

Abstract
With advancements in gene editing technologies, our ability to make precise and efficient modifications to the genome is increasing at a remarkable rate, paving the way for scientists and clinicians to uniquely treat a multitude of previously irremediable diseases. CRISPR-Cas9, short for clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9, is a gene editing platform with the ability to alter the nucleotide sequence of the genome in living cells. This technology is increasing the number and pace at which new gene editing treatments for genetic disorders are moving toward the clinic. The β-hemoglobinopathies are a group of monogenic diseases, which despite their high prevalence and chronic debilitating nature, continue to have few therapeutic options available. In this review, we will discuss our existing comprehension of the genetics and current state of treatment for β-hemoglobinopathies, consider potential genome editing therapeutic strategies, and provide an overview of the current state of clinical trials using CRISPR-Cas9 gene editing.
Keywords: sickle cell disease, sickle cell anemia, fetal hemoglobin, hemoglobinopathy, CRISPR, gene editing, genome engineering
Introduction
Hemoglobin (Hb) is a heterotetramer composed of two α-globin and two β-like globin subunits that transports oxygen throughout the body. β-hemoglobinopathies are a group of disorders characterized by quantitative or qualitative defects in β-globin synthesis. Quantitative defects, known as β-thalassemia, are caused due to deletions or point mutations in the HBB gene (which encodes β-globin) or its regulatory elements that result in decreased production of the β-globin chains. This imbalance in the production of the α- and β-globin chains results in accumulation and subsequently precipitation of the excess α-globin molecules within the erythroid precursors and their premature death. On the other hand, qualitative defects arise from mutations of HBB resulting in the production of an altered β-globin molecule. Sickle Hb (HbS) is one such structural variant of adult Hb (HbA) that is characterized by a single A-to-T transversion in the sixth codon of HBB, resulting in a p.Glu6Val substitution in the β-globin subunit of HbA. Sickle cell disease (SCD) is a collective term referring to any β-hemoglobinopathy containing the HbS allele. Homozygosity for this mutation results in sickle cell anemia (SCA) which is the most severe type of SCD[1]. Two other common β-globin structural variants are HbC[2] and HbE[3]. HbC (c.19G>A) is caused by a p.Glu6Lys substitution in the β-globin subunit. Individuals homozygous for the HbC mutation, a condition known as hemoglobin C disease, present with mild hemolytic anemia[2,4]. HbE (c.79G>A) is a structural variant resulting in a p.Glu26Lys substitution in the β-globin subunit. The mutation is found at the 3′ end of exon 1 ofHBB and introduces a cryptic donor sequence, which allows for aberrant splicing and causes a frameshift in the open reading frame. The frameshift results in decreased levels of β-globin and a mild β-thalassemia phenotype. A detailed catalog of various hemoglobin variants and mutations that cause thalassemia is maintained at the HbVar database[5]. Coinheritance of various quantitative and qualitative defects is possible and results in severe phenotypes in many cases.
Since SCA is one of the most common and potentially most devastating β-hemoglobinopathies, it will be discussed in detail in the next few sections. However, many of the concepts and ideas discussed here are applicable to all β-hemoglobinopathies. In SCA, under hypoxic conditions HbS rapidly polymerizes, reducing the deformability of red blood cells (RBCs) and turning them into the characteristic sickle shape (Fig. 1). Following multiple cycles of HbS polymerization/depolymerization, sickled RBCs either lyse during passage through capillaries, decreasing their half-life, or occlude small blood vessels causing a number of devastating clinical complications[6]. Chronic hemolysis and occlusion of the blood vessels by these deformed RBCs are responsible for the vasculopathy and co-morbidities that occur in virtually all body organs in a patient with SCA, including anemia, debilitating recurrent pain crises, acute chest syndrome, pulmonary hypertension, avascular necrosis, kidney failure, thromboses, liver disease and stroke[7–9]. Most of these patients experience a poor quality of life and significantly reduced lifespan[7–8,10].
Figure 1.
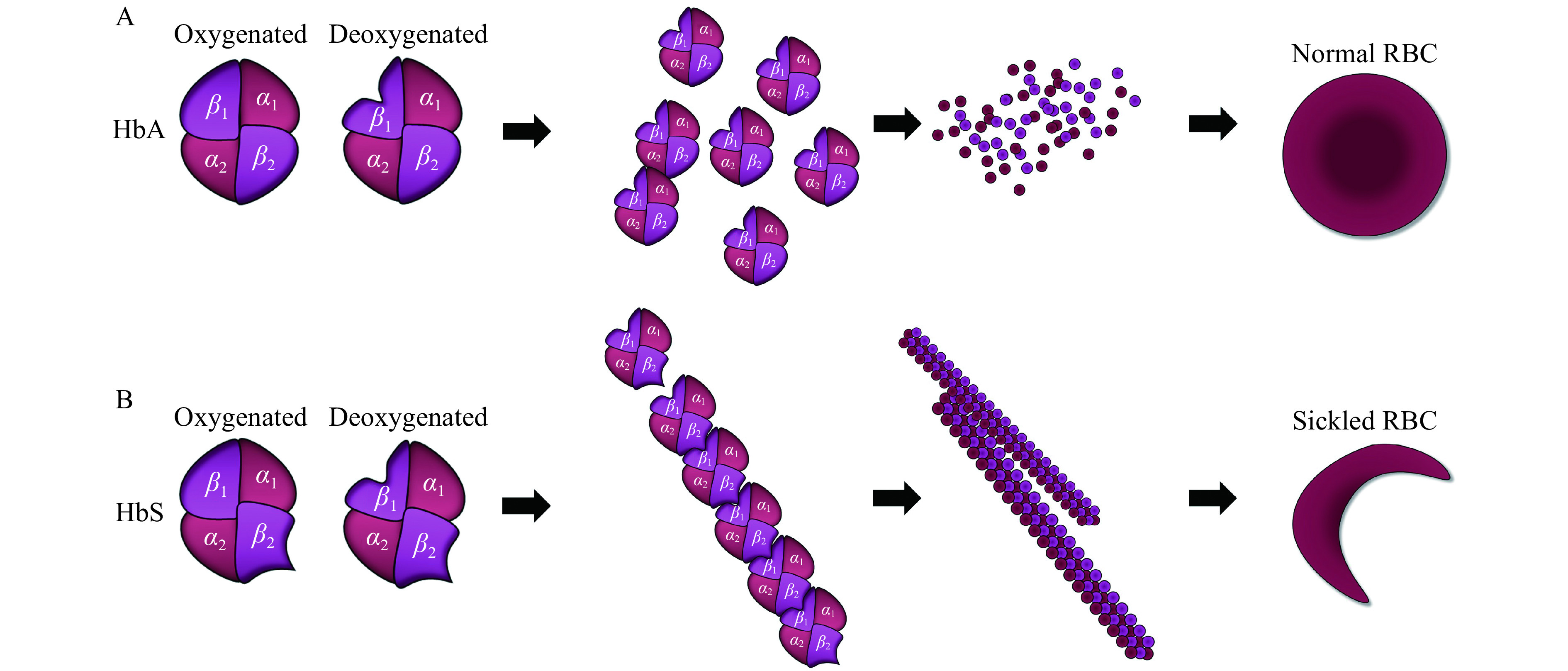
HbS fiber formation and red blood cell sickling.
A: Adult hemoglobin (HbA) molecules are heterotetramers composed of two α-and two β-globin subunits and normally exist as solitary molecules inside red blood cells. B: In sickle cell disease (SCD), a substitution in the sixth amino acid of the β-globin subunit causes a structural aberration (HbS) that leads to polymerization of tetramers upon deoxygenation. These tetramers then align to form rod-like fibers that cause the deformation of the red blood cells into the characteristic sickle shape. RBC: red blood cell.
Approximately 100 000 individuals in the United States and millions more worldwide are affected by SCD[7]. An absolute number of children born each year with SCD is unknown due to a lack of adequate screening and detection in resource-limited countries where the disease is most prevalent; however, estimations suggest that more than 300 000 children are born annually with SCD, including nearly 3000 in the United States[11]. In high-income countries, due to extensive newborn screening programs and comprehensive medical services, more than 95% of patients with SCD survive through childhood. However, in Africa and South Asia, the majority of children born with the disease often remain undiagnosed and die under the age of 5 years[12]. SCD puts a huge burden on the health-care systems and has a considerable impact on affected individuals. In the United States alone, the cost of direct medical care for individuals with SCD exceeds $1 billion per year[13].
More than a century after the disease was first described, and after decades of scientific research, gene therapy for SCD is only recently in clinical trials. In this review, we discuss the underlying genetics of SCD, how it is currently being treated, strategies for CRISPR-Cas9 gene editing therapies, and the current state of gene editing clinical trials.
Hemoglobin switching and hereditary persistence of fetal hemoglobin
Both the α- and β-like globin genes are developmentally regulated, but for the purpose of this review, only regulation of the β-like globin genes will be discussed. The β-globin locus consists of five β-like globin genes, which are expressed in a spatiotemporal manner (Fig. 2). ε-globin (HBE1) is expressed in the early embryonic yolk sac and heterodimerizes with ζ-globin (an α-like globin gene, HBZ) to form embryonic Hb (ζ2ε2). As embryonic development progresses, the site of erythropoiesis moves from the yolk sac to the fetal liver and spleen, where erythroid cells then produce Gγ-globin (HBG2) and Aγ-globin (HBG1), which, with α-globin, results in fetal hemoglobin (HbF, α2γ2). The final site of erythropoiesis is the bone marrow, where an adult pattern of Hb is established around birth. The expression of HBG1 and HBG2 becomes repressed around the time of birth, and the expression of HBB is turned on, which results in the production of β-globin. Adult Hb (HbA, α2β2), which is comprised of two α-globin and β-globin subunits, is thereafter expressed throughout adult life. The γ- to β-globin switch, also known as Hb switching, is an intriguing and complex paradigm of the developmental regulation of gene expression and is incompletely understood. However, it is clinically very important because β-hemoglobinopathies can be treated by inhibiting this switch. Only about 2% of total Hb following birth is composed of δ-globin (HBD), which produces the alternate adult Hb (HbA2, α2δ2). For more information on hemoglobin switching, see references[14–16].
Figure 2.

Hemoglobin switching.
The spatiotemporal expression of the globin genes is shown (top). The levels of expression are shown with colors corresponding to the various β-globin genes in a diagram of the β-globin locus (bottom), which includes the upstream locus control region (LCR).
Several studies have described a correlation between elevated HbF levels in adults and a reduction in SCD severity[8,17–20]. The expression of HbF varies naturally between individuals, including those with SCD. Some individuals have persistently elevated levels of HbF well into adulthood, which is a benign condition called hereditary persistence of fetal hemoglobin (HPFH)[21]. When mutations causing HPFH are co-inherited with SCD, the clinical severity of SCD is significantly reduced[8,22–23]. These findings have galvanized research aimed at understanding the molecular machinery that represses HbF and how it could be therapeutically manipulated.
BCL11A
To better understand the genetic determinants that regulate HbF expression, and to uncover potential therapeutic targets, two complementary methods have been used to identify the trans-regulatory proteins and cis-regulatory DNA elements controlling the expression of genes at the β-globin locus: 1) analysis of rare individuals and pedigrees with very high HbF levels (individuals with HPFH)[24]; and 2) genome-wide association studies (GWAS) identifying high-frequency polymorphisms associated with mild to moderately elevated HbF[25–26]. GWAS illuminated loci significantly associated with HbF levels in healthy individuals, SCD, and β-thalassemia patients. One study used healthy individuals who exhibited extremes of F-cell distribution (cells with immunoreactive HbF) to identify a quantitative trait locus in the second intron of the oncogene B-cell lymphoma/leukemia 11A (BCL11A)[27], while a separate report demonstrated an association between genetic variants within the HBS1L-MYB locus and increased HbF levels[28]. Additional GWAS highlighted single nucleotide polymorphisms (SNPs) found in BCL11A, HBS1L-MYB, and the extended β-globin locus that were associated with HPFH[25–26]. Subsequent molecular studies have validated BCL11A as a direct repressor of HbF[29–34].
