

Abstract
Gene regulatory divergence is thought to play a central role in determining human-specific traits. However, our ability to link divergent regulation to divergent phenotypes is limited. Here, we utilized human-chimpanzee hybrid induced pluripotent stem cells to study gene expression separating these species. The tetraploid hybrid cells allowed us to separate cis- from trans-regulatory effects, and to control for non-genetic confounding factors. We differentiated these cells into cranial neural crest cells (CNCCs), the primary cell type giving rise to the face. We discovered evidence of lineage-specific selection on the hedgehog signaling pathway, including a human-specific 6-fold down-regulation of EVC2 (LIMBIN), a key hedgehog gene. Inducing a similar down-regulation of EVC2 substantially reduced hedgehog signaling output. Mice and humans lacking functional EVC2 show striking phenotypic parallels to human-chimpanzee craniofacial differences, suggesting that the regulatory divergence of hedgehog signaling may have contributed to the unique craniofacial morphology of humans.
Introduction
Humans and their closest extant relatives, chimpanzees and bonobos, differ in many key morphological aspects. One of the most divergent anatomical regions between these groups is the craniofacial region; compared to other apes, humans have a retracted face, high braincase, and small jaws1. These changes have likely affected key aspects of human evolution, including brain expansion, feeding, and vocalization1. Thus, studying these morphological differences could illuminate the evolutionary processes that shaped human anatomy, and perhaps reveal the driving mechanisms behind human disorders associated with these changes.
Many of these anatomical changes are likely driven by divergent gene regulation2–4. However, very little is known about the regulatory differences that underlie human-specific morphology. Identifying such changes has been an elusive goal, since it is challenging to distinguish genetically-driven regulatory changes from those driven by differences in environment, cell-type composition, and batch effects. Particularly important are cis-regulatory changes, which are thought to underlie most morphological divergence5. However, distinguishing cis- from trans-regulatory changes between species is even more challenging, since it can only be achieved through hybridization5.
Interspecific hybrids have been a particularly powerful tool for studying cis-regulation5–10. In hybrid cells, both alleles experience the same environment, including trans-acting regulators. Therefore, any allele-specific expression (ASE) must be due to cis-regulatory changes between species, rather than trans- or environmental effects5–11. Thus, even without pinpointing the specific sequence that underlies ASE in a hybrid cell, one can conclude that it is cis-driven (epigenetic marks that are carried over from the parental cells to the hybrid could be an exception, but these are expected to be rare; see Methods).
Results
Generating hybrid cranial neural crest cells
To identify cis-regulatory divergence that separates humans and chimpanzees, we generated human-chimpanzee tetraploid hybrid cells. For details about hybrid generation see the accompanying report by Agoglia et al.12. Briefly, this was achieved by fusing human and chimpanzee induced pluripotent stem cells (iPSCs) using polyethylene glycol, resulting in hybrid cells where each nucleus contains the chromosomes of both species12. We generated three such lines from a male-male pair (hereafter, Hy1 lines) and two additional lines from a female-female pair (hereafter, Hy2 lines). PCR and karyotyping confirmed the presence of a full set of human and chimpanzee chromosomes that was stable over dozens of passages12.
To explore cis-regulatory divergence that may have contributed to human craniofacial evolution, we differentiated the iPSCs into cranial neural crest cells (CNCCs), which are the primary cell type that gives rise to craniofacial bones, cartilage, teeth and connective tissue, as well as epidermal melanocytes and cranial neurons and glia13. Specifically, we carried out three independent differentiations of one of the hybrid iPSC lines, as well as three independent differentiations of each of its parental lines, into mesenchymal CNCCs (Fig. 1a, Extended Data Fig. 1a,b, Methods). We then performed RNA-seq on the hybrid and parental iPSCs and CNCCs (two replicates for each of the iPSCs and three for each of the CNCCs). Together, the hybrid iPSCs and CNCCs provide a platform to explore divergent regulation in cell types representing two developmental stages.
Figure 1. Human-chimpanzee hybrid cells capture inter-specific cis-expression changes.
