bases or less is most efficient, 200–500 bases is moderately efficient, and the efficiency drops sharply for constructs larger than 500 bases. There are a large number of publications describing electroporation approaches and protocols. Because it would be difficult to extensively discuss several of the articles using a few questions in this article, we list some important references for reading[54–57].
I have heard that CRISPR reagents can be delivered into zygotes via electroporation within the mother. Can you introduce me to those approaches? A method called Genome Editing via Oviductal Nucleic Acids Delivery (GONAD) was developed in 2015, which can deliver genome editing reagents into the zygotes within the pregnant mother without taking them out of the mother. This is achieved by surgically opening the oviduct, introducing genome editing components into the oviduct through microinjection needle, followed by in situ electroporation. This method is described in several papers[58–61].
I have heard that the models generated using CRISPR approach are mosaic. What does that mean? A mouse consisting of cells with different allele sets is called a mosaic founder, typically consisting of cells with more than two alleles of the same gene. This is possible in a mouse generated using CRISPR technology, because cleavage of genomic DNA by Cas9 can continue to occur beyond the one-cell stage (if both the copies were not edited at one-cell stage). In other words, if Cas9 has not cleaved and edited both copies of the targeted gene in the one-cell zygote, then when the zygote divides into a two-cell zygote, there will be four copies of the gene, and Cas9 can create different types of edits in those four copies. The schematics of how some of the mice generated using the CRISPR-Cas tool can become mosaic are shown in Fig. 5 . Similarly, if any of the four copies in the two cell stage are not edited, the unedited copies can potentially be edited after the two cell divides into four cells, and so on. For example, such a single mosaic founder may be made up of cells with as many as six different versions of the same gene[62–63].
Figure 5.
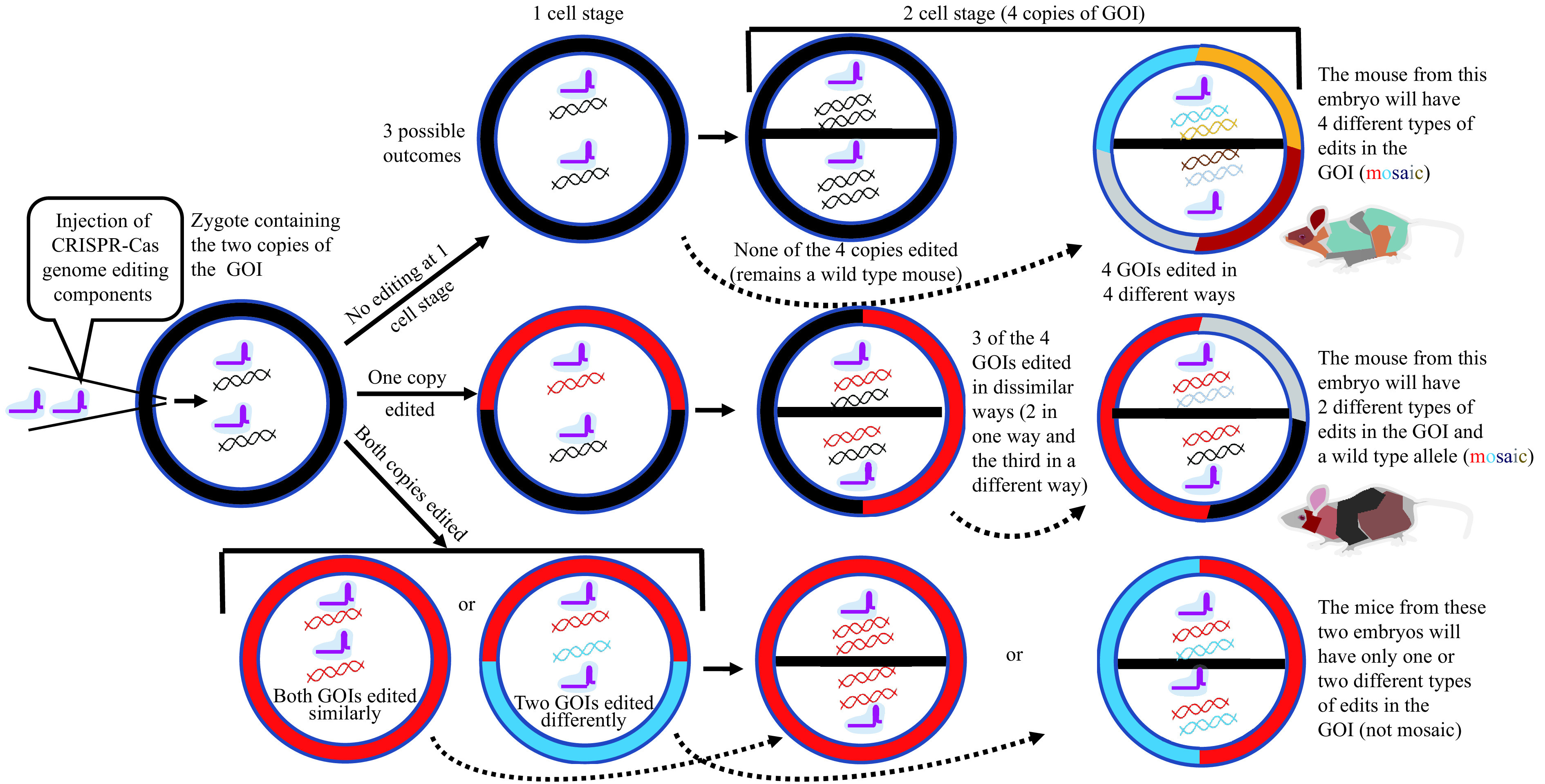
Schematics showing how some of the mice generated using the CRISPR-Cas tool can be mosaic.
GOI: gene of interest.
I have heard that mosaicism is bad but it is also good. What is bad and what is good? Bad: it is very hard to genotype the founder mouse lines (explained in the next question). Using the mosaic founder mice for phenotypic analysis experiments is not recommended because of mosaicism, and it is better to segregate the desired mutant allele by breeding and establishing the mouse line. Good: you can get many different mutant alleles in one mouse, and breeding of that founder can segregate different alleles. This feature provides many different mutant alleles useful for answering different biological questions. Mosaicism is also valuable because a live mouse can be obtained for genes that are embryonically lethal. Because a founder mosaic mouse can contain cells with both the lethal allele and the allele providing for survival[64], such models can be used for some studies.
