

Abstract
There are an estimated 10 000 monogenic diseases affecting tens of millions of individuals worldwide. The application of CRISPR/Cas genome editing tools to treat monogenic diseases is an emerging strategy with the potential to generate personalized treatment approaches for these patients. CRISPR/Cas-based systems are programmable and sequence-specific genome editing tools with the capacity to generate base pair resolution manipulations to DNA or RNA. The complexity of genomic insults resulting in heritable disease requires patient-specific genome editing strategies with consideration of DNA repair pathways, and CRISPR/Cas systems of different types, species, and those with additional enzymatic capacity and/or delivery methods. In this review we aim to discuss broad and multifaceted therapeutic applications of CRISPR/Cas gene editing systems including in harnessing of homology directed repair, non-homologous end joining, microhomology-mediated end joining, and base editing to permanently correct diverse monogenic diseases.
Keywords: gene editing, CRISPR-associated protein 9, CRISPR-Cas system, genetic disease, medical genetics, genetic therapy
Introduction
Monogenic diseases are single gene diseases which affect tens of millions of individuals across the globe[1]. Of the approximately 20 000 protein coding genes in the human genome, almost 4000 genes have been implicated as causal in a monogenic disease. Though each instance is rare, the sum of individuals suffering from monogenic diseases accumulates to a high mortality and disease burden[2]. Intrinsic characteristics of the patterns of inheritance have allowed founder mutations to persist in populations, such as the BRCA1 185delAG variant found in Ashkenazi Jews, thereby increasing the frequency of specific disease variants in specific populations[3]. Through genetic mapping, extensive efforts have been made to link causal pathogenic variants to disease. Genetic linkage of genotype to clinical phenotype is a crucial component in understanding pathophysiology to better develop treatment approaches, such as the development of statins for controlling cholesterol levels in individuals with monogenic hypercholesterolemia. With the increasing implementation of sequencing technology in the clinic, the detection of rare variants of known monogenic diseases and previously unknown ultrarare monogenic diseases have increased, outpacing treatment approaches identified through traditional interventions[2]. The heterogeneity of clinical phenotypes arising from sequence-specific pathogenic mutations further complicates our understanding of the role of individual genes in monogenic diseases[4]. In combination with drug resistant monogenic diseases, such as in epilepsy with Dravet Syndrome, these barriers to treatments underscore the necessity of novel and personalized therapeutic approaches[5]. In this review, we focus on the implementation of CRISPR/Cas genome editing systems to target pathogenic variants underlying monogenic disease at the basic, translational, and clinical stages of biomedical research. Gene editing is the technology through which precise modifications are made to specific DNA sequences. Gene editing approaches are utilized to enact corrective and permanent changes to a patient's genome in gene therapy.
CRISPR/Cas systems
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated proteins (Cas)-based genome editing has emerged as one of the most powerful tools for sequence-specific gene editing due to the programmable nature of its sequence specificity, broad targeting scope, efficacy of editing, and its functionality as a platform for engineering novel editing modalities[6]. Initially identified as an anti-viral adaptive immune system in prokaryotes, CRISPR/Cas systems have been repurposed for a multitude of functions including genome editing, transcriptional regulation, and visualization[7–9]. Present in approximately half of bacteria and nearly all archaea surveyed, CRISPR/Cas adaptive immune systems are broadly categorized as either Class 1 or Class 2. Class 1 CRISPR systems are comprised of a multi-subunit effector protein complex, while Class 2 CRISPR systems are comprised of a single effector protein[10–11]. The classification further delineates into type Ⅰ, Ⅲ, and Ⅳ systems categorized under Class 1, and type Ⅱ, Ⅴ, and Ⅵ categorized under Class 2. Despite the vast distinctions and evolutionary divergence between CRISPR/Cas types, CRISPR/Cas adaptive immunity shares the same core mechanistic steps: adaptation, CRISPR-RNA synthesis, and target elimination[11]. First, the genetic material of an invading viral infection is recognized and incorporated into the host CRISPR array. Second, when a subsequent viral infection from the same viral member is initiated and recognized by the host, the host uses the previously incorporated DNA sequence as a template to create a CRISPR-RNA (crRNA), which goes through type specific processes to form a mature crRNA. Third, the crRNA in association with the Cas effector protein or effector protein complex, recognizes and cleaves the invading viral DNA or RNA, thereby eliminating the viral threat[9–10,12]. By adopting the function of the crRNA and effector protein to target and cleave viral nucleic acids, researchers developed highly programmable and sequence-specific nucleases, with many breakthroughs generated through studies on the CRISPR/Cas9 system from Streptococcus pyogenes (SpCas9)[8,13–15].
SpCas9 is the prototypic and most frequently used CRISPR/Cas nuclease in basic and clinical research settings. The Cas9 protein is targeted to a locus of interest through a 20-bp homologous sequence mediated by a crRNA-trans-activating CRISPR RNA (tracrRNA) hybrid or an engineered single-guide RNA (sgRNA)[8,16]. The sgRNA confers sequence-specific targeting directly upstream of a protospacer adjacent motif (PAM) sequence, a unique string of nucleotides recognized by the Cas protein for effector functionality (Fig. 1A). SpCas9 recognizes a 5′-NGG-3′ PAM, while orthologous Cas9 proteins such as Cas9 from Staphylococcus aureus (SaCas9) and Neisseria meningitidis Cas9 (NmeCas9) require PAM sequences of 5′-NNGRRT-3′ and 5′-NNNRRT-3′ respectively[17–18]. Cas12a or Cpf1 (CRISPR from Prevotella and Francisella 1) is a type V Cas system that uses a 25-bp gRNA for sequence specificity which is downstream of a 5′-TTTV-3′ PAM sequence. As PAM sites are strict requirements for recognition by the Cas protein, PAMs serve as both a safeguard against off-target editing but also as a restriction limiting the scope of targetable sites. To address the limitations imposed by PAM sites, many groups have engineered Cas protein variants recognizing novel and flexible PAMs, such as xCas9 capable of recognizing the 5′-NG-3′, 5′-GAA-3′, and 5′-GAT-3′ PAMs and SpCas9-NG which recognizes the 5′-NG-3′ PAM[19–21]. Following sequence targeting and PAM recognition, SpCas9 generates a blunt double-strand break (DSB) in the DNA between the 4th and 3rd base pair upstream of the PAM site, while other Cas proteins, such as Cpf1, creates a staggered DSB[22–24]. DNA DSBs are a highly cytotoxic event, prompting a quick DNA damage response in a biochemical, temporal, and cell cycle dependent manner[25–26].
Figure 1.
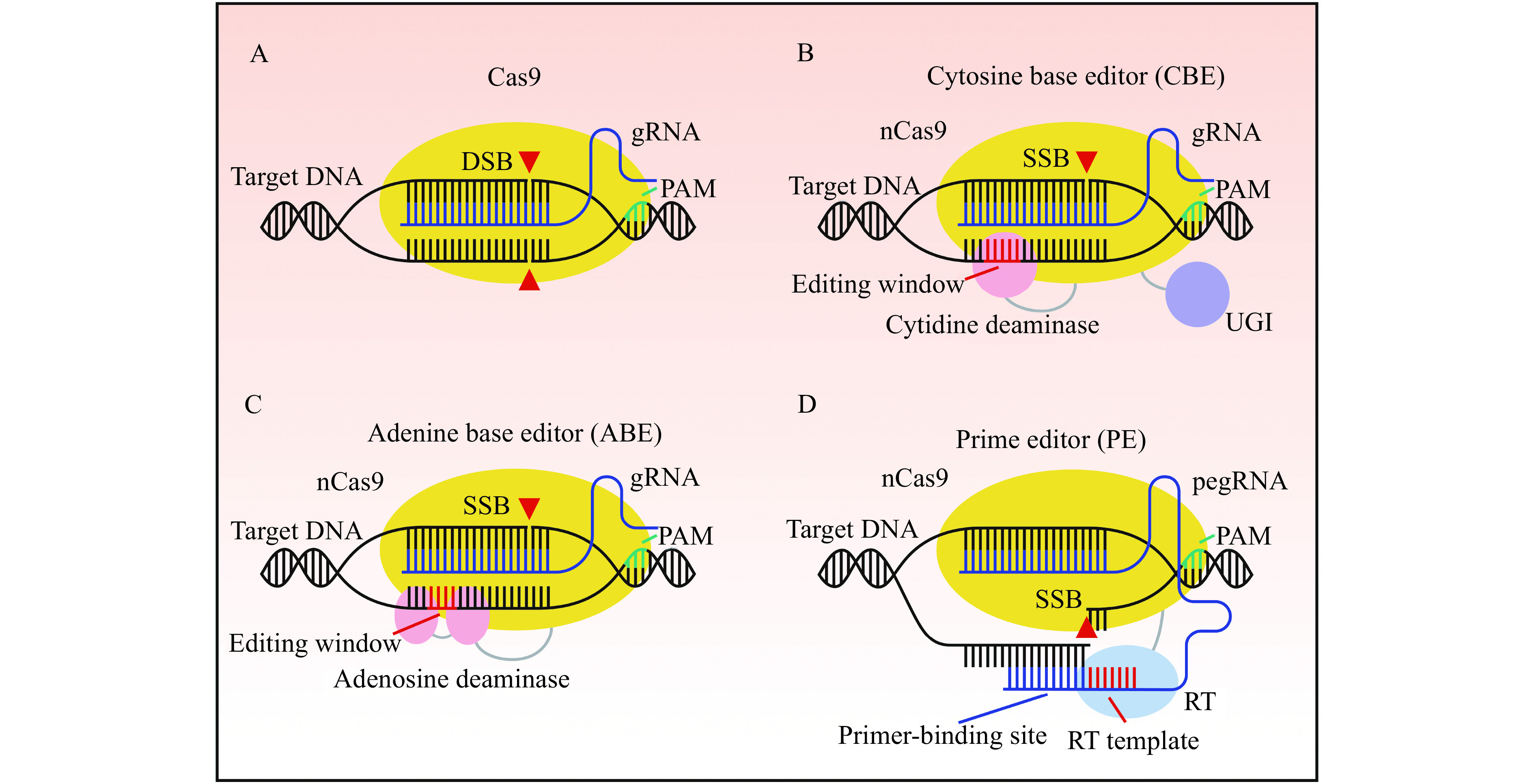
Genome editing strategies with Cas9, base editors, and prime editor.