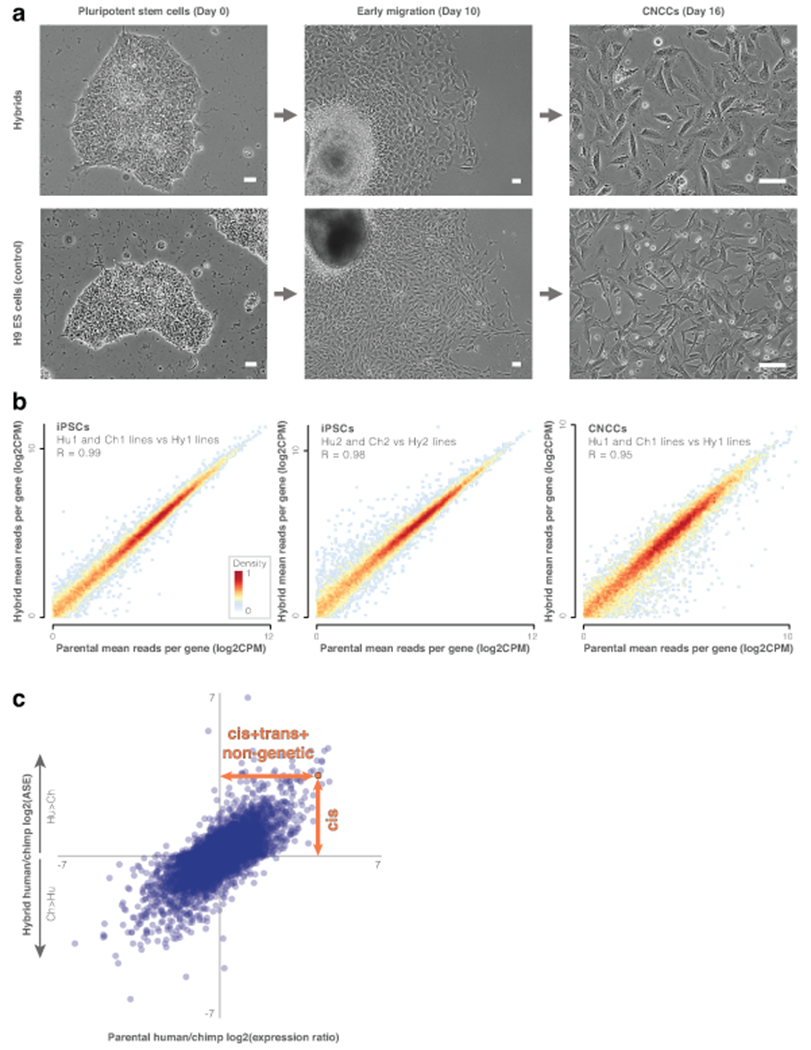
a. Phase contrast images of cranial neural crest cell (CNCC) derivation from human-chimpanzee hybrid iPSCs and positive control H9 human embryonic stem cells. Scale bars show 50μm. Three independent differentiations were conducted for each cell line. b. Heatmap of hybrid vs parental gene expression (e.g., mean expression of the three CNCC Hy1 samples vs the six CNCC Hu1 and Ch1 parental samples). Heatmaps show genes that are expressed in both (mean counts per million (CPM) > 1). The effect of tetraploidy on gene expression is likely minimal. c. Parental vs hybrid CNCC expression changes. Expression changes within the hybrid cells are driven by cis-regulatory changes (vertical orange arrow), while expression changes between the parental samples are driven by cis- and trans-regulatory changes and their combinatorial interaction, as well as by non-genetic factors, such as cell composition, environmental effects (e.g. response to cell culture), and batch effects (horizontal orange arrow). See Extended Data Fig. 1g for iPSCs. ASE, allele-specific expression.
In order to ensure that the tetraploid hybrid cells reflect diploid biology, we subjected them to several tests. First, we confirmed that tetraploidy did not affect differentiation by measuring the levels of iPSC and CNCC differentiation markers. We found that both hybrid cell types stably express their respective markers (Extended Data Fig. 1b; see Agoglia et al.12 for iPSC validation). Next, we compared gene expression between parental and hybrid cells to test if tetraploidy affected global gene expression levels. Specifically, if ploidy substantially impacts expression then we would expect the diploid parental lines to be more similar to one another than either is to the tetraploid hybrid cells. However, we observed the opposite: hybrid gene expression is highly correlated with both parents, even more than the parents are correlated with one another, and is similar to the mean of its two parents (Fig. 1b,c, Extended Data Fig. 1c, Supplementary Tables 1–3). In support of this, hybrid gene expression falls between the two parents in principal components analysis12. This modest effect of ploidy is perhaps not surprising, considering that although tetraploidy is not usually tolerated at the organismal level, it frequently occurs mosaically in vivo in many tissues14. Together, these results suggest that hybrid tetraploidy does not drastically affect expression patterns. Reproducibility between hybrid lines (Hy1 and Hy2) was also high, both at the level of expression (R = 0.97) and ASE (R = 0.90, Supplementary Tables 3–4). Finally, although tetraploid cells typically maintain their DNA content in culture15–17, aneuploidies are possible. However, we found no evidence of aneuploidy in the CNCCs. In the iPSCs, we identified chromosome 20 aneuploidy in three of the samples12 (a common aneuploidy in cultured iPSCs18). We therefore removed this chromosome from all analyses.