I have heard that the genotyping of the models generated using CRISPR approach is a bit complex, why is it so? As mentioned above, founder mice are often mosaic containing a mixture of alleles, including expected mutant allele, wild-type allele, and indel alleles. When you try to perform genotyping using Sanger sequencing, you will typically see mixed peaks in the electropherograms and it is hard to figure out whether the mouse contains your desired mutation[65–67]. In such a case, you can separate each allele by cloning and sequencing of PCR products that can produce cleaner sequencing reactions, but the presence of the desired allele is best confirmed by breeding the founder to wild-type mice to produce obligate heterozygous mutant mice. DNA sequencing of the offspring mice will reveal the sequence of the inherited mutant allele and the wild-type allele. It is not advisable to mate founders to each other for transmission of mutant alleles because the offspring will be compound heterozygotes for mutations in the gene, and it is ideal to keep the mutant lines separate until ensuring no off-target events. Also, two independent lines phenotyped separately will serve as a validation for the observed functional phenotype.
Is it always necessary to confirm the desired mutation by sequencing in the F1 generation? Yes, even if you have identified the desired allele in the founder mouse by sequencing, it is very critical to confirm it in the F1 generation because of mosaicism.
Is it necessary to sequence the entire region in my knock-in mouse? Yes. The insertion of donor sequences at the CRISPR cleavage sites depends entirely on endogenous DNA repair mechanism, and often this process is error-prone. The repair of chromosome breaks is a complex process. Similarly, repair of CRISPR-Cas induced breaks can introduce unanticipated complex rearrangements[68–69]. Thus, it is necessary to fully sequence the targeted region including the homology arms, and a few hundred bases beyond the homology arms, to ensure that the targeted region is accurate. Even if the junctions seem accurate, there can be single nucleotide error within the targeted region (in case kilobases long insertions). Although it is not shown by direct comparison of ssDNA and dsDNA donors, intuitively, ssDNA donors can elicit such errors more often than dsDNAs.
I often hear about the concerns of off-target gene editing events, and chances of off-target insertion of the donor DNAs. How do I ensure my mouse does not have off-target events?