A: Cas9 generates a DNA double-strand break (DSB) through guide RNA (gRNA) facilitated complementarity to a target DNA locus. Protospacer adjacent motif (PAM) recognition by the Cas9-gRNA complex is required for cleavage. B: Cytosine base editor (CBE) mediates C-G to T-A transition mutations at the editing window through a cytidine deaminase fused to a Cas9 nickase (nCas9). Following deamination, a fused uracil glycosylase inhibitor (UGI) inhibits base excision repair by uracil N-glycosylase, thereby increasing the desired T-A outcome. A DNA single-strand break (SSB) on the non-edited strand induces repair of the non-edited strand using the edited strand as a template. C: Adenine base editors (ABE) mediate A-T to G-C transition mutations at the editing window through an evolved adenosine deaminase fused to nCas9. An SSB on the non-edited strand induces repair of the non-edited strand using the edited strand as a template. D: Prime editors (PE) incorporate novel sequence through a reverse transcriptase (RT) fused to a nCas9 complexed to a prime editing guide RNA (pegRNA). The pegRNA is an extended gRNA with a primer-binding site and a RT template. The primer-binding site anneals to the nicked PAM strand, and novel DNA containing the edit is directly incorporated through the RT using the RT template.
HDR-mediated therapeutic genome editing
Homology directed repair (HDR) is an error-free DNA damage response initiated following a DSB. HDR is an accurate DSB repair process due to the high degree of sequence homology required for templating DNA synthesis[26–27]. Following a DSB, the 5′ ends of the DNA are resected creating a 3′ overhang and an adequate scaffold for DNA damage response proteins to engage. Strand invasion by a homologous template generates a double Holliday junction allowing for polymerase-mediated repair of lost DNA sequence. HDR is active during the G2/S phase of the cell cycle due to the preference for sister chromatids as a homologous template[28]. Additionally, exogenous templates can serve as homologous sequences for HDR, thereby resulting in a mechanism to generate sequence-specific targeted insertions or replacement[29]. Targeted insertions via exogenous template DNA have been mediated through various constructs including plasmid DNA, single-stranded oligodeoxynucleotides (ssODN), and double-stranded oligodeoxynucleotides (dsODN) donor (Fig. 2A)[30–31]. Comparative analysis of SpCas9, NmCas9, SaCas9, Acidaminococcus sp. BV3L6 Cpf1 (AsCpf1), and Lachnospiraceae bacterium ND2006 Cpf1 (LbCpf1) demonstrated distinct editing and HDR characteristic preferences including the use of 5′ asymmetrically extended ssODN donor templates for SpCas9 and LbCpf1[22,32]. HDR-mediated sequence insertions have become ubiquitous methods of generating research models of specific genotypes, as well as correcting pathogenic variants of disease[33–34].
Figure 2.
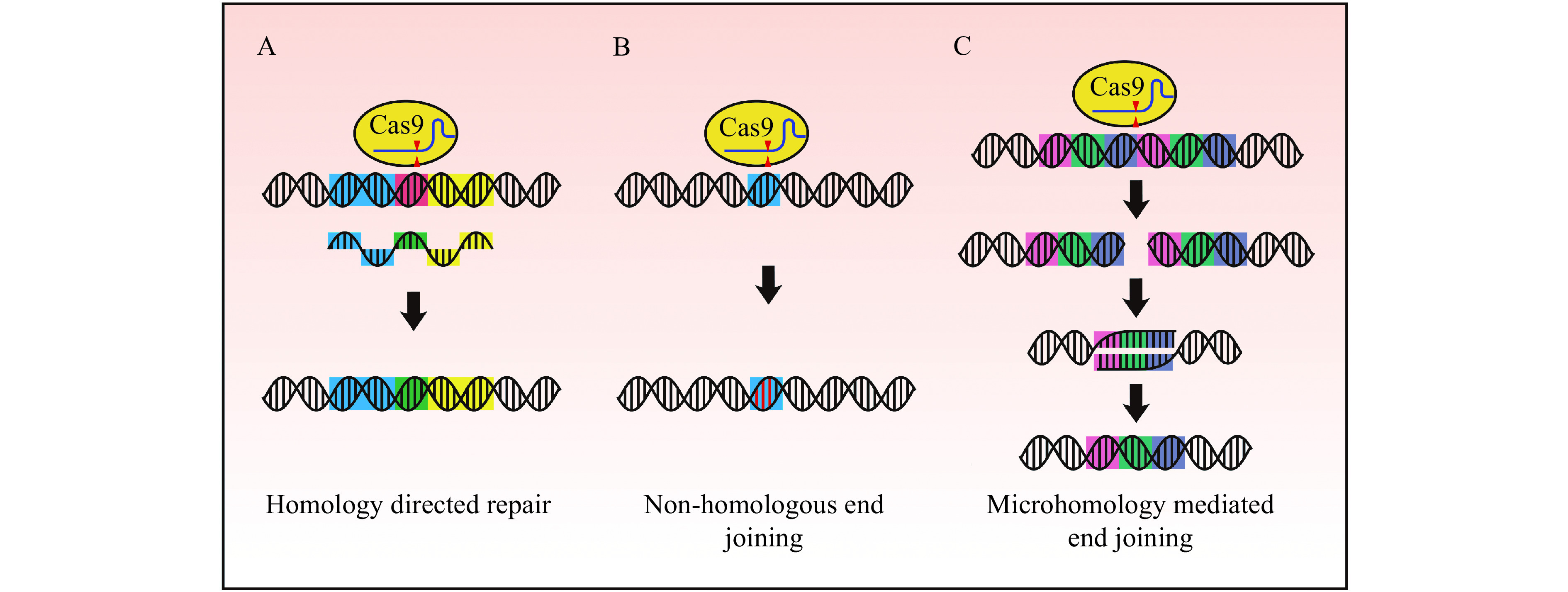
DNA repair pathways following a double-strand break.
A: Homology directed repair (HDR) can facilitate targeted sequence modification by providing a repair template with homology arms to the target sequence (indicated in blue and yellow). Repair templates can be single-stranded oligodeoxynucleotides (ssODN) as illustrated (sequence modification indicated in green), or double-stranded such as plasmid template donors or sister chromatids. B: Non-homologous end joining (NHEJ) is an error prone DNA damage response often resulting in insertions and deletions near the cleavage site (indels indicated in red). C: Microduplications can be precisely deleted by microhomology-mediated end joining (MMEJ). Cas9 generates a double-strand break between repeat sequences, and the subsequent processes of 5′ end resection followed by annealing of the homologous regions result in the deletion of the microduplication.