Identifying allele-specific expression
Next, we set out to analyze ASE between the species. To distinguish between human and chimpanzee alleles, we only retained reads that overlap genomic positions where human and chimpanzee sequences differ (48% of reads, covering 98% of expressed genes in iPSCs and 95% in CNCCs, Supplementary Tables 1–2). To minimize false signals of allelic imbalance, we (1) discarded reads that show mapping bias19, (2) compared only orthologous genes, and (3) required that genes show similar ASE when mapping to both the human and chimpanzee genomes (Extended Data Fig. 1d,e, Methods). Finally, we used DEseq2 to identify ASE20. We applied the same pipeline to parental lines to enable direct comparisons between samples.
We identified 6,009 genes with significant ASE (q-value < 0.05) in the hybrid iPSCs, of which 3,010 are up-regulated (hereafter Hu>Ch genes) and 2,999 are down-regulated in humans compared to chimpanzees (hereafter, Ch>Hu genes). In the hybrid CNCCs, we found 1,815 Hu>Ch genes and 1,797 Ch>Hu genes (Supplementary Table 5, Extended Data Fig. 1f,g). We also found that cis-regulation drives 49% and 40% of the overall expression change in iPSCs and CNCCs, respectively (Methods). This is higher than the cis-contribution estimates of human polymorphisms (12-37%), in agreement with previous reports of increased cis-contribution in comparisons between species (24-64%)5,21,22.
To investigate the extent to which ASE is associated with other types of regulatory divergence, we analyzed 28 datasets related to human-chimpanzee divergence in DNA sequence23,24,33,34,25–32, transcription factor binding35, DNA methylation36, chromatin accessibility37–40, 3D chromosomal interactions41,42, and histone modifications43. We found that ASE in the hybrid cells overlaps significantly with many different metrics of sequence and chromatin divergence (Supplementary Tables 6–8, Extended Data Fig. 2a).
Divergent expression is linked to divergent phenotypes
To date, thousands of loci with divergent regulation between humans and chimpanzees have been identified, and hundreds of divergent phenotypes have been described43–45. However, how these phenotypes are linked to these divergent loci remains largely unknown44. To bridge this gap, we investigated whether differentially expressed genes tend to be linked to divergent traits. We focused on the skeletal system, because of its highly divergent and uniquely defining features in humans, especially in the face1.
First, we examined whether ASE genes tend to affect some anatomical regions more than others. We used Gene ORGANizer, which utilizes phenotypes observed in Mendelian disorders to link genes to the body parts they affect, and then tests whether the examined group of genes is linked to some body parts more than expected by chance46. While controlling for cell type-specific expression (Methods), we found several significant body parts. These include the vocal tract, skull, face, joints and pelvis. Interestingly, these body parts are among the most phenotypically divergent regions between humans and chimpanzees1. We found the strongest enrichment within the voice box (larynx), with almost twice as many Ch>Hu than Hu>Ch genes linked to it (48 Ch>Hu vs. 25 Hu>Ch in CNCCs, false discovery rate (FDR) = 0.013, Fisher’s exact test), followed by the upper and lower jaws (1.23x and 1.22x, CNCCs, FDR = 0.017 and FDR = 0.036, respectively; Fig. 2a; Supplementary Tables 9–12). These results add to our previous findings that genes affecting the larynx and face became extensively hypermethylated in recent human evolution, and that this down-regulation might have contributed to the unique facial and vocal tract anatomy in modern humans36.
Figure 2. More divergent expression is more tightly associated with divergent traits.
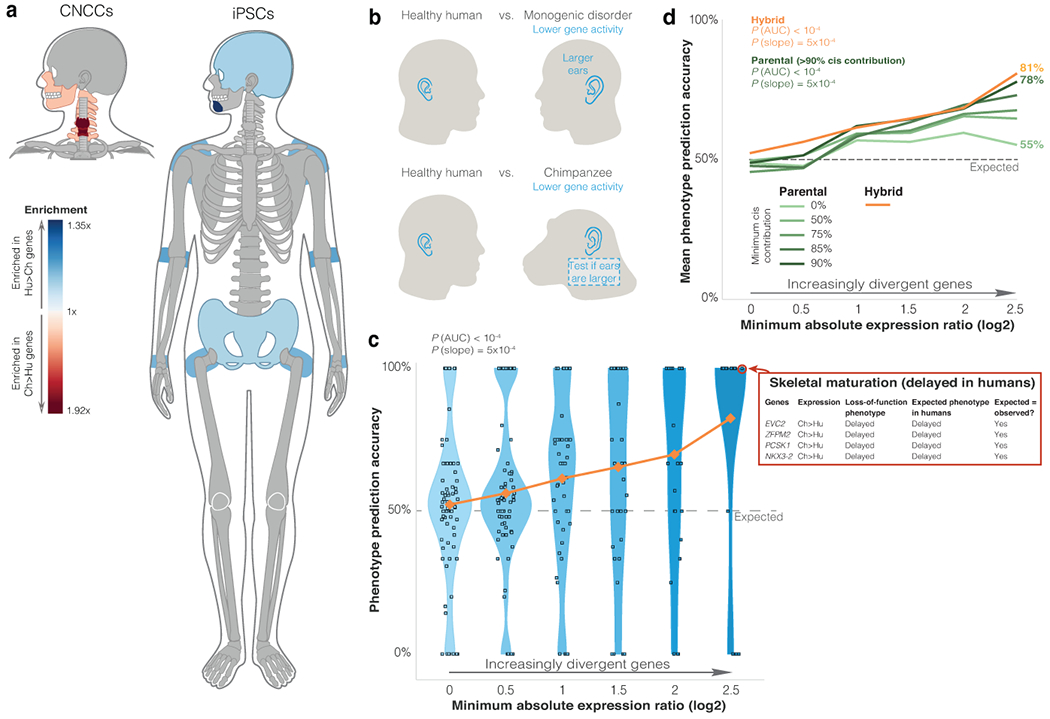