BCL11A expression is inversely correlated with HbF levels[29]. Knockout and knockdown approaches in both human and mouse models have demonstrated that BCL11A is a bona fide γ-globin repressor[32–34]. While the major role of BCL11A in erythroid precursors seems to be HbF repression, this multiple zinc-finger transcription factor is also known to function outside the erythroid lineage. BCL11A has been implicated in the development, maturation, or self-renewal of multiple lineages including B-lymphocytes, hematopoietic stem cells (HSCs), dendritic, breast, and pancreatic cells, as well as cells within the central nervous system[35–39]. Additionally, global loss of BCL11A results in perinatal lethality in mice[37], and humans who carry rare variants resulting in BCL11A haploinsufficiency, present with autism-like neurological disorders in addition to expressing high levels of HbF[40–42]. However, erythroid-specific deletion of BCL11A in mice de-represses γ-globin and reverses the symptoms of SCD without perturbing erythropoiesis[33]. Thus, disrupting the erythroid-specific function of BCL11A while maintaining its non-erythroid roles would be an ideal therapy to cure SCD.
A significant advancement was made when Bauer and colleagues subjected primary human erythroblasts to a DNase I sensitivity assay and discovered that HbF-inducing SNPs in the second intron of BCL11A overlap with areas of open chromatin[43]. The authors further demonstrated that these SNPs are embedded in an erythroid-specific enhancer bound by the transcription factors GATA1 and TAL1, which drives BCL11A specifically in the erythroid lineage. Deletion of the enhancer region in mouse erythroleukemia (MEL) cells resulted in a severe reduction in BCL11A expression with a corresponding induction of HbF. Conversely, when the enhancer was deleted in a pre-B lymphocyte cell line (a non-erythroid line), BCL11A expression was unchanged[43].
Other investigations into the molecular regulation of the β-globin locus have uncovered several BCL11A binding sites. Most notable is the upstream enhancer that consists of a cluster of erythroid-specific cis-DNA elements termed the locus control region (LCR) (Fig. 2), which is brought into close proximity to the globin promoters by DNA looping, providing robust expression of the globin genes[32,44–45]. In addition, a separate regulatory region bound by BCL11A, GATA1, and HDAC1 was identified upstream of the δ-globin gene by analyzing naturally-occurring, HPFH-associated deletions in the β-globin locus[46]. More recently, BCL11A was shown to directly bind and repress γ-globin production by binding a TGACCA motif located in the proximal promoters of the duplicated γ-globin genes[47]. This motif has been found to be mutated in some individuals with HPFH[48–49]. Furthermore, genetic manipulation of the TGACCA motif disrupts BCL11A binding and derepresses γ-globin[47]. These data provide evidence that hemoglobin switching is, in part, controlled by direct binding of BCL11A at the γ-globin promoters, a mechanism that can be exploited for the treatment of β-hemoglobinopathies. Overall, BCL11A is an attractive therapeutic target, and different gene editing approaches targeting BCL11A are covered below. It should be noted that BCL11A is a proto-oncogene that was so named because multiple B-lymphoid leukemias have translocations involving this gene. Therefore, manipulation of BCL11A or its binding sites as a strategy to treat SCD should be thoroughly vetted for safety as with any gene editing strategy.
Currently available treatments for sickle cell disease
Disease modifying therapies
Pharmacotherapy
Until recently, hydroxyurea (HU) was the only FDA-approved drug for treatment of SCD. HU was discovered as a SCD therapy while screening the effects of cytotoxic compounds for their ability to induce HbF in non-human primates[16,50–51]. HU is a ribonucleotide reductase inhibitor that evokes several effects and, at low doses, increases HbF. The exact pathway by which HU accomplishes HbF induction is not clearly elucidated, but multiple potential mechanisms have been described[52]. HU has proven to be safe and well-tolerated in short and long-term use in both children and adults and has been suggested to be made available to all SCD patients[53]. Despite its beneficial effects, HU is underutilized due to user-related compliance issues, dose-related hematological toxicities, potential for teratogenicity, fear of side effects, etc[54–55]. Recently, three new drugs have been approved for treatment of SCD: L-glutamine, voxelotor, and crizanlizumab.
L-glutamine is a conditionally essential amino acid, which in a phase 3 randomized trial was shown to reduce numbers of vaso-occlusive crises, acute chest syndrome incidences, and hospitalizations in SCD patients[56]. It has been described that a decrease in erythrocyte glutamine and glutathione leads to an abundance of reactive oxygen species, which contributes to the cytotoxic milieu of the sickled RBCs and is associated with hemolysis[57–58]. Glutamine acts as a precursor to glutathione, which reduces oxidative stress, thus reducing hemolysis[59–60]. The exact mechanism by which it reduces pain crises is, however, unknown. Crizanlizumab is a humanized monoclonal antibody that binds to P-selectin and blocks its interaction with P-selectin glycoprotein ligand 1. P-selectin is expressed on the surface of endothelial cells and mediates adhesion of sickle RBCs to the surface of vessels resulting in vascular occlusion[61–63]. P-selectin mediates binding of platelets to neutrophils in mice and humans with SCD[64]. Thus, blockade of P-selectin by crizanlizumab reduces the risk of vaso-occlusion and inflammation. Indeed, in a randomized, phase 2 trial, treatment with crizanlizumab halved the annual rate of sickle cell-related pain crises in comparison with placebo[65]. Lastly, voxelotor is an HbS polymerization inhibitor. It reversibly binds to hemoglobin and stabilizes it in its oxygenated state[66–67]. In a phase 3 randomized, clinical trial, voxelotor increased hemoglobin levels and reduced markers of hemolysis in individuals with SCD[68]. While all these drugs have been helpful in modifying the symptoms of SCD, none of them are curative. Moreover, many patients have a much higher severity of disease or do not respond to one or more of these drugs and need alternative therapies.
Blood transfusion therapy
One of the most severe and debilitating consequences of SCD is acute stroke[69]. Red blood cell exchange transfusions have been shown to significantly decrease the risk of stroke in patients with SCA[70]. The Stroke Prevention Trial in Sickle Cell Anemia (STOP I) randomly assigned children 2 to 16 years old, who were at a high risk of developing a stroke based on transcranial Doppler, to receive either chronic blood transfusions every 3 to 4 weeks or a standard care regimen. Children in the standard care cohort developed a stroke at an incidence of 10% per year, while the only stroke in the chronic transfusion group occurred after 26 months. The trial was terminated early after 12 children had strokes, 11 of whom were receiving standard care[70]. It has also been demonstrated that discontinuing regular transfusion treatments can result in an increased stroke risk[71]. Although now considered standard of care, regular blood transfusions are incompletely effective in preventing strokes in those who have already developed one. Recurrent strokes and silent cerebral infarcts continue to occur among children with silent cerebral infarcts who are currently receiving regular blood transfusion therapy[72]. Thus, chronic blood transfusion therapy can, at best, only be considered palliative for secondary prevention of strokes. Additionally, recurrent transfusions are complicated by the risk of alloimmunization to RBC antigens, delayed hemolytic transfusion reactions and iron overload[73–74]. Several patients develop alloantibodies to transfused antigens and then are no longer able to receive transfusions from donors to whom they are immunized[75].
Bone marrow transplantation
The only widely available curative treatment for patients with SCD is an allogeneic hematopoietic stem cell transplantation (HSCT). HSCT involves replacing the HSCs of an individual with SCA using HSCs from a human leukocyte antigen (HLA)-matched donor. However, due to immunological complications such as graft rejection, graft-versus-host disease as well as other issues associated with myeloablative chemotherapy exposure such as infertility and infections, including a low but non-zero risk of death, HSCT is generally reserved for the most severe cases and for those who are unresponsive to alternative treatments such as hydroxyurea[76–77]. At the same time, less than 20% of patients with SCA have an eligible donor available, making this therapy inaccessible to most patients who really need it.
Gene addition therapy
Several preclinical and clinical studies are currently attempting a gene transfer approach wherein a functional copy of the β-globin gene is transferred to the HSCs. The most successful of these has been the clinical trial of a drug product called LentiGlobin BB305 led by Bluebird Bio (NCT01745120 and NCT02151526)[78–83]. This therapy involves reinfusion of autologous CD34+ HSCs after lentiviral integration of a β-globin gene marked by an anti-sickling T87Q substitution[80]. Driven by cis-regulatory elements, this vector delivers erythroid lineage-specific expression of the therapeutic modified HBB encoding an anti-sickling variant (βA87Thr:Gln) in patients with β-thalassemia and SCD. Published reports indicate that study participants have increased circulating hemoglobin, reduced or eliminated need for red blood cell transfusions, and no signs of clonal dominance due to insertional mutagenesis of the therapeutic vector[83]. While this treatment is extremely promising and has received conditional marketing approval from the European Medicines Agency for patients older than 12 years of age[84], long-term follow-up of these patients is needed. Even with the self-inactivating lentiviral vectors, the risk of leukemogenesis from a wayward integration still remains, a pitfall that precise gene editing should be able to avoid. Additionally, the silencing of integrated elements and the fraction of transduced cells should be considered, as well as the cost and complexity to scale up lentiviral production to treat large numbers of patients.
Gene editing options with CRISPR-Cas9
Gene editing can be defined as creating a genomic modification in a targeted manner. Traditional genome editing strategies involve creating a targeted DNA double-strand break (DSB) in genomic DNA near the site of desired change. This can be achieved using several different nuclease platforms including, but not limited to, meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases, and CRISPR-Cas9. For the purposes of this review, we will focus on the CRISPR-Cas9 system from Streptococcus pyogenes. In its simplest form, CRISPR-Cas9 is a two-component system consisting of a Cas9 nuclease and a guide RNA (gRNA). In brief, the Cas9 protein requires a protospacer adjacent motif (PAM) of 5′-NGG-3′ in order to efficiently bind DNA. Additional sequence specificity comes from the gRNA, which is a short RNA molecule containing an approximately 20 base pair (bp) sequence that complexes with and "guides" Cas9 to a user-defined location in the genome by complementary base pairing to the DNA region of interest adjacent to the PAM, allowing Cas9 to cut at the appropriate place in the genome.
Because an unresolved DSB can be detrimental and even deadly, mammalian cells are quite efficient at repairing such breaks. After the nuclease-induced DSB is created, the endogenous cellular repair machinery will repair the DNA lesion by one of a few pathways including but not limited to non-homologous end joining (NHEJ), microhomology mediated end joining (MMEJ), or homology direct repair (HDR). NHEJ can be error-prone, and aberrant repair by this pathway leads to stochastic insertions and/or deletions (indels) at the site of the break and is useful for disrupting genes, binding sites, and specific DNA motifs. MMEJ relies on short microhomologies at or near the cut site and often results in the deletion of the intervening sequences between said microhomologies and can also disrupt genes, binding sites, and DNA motifs.
While NHEJ and MMEJ only require a targeted double-strand break to stimulate repair, HDR requires the addition of a donor template with homology to the cut site to be introduced into the nucleus of the targeted cell. Donor templates can be either circular or linearized double-stranded DNA (dsDNA), single-stranded DNA (ssDNA), single-stranded oligodeoxynucleotides (ssODNs), or viral donors. HDR is said to be "user-defined" because the donor template can be synthesized with the desired genomic modification.
As described above, SCA is caused by a single point mutation in the HBB gene and the disease phenotype is manifested in the red blood cells of the afflicted individual. Normal adult RBCs are terminally differentiated and have a limited lifespan with an average time to senescence of approximately 120 days, while sickled RBCs last around 20 days[85–87]. Additionally, RBCs have undergone enucleation, so there is no DNA to correct in the disease-causing cell type. Therefore, for a gene therapy approach to have the promise of being curative for SCA, the modification must occur in the hematopoietic stem cells (HSCs).
When considering gene editing therapy for β-hemoglobinopathies, approaches can be divided into two general strategies: 1) those developed to repair or modify the underlying genetic mutation (gene correction); and 2) those designed to elevate HbF levels. While methods to elevate HbF levels could ameliorate SCA as well as β-thalassemias, approaches aiming to correct the HbS mutation would be confined to SCA.