Leber congenital amaurosis (LCA) is an autosomal recessive disease, causing childhood-onset blindness, with 6% of cases linked to mutations in RPE65[35]. Current treatments are limited, with voretigene neparvovec, a gene therapy approach delivering an intact RPE65 gene by adeno-associated virus (AAV) to patient retinas, as the only treatment approved by the FDA[36]. However, though visual restoration was incomplete, voretigene neparvovec progressed retinal gene therapy approaches, and established practical methods for the delivery of genetic material to patient retinas. Building upon these delivery methods, 3-week-old rd12 mice, a rodent model of LCA, were treated with a dual AAV system containing a separate Cas9 and sgRNA-Rpe65 donor construct, for delivery of the Cas9 endonuclease and an HDR template. Following AAV delivery through subretinal injection, Rpe65 was permanently corrected in approximately 1% of targeted cells, with functional restoration of retinal function exceeding genetic correction. Treated rd12 mice responded to bright stimuli, and quantification of a- and b- wave amplitudes demonstrated significant increases toward wildtype control counterparts[37].
Mutations in ornithine transcarbamylase (OTC) in patients results in hyperammonemia leading to neurological dysfunction, and in neonatal males, is often fatal[38]. A mouse model for OTC deficiencies, spfash, has a G-to-A mutation resulting in aberrant splicing and a 20-fold reduction in OTC mRNA and protein expression. As genetic liver disease often presents in newborns, postnatal day 2 (p2) spfash mice were treated with AAV8 separately packaging SaCas9 and a sgRNA along with an OTC repair template for dual delivery via temporal vein injection. Deep sequencing analysis of the targeted OTC gene identified 6% to 20% templated gene correction, with insertions and deletions (indels) identified in approximately 30% of OTC alleles. Most treated mice had higher OTC protein levels than their untreated counterparts but failed to reach the levels of wildtype controls. However, by challenging the treated mice via a high-protein diet, restoration of OTC function was demonstrated by a reduction in indicators of pathology and toxicity, in addition to complete survival within the treated cohort, contrary to the 70% survival of untreated or untargeted control mice[39]. Due to HDR restrictions in adult liver, adult mice treated with the same dual AAV gene editing system had significantly lower corrective editing and harbored more large deletions resulting in a loss of the minimal expression of OTC which remained in pathogenic cells.
To address the limitations of in vivo HDR, many groups have utilized an ex vivo editing approach for targeted insertions. Fah−/− mice, a rodent model of hereditary tyrosinemia type I (HTI), lack expression of fumarylacetoacetate hydrolase (Fah), an enzyme responsible for the final step of tyrosine catabolism. Fah−/− hepatocytes harbor a G-to-A point mutation in the splice donor of exon 7 resulting in splicing defects and impaired Fah enzymatic activity[40]. Fah deficiency is fatal owing to the accumulation of toxic metabolites in the hepatic system resulting in cell death, and eventually liver failure[41]. Ex vivo templated gene correction was enhanced due to an upregulation of HDR-related genes in cultured hepatocytes compared to in vivo counterparts which was further evidenced by an increase in cellular proliferation marker, Ki-67[42]. Cultured Fah−/− hepatocytes were treated with a dual AAV gene editing strategy. Separate packages of an SaCas9 and gRNA, and a homologous repair template and gRNA resulted in a low fraction of HDR corrected hepatocytes. Once transplanted into the Fah−/− liver, engrafted cells demonstrated a proliferative advantage leading to functional restoration as indicated by a reduction in hallmark indicators of liver impairment[42]. As hepatocytes are polyploidy and only a single corrected Fah allele can restore enzymatic activity, the functional correction efficiencies are greater than editing efficiencies as quantified by sequencing bulk samples[43].
NHEJ-mediated therapeutic genome editing
Non-homologous end joining (NHEJ) is the primary mechanism responsible for repairing DNA DSBs in mammalian cells[44]. Unlike HDR, NHEJ is an error prone response, often resulting in indels due to the direct ligation of separated DNA strands following end resection (Fig. 2B)[45]. NHEJ has become a useful editing strategy in addition to HDR, with advantages including increased efficiency and being template free[15]. Because indels generated by CRISPR/Cas genome editing are site-specific, targeted indel formation by NHEJ is an efficacious mechanism for introducing frameshift mutations and premature stop codons. This strategy has been utilized in research and clinical contexts to disrupt gene activity or bypass mutated genomic regions.
The adaptive capacity of tumors to evade immune recognition and response can be in part facilitated through upregulation of programmed death-ligand 1 (PD-L1). PD-L1+ tumors are indicators of poor prognosis in multiple tumor types due to interactions with programmed death-1 receptors (PD-1) on activated T-cells to suppress immune response[46]. Clinical studies have demonstrated that inhibition of PD-1 by PD-1 inhibitors significantly enhances antitumor immunity[47]. Disruption of the PD-1 gene, PDCD-1, in mesothelin-targeted CAR T cells (Meso CAR T) by NHEJ generated a 60% reduction in PD-1hi cells. When PD-1 disrupted Meso CAR T cells were introduced into a xenograft mouse model, the in vivo tumor response of PD-1 disrupted Meso CAR T cells demonstrated a significant reduction in tumor burden, and increased genomic copy number, suggesting increased expansion of PD-1 disrupted Meso CAR T cells[48]. In accordance with this study, phase 1 clinical trial to evaluate the effect of CRISPR/Cas9 disrupted PD-1 TCR knock-out CAR T cells in patients with mesothelin positive multiple solid tumors has been initiated[49].
β-hemoglobinopathies are hematologic diseases, encompassing in part, the genetic diseases β-thalassemia and sickle cell disease (SCD)[50]. Treatments for patients with β-hemoglobinopathies are incomplete with allogeneic hematopoietic stem cell (HSC) transplants as the only definitive treatment[51]. Insufficient availability of HSCs for transplantation has focused efforts on genetic therapies for generating new treatment avenues. During human development, there exist two transitions in globin regulation: from embryonic to fetal globin (HbF) during gestation, and fetal to adult globin (HbA) following birth[52]. Owing to this regulatory change, symptoms of β-thalassemia and SCD begin to manifest in the first few months of life, due to the silencing of HbF expression (Fig. 3A). Individuals with hereditary persistence of fetal globin (HPFH), a condition in which HbF is not silenced following birth, are largely protected by the deleterious outcomes of SCD or β-thalassemia[53]. A strategy to reactivate endogenous expression at the HbF locus was developed by disrupting a transcriptional inhibitor, BCL11A (Fig. 3B)[54]. Human CD34+ hematopoietic stem and progenitor cells (HSPC) targeted for disruption of BCL11A showed increased levels of HbF in erythroid cells, and a phase 1/2 clinical trial transplanting Cas9-mediated BCL11A disrupted autologous CD34+ human HSPCs, termed CTX001, has been initiated[55–56]. Early evidence has shown a successful proof of concept with the first two patients independent of blood transfusions following CTX001 infusion[57].
Figure 3.
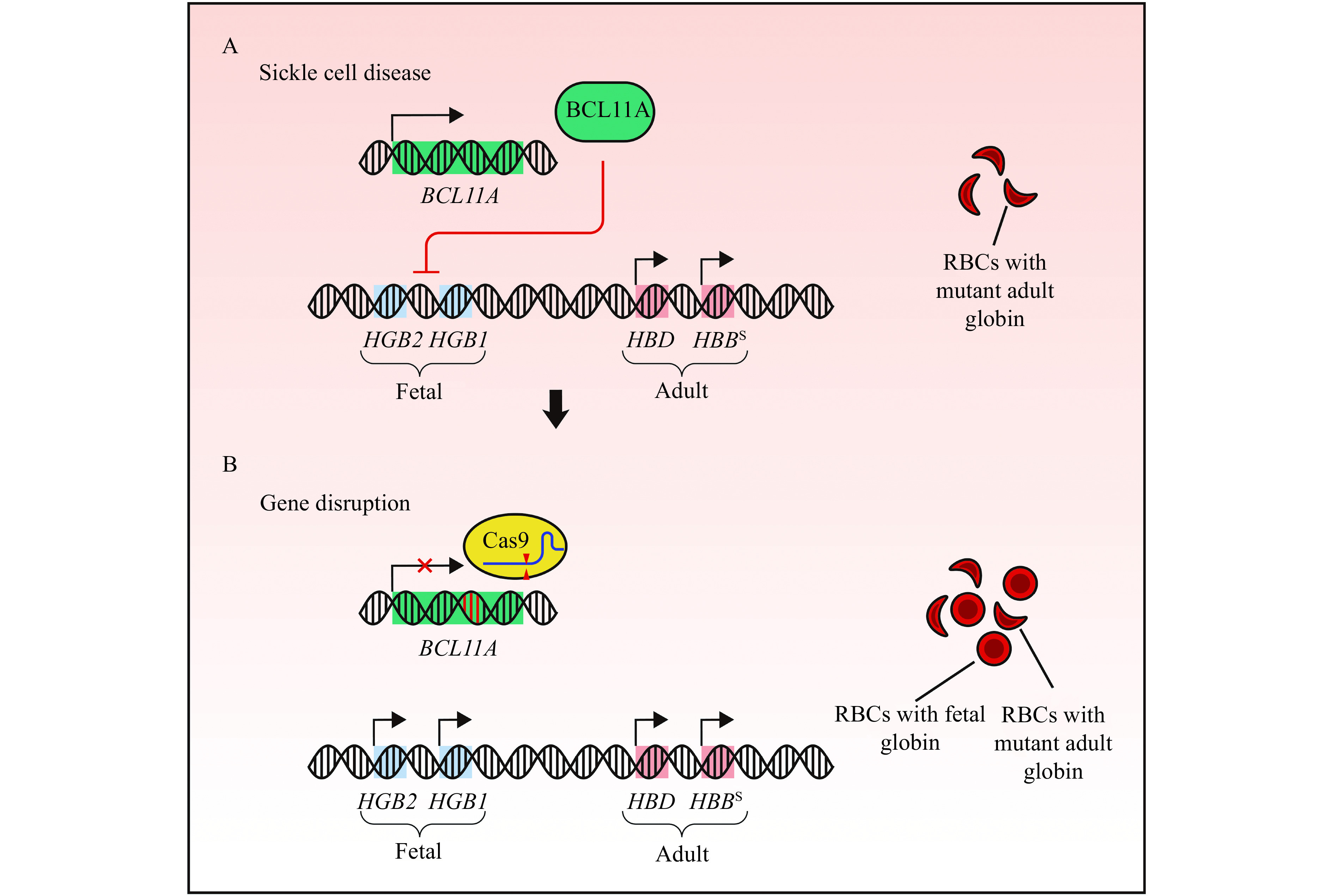
Rescue of fetal globin expression by non-homologous end joining-mediated gene disruption alleviates sickle cell disease.