a. Gene ORGANizer output of significantly enriched body parts (FDR < 0.05, one-sided hypergeometric test) within ASE genes in the hybrid cells. b. Workflow of linking expression changes to potential phenotypic effects. We used phenotypes in monogenic disorders as indicators of expected phenotypic direction when relevant genes are down-regulated. Then, we predicted this direction for the lineage with lower expression. Finally, we tested whether predicted phenotypes match known human-chimpanzee phenotypic differences. c. Phenotype prediction accuracy among genes with increasingly divergent ASE. Correct predictions are cases where the phenotype assigned to a gene based on its ASE matches the known phenotype between humans and chimpanzees (see skeletal maturation example). If a gene is not related to phenotypic divergence, there is a 50% likelihood that the phenotype assigned to the gene based on its ASE would match the human-chimpanzee phenotypic difference. Each phenotype is represented as a square. Y-axis shows for each phenotype the fraction of genes whose prediction was correct. Horizontal distribution of squares within each bin is for display purposes only. Orange shows mean accuracy. Randomization test P-values are shown for overall accuracy compared to random (PAUC), and accuracy increase compared to random (Pslope). d. Mean phenotype prediction accuracy in groups of genes with increasingly more divergent expression. Orange shows mean accuracy in hybrid cells (from panel c). Green shows mean accuracy for differentially expressed genes with various cis-contribution thresholds in the parental samples. P-values were computed as in panel c.
Next, we delved into the specific phenotypes associated with ASE genes. To this end, we used the Human Phenotype Ontology (HPO) database, where genes are linked to phenotypes based on the Mendelian disorders they underlie47. Most of these disorders are caused by loss-of-function of one or both gene copies, and could therefore provide a clue as to the direction of phenotypic change when gene activity decreases. We found five significantly over-represented phenotypes: forehead width, chin width, nasal bridge width, distance between the eyes, and skull length compared to width (FDR < 0.05, hypergeometric test, Supplementary Table 13). Interestingly, all five phenotypes are divergent between humans and chimpanzees, and in four out of these five phenotypes, the direction of phenotypic change in the species with the lower expression is the direction of the phenotypic change in human patients with loss-of-function. For example, genes whose loss-of-function results in a wider nasal bridge tend to be down-regulated in humans, which is consistent with humans having a wider nasal bridge compared to chimpanzees.
To test the link between divergent genes and divergent phenotypes more systematically, we used our previously published phenotype directionality prediction approach48. This approach is based on two hypotheses: 1) substantial regulatory changes are more likely to result in phenotypic changes than small regulatory changes, and 2) the direction of phenotypic change associated with down-regulation is expected to be the direction of phenotypic change associated with loss-of-function. More specifically, each differentially expressed gene was first linked to its HPO phenotypes47. Then, the phenotype of the disorder (e.g., larger ears) was predicted to occur in the lineage exhibiting the lower expression (e.g., chimpanzee). Next, each of these phenotypes was examined against known human-chimpanzee skeletal phenotypes to determine if their directions match (e.g., if chimpanzees have larger ears, representing a correct phenotype prediction, Supplementary Tables 14–15, Fig. 2b). Lastly, we computed overall accuracy by examining for each phenotype the fraction of linked genes with a correct phenotype prediction.
We began by applying the phenotype directionality prediction approach to subsets of genes with increasingly more extreme ASE. We found that: (1) phenotypes linked to genes with more extreme ASE are more likely to be divergent between humans and chimpanzees, and (2) more extreme ASE is more likely to correctly predict the direction of phenotypic change. These gene-phenotype associations are significantly stronger than expected by chance, both in their overall accuracy (PAUC < 10−4, randomization test) and the improvement in accuracy with more divergent ASE (Pslope = 5x10−4, Fig. 2c, Extended Data Fig. 2b–d). Within the most divergent genes, 100% of linked traits are divergent, and these genes are 4.3x more likely to be associated with the correct, rather than incorrect, phenotypic direction (i.e., 81% accuracy).