Gene correction
Theoretically, the most logical method to cure a genetic disease is to directly modify or correct the disease-causing variant. This approach would ensure the preservation of all cis-regulatory elements that might control gene function as well as ameliorate any detrimental effects of the mutant gene. Traditional gene correction strategies rely on HDR which has some technical challenges. First, HDR approaches do not preclude repair by the NHEJ pathway. In fact, the NHEJ pathway is the preferred pathway of repair in mammalian cells. Moreover, because the disease-causing mutation for SCA is in the coding region, an HDR strategy will also result in indels within the HBB gene of some cells leading to gene knockout and potentially producing β-thalassemia (Fig. 3). Second, HDR is limited to the S or G2 phase of the cell cycle, which may be limiting when trying to target stem cells that are not cycling. Also, HDR requires the additional obligate delivery of an exogenous donor template, which increases manufacturing costs, can increase toxicity, and has more potential for an immunological response.
Figure 3.
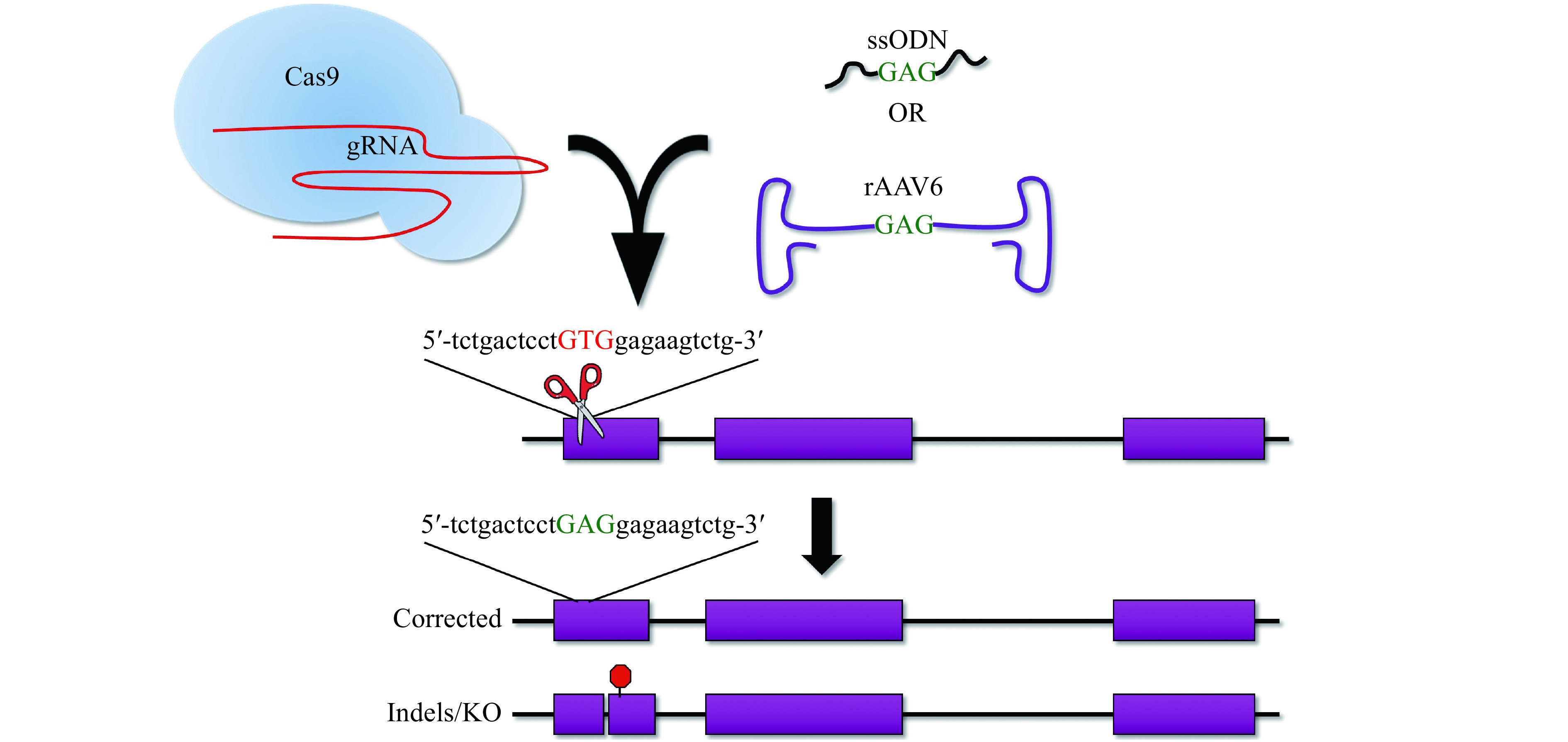
Gene correction of the HbS mutation with donor templates via homology direct repair (HDR).
Gene correction of the HbS mutation requires a Cas9:gRNA complex to create a DSB (scissors) and a donor template to specify the desired change (ssODN or rAAV6 donors shown). The resultant outcomes show the correct HDR integration with the wild type GAG (green text) substituted for the pathogenic GTG (red text). Additionally, repair via the error-prone and dominant DNA repair pathway in mammalian cells will result in indels and knockout (KO) of the HBB gene. DSB: double-strand break; ssODN: single-stranded oligodeoxynucleotides.
Several groups have shown profound levels of correction using HDR in induced pluripotent stem cell (iPSC)-derived HSCs and CD34+ (a marker for hematopoietic stem and progenitor [HSPC]) cells. Although the level of correction has been favorable in some iPSC-derived HSPC experiments[88–91], currently, the differentiation of iPSCs toward HSCs leads to primitive rather than definitive hematopoietic cells, which cannot engraft into a xenograft mouse model, leaving the investigations lacking the ultimate functional read-out. Thus, until improved iPSC differentiation protocols are developed for the HSC lineage, bone marrow (BM) or mobilized CD34+ cells, although more difficult and expensive to obtain, will likely be the preferred cellular source for proof-of-principle studies.
In 2016, DeWitt and colleagues optimized a selection-free strategy to edit CD34+ HSPCs using ribonucleoproteins (RNPs) consisting of an unmodified single gRNA and Cas9 protein along with a ssODN donor[92]. In these studies, this group was able to achieve 11.8%±3.7% correction in the input HSPC population just prior to injection into an immunodeficient mouse model. Sixteen weeks following engraftment, however, only 2.3% (BM) and 3.7% (spleen) of engrafted cells maintained the HDR-mediated editing at the SCD mutation, suggesting that the long term repopulating HSCs which constitute a minority of the heterogeneous CD34+ cell population may be relatively refractory to HDR-mediated events or donor delivery in comparison to the more-committed hematopoietic progenitors[93]. Although it is not definitively known how much correction is required for such a treatment to be curative for SCA, one might speculate that even a low level of long-term edited cells may be clinically relevant. Previous observations after allogeneic hematopoietic stem cell transplantation for SCA have shown mixed chimerism at low levels in the bone marrow and have translated to a clinically significant increase in the number of non-sickling RBCs[94–96]. This increase could be due to a longer life span of normal RBCs vs. HbS RBCs.
To enrich for HDR-edited HSPCs, Dever et al developed a strategy using RNPs and an adeno-associated viral donor containing a selectable marker, which after selection, resulted in a collection of cells with more than 85% of clones with the targeted integration event[97]. This selection method increased the long-term engraftment retention to between 10% and 75% (n=3) of the engrafted human cells at 16 weeks post xeno-transplantation. While the enrichment strategy increased the overall frequency of edited cells, it also led to an eightfold decrease in the total number of HSCs in the transplanted population. It is possible that a substantial ex vivo expansion phase would need to be introduced in order to scale up the recovery of larger numbers of edited HSCs prior to transplantation for clinical translation. Dever and colleagues also noted that in the unenriched population, they saw a decrease in editing frequencies between the input cells and the long-term engrafted cells, which further supports the notion that HSCs are more refractory to HDR than the bulk CD34+ population[97].
Until recently, strategies used to directly correct a genetic variant were restricted to HDR-mediated gene editing, which is encumbered by low efficiency and unwanted indels. However, recent developments of two new classes of genome editors have created exciting new approaches for gene modification. Cas9 base editors[98] and prime editors[99] provide a method for correcting disease-causing SNPs without the requirement of creating a potentially dangerous DSB or the need to deliver a separate donor template. Base editors eliminate the requirement of a DSB by utilizing a catalytically impaired Cas9 fused to either an adenine deaminase (adenine base editor, ABE) or cytidine deaminase (cytosine base editor, CBE)[100]. Like traditional CRISPR-Cas systems, base editors are targeted to specific DNA sequences with a gRNA, but instead of generating a DSB, they have the ability to directly install single base pair substitutions.
CBEs were created by fusing a cytosine deaminase and uracil glycosylase inhibitor (UGI) to a Cas9 nickase. While the gRNA/Cas9 complex targets and binds DNA, the cytosine deaminase converts the targeted cytosine to a uracil, which is read as thymidine. The addition of the UGI helps evade the base excision repair pathway, allowing the newly installed uracil to remain. The resulting U-G mismatch is then resolved to a T-A base pair by the DNA repair machinery[101–102]. Thus, CBEs can create C-to-T (or G-to-A) point mutations.
ABEs were created by fusing an evolved transfer RNA adenosine deaminase heterodimer to a Cas9-nickase. When targeted to a particular adenine, ABEs will catalyze the deamination of adenine to inosine, which is read as guanine by the polymerase. Following DNA repair/replication, the A-T base pair is converted to a G-C base pair[103]. A limitation of base editors is the requirement of the targeted adenine/cytosine to fall within a narrow activity window in relation to the NGG PAM. For example, ABE7.10 requires the target nucleotide to be within positions 4 to 7, with the NGG PAM being positions 21 to 23[103]. However, if more than one adenine/cytosine are in the activity window, substituting either or both nucleotides is possible and is referred to as a bystander mutation. Depending on the sequence context, a bystander mutation could be a silent or benign mutation, or a more deleterious, structure-altering amino acid change. Base editors are continually being developed that combine catalytically impaired Cas variants and orthologs with different deaminases to produce divergent base editing capabilities, including off-target reductions and broader PAM compatibility[100]. Another limitation is that base editors only have the capacity to catalyze transitions, that is, of the 12 possible base changes, current base editors can catalyze C>T, G>A, A>G, and T>C. In the interest of this review, atransversion is required to correct the SCA mutation (T>A), and therefore cannot be directly corrected by current base editing technology. For more information on base editors, please see Eidet al and Molla et al[98,104].
CBEs were created by fusing a cytosine deaminase and uracil glycosylase inhibitor (UGI) to a Cas9 nickase. While the gRNA/Cas9 complex targets and binds DNA, the cytosine deaminase converts the targeted cytosine to a uracil, which is read as thymidine. The addition of the UGI helps evade the base excision repair pathway, allowing the newly installed uracil to remain. The resulting U-G mismatch is then resolved to a T-A base pair by the DNA repair machinery[101–102]. Thus, CBEs can create C-to-T (or G-to-A) point mutations.
Despite requiring a transversion, the SCA mutation (c.20A>T; p.Glu6Val) can theoretically be targeted by ABEs on the noncoding strand. This, however, would not directly correct the SCA mutation. Instead, by converting the adenine on the opposite strand to a guanine, a cytosine would result in the coding strand, converting the pathogenic valine to an alanine. Alanine at p.6 has been identified in the human population as HbG–Makassar, and while these individuals appear to have a much less severe clinical phenotype[105–106], symptoms arising from a compound heterozygous HbG/HbS genotype is not well documented. However, the most desirable SpCas9 variant PAM (Cas9-NGG) is found 18 bps upstream, which would place the target adenine in position 2. Since the activity window for SpCas9-NGG ABE consists of positions 4 to 7, base editing the SCA mutation may not be practical until base editors are further evolved. It should also be noted that two additional adenines fall within the activity window in this particular locus; however, synonymous mutations result when these bystanders are substituted (Fig. 4).
Figure 4.