Off-target editing events: these are generally rare among genetically engineered mice[70]. If the guide RNA(s) used are quite specific (i.e., if there are no high probability of off-target recognition sequences in the genome), it is rare that the Cas9 would lead to off-target editing events. Resources permitting, it would be ideal to make sure that your mouse line does not have off-target editing events—preferably by using methods such as whole genome sequencing—but that may not be practical for many researchers. One inexpensive approach is to check a few highly likely off-target sites by targeted PCR amplification and sequencing. This approach is quite useful in cases where your guide(s) have highly likely off-target recognition sequences in the genome. The guide RNAs with high specificity scores[71–72] in combination with a Cas9 protein that has been engineered to reduce off-target hits[73] can reduce the chances of off-target hits.
Off-target donor insertion events: these can occur in mice generated by using both CRISPR and non-CRISPR-based approaches. There is no systematic study, however, showing if CRISPR-based methods produce higher frequency of off-target donor insertion events. In general, it is thought that off-target donor insertion events could be higher among the linear dsDNA donors, compared to other types of DNAs, such as linear ssDNAs and circular dsDNAs (plasmids).
What do I do if I have off-target editing and/or off-target donor insertion events? If you have identified off-target effects in your mouse and if they are sufficiently far away from your targeted mutation site (or better, if they are in a different chromosome), they can be easily segregated by breeding. This is achieved by checking those off-target events (by genotyping and sequencing) in the offspring of founders mated to wild-type mice, and use only those offspring mice that do not contain the same off-target events as the breeder stock for establishing the line.
My CRISPR-based model generation project failed. What could be the reason? Can you provide some alternative solutions? Most CRISPR-based projects work if the design is accurate and if the experiments are followed, as per standard protocols. About 5% to 10% of projects fail. One of the main reasons for this could be inefficient guide RNA(s), assuming that the design is correct and the experiments were performed well[74]. This can be addressed if there is an option to use alternative guide(s). The efficiencies of the guides can also be tested beforehand, by in vitro digestion of PCR product containing target site, and/or by in vivo testing using cell lines or microinjecting a few mouse zygotes and culturing and analyzing them at blastocyst stage for the presence of indels at the target sites. In the case of point mutation knock-ins, if there are no guides close to the desired site, you can consider using two guides flanking the target region, making sure that the two guides are at least 30 bases apart from their facing ends[45]. If you are knocking a fusion cassette (for example Cre or rtTA or a reporter) into one of the ends of your GOI, and the project is unsuccessful, you can consider fusing it to the other end of GOI (provided it is OK in regard to the structure of your protein).
My CRISPR-based model generation was successful, but I realized I can use the same model for re-engineering its locus to add some additional expression cassettes (or swap a new cassette with the previously inserted cassette). Can I re-engineer my model to achieve this? This is quite possible. This is one of the best things about CRISPR technology: that new cassettes or additional pieces of DNA can be inserted into the locus that was previously modified. Similarly, some widely popular mouse models having reporter or tetracycline inducible cassettes inserted into some safe harbor loci like ROSA[75] and TIGRE[76–77] can be used for re-engineering their loci. The well-characterized expression cassettes in those models can be swapped (or fused) with your GOI. A couple of examples of re-engineering model designs can be found in a recent perspective article on COVID-19 mouse models[78].
I am interested in learning more. Can you list a few important review articles and research articles about the state-of-the-art methods, and the current practices of creating genetically engineered mice? Relevant articles are cited in the respective questions. Here we list them again, and a few more, under different headings. Note that this is not a comprehensive list. We mention here only a few articles for you to get started. We apologize to the authors of other excellent reviews and research articles that we are unable to cite, due to space constraints.
Reviews:
For "traditional transgenic technologies", please see refs.[79–80]; For "history of transgenic technologies", please see refs.[2,81]; For "Cre-loxP Technology", please see refs.[24,82–83]; For "CRISPR technologies to generate mouse models", please see refs.[84–85]; For "CRISPR technologies to generate mouse models", please see refs.[84–85].
Research articles:
For "first transgenic mouse paper", please see ref.[13]; For "first knockout mouse papers", please see refs.[86–87]; For "first CRISPR mouse paper", please see ref.[62]; For "first CRISPR electroporation papers", please see refs.[53–54]; For "first CRISPR in vivo electroporation paper", please see ref.[58]; For "most promising (or more commonly used) method for developing conditional and knock-in alleles (up to 2 kb)", please see refs.[47–48]; For "CRISPR strategies/approaches that can be promising for larger cassette insertions (>2 kb)", please see refs.[49–52].