Next, we compared the phenotypic prediction accuracy of ASE compared to parental differential expression. We found that ASE is more strongly associated with phenotypic divergence than is parental differential expression (81% accuracy for ASE vs 55% for parental, genes with ≥ 2.5 log2(fold-change), Fig. 2d, Extended Data Fig. 2c). However, divergent parental expression becomes more tightly linked with divergent phenotypes when taking into consideration cis-contribution; genes with higher cis-contribution to their differential expression are more tightly linked to divergent phenotypes (Fig. 2d). These observations could be due to the fact that hybrid ASE is solely cis-regulatory, or alternatively, that it controls for confounding factors such as environmental and batch effects. In summary, we propose that (1) genes with more extreme expression changes are more likely to be associated with divergent traits; and (2) using ASE data from hybrid cells improves the ability to infer phenotypic information from differential expression.
Hedgehog signaling shows evidence of selection
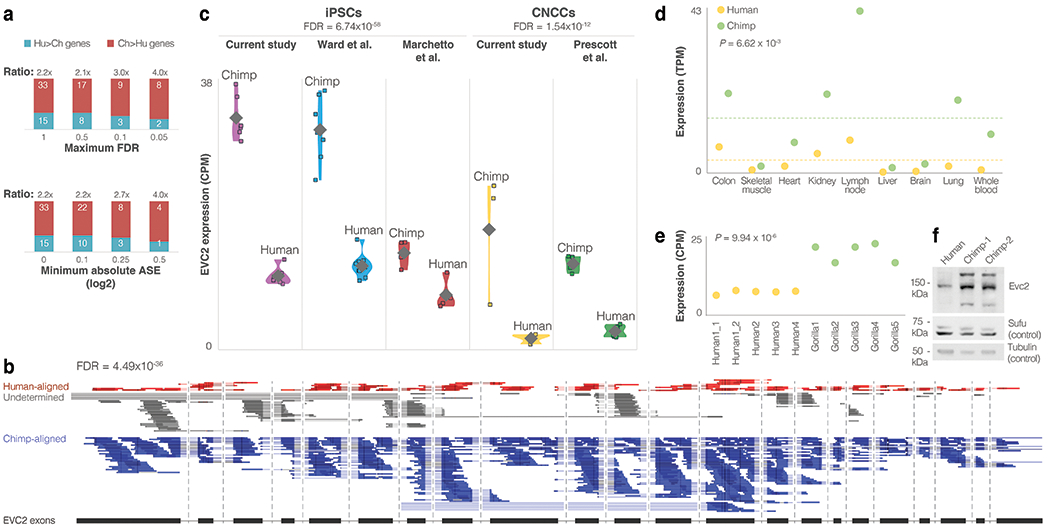
After exploring the links between single genes and phenotypes, we turned to analyze the pathway level. Our genome-wide catalog of cis-regulatory divergence allowed us to apply a test of lineage-specific selection known as the sign test9,49. In this test, we search for gene sets (such as pathways) that show an excess of cis-regulatory changes in one direction (e.g., an excess of genes with higher expression of the human alleles). If any pathway deviates significantly from the random expectation of a roughly equal number of independent up- and down-regulatory changes, then the null hypothesis of neutrality can be rejected in favor of polygenic selection9,49. Performing this test on the 134 pathways in KEGG50, we found three pathways that show significant deviation from neutrality. The strongest imbalance was observed in the hedgehog (Hh) signaling pathway, a key regulator of skeletal patterning51, with more than twice as many down- as up-regulated genes (33 Ch>Hu vs 15 Hu>Ch genes in CNCCs, FDR = 0.03, binomial test, Fig. 3a, Supplementary Tables 16–17). A similar down:up skew is observed when upstream regulators of Hh ligand production52 are included (54:27 down:up, P = 2.6x10−3), and becomes more pronounced with increasingly more stringent thresholds (Extended Data Fig. 3a). We found a similar pattern at the level of translation, with 23 of the 34 Hh-related mRNAs with translation rate data53 having lower translation levels in human compared to chimpanzee lymphoblastoid cells (P = 0.025, binomial test). These results suggest that the cis-regulation of Hh pathway genes has likely been subject to differential selection in the human vs. chimpanzee lineage. The preponderance of human down-regulation was present among both positive and negative regulators of Hh signaling, suggesting that the effects may be more complex than simply reducing Hh signaling across the many cell types where it functions.
Figure 3. EVC2 down-regulation is likely to have reduced Hh signaling output in humans.