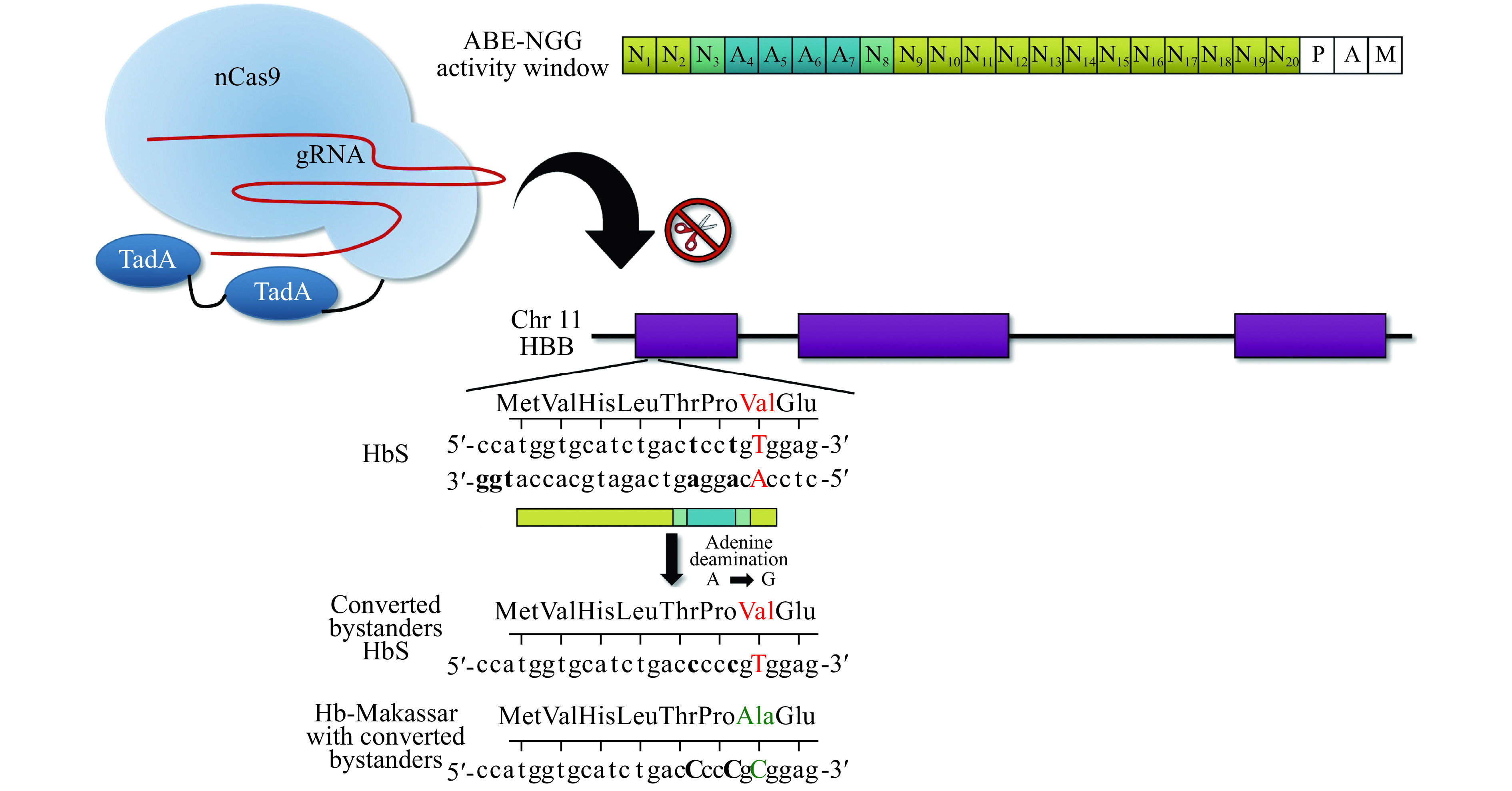
Adenine base editor (ABE) approach to generate Hb-Makassar for HbS therapy.
The activity window of the ABE-NGG protospacer is shown in blue (top right). Notice the pathogenic T: A falls outside the activity window (red text), while two neighboring adenines are within the window (bold text). This strategy is likely to result in primarily converting the adenine bystanders, which are synonymous mutations (Thr>Thr and Pro>Pro), and to a lesser degree, converting the bystanders along with the HbS Val to the Hb-Makassar Ala (green text). nCas9: Cas9 nickase; TadA: tRNA-specific adenine deaminase.
Other Hb SNPs that can be resolved with a transition are found at the HbC and HbE mutations. Given HbSC is the second most common form of SCD with 80 000 births per year worldwide[4], establishing a therapeutic intervention is critical and could be targeted via traditional HDR or base editing. Individuals who are compound heterozygous for HbE and β-thalassemia (HbE/β-thal) comprise 50% of the clinically severe β-thalassemia cases[107]. Using a base editor to correct HbE in the HbE/β-thal background would result in the genotype β-thal/WT, which is clinically benign or mildly symptomatic.
In addition to base editing, a new DSB-free genome editing platform has recently been developed called prime editing[99]. Briefly, the prime editor consists of a Cas9-nickase conjugated to a reverse transcriptase that is targeted to a particular genomic locus via a prime editing gRNA (pegRNA). The pegRNA consists of three components: a traditional gRNA allowing for Cas9 targeting, a primer binding site for reverse transcriptase initiation, and a reverse transcriptase template containing the user-defined genetic modification for genomic incorporation (Fig. 5). Anzalone and colleagues were able to successfully create small (<100 bp) targeted insertions and deletions as well as all possible transitions and transversions. As a proof-of-concept study, Anzaloneet al created the SCA mutation in HEK293 cells and achieved up to 58% correction with only 1.4% indels using prime editing[99]. The potential of prime editing is immense; however, its implementation is still a bit laborious. For example, employing prime editing requires several considerations, such as the primer binding site length, reverse transcriptase template length, where to place a second nicking gRNA, and which DNA strand to edit. Additionally, genome wide off-target studies at both the DNA and RNA level are needed in order to better understand the specificity of prime editors. Although prime editing grants flexibility in editing options, it seems that large insertions or deletions (>100 bp) may not be feasible with a pegRNA.
Figure 5.
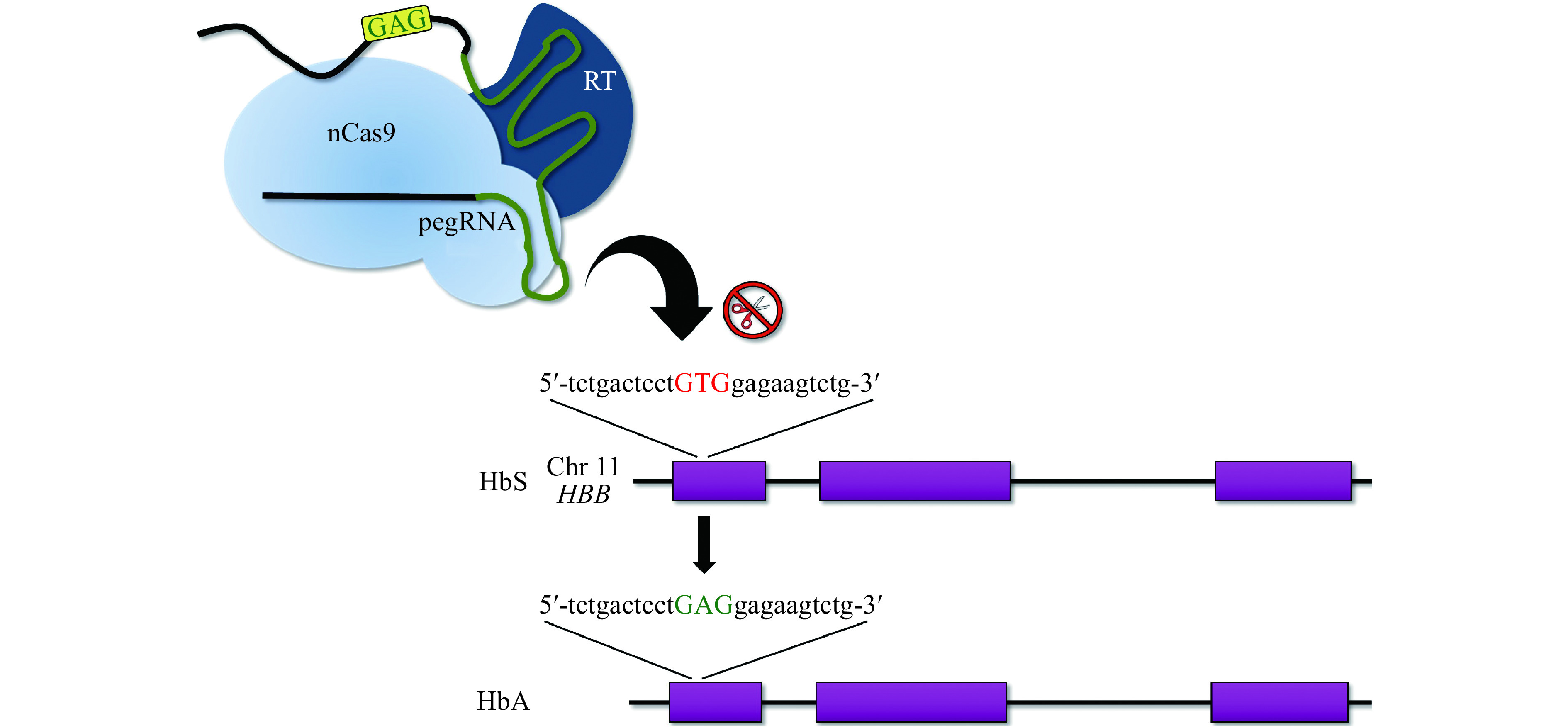
HbS correction with prime editing.
Schematic representing the gene editing approach by Anzalone et al[100] used to correct the HbS mutation in HEK293 cells. This method is a direct correction of the pathogenic T:A base pair to A:T (a transversion) that does not require the use of an exogenous donor template or DSB. nCas9: Cas9 nickase; RT: reverse transcriptase; pegRNA: prime editing gRNA.
Gene editing to elevate HbF levels
Since the 1950s, clinicians and scientists have recognized that levels of HbF modify disease severity in individuals with β-globin disorders[108]. As mentioned above, the protective effect of high levels of HbF has inspired a tremendous effort to elucidate the causes of HPFH[24] and to create therapeutic genome editing strategies that mimic these naturally occurring mutations[109]. Human genetic studies have provided a roadmap of the many variants causing HPFH[25,27–28,40,43,110], which can be used when designing personalized gene editing therapies.
HPFH is caused by two different types of mutations. First, there are large deletions (10s to 100s of kilobases) called deletional HPFH mutations that delete the β- and δ-globin genes but retain either one or both γ-globin genes. Re-creating deletional HPFH mutations would require using two gRNAs to generate two DSBs simultaneously. While this approach has demonstrated increases in HbF[111], it is less efficient than using one gRNA and is accompanied with the possibility of creating large inversions as well as an increased off-target potential from introducing two gRNAs. Second, there are non-deletional HPFH mutations that prevent trans-regulatory proteins from repressing γ-globin by 1) preventing transcriptional repressors from binding to cis-regulatory DNA elements within the γ-globin promoter[112–115] (Fig. 6A and B); 2) creating de novo binding sites for transcriptional activators[116–117] (Fig. 7); or 3) inhibiting BCL11A expression specifically in the erythroid lineage[43] (Fig. 6A and C). Using non-deletional HPFH mutations as a guide, disruption of recognition sequences or creation of new transcriptional activator sites, either by traditional genome editing using a DSB or the newer base editing and prime editing strategies, can be achieved.
Figure 6.