A: β-hemoglobinopathies are caused by pathogenic variants in the adult β-globin gene. HBBS, the variant underlying sickle cell disease, produces mutant beta subunits of adult globin resulting in sickle shaped red blood cells (RBCs). B: Non-homologous end joining-mediated disruption of BCL11A derepresses expression of fetal globin (HGB2 and HGB1). Activation of fetal globin expression can alleviate the disease severity of β-hemoglobinopathies.
Mutations that disrupt normal RNA splicing patterns are known contributors to disease, with the most prevalent pathogenic variants altering cis-elements including the 5′ splice site, 3′ splice site, branch point or regulatory sites that facilitate spliceosome assembly[58–59]. Pathogenic mis-splicing has been implicated in β-thalassemia, cystic fibrosis, and LCA arising in the aberrant inclusion or exclusion of coding sequence[58]. LCA type 10 (LCA10) is a form of autosomal recessive retinal dystrophy caused by mutations in CEP290. IVS26, the most common mutation in LCA10, creates a novel splice donor site due to an A-to-G mutation in intron 26. Aberrant splicing generates a cryptic pseudoexon, resulting in an mRNA with a premature stop codon[60]. By targeting the IVS26 site with a pair of gRNAs flanking the mutation, a corrective inversion or deletion event is possible such that normal mRNA splicing is restored. The editing components were packaged in an AAV5 vector containing the pair of gRNAs and a SaCas9 expressed under a retina-specific promoter. Editing in fully mature retinal explant cultures resulted in a corrective editing rate of 16.6%±6.5%. Further experiments in a humanized CEP290 IVS26 knock-in mouse model and non-human primate models showed dosage responses exceeding the targeted 10% minimal threshold of corrected foveal cone photoreceptors needed to restore normal vision[61]. This proof of concept study has led to the breakthrough phase 1/2 clinical trial titled EDIT-101 leading to the direct administration of CRISPR/Cas genome editing systems into a human patient for the first time[62].
Duchenne muscular dystrophy (DMD) is a fatal disease of the muscle affecting approximately 1 in every 5000 males at birth. DMD is caused by mutations in dystrophin, a protein responsible for linking the dystroglycan complex with the cytoskeleton to maintain structure during muscle cell contraction[63]. Despite the heterogeneity of mutations causal in DMD, it has been estimated that 80% of individuals with DMD could be treated with exon-skipping strategies to restore partial dystrophin expression[64]. Myoediting is a strategy to correct Dmd mutations by NHEJ-mediated exon skipping. The strategy uses paired gRNAs to target the 5′ and 3′ ends of exon 23 to remove the premature termination codon present in the mdx mouse, an animal model of DMD. Notably, deletion of exon 23 rescued normal open reading frame (exon 22 to exon 24), thereby generating a partial restoration of dystrophin protein expression following intraperitoneal, intramuscular, or retro-orbital injection of AAV9 package gene editing systems[65]. As partial restoration of dystrophin expression can improve patient cardiomyopathy and protect against contraction induced injury to skeletal muscle, myoediting has the potential to offer therapeutic benefit to majority of DMD patients.
Strikingly, recent publications have demonstrated that the indels produced following NHEJ are a predictable event, dictated largely by the Cas9 cleavage site and the surrounding local sequence[66–68]. Single base duplications, short deletions, and microhomology-mediated deletions are among the most common indel occurrences and can be predicted with small cell type-specific variations. Fanconi anemia (FA) is a genetic disorder of the hematopoietic system caused by mutations in the FANC genes, key components maintaining genome stability through involvement in DNA repair and cell division processes[69–70]. HDR is impaired in FA due to the intrinsic inefficiency of HDR in primitive HSPCs, and increased efficiency of NHEJ in FA cells. Patients with mutations in FANC genes occasionally harbor compensatory mutations restoring the pathogenic frameshift mutations, demonstrating a therapeutic modality generated by corrective indels. NHEJ-mediated repair of prevalent patient-specific mutations of FANCA including the c.295C>T point mutation and the c.3558insG indel variant, produced healthy donor like cells with anin vitro and in vivo rodent transplantation model proliferative advantage[71]. These findings demonstrate an efficient and precise method of targeted sequence reversion despite limitations imposed by disrupted DNA repair pathways.
MMEJ-mediated therapeutic genome editing
Microhomology-mediated end joining (MMEJ), also called alternative end joining, is a DNA damage response occurring following DNA DSBs[72]. MMEJ is an alternative repair pathway to HDR, initiated following DNA end resection[72]. Based on a sufficient region of sequence homology flanking a DSB, approximately 5 to 25 bp, a DSB is repaired through annealing the homologous regions together, thereby deleting one repeat and the intermediate sequence (Fig. 2C)[73]. Microduplications and sequence repeats are a common DNA replication error resulting in nascent genetic disease[74]. Inducing targeted DSB at a site flanked by these repeats meets the criteria to initiate the MMEJ DNA damage response, thereby having the potential to revert pathogenic microduplications and sequence repeats into a wild-type allele.
Limb-girdle muscular dystrophy type 2G (LGMD2G) is a systemic disease affecting multiple tissues including skeletal and cardiac muscle[75]. A pathogenic 8-bp duplication in the TCAP gene is found in approximately 1 in 1000 alleles in Eastern Asian populations. Patient derived induced pluripotent stem cells (iPSC) homozygous for the pathogenic microduplication at the TCAP gene were targeted for MMEJ facilitated gene editing. By targeting ribonucleoprotein complex of SpCas9 and gRNA to the mutant allele, a double-strand cleavage was induced at the boundary of the 8-bp duplication resulting in a precise 8-bp deletion in 57% of all cells targeted for editing. The same study demonstrated an MMEJ based gene editing approach for correction of a pathogenic 16-bp microduplication in HPS1, in which pathogenic variants are associated with Hermansky-Pudlak syndrome type 1 (HPS1). PAM site restrictions prevented cleavage at the center of the microduplication, but an array of gRNAs designed to induce cleavage at staggered locations along the microduplication resulted in variable wild-type reversion with the desired 16-bp deletion occurring at higher frequencies based on proximity of the cleavage to the center of the microduplication[76].
MMEJ gives rise to the ability to generate precise DNA integration via providing an exogenous MMEJ-plasmid construct. MMEJ-plasmids contain donor templates for MMEJ-mediated targeted insertions where microhomology arms span approximately 5 to 25 bp[77]. Eight-week-old mice homozygous for a pathogenic frameshift insertion in exon 5 of the Fah gene were treated with an MMEJ plasmid containing cDNA for exons 5 through 14 of the Fah gene and an sgRNA targeting intron 4 of Fah, in tandem with a plasmid encoding SpCas9, delivered through hydrodynamic tail-vein injection. A week after in vivo editing, NTBC, a pharmacological treatment for HTI, was withdrawn from treated mice. Targeted integration was evidenced by sequencing analysis, FAH protein expression, and drastically increased survival following 14 weeks without NTBC. The study further demonstrated an increase in MMEJ-mediated sequence integration 10-fold greater than HDR-based knock-in strategies, likely owing to differences in cell cycle status[78].