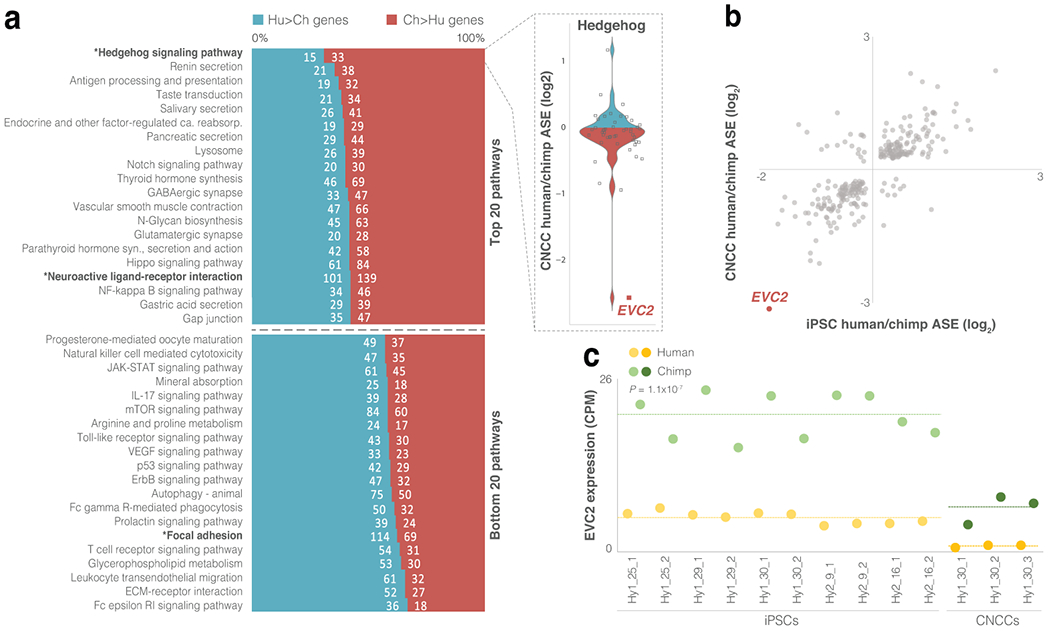
a. For each KEGG pathway, the ratio of Hu>Ch to Ch>Hu genes was tested. Asterisks mark pathways with FDR < 0.05 (binomial test). b. Chimpanzee to human expression ratio in hybrid iPSCs and CNCCs for skeleton-related genes that are differentially expressed in both cell types. EVC2 is the most down-regulated gene in humans compared to chimpanzees. c. EVC2 expression across all hybrid cell samples showing a ~4-fold mean decrease in human compared to chimpanzee in iPSCs and a ~6-fold mean decrease in CNCCs. Dashed line shows mean expression. Paired t-test P-values are shown.
To gain further insight into the underlying regulatory divergence of Hh genes, we explored human and chimpanzee CNCC chromatin accessibility (ATAC-seq) data43. In each Hh gene, we compared the ratio of ATAC-seq peaks between the species to the ratio of expression change and found that increased expression in a species is associated with an increased number of ATAC-seq peaks in that species (Pearson’s R = 0.56 and P = 3.11x10−5). This is consistent with species-specific chromatin accessibility contributing to the divergence of Hh genes.
The role of EVC2 in human craniofacial morphology
Interestingly, the skeleton-related gene with the strongest cis-acting down-regulation in humans, EVC2, is part of the Hh pathway (Fig. 3b). Considering the strong link that we found between ASE and phenotypic divergence (Fig. 2), as well as the likely lineage-specific selection on Hh signaling, this gene was a promising candidate for further investigation. EVC2 (also known as LIMBIN) is a transmembrane protein that forms a complex with EVC at the base of the primary cilia. The EVC-EVC2 complex functions as a scaffold to directly bind and facilitate signaling by Smoothened (SMO), the protein that transmits the Hh signal across the membrane in all metazoans54. Loss of EVC2 was shown to reduce Hh signaling in mice by 40-60%55. We present our investigation of EVC2 in three parts: 1) its expression divergence in humans; 2) the effects this divergence may have on Hh signaling; and 3) the effects this divergence may have on craniofacial phenotypes.
Compared to the levels of the chimpanzee EVC2 alleles in the hybrid cells, the human alleles are expressed at only 17% in CNCCs (FDR = 2.1x10−36) and 27% in iPSCs (2.8x10−69, Fig. 3c, Extended Data Fig. 3b). This pattern is consistent across all hybrid cells (P = 1.1x10−7, paired t-test, Fig. 3c). The hybrid and parental samples show similar human down-regulation of EVC2 (19% and 39% of the chimpanzee levels in the parental CNCCs and iPSCs, FDR = 2.7x10−12 and 1.1x10−56, respectively, Extended Data Fig. 3c), suggesting that EVC2 down-regulation is mainly driven by cis-regulatory changes. Additionally, EVC2 is the only Hh gene that is detectable by ribosome profiling in chimpanzee but not in human lymphoblastoid cells53. We also examined whether other tissues show a similar pattern of EVC2 down-regulation. We found that across all nine tissues in which data for both species are available56, EVC2 is down-regulated in humans, ranging from only 4% of the chimpanzee expression level in whole blood to 38% in colon, with a mean of 17% (P = 0.012, t-test, Supplementary Tables 18–19, Extended Data Fig. 3d). To identify the lineage in which the differential expression emerged, we examined gorilla iPSC expression data57. We found that across five human and five gorilla samples, EVC2 is expressed at significantly lower levels in humans, with similar ratios to the ones observed between human and chimpanzee iPSCs (mean = 35%, P = 9.9x10−6, t-test, Extended Data Fig. 3e). This suggests that the cis-regulatory down-regulation of EVC2 likely emerged in the human lineage.