Acknowledgments
We thank D. D. Meigs (University of Nebraska Medical Center) and Tonya Cejka (freelance English editor) for editing assistance. C.B.G. is funded by NIH grants R35HG010719, R21GM129559, R21AI143394 and R21DA046831. M.O. is funded by 2016–2017 Tokai University School of Medicine Project Research, the Research Aid from the Institute of Medical Sciences in Tokai University, Grant-in-Aid for Scientific Research (25290035) from MEXT, and a Grant-in-Aid for Challenging Exploratory Research (15K14371) from JSPS.
Contributor Information
Channabasavaiah B. Gurumurthy, Email: cgurumurthy@unmc.edu.
Thomas L. Saunders, Email: tsaunder@umich.edu.
Masato Ohtsuka, Email: masato@is.icc.u-tokai.ac.jp.
References
- 1.Palmiter RD, Brinster RL Transgenic mice. Cell. 1985;41(2):343–345. doi: 10.1016/s0092-8674(85)80004-0. [DOI] [PubMed] [Google Scholar]
- 2.Saunders TL. The history of transgenesis[M]//Larson MA. Transgenic Mouse: Transgenic Mouse. New York: Humana, 2020: 1–26, doi: 10.1007/978-1-4939-9837-1_1.
- 3.Brinster RL, Braun RE, Lo D, et al Targeted correction of a major histocompatibility class II E alpha gene by DNA microinjected into mouse eggs. Proc Natl Acad Sci U S A. 1989;86(18):7087–7091. doi: 10.1073/pnas.86.18.7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cain-Hom C, Splinter E, van Min M, et al Efficient mapping of transgene integration sites and local structural changes in Cre transgenic mice using targeted locus amplification. Nucleic Acids Res. 2017;45(8):e62. doi: 10.1093/nar/gkw1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang C, Jacobsen JC, Ernst C, et al Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44(4):390–397. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubose AJ, Lichtenstein ST, Narisu N, et al Use of microarray hybrid capture and next-generation sequencing to identify the anatomy of a transgene. Nucleic Acids Res. 2013;41(6):e70. doi: 10.1093/nar/gks1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin LO, Splinter E, Davis TL, et al Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 2019;29(3):494–505. doi: 10.1101/gr.233866.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meisler MH Insertional mutation of 'classical' and novel genes in transgenic mice. Trends Genet. 1992;8(10):341–344. doi: 10.1016/0168-9525(92)90278-C. [DOI] [PubMed] [Google Scholar]
- 9.Clark AJ, Bissinger P, Bullock DW, et al Chromosomal position effects and the modulation of transgene expression. Reprod Fertil Dev. 1994;6(5):589–598. doi: 10.1071/RD9940589. [DOI] [PubMed] [Google Scholar]
- 10.Gödecke N, Zha LS, Spencer S, et al Controlled re-activation of epigenetically silenced Tet promoter-driven transgene expression by targeted demethylation. Nucleic Acids Res. 2017;45(16):e147. doi: 10.1093/nar/gkx601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin TP Microinjection of mouse eggs. Science. 1966;151(3708):333–337. doi: 10.1126/science.151.3708.333. [DOI] [PubMed] [Google Scholar]
- 12.Wilson IB, Bolton E, Cuttler RH Preimplantation differentiation in the mouse egg as revealed by microinjection of vital markers. https://dev.biologists.org/content/27/2/467.long. J Embryol Exp Morphol. 1972;27(2):467–469. [PubMed] [Google Scholar]
- 13.Gordon JW, Scangos GA, Plotkin DJ, et al Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci U S A. 1980;77(12):7380–7384. doi: 10.1073/pnas.77.12.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohtsuka M, Ogiwara S, Miura H, et al Pronuclear injection-based mouse targeted transgenesis for reproducible and highly efficient transgene expression. Nucleic Acids Res. 2010;38(22):e198. doi: 10.1093/nar/gkq860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohtsuka M, Miura H, Mochida K, et al One-step generation of multiple transgenic mouse lines using an improved Pronuclear Injection-based Targeted Transgenesis (i-PITT) . BMC Genomics. 2015;16(1):274. doi: 10.1186/s12864-015-1432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tasic B, Hippenmeyer S, Wang C, et al Site-specific integrase-mediated transgenesis in mice via pronuclear injection . Proc Natl Acad Sci U S A. 2011;108(19):7902–7907. doi: 10.1073/pnas.1019507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capecchi MR Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6(6):507–512. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- 18.Sauer B, Henderson N Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A. 1988;85(14):5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skarnes WC, Rosen B, West AP, et al A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474(7351):337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White JK, Gerdin AK, Karp NA, et al Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell. 2013;154(2):452–464. doi: 10.1016/j.cell.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meier ID, Bernreuther C, Tilling T, et al Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J. 2010;24(6):1714–1724. doi: 10.1096/fj.09-140749. [DOI] [PubMed] [Google Scholar]