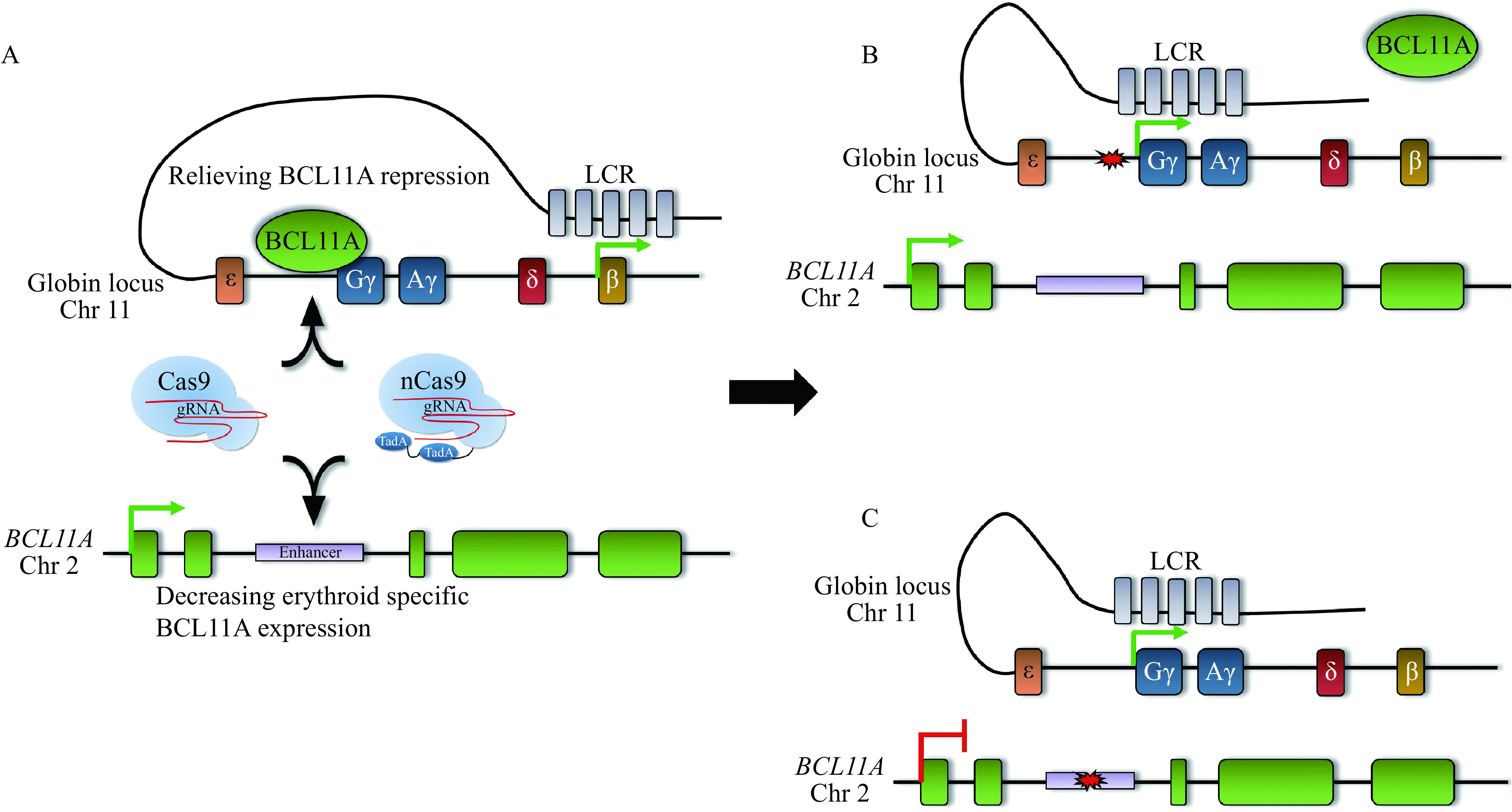
Elevating HbF levels by distinct genome editing approaches.
A: Traditional Cas9 or base editors can be used to either disrupt the BCL11A binding motif upstream of the γ-globin promoters (top) or the erythroid-specific BCL11A enhancer (bottom). B: Disruption of the BCL11A binding motif displaces BCL11A, allowing the activating locus control region (LCR) access to the γ-globin promoters. C: Creating indels or base substitutions in the BCL11A enhancer decreases expression of BCL11A specifically in the erythroid lineage, which also allows the LCR access to the γ-globin promoters.
Figure 7.

Creating de novo activating motifs to elevate HbF.
Adenine base editors can be used to recapitulate an HPFH variant upstream of the γ-globin promoters. Adenine deamination at-113A creates a de novo GATA1 binding motif, which displaces BCL11A and allows for γ-globin upregulation.
Disrupting transcriptional repressor binding, and subsequent de-repression of HbF, has been demonstrated by re-creating a naturally-occurring 13-bp deletion[49] in the γ-globin promoters[114] (Fig 6A and B). Following the creation of indels in the −102 to −114 region of the γ-globin promoters in SCA patient-derived CD34+ HSPCs, control (mock-transduced) and edited HSPCs were differentiated toward the erythroid lineage and subjected to hypoxic conditions to induce sickling. Gene editing increased the percentage of F-cells from approximately 65% to 90% and reduced sickling from 24% to 4%[114], providing proof-of-principle that re-creating a naturally-occurring 13-bp deletion is sufficient to inhibit sickling in cultured SCA patient-derived HSPCs. A caveat to this approach is the possibility of generating large deletions or inversions owing to the gRNA being simultaneously targeted to the highly homologous Aγ-globin and Gγ-globin promoters. Indeed, an approximately 5-kb deletion in human CD34+ HSPCs was observed at rates of 30% prior to transplantation into non-obese diabetic/severe combined immunodeficiency mice, which decreased to 11% at 17 weeks following transplantation; however, despite the occurrence of this deletion, the observed increase in %HbF was not significantly altered[113]. The possibility of an inversion of the intervening DNA is also possible, eliminating expression of both γ-globin genes; however, inversions were detected at very low frequencies (approximately 1%) in bulk edited CD34+ HSPCs[113]. Additionally, no off-target editing was identified in transplanted CD34+ HSPCs after targeted deep sequencing of the top potential off-target candidate sites predicted by a sensitive in vitro method called circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq)[118]. These reports and others[112,115] have demonstrated that re-creating naturally occurring HPFH variants may be an effective therapeutic gene editing strategy to increase HbF levels.
Other non-deletional HPFH variants exist as SNPs around the −114 region of the γ-globin promoter, including −117G>A, −114C>A, −114C>T, and −114C>G, which disrupt the BCL11A or ZBTB7A binding motifs (ZBTB7A being another major transcriptional repressor of γ-globin)[47,112,119]. Recently, a −113A>G substitution, which creates a GATA1 consensus motif, was identified in an individual with HPFH, boosting HbF levels to 6.5%[24]; normal HbF levels in healthy adults are lower than 1%[120]. To determine if the −113A>G substitution is sufficient to cause the HPFH phenotype, CRISPR-Cas9 was used to introduce the −113A>G HPFH mutation, an artificial GATA site, or a control −113A>C substitution in modified HUDEP2 cells using HDR and a donor template. The results showed that both the −113A>G substitution and the artificial GATA site, but not the −113A>C substitution, are sufficient to elevate HbF levels[117]. Furthermore, rather than disrupting BCL11A binding, in vitro experiments suggested the −113A>G variant creates ade novo GATA1 consensus motif that allows GATA1 to outcompete BCL11A for binding, therefore activating γ-globin expression[117] (Fig. 7). These findings suggest that re-creation of HPFH SNPs or insertion of de novo activating motifs upstream of the γ-globin gene could be alternative approaches to treating β-hemoglobinopathies by increasing HbF levels.
The ability to ameliorate symptoms by inserting a de novo activating motif is possible with base editors. In the example of the −113A>G HPFH variant, the adenine would fall into position 8 using an NGG-ABE, which is less than ideal. However, new versions of base editors are continually being evolved with more relaxed PAMs that expand the breadth of base editing to loci that were once inaccessible[121–122]. The Cas9-NG ABE developed by Hu and colleagues[121] would put the −113A in position 7 instead of 8, which is now within the activity window.
A similar HPFH-associated variant that creates a de novo consensus motif is −175T>C. This variant creates an E-Box consensus motif for the transcriptional activator T-cell acute lymphocytic leukemia protein 1 (TAL1) and has been shown to increase HbF levels in humans to 41% of total hemoglobin[116,123]. When using Cas9-NG ABE, the −175A (the adenine being on the opposite strand) would be placed at the protospacer position 5, an ideal placement to use with ABE[103]. An additional HPFH-causing variant (−198T>C), known as the British-type HPFH[124–125], can also be created using ABEs. The British-type HPFH variant has been shown to introduce a de novo Krüppel-like factor 1 (KLF1/erythroid KLF) consensus motif, allowing KLF1 to upregulate γ-globin expression[126].
Disrupting the BCL11A enhancer can also be achieved with base editing[127–128]. Evolution of ABEs led to the generation of ABE8e, which was used in proof-of-concept experiments to target two adenine residues within the BCL11A enhancer that both fall within the protospacer activity window (positions 4 and 7). Treatment of HEK293 cells with ABE8e produced simultaneous editing of both adenine residues in 54.4% of alleles compared to 7.9% with ABE7.10[128]. Additionally, a new CBE was recently purified (A3A [N57Q]-BE3) that reduces bystander mutations and was used for multiplex base editing the BCL11A enhancer and a common Chinese β-thalassemia variant (−28A>G) in patient-derived CD34+ HSPCs[127]. The synergistic effects of multiplex editing led to higher β-globin expression than targeting the −28A>G variant alone, and higher γ-globin expression compared to targeting only theBCL11A enhancer[127]. These data highlight base editing as a potential therapeutic alternative to nuclease interventions. Moreover, re-creating HPFH mutations is an especially attractive strategy as the effects of these mutations are known.
Delivery
Although there are a number of potential genome editing strategies and tools to treat β-hemoglobinopathies, each method requires efficient and safe delivery of the editing components to HSCs. The most practical treatment platform for β-globin disorders is autologous hematopoietic stem cell transplantation of genome-edited cells. Using the patient's cells for ex vivo genome editing offsets the necessity to identify compatible donors and practically abolishes the risk of graft-versus-host disease. Compared to other tissues, such as the heart or brain, accessing the patient's target cells, CD34+ HSPCs, whether from bone marrow or mobilized peripheral blood, is a routine procedure that collects the cellular material needed for ex vivo delivery of the gene editing components. The structure of the components being delivered depends on the therapeutic approach. Therapies utilizing NHEJ, base editing, and prime editing are operationally simpler in that they only require the delivery of the RNA or RNP complex, whereas HDR-based approaches require the co-delivery of the donor template, for example, a ssODN, that could increase costs and possibly trigger an immune response. It has become increasingly clear that while nuclease-encoded plasmid DNA or mRNA will evoke a toxic, antiviral type I interferon response in human primary cells, delivery of Cas9-gRNA as an RNP complex is well-tolerated[129–131]. Delivery of RNPs also ensures short-lived expression of the editing components, which can mitigate off-target effects[132] and deleterious cellular responses, such as activation of the p53 pathway[133]. When the addition of template DNA is required for HDR-mediated editing, recombinant adeno-associated viral vectors, which have evolved to evade cellular detection, is an efficient vehicle to deliver DNA templates[134–137]. However, generating a GMP-grade viral delivery system augments workload and expenses, and there is still debate about whether viral systems lead to insertional mutagenesis[138]. There have been few genotoxic events reported in patients treated with lentiviral-based methods[139–140]; however, careful consideration should be given when applying these strategies. Long-term follow-up is also recommended out of concern for random lentiviral integration[138,141] and production of aberrant transcripts[140,142]. Additional considerations to optimize efficiency include using chemically-modified gRNAs, which have demonstrated more stability and increased specificity[143].
Electroporation is a non-selective delivery system that has been in use for many years as a vehicle for transferring DNA, RNA, and/or protein into cells by enlarging pores on the cell membrane via an electric pulse, through which the editing components can be shuttled into the cell. Many studies report high delivery efficiency using electroporation, specifically in HSPCs[97,144–146]. Regardless of the structure of the editing components and/or delivery vehicle, a sufficient amount of highly active and specific Cas9 must be used to achieve efficient editing at a clinical scale, which has been suggested to be more than 108 CD34+ HSPCs[147].