To measure EVC2 protein abundance in primary samples, rather than in in vitro differentiated cells, we obtained human and chimpanzee dental pulp stem cells (DPSCs). These primary cells develop from CNCCs and are central in the formation of teeth. Both EVC2 and Hh signaling play key roles in dental development, and dental abnormalities are a hallmark of EVC2 loss-of-function. Consistent with our prior protein and RNA measurements in cell lines, the abundance of EVC2 protein in human DPSCs is only 29% of that in chimpanzee DPSCs (P = 0.02, t-test, Fig. 4a, Extended Data Fig. 3f).
Figure 4. EVC2 down-regulation in humans is likely to have reduced Hh signaling output.
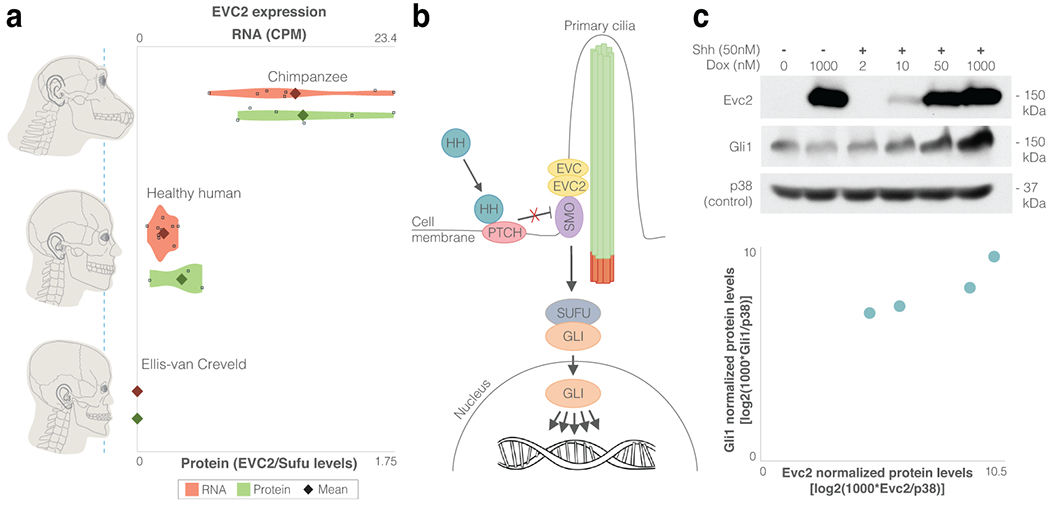
a. EVC2 RNA expression levels (parental CNCCs from the current study and from Prescott et al.43) and protein expression levels (DPSCs from the current study). For EVC2 loss-of-function (Ellis-van Creveld patients), functional RNA and protein levels are presented. b. HH ligands bind and inhibit their receptor (PTCH), thereby allowing SMO to accumulate in primary cilia and engage the EVC-EVC2 complex at the cilia base. The EVC-EVC2 complex acts as a scaffold to facilitate SMO signaling to the GLI family of transcription factors. c. GLI1, the product of a direct Hh target gene, was measured across four induction levels by immunoblotting as a metric of signaling strength induced by the ligand Sonic Hedgehog (Shh) in cells expressing different levels of EVC2 protein. The samples derive from the same experiment and blots were processed in parallel.
Finally, using chromatin accessibility and transcription factor binding data from CNCCs43, we identified regions within intron 6 and intron 19 that show higher chimpanzee accessibility and transcription factor binding compared to human. We tested these sequences using a reporter assay and found that in both introns, the human allele drove weaker expression (P = 8.2x10−4, P = 9.4x10−4, t-test, Extended Data Fig.4, Methods).