- 22.Bult CJ, Blake JA, Smith CL, et al Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2019;47(D1):D801–D806. doi: 10.1093/nar/gky1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurumurthy CB, Joshi PS, Kurz SG, et al Validation of simple sequence length polymorphism regions of commonly used mouse strains for marker assisted speed congenics screening. Int J Genomics. 2015;2015:735845. doi: 10.1155/2015/735845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song AJ, Palmiter RD Detecting and avoiding problems when using the Cre–lox system. Trends Genet. 2018;34(5):333–340. doi: 10.1016/j.tig.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madisen L, Zwingman TA, Sunkin SM, et al A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández-Chacón M, Casquero-García V, Luo W, et al iSuRe-Cre is a genetic tool to reliably induce and report Cre-dependent genetic modifications. Nat Commun. 2019;10(1):2262. doi: 10.1038/s41467-019-10239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soriano P Generalized lacZ expression with the ROSA26 Cre reporter strain . Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 28.Niwa H, Yamamura K, Miyazaki J Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 29.Sakai N. Principles for the use of in vivo transgene techniques: overview and an introductory practical guide for the selection of tetracycline-controlled transgenic mice[M]//Shiozawa S. Arthritis Research: Methods and Protocols. New York: Humana Press, 2014: 33–40, doi: 10.1007/978-1-4939-0404-4_4.
- 30.Chen XY, Zaro JL, Shen WC Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan MD, King AMQ, Thomas GP Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J Gen Virol. 1991;72(11):2727–2732. doi: 10.1099/0022-1317-72-11-2727. [DOI] [PubMed] [Google Scholar]
- 32.Kim JH, Lee SR, Li LH, et al High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011;6(4):e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hosur V, Low BE, Li D, et al Genes adapt to outsmart gene-targeting strategies in mutant mouse strains by skipping exons to reinitiate transcription and translation. Genome Biol. 2020;21(1):168. doi: 10.1186/s13059-020-02086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Brolosy MA, Kontarakis Z, Rossi A, et al Genetic compensation triggered by mutant mRNA degradation. Nature. 2019;568(7751):193–197. doi: 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bendriem RM, Singh S, Aleem AA, et al Tight junction protein occludin regulates progenitor Self-Renewal and survival in developing cortex. eLife. 2019;8:e49376. doi: 10.7554/eLife.49376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Popp MW, Maquat LE Leveraging rules of nonsense-mediated Mrna decay for genome engineering and personalized medicine. Cell. 2016;165(6):1319–1322. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyu Q, Dhagia V, Han Y, et al CRISPR-Cas9-mediated epitope tagging provides accurate and versatile assessment of myocardin-brief report. Arterioscler Thromb Vasc Biol. 2018;38(9):2184–2190. doi: 10.1161/ATVBAHA.118.311171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi M, Lu YW, Zhao JJ, et al Transcriptional control of a novel long noncoding RNA Mymsl in smooth muscle cells by a single Cis-element and its initial functional characterization in vessels . J Mol Cell Cardiol. 2020;138:147–157. doi: 10.1016/j.yjmcc.2019.11.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miano JM, Long XC The short and long of noncoding sequences in the control of vascular cell phenotypes. Cell Mol Life Sci. 2015;72(18):3457–3488. doi: 10.1007/s00018-015-1936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miano JM, Long XC, Lyu Q CRISPR links to long noncoding RNA function in mice: a practical approach. Vascul Pharmacol. 2019;114:1–12. doi: 10.1016/j.vph.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isakova A, Fehlmann T, Keller A, et al A mouse tissue atlas of small noncoding RNA. Proc Natl Acad Sci U S A. 2020;117(41):25634–25645. doi: 10.1073/pnas.2002277117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ratnere I, Dubchak I Obtaining comparative genomic data with the VISTA family of computational tools. Curr Protoc Bioinformatics. 2009;26(1):10.6.1–10.6.17. doi: 10.1002/0471250953.bi1006s26. [DOI] [PubMed] [Google Scholar]