Current genome editing trials
Currently, a few clinical trials are underway to study CRISPR-based treatments in β-thalassemia and SCD patients. One such method of treatment is currently in phase 1/2 clinical trials, showing promising initial findings. The investigational treatment CTX001 being developed by CRISPR Therapeutics and Vertex Pharmaceuticals was developed based on the discovery of the BCL11A erythroid-specific enhancer[43]. CTX001 is a therapeutic CRISPR-Cas9 RNP complex that creates indels at the GATA1/TAL1 binding sites in the BCL11A erythroid specific enhancer, which significantly diminishes BCL11A expression exclusively in the erythroid lineage, elevating HbF levels (Fig. 6A and C). It has been demonstrated that a Cas9:gRNA RNP complex targeting the BCL11A enhancer in CD34+ HSPCs can achieve up to 90% indels, repressing BCL11A expression by more than 50%[148]. Thus far, three patients, two with transfusion-dependent β-thalassemia (NCT03655678) and one with SCA (NCT03745287) who were treated with this therapy, have discontinued RBC transfusions. Moreover, treated patients have been expressing high amounts of fetal hemoglobin for more than 6 months following treatment[149]. CTX001 treatment consists of mobilizing and collecting patients' CD34+ HSPCs, which are transiently treated with CRISPR-Cas9 ex vivo, and following busulfan myeloablative conditioning, are reinfused. This approach is widely applicable in that a consensus motif is targeted rather than patient-specific variants, which allows for the production of only one gRNA as a universal treatment.
Sangamo Biosciences and Sanofi are conducting a similar clinical trial using ZFNs for genome editing. Like the previous study, this phase 1/2 trial also targets the disruption of the erythroid lineage-specific BCL11A enhancer in autologous CD34+ cells to increase expression of HbF in their erythroid progeny (NCT03432364). Alternative strategies to treat β-thalassemia and SCD, including via non-nuclease gene delivery, have been reviewed elsewhere[150–154].
Off-target editing prediction and identification
All gene editing approaches hold the possibility of creating unwanted genomic alterations by acting at unintended sites. Off-target alterations to the genome may result from similarity to the target sequence, with or without mismatches, bulges, or non-canonical PAMs. Off-target sites will differ for each gene editing tool. Moreover, each gRNA and Cas9 combination has a unique off-target profile. Similarly, gRNAs used with different Cas9 variants or fusions will have a unique off-target potential. As mentioned above, transient expression of CRISPR reagents via RNP delivery has been shown to reduce off-target editing[155]. Additionally, if a donor is required for a given strategy, off-target integration is a possibility. One of the more concerning outcomes of off-target mutagenesis with a CRISPR-Cas9 therapy applied to HSPCs is the possibility of clonal expansion and leukemogenesis.
In order to identify bona-fide off-target sites within the vast genome, an iterative process is often undertaken (Fig. 8). First, in silico and/or in vitro methods are used to predict off-target sites, which are then verified by more in-depth methods, such as targeted amplification or a multiplexed targeted amplification approach called RNAse H-dependent (rhAmp) PCR[156], followed by next generation sequencing (NGS). There are a number of programs and protocols for identifying and validating potential off-target sites, each with their own strengths and weaknesses, reviewed in depth elsewhere[157–159].
Figure 8.

Off-target discovery and validation process.
The first step in the off-target identification process is discovery, either through in silico prediction of similar sequences, in vitro activity of the nuclease on naked or genomic-context DNA, or a combination of both in silico and in vitro methods. These methods produce a list of putative off-target sites that can then be further interrogated by targeted PCR amplification of the candidate sites in the cell type of interest, followed by next generation sequencing. Sites containing indels are considered verified off-targets.
In silico off-target prediction
There are several in silico programs available to identify potential off-target sites in the genome, based on sequence similarity to the target alone or also considering mismatches and extra base pairs (bulges)[160–166]. Some programs use machine learning to predict off-targets[167–168], while Doench and colleagues developed an algorithm for cutting frequency determination (CFD) through intensive screens of perfect match and single base mutated guides[169–170]. CFD was developed to maximize on-target activity and predict off-target behavior of a guide, allowing for the selection of guides with low off-target potential.
In vitro off-target detection
Independently or in conjunction with in silico prediction, off-targets can be identified by in vitro methods. CIRCLE-seq[118] and Digenome-seq[171] are ultra-sensitive methods to detect nuclease activity on cell-free, i.e., "naked" DNA. This sensitivity likely over-represents the number of bona-fide off-targets identified by targeted deep sequencing of the candidates.
Alternatively, many protocols rely on gRNA binding or Cas9 nuclease activity on genomic DNA in a cellular context, which may more closely resemble off-targets in the experimental environment. GUIDE-seq[172] captures DSBs with a short dsDNA fragment via NHEJ. This process will identify any site of DNA breakage, regardless of cause, and requires the cells be amenable to the introduction of the DNA fragments, which can be a limitation especially in primary cells. DNA DSB labeling is also the mode of action of the BLESS protocol[173], which uses barcoded, biotinylated linkers to mark breaks. A method called end-sequencing[174] is similar to BLESS, but immobilizes cells in agarose to minimize DNA damage from fixation steps. Most recently, the BLISS technique[175] has been developed which mobilizes T7 promoter sequences into breaks, giving the investigator the ability to use T7 transcription to sequence sites. UDiTaS[176] utilizes uni-directional targeted sequencing to detect indels, as well as larger chromosomal rearrangements such as translocations and inversions. DISCOVER-Seq[177] identifies DNA damage via detection of DNA repair factors that are recruited to the sites of breaks in live cells.
The above assays are useful in detecting off-target edits after the creation of a DSB, but new methods are still being developed to fully interrogate genome-wide off-target potential of base editors and prime editors. A modified version of Digenome-seq was developed to evaluate the off-target potential of base editors by using enzymes that create DSBs at the products of adenine or cytosine deamination: inosine or uracil, respectively[178]. A novel method employed for off-target identification with base editor technology involved injecting the editor into one cell of a two-cell embryo[179]. The edited cell and its control sister cell were then compared, and the differences observed in the genomes of the two cells were attributed to the activity of the base editor. While this work identified many putative off-target sites, its robustness and reproducibility will need to be further assessed.
Targeted deep sequencing of putative off-targets
Once candidate off-targets have been identified, these sites can be interrogated via targeted deep sequencing in the experimental context. Sites are amplified by classic targeted PCR or rhAmp PCR[156] followed by NGS. The starting material and read coverage will determine the sensitivity and detection level of the off-target validation.
Conclusion
In this review, we have discussed several strategies for treating β-hemoglobinopathies, specifically SCD, and the associated technical considerations. We believe that these technical limitations will be overcome, and in fact, the first U.S. human trials for a gene editing approach for SCD are showing great promise. As we deepen our understanding of gene editing technologies and the genetic factors involved in these diseases, even less restrictive and more accurate and efficient genome editing strategies will be developed. Indeed, we are optimistic that several gene editing therapies for the treatment of β-globin disorders will prove beneficial, giving patients and doctors a portfolio of treatment options. As these curative therapies continue to advance, perhaps the greatest burden will be making these treatments available in resource-limited areas in which there is the largest disease burden.
The cost of producing gene editing-based therapies will be high because of the costs associated with the development, manufacturing, distribution, and approval of a personalized cell product. Additionally, highly specialized equipment, facilities, and trained physicians are needed in order to successfully perform ex vivo editing and reimplantation of the modified and quality-controlled cell product. This will ultimately translate to limited access of these therapies to a population which is more likely to live in low-income and developing regions, such as Africa and Southeast Asia that lack the advanced equipment and facilities required for isolating, editing, culturing and delivering cellular material to humans. It is essential that as these therapies are being developed, we continually consider and evaluate strategies that will make them available to resource-constrained regions where hemoglobin disorders are devastatingly prevalent. Advances in in vivo delivery of gene therapy or genome editing treatments would likely allow a broader uptake of these approaches across the globe.
Although SCD was first described over a century ago and the genetic basis determined more than 60 years ago, development of novel treatment options has been sluggish and limited. With advances in multiple fields, we no longer hang our hopes on just another treatment for SCD: we envision a cure.
References
- 1.Modell B, Darlison M Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. doi: 10.2471/blt.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagel RL, Fabry ME, Steinberg MH The paradox of hemoglobin SC disease. Blood Rev. 2003;17(3):167–178. doi: 10.1016/S0268-960X(03)00003-1. [DOI] [PubMed] [Google Scholar]
- 3.Orkin SH, Kazazian Jr HH, Antonarakis SE, et al Abnormal RNA processing due to the exon mutation of βE-globin gene . Nature. 1982;300(5894):768–769. doi: 10.1038/300768a0. [DOI] [PubMed] [Google Scholar]
- 4.Hannemann A, Weiss E, Rees DC, et al The properties of red blood cells from patients heterozygous for HbS and HbC (HbSC genotype) Anemia. 2011;2011:248527. doi: 10.1155/2011/248527.Epub2010Oct13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giardine B, Borg J, Viennas E, et al Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42(D1):D1063–D1069. doi: 10.1093/nar/gkt911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulukonis ST, Eckman JR, Snyder AB, et al Defining sickle cell disease mortality using a population-based surveillance system, 2004 through 2008. Public Health Rep. 2016;131(2):367–375. doi: 10.1177/003335491613100221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassell KL Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512–S521. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 8.Platt OS, Brambilla DJ, Rosse WF, et al Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 9.Powars DR, Chan LS, Hiti A, et al Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84(6):363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 10.Hulbert ML, McKinstry RC, Lacey JL, et al Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood. 2011;117(3):772–779. doi: 10.1182/blood-2010-01-261123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piel FB, Patil AP, Howes RE, et al Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grosse SD, Odame I, Atrash HK, et al Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6 Suppl 4):S398–S405. doi: 10.1016/j.amepre.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kauf TL, Coates TD, Liu HZ, et al The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84(6):323–327. doi: 10.1002/ajh.21408. [DOI] [PubMed] [Google Scholar]
- 14.Vinjamur DS, Bauer DE, Orkin SH Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br J Haematol. 2018;180(5):630–643. doi: 10.1111/bjh.15038. [DOI] [PubMed] [Google Scholar]
- 15.Basak A, Sankaran VG Regulation of the fetal hemoglobin silencing factor BCL11A. Ann N Y Acad Sci. 2016;1368(1):25–30. doi: 10.1111/nyas.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamatoyannopoulos G Control of globin gene expression during development and erythroid differentiation. Exp Hematol. 2005;33(3):259–271. doi: 10.1016/j.exphem.2004.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serjeant GR, Serjeant BE, Mason K Heterocellular hereditary persistence of fetal haemoglobin and homozygous sickle-cell disease. Lancet. 1977;309(8015):795–796. doi: 10.1016/s0140-6736(77)92976-2. [DOI] [PubMed] [Google Scholar]