The 5 to 25 bp of sequence homology required between a target sequence and an MMEJ donor is advantageous considering the limitations in packaging size of common viral delivery methods[79]. Recombinant AAVs are especially promising genetic delivery tools due to the safe and stable nature of transgene expression and variant tropism in tissues of biomedical interest[80–81]. As the packaging load of recombinant AAVs is limited to approximately 4.7 kb, a dual AAV delivery system is often utilized, which narrows the cells recipient of all gene editing components in appropriate stoichiometry[39,82]. In a mouse model of blindness caused by the pathogenic Gnat1IRD2/IRD2 and Pde6ccpfl1/cpfl1 mutations, a single AAV vector utilizing an MMEJ-mediated repair gene editing approach was developed. The MMEJ donor was packaged along with an SaCas9, two gRNAs targeting the IRD2 mutation in Gnat1, and appropriate regulatory elements including the neural retina-specific promoter GRK1 driving SaCas9 expression, all in a single AAV8 plasmid vector. Subretinal injections of 6-month old mice resulted in Gnat1 expression in photoreceptors at both the protein and mRNA levels, with sequencing of in vivo integration showing 11%[83].
Base editor-mediated therapeutic genome editing
Base editing systems are a precise editing modality mediated by CRISPR/Cas gene editing tools, capable of generating single nucleotide transitions in DNA. Cytosine base editors (CBE) convert C-G to T-A base pairs, while adenine base editors (ABE) generate A-T to G-C base pair edits (Fig. 1B and C)[84–85]. These precise base pair edits occur within the editing window, a defined region corresponding to positions along the gRNA sequence, which is intrinsic to each base editing system.
CBE is an engineered gene editing tool built upon a modified enzymatically inactive Cas9 (dCas9). The APOBEC1 cytosine deaminase protein was fused to dCas9 which facilitates a C-to-U conversion, which through the endogenous mismatch repair pathway, is repaired to T-A, or reverted to the original C-G. To facilitate the C-to-T transitions, an uracil glycosylase inhibitor (UGI), which impairs the uracil base excision repair pathway, was fused onto the C-terminal of the dCas9 platform, resulting in the optimized system, to facilitate double-stranded edits, in optimized BE3 system, dCas9 was replaced with a Cas9 nickase (D10A), which biases the genomic repair machinery to preferentially generate the T-A edited base pair by generating a single-stranded cut on the non-edited strand[84]. To increase efficiency and reduce indel formation, a bacteriophage Mu protein, Gam, which binds to DSBs was added, resulting in the formation of BE4, with further optimization steps to improve expression and activity resulted in BE4max[86–87].
The generation of ABEs was initially met with difficulty due to the absence of an endogenous ssDNA adenosine deaminase. Targeted evolution of an Escherichia coli tRNA adenosine deaminase, known as TadA, led to the generation of mutant TadA proteins, which when fused to a dCas9, can convert A to deoxyinosine (I), which are subsequently recognized as G in the RNA[85]. Initial engineering reports indicated ABE7.10 as the most efficient and specific ABE editing system, which contains a wildtype noncatalytic TadA monomer and the evolved TadA adenosine deaminase on the dCas9 platform. Similar to BE4max, ABEmax was generated with further optimizations to improve expression and activity[87].
Base editing strategies are a promising therapeutic approach for correcting pathogenic variants due to the possibility of generating precise base pair resolution edits without potentially harmful DSBs. Marfan syndrome (MFS) is an autosomal dominant disorder of the connective tissue arising from mutations in FBN1, a gene coding for the protein, fibrillin 1[88]. Fibrillin 1 is a critical component of microfibrils, with pathogenic variants in the FBN1 causing skeletal abnormalities and cardiovascular manifestations resulting in major morbidity and mortality[89]. Embryos harboring a patient-specific heterozygous MFS mutation, FBN1T7498C, were generated by in vitro fertilization of an immature oocyte with sperm from the corresponding MFS patient. FBN1T7498C is an eligible candidate for CBE targeted gene correction with an appropriate SpCas9 PAM 5′-NGG-3′ to ensure that only the causal mutation resides within the 4th to 8th gRNA position corresponding to the editing window of BE3, enabling a corrective edit while reducing the potential for deleterious bystander mutations[90]. BE3 mRNA and locus-specific sgRNA were microinjected into the zygotes. 6 of 7 injected embryos corrected to the wildtype FBN1 sequence, while 1 of 7 injected embryos harboring an unintended transition mutation. Furthermore, deep sequencing of potential gRNA dependent off-target sites indicated no detectable off-target editing events. A subsequent whole-genome analysis of 419 possible off-target sites as determined by a tolerance of 5 mismatches against the corrective gRNA resulted in no off-targets identified[91].
In addition to correcting pathogenic alleles by reverting disease-causing variants to wildtype sequences, expression of pathogenic transcripts can be halted through nonsense mediated decay by generating stop codons in a coding sequence. CBEs can generate stop codons within a coding sequence through C-to-T conversions of CAG, CGA, or CAA codons[92–93]. Approximately 20% of amyotrophic lateral sclerosis (ALS) cases are caused by mutations in Cu-Zn superoxide dismutase 1 (SOD1). In these cases, pathogenic SOD1 expression in motor neuron and non-neural cells are thought to underlie disease progression. ALS is a complex and fatal disorder affecting multiple tissues due in part to a loss of motor neurons in the spinal cord and brain. The G93A-SOD1 mouse model of ALS harbors 25 copies of a pathogenic human SOD1G93A transgene, thereby resulting in an aggressive neurodegenerative disorder characterized by the accumulation of mutant SOD1 protein, and ALS symptoms including motor neuron loss and muscular atrophy[94]. Targeted conversions of SOD1G93A coding sequence to generate stop codons can disrupt pathogenic SOD1 expression. The sequence length of CBE systems requires additional considerations when developing in vivo delivery approaches as they exceed the packaging capacity of AAVs. Therefore, to target SOD1G93A, a split-intein approach that facilitates trans-splicing of two halves of a split CBE system was developed which allows BE3 and a SOD1-specific gRNA to be separately packaged into a dual AAV9 system. An intrathecal injection of the AAVs into the SOD1G93A mice resulted in improved survival, and alleviation of symptoms including motor coordination, hindlimb grip strength, and increased weight. Molecular analyses indicated a reduction in SOD1 reactive inclusions, and increased motor neurons and decreased denervated neuromuscular junctions in treated mice, thereby demonstrating the potential of CBE-mediated correction strategies to prevent pathogenesis[94]. Mirroring the generation of a stop codon to disrupt pathogenic transcript expression, ABE systems can convert ATG start codons to GTG or ACG in a strategy called i-Silence[95]. ClinVar analysis reveals 247 human diseases with aberrant start codon mutations, with 147 of these mutations targetable by the i-Silence strategy.
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which affects multiple organs with primary morbidity and mortality associated with the lungs[96]. A biobank of 664 patients, representing approximately half of the Dutch CF population, was described and surveyed revealing that approximately 20% of mutations could be corrected with base editors[97]. To test the efficacy of base editors on CF mutations, organoids from patient derived tissues were generated and assessed for molecular signatures of correction through CFTR gene, transcript, and protein analysis as well as restoration of organoid function. Intestinal organoids from a patient harboring a homozygous CFTR c.2353C>T were generated from a rectal biopsy. The organoids were dissociated into single cells and treated with an ABEmax with the pathogenic C-to-T (G-to-A) mutation. Editing efficiency by ABEmax was 9.3%, 5-fold greater than targeted correction by HDR-mediated correction, and a forskolin induced swelling assay to assess functional rescue of CFTR function in the intestinal organoids showed restoration to wildtype organoid levels. Patient derived intestinal organoids were generated from a patient harboring a homozygous c.3846G>A mutation, in addition to a patient heterozygous for the third most prevalent nonsense CF mutation, c.1657C>T. The c.3846G>A and c.1657C>T mutations did not have appropriate 5′-NGG-3′ PAMs, but could be targeted using mutation-specific gRNAs and a xABEmax plasmid built upon the xCas9 system which recognizes more flexible 5′-NGN-3′ PAM. After the dissociated organoids were treated with the xABEmax system and were expanded into intestinal organoids where forskolin induced swelling assays demonstrated rescue of organoid function to wildtype levels, despite correction efficiencies of about 1.5%. As 10% of residual CFTR function is associated with a milder form of disease, the editing efficiencies demonstrated, specifically of the ABEmax correction strategy for the c.2353C>T mutation, has the potential to be an impactful therapeutic approach[98–99].