- 43.Economides AN, Frendewey D, Yang P, et al Conditionals by inversion provide a universal method for the generation of conditional alleles. Proc Natl Acad Sci U S A. 2013;110(34):E3179–E3188. doi: 10.1073/pnas.1217812110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guzzardo PM, Rashkova C, Dos Santos RL, et al A small cassette enables conditional gene inactivation by CRISPR/Cas9. Sci Rep. 2017;7(1):16770. doi: 10.1038/s41598-017-16931-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie F, Zhou XY, Lin TT, et al Production of gene-edited pigs harboring orthologous human mutations via double cutting by CRISPR/Cas9 with long single-stranded DNAs as homology-directed repair templates by zygote injection . Transgenic Res. 2020;29(5-6):587–598. doi: 10.1007/s11248-020-00218-7. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura Y, Gojobori T, Ikemura T Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28(1):292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quadros RM, Miura H, Harms DW, et al Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins . Genome Biol. 2017;18(1):92. doi: 10.1186/s13059-017-1220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miura H, Quadros RM, Gurumurthy CB, et al Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors . Nat Protoc. 2018;13(1):195–215. doi: 10.1038/nprot.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chu VT, Weber T, Graf R, et al Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes . BMC Biotechnol. 2016;16:4. doi: 10.1186/s12896-016-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu B, Posfai E, Rossant J Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat Biotechnol. 2018;36(7):632–637. doi: 10.1038/nbt.4166. [DOI] [PubMed] [Google Scholar]
- 51.Abe T, Inoue KI, Furuta Y, et al Pronuclear microinjection during s-phase increases the efficiency of CRISPR-Cas9-assisted knockin of large DNA donors in mouse zygotes. Cell Rep. 2020;31(7):107653. doi: 10.1016/j.celrep.2020.107653. [DOI] [PubMed] [Google Scholar]
- 52.Yoshimi K, Oka Y, Miyasaka Y, et al Combi-CRISPR: combination of NHEJ and HDR provides efficient and precise plasmid-based knock-ins in mice and rats . Hum Genet. 2021;140(2):277–287. doi: 10.1007/s00439-020-02198-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashimoto M, Takemoto T Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci Rep. 2015;5:11315. doi: 10.1038/srep11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin WN, Dion SL, Kutny PM, et al Efficient CRISPR/Cas9-mediated genome editing in mice by zygote electroporation of nuclease. Genetics. 2015;200(2):423–430. doi: 10.1534/genetics.115.176594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang WB, Kutny PM, Byers SL, et al Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. J Genet Genomics. 2016;43(5):319–327. doi: 10.1016/j.jgg.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen SA, Lee B, Lee AYF, et al Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J Biol Chem. 2016;291(28):14457–14467. doi: 10.1074/jbc.M116.733154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tröder SE, Ebert LK, Butt L, et al An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS One. 2018;13(5):e0196891. doi: 10.1371/journal.pone.0196891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takahashi G, Gurumurthy CB, Wada K, et al GONAD: genome-editing via Oviductal nucleic acids delivery system: a novel microinjection independent genome engineering method in mice . Sci Rep. 2015;5:11406. doi: 10.1038/srep11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gurumurthy CB, Takahashi G, Wada K, et al GONAD: a novel CRISPR/Cas9 genome editing method that does not require ex vivo handling of embryos. Curr Protoc Hum Genet. 2016;88(1):15.8.1–15.8.12. doi: 10.1002/0471142905.hg1508s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohtsuka M, Sato M, Miura H, et al i-GONAD: a robust method for in situ germline genome engineering using CRISPR nucleases . Genome Biol. 2018;19(1):25. doi: 10.1186/s13059-018-1400-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gurumurthy CB, Sato M, Nakamura A, et al Creation of CRISPR-based germline-genome-engineered mice without ex vivo handling of zygotes by i-GONAD . Nat Protoc. 2019;14(8):2452–2482. doi: 10.1038/s41596-019-0187-x. [DOI] [PubMed] [Google Scholar]
- 62.Shen B, Zhang J, Wu HY, et al Generation of gene-modified mice via Cas9/RNA-mediated gene targeting . Cell Res. 2013;23(5):720–723. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yen ST, Zhang M, Deng JM, et al Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev Biol. 2014;393(1):3–9. doi: 10.1016/j.ydbio.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y, Zhang J, Peng BY, et al Generating viable mice with heritable embryonically lethal mutations using the CRISPR-Cas9 system in two-cell embryos. Nat Commun. 2019;10(1):2883. doi: 10.1038/s41467-019-10748-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quadros RM, Harms DW, Ohtsuka M, et al Insertion of sequences at the original provirus integration site of mouse ROSA26 locus using the CRISPR/Cas9 system . FEBS Open Bio. 2015;5(1):191–197. doi: 10.1016/j.fob.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mianné J, Codner GF, Caulder A, et al Analysing the outcome of CRISPR-aided genome editing in embryos: Screening, genotyping and quality control. Methods. 2017;121-122:68–76. doi: 10.1016/j.ymeth.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 67.Parker-Thornburg J. Breeding strategies for genetically modified mice[M]//Larson MA. Transgenic Mouse: Methods and Protocols. New York: Humana, 2020: 163–169, doi: 10.1007/978-1-4939-9837-1_14.