- 18.Perrine RP, Brown MJ, Clegg JB, et al Benign sickle-cell anaemia. Lancet. 1972;300(7788):1163–1167. doi: 10.1016/S0140-6736(72)92592-5. [DOI] [PubMed] [Google Scholar]
- 19.Nuinoon M, Makarasara W, Mushiroda T, et al A genome-wide association identified the common genetic variants influence disease severity in β0-thalassemia/hemoglobin E . Hum Genet. 2010;127(3):303–314. doi: 10.1007/s00439-009-0770-2. [DOI] [PubMed] [Google Scholar]
- 20.Galanello R, Sanna S, Perseu L, et al Amelioration of Sardinianβ0 thalassemia by genetic modifiers . Blood. 2009;114(18):3935–3937. doi: 10.1182/blood-2009-04-217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forget BG Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. 1998;850(1):38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- 22.Fessas P, Stamatoyannopoulos G Hereditary persistence of fetal hemoglobin in Greece. A study and a Comparison. Blood. 1964;24(3):223–240. doi: 10.1182/blood.V24.3.223.223. [DOI] [PubMed] [Google Scholar]
- 23.Weatherall DJ Phenotype—genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–255. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]
- 24.Amato A, Cappabianca MP, Perri M, et al Interpreting elevated fetal hemoglobin in pathology and health at the basic laboratory level: new and known γ- gene mutations associated with hereditary persistence of fetal hemoglobin. Int J Lab Hematol. 2014;36(1):13–19. doi: 10.1111/ijlh.12094. [DOI] [PubMed] [Google Scholar]
- 25.Uda M, Galanello, R, Sanna S, et al Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia . Proc Natl Acad Sci U S A. 2008;105(5):1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lettre G, Sankaran VG, Bezerra MAC, et al DNA polymorphisms at the BCL11A, HBS1L-MYB, and β-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease . Proc Natl Acad Sci U S A. 2008;105(33):11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menzel S, Garner C, Gut I, et al A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 28.Thein SL, Menzel S, Peng X, et al Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults . Proc Natl Acad Sci U S A. 2007;104(27):11346–11351. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sankaran VG, Menne TF, Xu J, et al Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A . Science. 2008;322(5909):1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 30.Sankaran VG, Xu J, Ragoczy T, et al Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460(7259):1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Bauer DE, Kerenyi MA, et al Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci U S A. 2013;110(16):6518–6523. doi: 10.1073/pnas.1303976110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu J, Sankaran VG, Ni M, et al Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24(8):783–798. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Peng C, Sankaran VG, et al Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334(6058):993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esteghamat F, Gillemans N, Bilic I., et al Erythropoiesis and globin switching in compound Klf1::Bcl11a mutant mice . Blood. 2013;121(13):2553–2562. doi: 10.1182/blood-2012-06-434530. [DOI] [PubMed] [Google Scholar]
- 35.Luc S, Huang JL, McEldoon JL, et al Bcl11a deficiency leads to hematopoietic stem cell defects with an aging-like phenotype . Cell Rep. 2016;16(12):3181–3194. doi: 10.1016/j.celrep.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ippolito GC, Dekker JD, Wang YH, et al Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci U S A. 2014;111(11):E998–E1006. doi: 10.1073/pnas.1319228111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu PT, Keller JR, Ortiz M, et al Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4(6):525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 38.Tsang JCH, Yu Y, Burke S, et al Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells . Genome Biol. 2015;16:178. doi: 10.1186/s13059-015-0739-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greig LC, Woodworth MB, Greppi C, et al Ctip1 controls acquisition of sensory area identity and establishment of sensory input fields in the developing neocortex . Neuron. 2016;90(2):261–277. doi: 10.1016/j.neuron.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basak A, Hancarova M, Ulirsch JC, et al BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations . J Clin Invest. 2015;125(6):2363–2368. doi: 10.1172/JCI81163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dias C, Estruch SB, Graham SA, et al BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription . Am J Hum Genet. 2016;99(2):253–274. doi: 10.1016/j.ajhg.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Funnell APW, Prontera P, Ottaviani V, et al 2p15-p16.1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment . Blood. 2015;126(1):89–93. doi: 10.1182/blood-2015-04-638528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bauer DE, Kamran SC, Lessard S, et al An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level . Science. 2013;342(6155):253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grosveld F, Van Assendelft GB, Greaves DR, et al Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell. 1987;51(6):975–985. doi: 10.1016/0092-8674(87)90584-8. [DOI] [PubMed] [Google Scholar]
- 45.Tuan D, Solomon W, Li Q, et al The "beta-like-globin" gene domain in human erythroid cells. Proc Natl Acad Sci U S A. 1985;82(19):6384–6388. doi: 10.1073/pnas.82.19.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sankaran VG, Xu J, Byron R, et al A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365(9):807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu N, Hargreaves VV, Zhu Q, et al Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173(2):430–442. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collins FS, Metherall JE, Yamakawa M, et al A point mutation in the Aγ-globin gene promoter in Greek hereditary persistence of fetal haemoglobin . Nature. 1985;313(6000):325–326. doi: 10.1038/313325a0. [DOI] [PubMed] [Google Scholar]
- 49.Gilman JG, Mishima N, Wen XJ, et al Distal CCAAT box deletion in the Aγ globin gene of two black adolescents with elevated fetal Aγ globin . Nucleic Acids Res. 1988;16(22):10635–10642. doi: 10.1093/nar/16.22.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Letvin NL, Linch DC, Beardsley GP, et al Augmentation of fetal-hemoglobin production in anemic monkeys by hydroxyurea. N Engl J Med. 1984;310(14):869–873. doi: 10.1056/NEJM198404053101401. [DOI] [PubMed] [Google Scholar]
- 51.Platt OS, Orkin SH, Dover G, et al Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984;74(2):652–656. doi: 10.1172/JCI111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pule GD, Mowla S, Novitzky N, et al A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Rev Hematol. 2015;8(5):669–679. doi: 10.1586/17474086.2015.1078235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 54.Brandow AM, Jirovec DL, Panepinto JA Hydroxyurea in children with sickle cell disease: practice patterns and barriers to utilization. Am J Hematol. 2010;85(8):611–613. doi: 10.1002/ajh.21749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brandow AM, Panepinto JA Hydroxyurea use in sickle cell disease: the battle with low prescription rates, poor patient compliance and fears of toxicities. Expert Rev Hematol. 2010;3(3):255–260. doi: 10.1586/ehm.10.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niihara Y, Miller ST, Kanter J, et al A phase 3 trial of L-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226–235. doi: 10.1056/NEJMoa1715971. [DOI] [PubMed] [Google Scholar]
- 57.Li SD, Su YD, Li M, et al Hemin-mediated hemolysis in erythrocytes: effects of ascorbic acid and glutathione. Acta Biochim Biophys Sin (Shanghai) 2006;38(1):63–69. doi: 10.1111/j.1745-7270.2006.00127.x. [DOI] [PubMed] [Google Scholar]
- 58.Kato GJ, Steinberg MH, Gladwin MT Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–760. doi: 10.1172/JCI89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones DP Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/S0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 60.Townsend DM, Tew KD, Tapiero H The importance of glutathione in human disease. Biomed Pharmacother. 2003;57(3-4):145–155. doi: 10.1016/S0753-3322(03)00043-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsui NM, Borsig L, Rosen SD, et al P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98(6):1955–1962. doi: 10.1182/blood.V98.6.1955. [DOI] [PubMed] [Google Scholar]
- 62.Matsui NM, Varki A, Embury SH Heparin inhibits the flow adhesion of sickle red blood cells to P-selectin. Blood. 2002;100(10):3790–3796. doi: 10.1182/blood-2002-02-0626. [DOI] [PubMed] [Google Scholar]
- 63.Embury SH, Matsui NM, Ramanujam S, et al The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo . Blood. 2004;104(10):3378–3385. doi: 10.1182/blood-2004-02-0713. [DOI] [PubMed] [Google Scholar]
- 64.Polanowska-Grabowska R, Wallace K, Field JJ, et al P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler Thromb Vasc Biol. 2010;30(12):2392–2399. doi: 10.1161/ATVBAHA.110.211615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ataga KI, Kutlar A, Kanter J, et al Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376(5):429–439. doi: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Metcalf B, Chuang C, Dufu K, et al Discovery of GBT440, an orally bioavailable R-state stabilizer of sickle cell hemoglobin. ACS Med Chem Lett. 2017;8(3):321–326. doi: 10.1021/acsmedchemlett.6b00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oksenberg D, Dufu K, Patel MP, et al GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol. 2016;175(1):141–153. doi: 10.1111/bjh.14214. [DOI] [PubMed] [Google Scholar]
- 68.Vichinsky E, Hoppe CC, Ataga KI, et al A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509–519. doi: 10.1056/NEJMoa1903212. [DOI] [PubMed] [Google Scholar]
- 69.Platt OS Preventing stroke in sickle cell anemia. N Engl J Med. 2005;353(26):2743–2745. doi: 10.1056/NEJMp058274. [DOI] [PubMed] [Google Scholar]
- 70.Adams RJ, McKie VC, Hsu L, et al Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 71.The Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial Investigators Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005;353(26):2769–2778. doi: 10.1056/NEJMoa050460. [DOI] [PubMed] [Google Scholar]
- 72.Gladwin MT, Sachdev V, Jison ML, et al Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 73.Chou ST Transfusion therapy for sickle cell disease: a balancing act. Hematol Am Soc Hematol Educ Program. 2013;2013(1):439–446. doi: 10.1182/asheducation-2013.1.439. [DOI] [PubMed] [Google Scholar]
- 74.Vichinsky E, Torres M, Minniti CP, et al Efficacy and safety of deferasirox compared with deferoxamine in sickle cell disease: two-year results including pharmacokinetics and concomitant hydroxyurea. Am J Hematol. 2013;88(12):1068–1073. doi: 10.1002/ajh.23569. [DOI] [PubMed] [Google Scholar]
- 75.Yazdanbakhsh K, Ware RE, Noizat-Pirenne F Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120(3):528–537. doi: 10.1182/blood-2011-11-327361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chakravorty S, Williams TN Sickle cell disease: a neglected chronic disease of increasing global health importance. Arch Dis Child. 2015;100(1):48–53. doi: 10.1136/archdischild-2013-303773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kassim AA, Sharma D Hematopoietic stem cell transplantation for sickle cell disease: The changing landscape. Hematol Oncol Stem Cell Ther. 2017;10(4):259–266. doi: 10.1016/j.hemonc.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 78.ClinicalTrials.gov. Gene transfer for patients with sickle cell disease[EB/OL]. [2019-05-02]. https://clinicaltrials.gov/ct2/show/NCT02186418.
- 79.ClinicalTrials.gov. Stem cell gene therapy for sickle cell disease[EB/OL]. [2019-10-18].https://clinicaltrials.gov/ct2/show/NCT02247843?cond=NCT02247843&draw=2&rank=1.
- 80.Negre O, Bartholomae C, Beuzard Y, et al Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease. Curr Gene Ther. 2015;15(1):64–81. doi: 10.2174/1566523214666141127095336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ribeil JA, Hacein-Bey-Abina S, Payen E, et al Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 82.Negre O, Eggimann AV, Beuzard Y, et al Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the βA(T87Q)-Globin gene . Hum Gene Ther. 2016;27(2):148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thompson AA, Walters MC, Kwiatkowski J, et al Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 84.European Medicines Agency. Zynteglo[EB/OL].[2019-05-29]. https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo.