Recent innovations in base editing technologies have led to the generation of the dual-deaminases, a CRISPR/Cas gene editing platform capable of both A>G and C>T transition mutations within the editing window[100–102]. Dual-deaminases are built by combining ABE and CBE components onto the same nCas9 to target a locus for dual-deamination with a single gRNA. Dual-deaminases have the capacity to model and correct multinucleotide variants, as well as generate novel codon changes previously outside the capacity of ABE or CBE, with a single gRNA targeted process[100–102]. Within the promoter of the γ-globin genes are two mutations known to alleviate symptoms of β-hemoglobinopathies through increasing HbF production. The −113A-to-G mutation creates a GATA1 binding site, activating HBG1 transcription, while the -114C-to-T mutation disrupts binding of the transcriptional repressor, BCL11A[103–104]. By using the dual-deaminase A&C-BEmax, it was demonstrated that the simultaneous −113A-to-G and −114C-to-T mutations generated erythroid precursor cell lines with the greatestHBG mRNA expression[101]. Further analysis of edited clones revealed that additional mutations disrupting the promoter region did not increase induction of HBG, and that the single −114C-to-T did not match the restoration of the combined −113A-to-G and −114C-to-T mutations. This proof of principle experiment demonstrates single nucleotide resolution dissection of gene sequence in a single experiment, and the potential application of a dual-deaminase system in therapeutic gene editing.
Prime editing
Prime editors (PE) are a gene editing system capable of generating targeted insertions, deletions, and all single base pair changes without a DSB or donor DNA template (Fig. 1D)[105]. PEs were generated by fusing a reverse transcriptase (RT) to a nCas9, and by utilizing a modified gRNA, known as a prime editing guide RNA (pegRNA), which includes a distinct primer-binding region followed by RT template which includes the desired edit. Once localized to the target site, the PAM strand of the DNA is nicked, followed by hybridization of the primer-binding region of the pegRNA to the corresponding region in the PAM strand. New DNA is synthesized by the RT templated by the pegRNA RT template resulting in a 5′ flap containing the original unedited sequence, and a 3′ flap harboring the edited sequence. 5′ flap cleavage is mediated through endonucleases including flap endonuclease 1, leading to ligation of newly synthesized DNA. In the optimized PE variant PE3, the unedited strand is nicked by a nCas9 to preferentially repair the unedited strand to match the edited strand. Initial results indicate that prime editors are a robust editing modality with situational advantageous uses over base editors and traditional CRISPR/Cas systems. Base editors can be constrained by multiple substrates within an editing window leading to bystander activity, or a lack of PAM sites to place intended targets within an editing window. PE can circumvent these limitations due to the templated nature of PE edits and the additional distance of possible edits due to the extended length supplied by the pegRNA. PEs can generate targeted insertions with comparable efficiencies to HDR with fewer unintended indels, though the size of the insertion can be a preventative factor for the current generation of PEs. The multiple mechanistic steps underlying the editing capacity of PE and the large genomic size of the PE system limits in vivo editing. PE is a promising technology to expand the catalog of CRISPR/Cas gene editing tools for therapeutic purposes with further optimization.
Discussion and perspectives
CRISPR/Cas gene editing systems have the potential to correct thousands of monogenic diseases. Functional correction, partial restoration, and conversion of debilitating disease to milder variants is regulated, in part, at two stages: the corrective capacity of the specific gene editing strategy, and the delivery of the gene editing components to targeted tissues. A suitable CRISPR/Cas gene editing strategy can be chosen based on characteristics underlying the mutant variant including the type of mutation and the cell cycle status of the target cells, as outlined in a decision tree (Fig. 4). Following the path of the decision tree results in one of seven possible gene editing strategies. For many mutations, multiple gene editing strategies are possible and should be considered to identify the optimal correction strategy. The practical scope of gene editing is dictated by concerns regarding efficiency, which is inherent to each scenario and requires consideration of corrective doses. For example, 10% of corrected foveal cone photoreceptors restores functional vision in LCA, as does correction of 30% to 50% of DMD cardiomyocytes for rescue of contractile phenotype[61,65,106].
Figure 4.

Decision tree for choosing gene editing strategy for diverse mutations.
CRISPR/Cas gene editing strategies can be chosen based on characteristics underlying the mutant variant. Following the decision tree will result in one of seven possible editing strategies. Homology directed repair (HDR) knock-in can produce a diverse range of modifications through a templated repair process, e.g., correction of leber congenital amaurosis through providing a donor oligo for templated repair. Non-homologous end joining (NHEJ) predictable indel generation is a method of introducing specific indels to restore the sequence or open reading frame, e.g., restoring the disrupted reading frame in Fanconi anemia. Microhomology-mediated end joining (MMEJ) knock-in and prime editor-mediated sequence editing can facilitate sequence modification in post-mitotic cells, e.g., correction of hereditary tyrosinemia type I through targeted insertion of a MMEJ plasmid. MMEJ repeat deletion can precisely delete short sequence repeats and microduplications through annealing of repeat sequences following MMEJ double-strand break repair, e.g., precise deletion of a 8-bp duplication in limb-girdle muscular dystrophy type 2G. Cytosine base editors can mediate C-to-T transition mutations in defined editing windows, e.g., correction of patient-specific Marfan syndrome mutations in patient embryos. Adenosine base editors can mediate A-to-G transition mutations in defined editing windows, e.g., correction of diverse cystic fibrosis mutations in intestinal organoids. Dual-deaminases can mediate both C-to-T and A-to-G transition mutations in a defined editing window, e.g., simultaneous generation of an activator binding site and disruption of an inhibitor binding site to alleviate symptoms of β-thalassemia.
Delivery of CRISPR/Cas genome editing systems into target cells and tissues is critical to successful therapeutic genome editing. Adeno associated viruses (AAV) are the leading approach for delivery of gene editing systems, gRNAs, and donor templates. AAVs are single-stranded viral vectors capable of delivering genomic payloads to multiple tissues, with minimal immunogenicity[107]. Despite the prevalence of AAVs, there are considerable obstacles limiting their widespread use, namely genomic packaging limitations (approximately 4.7 kb). Additionally, integration of AAV vectors into Cas9 induced DSB sites can occur at high frequencies across pathologically relevant tissue types including in the mouse brain, skeletal muscle, and cochlea[108]. Integration occurred in multiple sites across the genome, enriched at the Cas9 cleavage site, as well as at favored sites independent of gRNA homology. To address limitations imposed by AAVs, multiple groups have bioengineered delivery approaches using polymers, nanoparticles, and modified viral platforms[109–111].
Off-target edits are unintended mutations generated at any region not specified by a gene editing modality[112]. These unintended edits are a major concern when considering the potential applications of CRISPR/Cas systems in basic, translational, and clinical settings due to the uncertain consequences of off-target mutations. Efforts to reduce off-target edits have focused on both gRNA-based and Cas protein-based interactions. Multiple groups have developed tools to predict gRNA efficiency and specificity building on an understanding of gRNA and target characteristics including base pair composition, gRNA length, similarity of on- and off-target sequences, presence of PAMs, and mismatch locality[113–115]. High-fidelity variants of SpCas9 such as SpCas9-HF1 and eSpCas9 with comparable on-target editing efficiency but reduced or undetectable off-target edits have been generated through sequence and structurally driven engineering approaches[116–117].
The additional components of the CBE and ABE systems result in different off-target profiles to Cas9 alone[118]. CBEs have been demonstrated to have increased off-target DNA editing, in addition to RNA editing, due in part to gRNA independent off-target mutagenesis by APOBEC1[119–121]. Engineered CBE variants have been generated to drastically reduce off-target editing, with no minimal differences in on-target editing demonstrating strategies to increase the on- to off-target differential[121–123]. Strikingly, off-target analysis of ABEs reveals far fewer off-target edits, with identified mutations at similar frequencies to background[119,124–125].
As the numbers and types of gene editing technologies built upon CRISPR/Cas systems expands, our understanding of their applications follows. Each gene editing approach employs strategies unique to the architecture of the disease, and through collaborative efforts among the scientific and medical communities, our ability to generate the appropriate tools and develop the right understanding flourishes. The test to prove the potential of CRISPR/Cas systems to correct pathogenic mutations and alleviate disease has begun with multiple FDA approved clinical trials underway. With the pace of development and the ingenuity of the field, we are certain of a bright future for CRISPR/Cas gene editing approaches for the treatment of monogenic diseases.
Acknowledgments
We thank Dr. Joseph Miano for constructive criticism of the manuscript. This work was supported by grants from DHPS Foundation and Kids Connect Charitable Fund.
References
- 1.WHO. Genes and human diseases[EB/OL]. [2019-03-21]. http://www.who.int/genomics/public/geneticdiseases/en/.