- 68.Boroviak K, Fu BY, Yang FT, et al Revealing hidden complexities of genomic rearrangements generated with Cas9. Sci Rep. 2017;7(1):12867. doi: 10.1038/s41598-017-12740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kosicki M, Tomberg K, Bradley A Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iyer V, Shen B, Zhang WS, et al Off-target mutations are rare in Cas9-modified mice. Nat Methods. 2015;12(6):479. doi: 10.1038/nmeth.3408. [DOI] [PubMed] [Google Scholar]
- 71.Anderson KR, Haeussler M, Watanabe C, et al CRISPR off-target analysis in genetically engineered rats and mice. Nat Methods. 2018;15(7):512–514. doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doench JG, Fusi N, Sullender M, et al Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vakulskas CA, Dever DP, Rettig GR, et al A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med. 2018;24(8):1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McBeath E, Parker-Thornburg J, Fujii Y, et al Rapid evaluation of CRISPR guides and donors for engineering mice. Genes. 2020;11(6):628. doi: 10.3390/genes11060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muzumdar MD, Tasic B, Miyamichi K, et al A global double-fluorescent Cre reporter mouse. Genes. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 76.Madisen L, Garner AR, Shimaoka D, et al Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron. 2015;85(5):942–958. doi: 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daigle TL, Madisen L, Hage TA, et al A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell. 2018;174(2):465–480. doi: 10.1016/j.cell.2018.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gurumurthy CB, Quadros RM, Richardson GP, et al Genetically modified mouse models to help fight COVID-19. Nat Protoc. 2020;15(12):3777–3787. doi: 10.1038/s41596-020-00403-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Müller U Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis. Mech Dev. 1999;82(1-2):3–21. doi: 10.1016/s0925-4773(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 80.Palmiter RD, Brinster RL Germ-line transformation of mice. Annu Rev Genet. 1986;20:465–499. doi: 10.1146/annurev.ge.20.120186.002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu WH. Microinjection and micromanipulation: a historical perspective[M]//Liu CY, Du YB. Microinjection: Methods and Protocols. New York: Humana Press, 2019: 1–16, doi: 10.1007/978-1-4939-8831-0_1.
- 82.Anastassiadis K, Glaser S, Kranz A, et al A practical summary of site-specific recombination, conditional mutagenesis, and tamoxifen induction of CreERT2. Methods Enzymol. 2010;477:109–123. doi: 10.1016/S0076-6879(10)77007-5. [DOI] [PubMed] [Google Scholar]
- 83.Nagy A Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26(2):99–109. doi: 10.1002/(SICI)1526-968X(200002)26:2<99::AID-GENE1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 84.Gurumurthy CB, Lloyd KCK Generating mouse models for biomedical research: technological advances. Dis Model Mech. 2019;12(1):dmm029462. doi: 10.1242/dmm.029462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miano JM, Zhu QM, Lowenstein CJ A CRISPR path to engineering new genetic mouse models for cardiovascular research. Arterioscler Thromb Vasc Biol. 2016;36(6):1058–1075. doi: 10.1161/ATVBAHA.116.304790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doetschman T, Gregg RG, Maeda N, et al Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature. 1987;330(6148):576–578. doi: 10.1038/330576a0. [DOI] [PubMed] [Google Scholar]
- 87.Thomas KR, Capecchi MR Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51(3):503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]