- 85.Eadie GS, Brown Jr IW, Curtis WG The potential life span and ultimate survival of fresh red blood cells in normal healthy recipients as studied by simultaneous Cr51 tagging and differential hemolysis . J Clin Invest. 1955;34(4):629–636. doi: 10.1172/JCI103112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cline MJ, Berlin NI Red blood cell life span using DFP32 as a cohort label . Blood. 1962;19(6):715–723. doi: 10.1182/blood.V19.6.715.715. [DOI] [PubMed] [Google Scholar]
- 87.Shemin D, Rittenberg D The life span of the human red blood cell. https://www.sciencedirect.com/science/article/pii/S0021925817352018?via%3Dihub. J Biol Chem. 1946;166(2):627–636. [PubMed] [Google Scholar]
- 88.Huang XS, Wang Y, Yan W, et al Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells. 2015;33(5):1470–1479. doi: 10.1002/stem.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park S, Gianotti-Sommer A, Molina-Estevez FJ, et al A comprehensive, ethnically diverse library of sickle cell disease-specific induced pluripotent stem cells. Stem Cell Rep. 2017;8(4):1076–1085. doi: 10.1016/j.stemcr.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li C, Ding L, Sun CW, et al Novel HDAd/EBV reprogramming vector and highly efficient Ad/CRISPR-Cas sickle cell disease gene correction. Sci Rep. 2016;6:30422. doi: 10.1038/srep30422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin RM, Ikeda K, Cromer MK, et al Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell. 2019;24(5):821–828. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 92.DeWitt MA, Magis W, Bray NL, et al Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8(360):360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Genovese P, Schiroli G, Escobar G, et al Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walters MC, Patience M, Leisenring W, et al Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7(12):665–673. doi: 10.1053/bbmt.2001.v7.pm11787529. [DOI] [PubMed] [Google Scholar]
- 95.Wu CJ, Gladwin M, Tisdale J, et al Mixed haematopoietic chimerism for sickle cell disease prevents intravascular haemolysis. Br J Haematol. 2007;139(3):504–507. doi: 10.1111/j.1365-2141.2007.06803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iannone R, Casella JF, Fuchs EJ, et al Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and β-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519–528. doi: 10.1016/S1083-8791(03)00192-7. [DOI] [PubMed] [Google Scholar]
- 97.Dever DP, Bak RO, Reinisch A, et al CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539(7629):384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eid A, Alshareef S, Mahfouz MM CRISPR base editors: genome editing without double-stranded breaks. Biochem J. 2018;475(11):1955–1964. doi: 10.1042/BCJ20170793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anzalone AV, Randolph PB, Davis JR, et al Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rees HA, Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Komor AC, Kim YB, Packer MS, et al Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shimatani Z, Kashojiya S, Takayama M, et al Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol. 2017;35(5):441–443. doi: 10.1038/nbt.3833. [DOI] [PubMed] [Google Scholar]
- 103.Gaudelli NM, Komor AC, Rees HA, et al Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Molla KA, Yang YN CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol. 2019;37(10):1121–1142. doi: 10.1016/j.tibtech.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 105.Blackwell RQ, Oemijati S, Pribadi W, et al Hemoglobin G makassar: β6 Glu→Ala . Biochim Biophys Acta (BBA) - Protein Struct. 1970;214(3):396–401. doi: 10.1016/0005-2795(70)90297-7. [DOI] [PubMed] [Google Scholar]
- 106.Viprakasit V, Wiriyasateinkul A, Sattayasevana B, et al Hb G-makassar[β6(A3)Glu→Ala; CODON 6 (G A G→G C G)]: molecular characterization, clinical, and hematological effects . Hemoglobin. 2002;26(3):245–253. doi: 10.1081/HEM-120015028. [DOI] [PubMed] [Google Scholar]
- 107.Fucharoen S, Weatherall DJ The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2(8):a011734. doi: 10.1101/cshperspect.a011734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jacob GF, Raper AB Hereditary persistence of foetal haemoglobin production, and its interaction with the sickle-cell trait. Br J Haematol. 1958;4(2):138–149. doi: 10.1111/j.1365-2141.1958.tb03844.x. [DOI] [PubMed] [Google Scholar]
- 109.Lettre G, Bauer DE Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet. 2016;387(10037):2554–2564. doi: 10.1016/S0140-6736(15)01341-0. [DOI] [PubMed] [Google Scholar]
- 110.Borg J, Papadopoulos P, Georgitsi M, et al Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42(9):801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ye L, Wang JM, Tan YT, et al Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci U S A. 2016;113(38):10661–10665. doi: 10.1073/pnas.1612075113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martyn GE, Wienert B, Yang L, et al Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding . Nat Genet. 2018;50(4):498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 113.Métais JY, Doerfler PA, Mayuranathan T, et al Genome editing of HBG1 and HBG2 to induce fetal hemoglobin . Blood Adv. 2019;3(21):3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Traxler EA, Yao Y, Wang YD, et al A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016;22(9):987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Antoniani C, Meneghini V, Lattanzi A, et al Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood. 2018;131(17):1960–1973. doi: 10.1182/blood-2017-10-811505. [DOI] [PubMed] [Google Scholar]
- 116.Wienert B, Funnell APW, Norton LJ, et al Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat Commun. 2015;6:7085. doi: 10.1038/ncomms8085. [DOI] [PubMed] [Google Scholar]
- 117.Martyn GE, Wienert B, Kurita R, et al A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site . Blood. 2019;133(8):852–856. doi: 10.1182/blood-2018-07-863951. [DOI] [PubMed] [Google Scholar]
- 118.Tsai SQ, Nguyen NT, Malagon-Lopez J, et al CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets . Nat Methods. 2017;14(6):607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Masuda T, Wang X, Maeda M, et al Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351(6270):285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rochette J, Craig JE, Thein SL Fetal hemoglobin levels in adults. Blood Rev. 1994;8(4):213–224. doi: 10.1016/0268-960X(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 121.Hu JH, Miller SM, Geurts MH, et al Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nishimasu H, Shi X, Ishiguro S, et al Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361(6408):1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martin DI, Tsai SF, Orkin SH Increased γ-globin expression in a nondeletion HPFH mediated by an erythroid-specific DNA-binding factor . Nature. 1989;338(6214):435–438. doi: 10.1038/338435a0. [DOI] [PubMed] [Google Scholar]
- 124.Weatherall DJ, Cartner R, Clegg JB, et al A form of hereditary persistence of fetal haemoglobin characterized by uneven cellular distribution of haemoglobin F and the production of haemoglobins A and A2 in homozygotes . Br J Haematol. 1975;29(2):205–220. doi: 10.1111/j.1365-2141.1975.tb01815.x. [DOI] [PubMed] [Google Scholar]
- 125.Wood WG, MacRae IA, Darbre PD, et al The British type of non-deletion HPFH: characterization of developmental changes in vivo and erythroid growth in vitro . Br J Haematol. 1982;50(3):401–414. doi: 10.1111/j.1365-2141.1982.tb01935.x. [DOI] [PubMed] [Google Scholar]
- 126.Wienert B, Martyn GE, Kurita R, et al KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood. 2017;130(6):803–807. doi: 10.1182/blood-2017-02-767400. [DOI] [PubMed] [Google Scholar]
- 127.Zeng J, Wu YX, Ren CY, et al Therapeutic base editing of human hematopoietic stem cells. Nat Med. 2020;26(4):535–541. doi: 10.1038/s41591-020-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Richter MF, Zhao KT, Eton E, et al Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol. 2020;38(7):883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hendel A, Bak RO, Clark JT, et al Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33(9):985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.DiGiusto DL, Cannon PM, Holmes MC, et al Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol Ther Methods Clin Dev. 2016;3:16067. doi: 10.1038/mtm.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cromer MK, Vaidyanathan S, Ryan DE, et al Global transcriptional response to CRISPR/Cas9-AAV6-based genome editing in CD34+ hematopoietic stem and progenitor cells . Mol Ther. 2018;26(10):2431–2442. doi: 10.1016/j.ymthe.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pattanayak V, Lin S, Guilinger JP, et al High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ihry RJ, Worringer KA, Salick MR, et al p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24(7):939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 134.Wang JB, Exline CM, DeClercq JJ, et al Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015;33(12):1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sather BD, Romano Ibarra GS, Sommer K, et al Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template . Sci Transl Med. 2015;7(307):307ra156. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.De Ravin SS, Reik A, Liu PQ, et al Targeted gene addition in human CD34+ hematopoietic cells for correction of X-linked chronic granulomatous disease . Nat Biotechnol. 2016;34(4):424–429. doi: 10.1038/nbt.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bak RO, Dever DP, Porteus MH CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc. 2018;13(2):358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hoban MD, Orkin SH, Bauer DE Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016;127(7):839–848. doi: 10.1182/blood-2015-09-618587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.David RM, Doherty AT Viral vectors: the road to reducing genotoxicity. Toxicol Sci. 2017;155(2):315–325. doi: 10.1093/toxsci/kfw220. [DOI] [PubMed] [Google Scholar]
- 140.Cavazzana-Calvo M, Payen E, Negre O, et al Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia . Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zheng CX, Wang SM, Bai YH, et al Lentiviral vectors and adeno-associated virus vectors: useful tools for gene transfer in pain research. Anat Rec (Hoboken) 2018;301(5):825–836. doi: 10.1002/ar.23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moiani A, Paleari Y, Sartori D, et al Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J Clin Invest. 2012;122(5):1653–1666. doi: 10.1172/JCI61852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cromwell CR, Sung K, Park J, et al Incorporation of bridged nucleic acids into CRISPR RNAs improves Cas9 endonuclease specificity. Nat Commun. 2018;9(1):1448. doi: 10.1038/s41467-018-03927-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hoban MD, Lumaquin D, Kuo CY, et al CRISPR/Cas9-mediated correction of the sickle mutation in human CD34+ cells . Mol Ther. 2016;24(9):1561–1569. doi: 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hoban MD, Cost GJ, Mendel MC, et al Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125(17):2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mandal PK, Ferreira LMR, Collins R, et al Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15(5):643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Canver MC, Orkin SH Customizing the genome as therapy for the β-hemoglobinopathies. Blood. 2016;127(21):2536–2545. doi: 10.1182/blood-2016-01-678128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wu YX, Zeng J, Roscoe BP, et al Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25(5):776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Corbacioglu S, Cappellini MD, Chapin J, et al. Initial safety and efficacy results with a single dose of autologous CRISPR-Cas9-modified CD34+ hematopoietic stem and progenitor cells in transfusion-dependent β-thalassemia and sickle cell disease[C]//Proceedings of the 25th Congress of the European Hematology Association. Frankfurt, Germany: EHA, 2020.
- 150.Demirci S, Uchida N, Tisdale JF Gene therapy for sickle cell disease: an update. Cytotherapy. 2018;20(7):899–910. doi: 10.1016/j.jcyt.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cavazzana M, Mavilio F Gene therapy for hemoglobinopathies. Hum Gene Ther. 2018;29(10):1106–1113. doi: 10.1089/hum.2018.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hirakawa MP, Krishnakumar R, Timlin JA, et al Gene editing and CRISPR in the clinic: current and future perspectives. Biosci Rep. 2020;40(4):BSR20200127. doi: 10.1042/BSR20200127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ikawa Y, Miccio A, Magrin E, et al Gene therapy of hemoglobinopathies: progress and future challenges. Hum Mol Genet. 2019;28(R1):R24–R30. doi: 10.1093/hmg/ddz172. [DOI] [PubMed] [Google Scholar]
- 154.Magrin E, Miccio A, Cavazzana M Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. 2019;134(15):1203–1213. doi: 10.1182/blood.2019000949. [DOI] [PubMed] [Google Scholar]
- 155.Kim S, Kim D, Cho SW, et al Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins . Genome Res. 2014;24(6):1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dobosy JR, Rose SD, Beltz KR, et al RNase H-dependent PCR (rhPCR): improved specificity and single nucleotide polymorphism detection using blocked cleavable primers. BMC Biotechnol. 2011;11:80. doi: 10.1186/1472-6750-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Porter SN, Levine RM, Pruett-Miller SM A practical guide to genome editing using targeted nuclease technologies. Compr Physiol. 2019;9(2):665–714. doi: 10.1002/cphy.c180022. [DOI] [PubMed] [Google Scholar]
- 158.Zischewski J, Fischer R, Bortesi L Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv. 2017;35(1):95–104. doi: 10.1016/j.biotechadv.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 159.Li JJ, Hong SY, Chen WJ, et al Advances in detecting and reducing off-target effects generated by CRISPR-mediated genome editing. J Genet Genomics. 2019;46(11):513–521. doi: 10.1016/j.jgg.2019.11.002. [DOI] [PubMed] [Google Scholar]
- 160.Cancellieri S, Canver MC, Bombieri N, et al CRISPRitz: rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics. 2020;36(7):2001–2008. doi: 10.1093/bioinformatics/btz867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bae S, Park J, Kim JS Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30(10):1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haeussler M, Schönig K, Eckert H, et al Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hsu PD, Scott DA, Weinstein JA, et al DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Labun K, Montague TG, Gagnon JA, et al CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44(W1):W272–W276. doi: 10.1093/nar/gkw398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Listgarten J, Weinstein M, Kleinstiver BP, et al Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng. 2018;2(1):38–47. doi: 10.1038/s41551-017-0178-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Moreno-Mateos MA, Vejnar CE, Beaudoin JD, et al CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo . Nat Methods. 2015;12(10):982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lin JC, Wong KC Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics. 2018;34(17):I656–I663. doi: 10.1093/bioinformatics/bty554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Abadi S, Yan WX, Amar D, et al A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13(10):e1005807. doi: 10.1371/journal.pcbi.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Doench JG, Fusi N, Sullender M, et al Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Doench JG, Hartenian E, Graham DB, et al Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32(12):1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim D, Bae S, Park J, et al Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12(3):237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 172.Tsai SQ, Zheng ZL, Nguyen NT, et al GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33(2):187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Crosetto N, Mitra A, Silva MJ, et al Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods. 2013;10(4):361–365. doi: 10.1038/nmeth.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Canela A, Sridharan S, Sciascia N, et al DNA breaks and end resection measured genome-wide by end sequencing. Mol Cell. 2016;63(5):898–911. doi: 10.1016/j.molcel.2016.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yan WX, Mirzazadeh R, Garnerone S, et al BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun. 2017;8:15058. doi: 10.1038/ncomms15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Giannoukos G, Ciulla DM, Marco E, et al UDiTaSTM, a genome editing detection method for indels and genome rearrangements . BMC Genomics. 2018;19:212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wienert B, Wyman SK, Richardson CD, et al Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq . Science. 2019;364(6437):286–289. doi: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kim D, Kim DE, Lee G, et al Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol. 2019;37(4):430–435. doi: 10.1038/s41587-019-0050-1. [DOI] [PubMed] [Google Scholar]
- 179.Zuo EW, Sun YD, Wei W, et al Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364(6437):289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]