- 2.Prakash V, Moore M, Yáñez-Muñoz RJ Current progress in therapeutic gene editing for monogenic diseases. Mol Ther. 2016;24(3):465–474. doi: 10.1038/mt.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferla R, Calò V, Cascio S, et al Founder mutations in BRCA1 and BRCA2 genes . Ann Oncol. 2007;18 Suppl 6:vi93–vi98. doi: 10.1093/annonc/mdm234. [DOI] [PubMed] [Google Scholar]
- 4.Strehlow V, Heyne HO, Vlaskamp DRM, et al GRIN2A-related disorders: genotype and functional consequence predict phenotype . Brain. 2019;142(1):80–92. doi: 10.1093/brain/awy304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dravet C, Oguni H Dravet syndrome (severe myoclonic epilepsy in infancy) Handbook Clin Neurol. 2013;111:627–633. doi: 10.1016/B978-0-444-52891-9.00065-8. [DOI] [PubMed] [Google Scholar]
- 6.Khan SH Genome-editing technologies: concept, pros, and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol Ther - Nucleic Acids. 2019;16:326–334. doi: 10.1016/j.omtn.2019.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rath D, Amlinger L, Rath A, et al The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie. 2015;117:119–128. doi: 10.1016/j.biochi.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Jinek M, Chylinski K, Fonfara I, et al A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedenheft B, Sternberg SH, Doudna JA RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 10.Barrangou R, Marraffini LA CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell. 2014;54(2):234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarova KS, Haft DH, Barrangou R, et al Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol. 2011;9(6):467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terns MP, Terns RM CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14(3):321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasiunas G, Barrangou R, Horvath P, et al Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109(39):E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cong L, Ran FA, Cox D, et al Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mali P, Yang LH, Esvelt KM, et al RNA-guided human genome engineering via Cas9 . Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deltcheva E, Chylinski K, Sharma CM, et al CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III . Nature. 2011;471(7340):602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CM, Cradick TJ, Bao G The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells . Mol Ther. 2016;24(3):645–654. doi: 10.1038/mt.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gleditzsch D, Pausch P, Müller-Esparza H, et al PAM identification by CRISPR-Cas effector complexes: diversified mechanisms and structures. RNA Biol. 2019;16(4):504–517. doi: 10.1080/15476286.2018.1504546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu JH, Miller SM, Geurts MH, et al Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556(7699):57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinstiver BP, Prew MS, Tsai SQ, et al Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition . Nat Biotechnol. 2015;33(12):1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimasu H, Shi X, Ishiguro S, et al Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361(6408):1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang YM, Liu KI, Sutrisnoh NAB, et al Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biol. 2018;19:62. doi: 10.1186/s13059-018-1445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zetsche B, Gootenberg JS, Abudayyeh OO, et al Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishimasu H, Ran FA, Hsu PD, et al Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lieber MR The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapman JR, Taylor MRG, Boulton SJ Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Scully R, Panday A, Elango R, et al DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20(11):698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takata M, Sasaki MS, Sonoda E, et al Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17(18):5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu MJ, Rehman S, Tang XD, et al Methodologies for improving HDR efficiency. Front Genet. 2019;9:691. doi: 10.3389/fgene.2018.00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li K, Wang G, Andersen T, et al Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS One. 2014;9(8):e105779. doi: 10.1371/journal.pone.0105779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song F, Stieger K Optimizing the DNA donor template for homology-directed repair of double-strand breaks. Mol Ther - Nucleic Acids. 2017;7:53–60. doi: 10.1016/j.omtn.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson CD, Ray GJ, DeWitt MA, et al Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34(3):339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 33.Schumann K, Lin S, Boyer E, et al Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112(33):10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang JP, Li XL, Li GH, et al Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017;18(1):35. doi: 10.1186/s13059-017-1164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cideciyan AV Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy . Prog Retin Eye Res. 2010;29(5):398–427. doi: 10.1016/j.preteyeres.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maguire AM, Russell S, Wellman JA, et al Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation–associated inherited retinal dystrophy: results of phase 1 and 3 trials . Ophthalmology. 2019;126(9):1273–1285. doi: 10.1016/j.ophtha.2019.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Jo DH, Song DW, Cho CS, et al CRISPR-Cas9–mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis . Sci Adv. 2019;5(10):eaax1210. doi: 10.1126/sciadv.aax1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon N Ornithine transcarbamylase deficiency: a urea cycle defect. Eur J Paediatr Neur. 2003;7(3):115–121. doi: 10.1016/S1090-3798(03)00040-0. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Wang LL, Bell P, et al A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34(3):334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aponte JL, Sega GA, Hauser LJ, et al Point mutations in the murine fumarylacetoacetate hydrolase gene: animal models for the human genetic disorder hereditary tyrosinemia type 1. Proc Natl Acad Sci U S A. 2001;98(2):641–645. doi: 10.1073/pnas.98.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulk NK, Wursthorn K, Wang ZY, et al Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo . Hepatology. 2010;51(4):1200–1208. doi: 10.1002/hep.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.VanLith CJ, Guthman RM, Nicolas CT, et al Ex vivo hepatocyte reprograming promotes homology-directed DNA repair to correct metabolic disease in mice after transplantation . Hepatol Commun. 2019;3(4):558–573. doi: 10.1002/hep4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin H, Song CQ, Dorkin JR, et al Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo . Nat Biotechnol. 2016;34(3):328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis AJ, Chen DJ DNA double strand break repair via non-homologous end-joining . Transl Cancer Res. 2013;2(3):130–143. doi: 10.3978/j.issn.2218-676X.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brandsma I, Van Gent DC Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr. 2012;3(1):9. doi: 10.1186/2041-9414-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riley JL PD-1 signaling in primary T cells. Immunol Rev. 2009;229(1):114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keir ME, Butte MJ, Freeman GJ, et al PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu WH, Zi ZG, Jin YL, et al CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365–377. doi: 10.1007/s00262-018-2281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chinese PLA General Hospital. Study of CRISPR-Cas9 mediated PD-1 and TCR gene-knocked out mesothelin-directed CAR-T cells in patients with mesothelin positive multiple solid tumors[EB/OL]. [2018-06-04]. https://clinicaltrials.gov/ct2/show/record/NCT03545815.
- 50.Antony JS, Haque AKMA, Lamsfus‐Calle A, et al CRISPR/Cas9 system: a promising technology for the treatment of inherited and neoplastic hematological diseases. Adv Cell Gene Ther. 2018;1(1):e10. doi: 10.1002/acg2.10. [DOI] [Google Scholar]
- 51.Cavazzana M, Antoniani C, Miccio A Gene therapy for β-hemoglobinopathies. Mol Ther. 2017;25(5):1142–1154. doi: 10.1016/j.ymthe.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sankaran VG, Orkin SH The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sokolova A, Mararenko A, Rozin A, et al Hereditary persistence of hemoglobin F is protective against red cell sickling. A case report and brief review. Hematol/Oncol Stem Cell Ther. 2019;12(4):215–219. doi: 10.1016/j.hemonc.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 54.Xu J, Peng C, Sankaran VG, et al Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science. 2011;334(6058):993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bjurström CF, Mojadidi M, Phillips J, et al Reactivating fetal hemoglobin expression in human adult erythroblasts through BCL11A knockdown using targeted endonucleases. Mol Ther - Nucleic Acids. 2016;5:e351. doi: 10.1038/mtna.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vertex Pharmaceuticals Incorporated. A safety and efficacy study evaluating CTX001 in subjects with severe sickle cell disease[EB/OL]. [2018-11-19]. https://clinicaltrials.gov/ct2/show/NCT03745287.
- 57.CRISPR therapeutics and vertex announce positive safety and efficacy data from first two patients treated with investigational CRISPR/Cas9 gene-editing therapy CTX001® for severe hemoglobinopathies[EB/OL]. [2019-11-19]. https://investors.vrtx.com/news-releases/news-release-details/crispr-therapeutics-and-vertex-announce-positive-safety-and.
- 58.Scotti MM, Swanson MS RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faustino NA, Cooper TA Pre-mRNA splicing and human disease. Genes Dev. 2003;17(4):419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 60.Maeder ML, Stefanidakis M, Wilson CJ, et al Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25(2):229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 61.Geller AM, Sieving PA Assessment of foveal cone photoreceptors in Stargardt's macular dystrophy using a small dot detection task. Vision Res. 1993;33(11):1509–1524. doi: 10.1016/0042-6989(93)90144-L. [DOI] [PubMed] [Google Scholar]
- 62.Allergan. Single ascending dose study in participants with LCA10[EB/OL]. [2019-03-13]. https://clinicaltrials.gov/ct2/show/NCT03872479.
- 63.Campbell KP, Kahl SD Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338(6212):259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 64.Yokota T, Duddy W, Partridge T Optimizing exon skipping therapies for DMD. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2949311/ Acta Myol. 2007;26(3):179–184. [PMC free article] [PubMed] [Google Scholar]
- 65.Long CZ, Li H, Tiburcy M, et al Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci Adv. 2018;4(1):eaap9004. doi: 10.1126/sciadv.aap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen MW, Arbab M, Hsu JY, et al Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 2018;563(7733):646–651. doi: 10.1038/s41586-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chakrabarti AM, Henser-Brownhill T, Monserrat J, et al Target-specific precision of CRISPR-mediated genome editing. Mol Cell. 2019;73(4):699–713. doi: 10.1016/j.molcel.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allen F, Crepaldi L, Alsinet C, et al Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat Biotechnol. 2019;37(1):64–72. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nalepa G, Clapp DW Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. 2018;18(3):168–185. doi: 10.1038/nrc.2017.116. [DOI] [PubMed] [Google Scholar]
- 70.Ceccaldi R, Sarangi P, D'Andrea AD The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 71.Román-Rodríguez FJ, Ugalde L, Álvarez L, et al NHEJ-mediated repair of CRISPR-Cas9-induced DNA breaks efficiently corrects mutations in HSPCs from patients with fanconi anemia. Cell Stem Cell. 2019;25(5):607–621. doi: 10.1016/j.stem.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 72.Sfeir A, Symington LS Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci. 2015;40(11):701–714. doi: 10.1016/j.tibs.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Truong LN, Li YJ, Shi LZ, et al Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110(19):7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ottaviani D, LeCain M, Sheer D The role of microhomology in genomic structural variation. Trends Genet. 2014;30(3):85–94. doi: 10.1016/j.tig.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 75.Moreira ES, Wiltshire TJ, Faulkner G, et al Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24(2):163–166. doi: 10.1038/72822. [DOI] [PubMed] [Google Scholar]
- 76.Iyer S, Suresh S, Guo DS, et al Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nature. 2019;568(7753):561–565. doi: 10.1038/s41586-019-1076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakade S, Tsubota T, Sakane Y, et al Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat Commun. 2014;5:5560. doi: 10.1038/ncomms6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao X, Wang X, Liu JL, et al CRISPR/Cas9 – mediated precise targeted integration in vivo using a double cut donor with short homology arms . EBioMedicine. 2017;20:19–26. doi: 10.1016/j.ebiom.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lau CH, Suh Y In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease . F1000Res. 2017;6:2153. doi: 10.12688/f1000research.11243.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zincarelli C, Soltys S, Rengo G, et al Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16(6):1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 81.Samulski RJ, Muzyczka N AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol. 2014;1:427–451. doi: 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]
- 82.Moreno AM, Fu X, Zhu J, et al In situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation . Mol Ther. 2018;26(7):1818–1827. doi: 10.1016/j.ymthe.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishiguchi KM, Fujita K, Miya F, et al Single AAV-mediated mutation replacement genome editing in limited number of photoreceptors restores vision in mice. Nat Commun. 2020;11(1):482. doi: 10.1038/s41467-019-14181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Komor AC, Kim YB, Packer MS, et al Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaudelli NM, Komor AC, Rees HA, et al Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Komor AC, Zhao KT, Packer MS, et al Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv. 2017;3(8):eaao4774. doi: 10.1126/sciadv.aao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koblan LW, Doman JL, Wilson C, et al Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol. 2018;36(9):843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramirez F, Dietz HC Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev. 2007;17(3):252–258. doi: 10.1016/j.gde.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 89.Pepe G, Giusti B, Sticchi E, Abbate R, Gensini GF, Nistri S Marfan syndrome: current perspectives. Appl Clin Genet. 2016;9:55–65. doi: 10.2147/TACG.S96233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rees HA, Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeng YT, Li JN, Li GL, et al Correction of the Marfan syndrome pathogenic FBN1 mutation by base editing in human cells and heterozygous embryos . Mol Ther. 2018;26(11):2631–2637. doi: 10.1016/j.ymthe.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Billon P, Bryant EE, Joseph SA, et al CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP Codons. Mol Cell. 2017;67(6):1068–1079. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuscu C, Parlak M, Tufan T, et al CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat Methods. 2017;14(7):710–712. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- 94.Lim CKW, Gapinske M, Brooks AK, et al Treatment of a mouse model of ALS by in vivo base editing . Mol Ther. 2020;28(4):1177–1189. doi: 10.1016/j.ymthe.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang XJ, Liu ZW, Li GL, et al Efficient gene silencing by adenine base editor-mediated start codon mutation. Mol Ther. 2020;28(2):431–440. doi: 10.1016/j.ymthe.2019.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ratjen F, Bell SC, Rowe SM, et al Cystic fibrosis. Nat Rev Dis Primers. 2015;1(1):15010. doi: 10.1038/nrdp.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Geurts MH, De Poel E, Amatngalim GD, et al CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell. 2020;26(4):503–510. doi: 10.1016/j.stem.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 98.Green DM, McDougal KE, Blackman SM, et al Mutations that permit residual CFTR function delay acquisition of multiple respiratory pathogens in CF patients. Respir Res. 2010;11(1):140. doi: 10.1186/1465-9921-11-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ferec C, Cutting GR Assessing the disease-liability of mutations in CFTR. Cold Spring Harb Perspect Med. 2012;2(12):a009480. doi: 10.1101/cshperspect.a009480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grünewald J, Zhou RB, Lareau CA, et al A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol. 2020;38(7):861–864. doi: 10.1038/s41587-020-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang XH, Zhu BY, Chen L, et al Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol. 2020;38(7):856–860. doi: 10.1038/s41587-020-0527-y. [DOI] [PubMed] [Google Scholar]
- 102.Sakata RC, Ishiguro S, Mori H, et al Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat Biotechnol. 2020;38(7):865–869. doi: 10.1038/s41587-020-0509-0. [DOI] [PubMed] [Google Scholar]
- 103.Wienert B, Martyn GE, Funnell APW, et al Wake-up sleepy gene: reactivating fetal globin for β-hemoglobinopathies. Trends Genet. 2018;34(12):927–940. doi: 10.1016/j.tig.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 104.Martyn GE, Wienert B, Kurita R, et al A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood. 2019;133(8):852–856. doi: 10.1182/blood-2018-07-863951. [DOI] [PubMed] [Google Scholar]
- 105.Anzalone AV, Randolph PB, Davis JR, et al Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bostick B, Yue YP, Long C, et al Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102(1):121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]
- 107.Verdera HC, Kuranda K, Mingozzi F AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther. 2020;28(3):723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hanlon KS, Kleinstiver BP, Garcia SP, et al High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun. 2019;10(1):4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mangeot PE, Risson V, Fusil F, et al Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun. 2019;10(1):45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang LM, Wang P, Feng Q, et al Lipid nanoparticle-mediated efficient delivery of CRISPR/Cas9 for tumor therapy. NPG Asia Mater. 2017;9(10):e441. doi: 10.1038/AM.2017.185. [DOI] [Google Scholar]
- 111.Cheng WJ, Chen LC, Ho HO, et al Stearyl polyethylenimine complexed with plasmids as the core of human serum albumin nanoparticles noncovalently bound to CRISPR/Cas9 plasmids or siRNA for disrupting or silencing PD-L1 expression for immunotherapy. Int J Nanomedicine. 2018;13:7079–7094. doi: 10.2147/IJN.S181440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang XH, Tee LY, Wang XG, et al Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther - Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hsu PD, Scott DA, Weinstein JA, et al DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cho SW, Kim S, Kim Y, et al Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim D, Bae S, Park J, et al Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12(3):237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 116.Kleinstiver BP, Pattanayak V, Prew MS, et al High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Slaymaker IM, Gao LY, Zetsche B, et al Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim D, Kim DE, Lee G, et al Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol. 2019;37(4):430–435. doi: 10.1038/s41587-019-0050-1. [DOI] [PubMed] [Google Scholar]
- 119.Jin S, Zong Y, Gao Q, et al Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science. 2019;364(6437):292–295. doi: 10.1126/science.aaw7166. [DOI] [PubMed] [Google Scholar]
- 120.Zuo EW, Sun YD, Wei W, et al Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364(6437):289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grünewald J, Zhou RH, Iyer S, et al CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat Biotechnol. 2019;37(9):1041–1048. doi: 10.1038/s41587-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grünewald J, Zhou RH, Garcia SP, et al Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569(7756):433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou CY, Sun YD, Yan R, et al Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571(7764):275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 124.Lee HK, Willi M, Miller SM, et al Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat Commun. 2018;9(1):4804. doi: 10.1038/s41467-018-07322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liang PP, Xie XW, Zhi SY, et al Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat Commun. 2019;10(1):67. doi: 10.1038/s41467-018-07988-z. [DOI] [PMC free article] [PubMed] [Google Scholar]