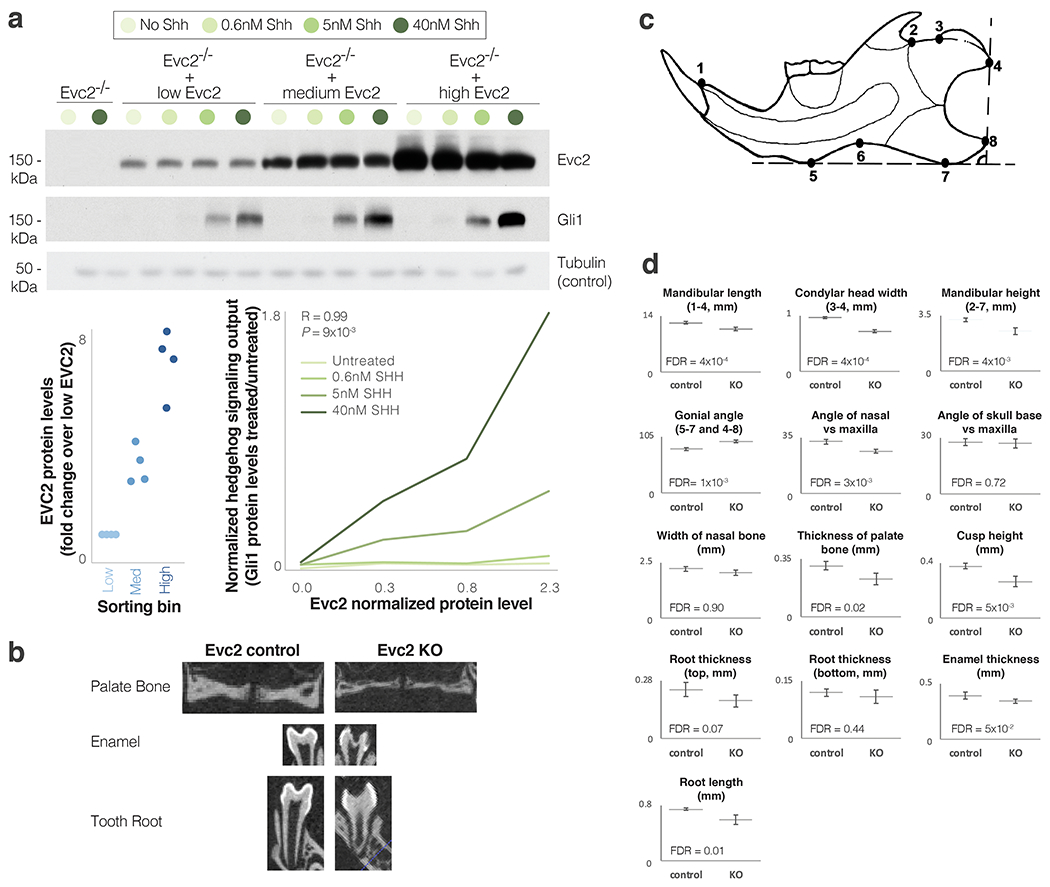
As classical morphogens, Hh ligands are known to pattern tissues by signaling in a graded fashion (Fig. 4b). Alterations in Hh signaling can result in markedly different developmental outcomes58,59, and have been implicated in CNCC survival, differentiation and proliferation, as well as in various craniofacial disorders51,60–62. Following evidence in mice that Evc2 loss-of-function results in reduction in Hh signaling output55, we sought to test the link between EVC2 protein levels and Hh signaling output. We stably introduced a cDNA encoding Evc2 fused to Yellow Fluorescent Protein (YFP) into Evc2−/− mouse NIH/3T3 fibroblast cells using retroviral infection and divided them into three groups based on their Evc2-YFP expression levels (low, medium and high). Importantly, the low-to-high ratio of Evc2-YFP expression (12%) is close to the human-to-chimpanzee ratio observed in CNCCs (17%). We found that when EVC2 is expressed at 12% of its maximum level, Hh signaling output is reduced by 3.7-fold (Extended Data Fig. 5a). To further test the link between EVC2 levels and Hh signaling output, we generated an NIH/3T3 cell line where Evc2 expression could be induced to different levels by exposing them to different concentrations of doxycycline (Dox). Again, we observed that increasing Evc2 expression increased the strength of Hh signaling, with maximum induction of Evc2 expression leading to a 6.2-fold increase in expression of the Hh target gene Gli1 (Fig. 4c).
Finally, we turned to investigate the potential phenotypic effects of EVC2 down-regulation. Specifically, we sought to examine to what extent EVC2 loss-of-function phenotypes resemble human-chimpanzee divergent phenotypes. To do so, we generated CNCC-specific Evc2 knockout (KO) mice (Evc2fx/fx;Wnt1-Cre, Methods) and measured their craniofacial phenotypes using microCT at postnatal day P28. For each known human-chimpanzee divergent phenotype, we tested whether it appears in Evc2 KO mice, and in what direction compared to control (Evc2fx/+;Wnt1-Cre) mice. We measured 13 phenotypes, and combined this with previous Evc2 KO measurements63–65. We found that 14 out of 16 phenotypes show the same directionality between control and KO mice as they do between chimpanzees and humans (88% compared to 50% expected by chance, P = 4.2x10−3, binomial test, Fig. 5, Extended Data Fig. 5b–d, Supplementary Table 20). In other words, Evc2 KO mice phenotypes resemble human phenotypes, including our retracted face.
Figure 5. Phenotypes driven by EVC2 KO are observed between humans and chimpanzees.
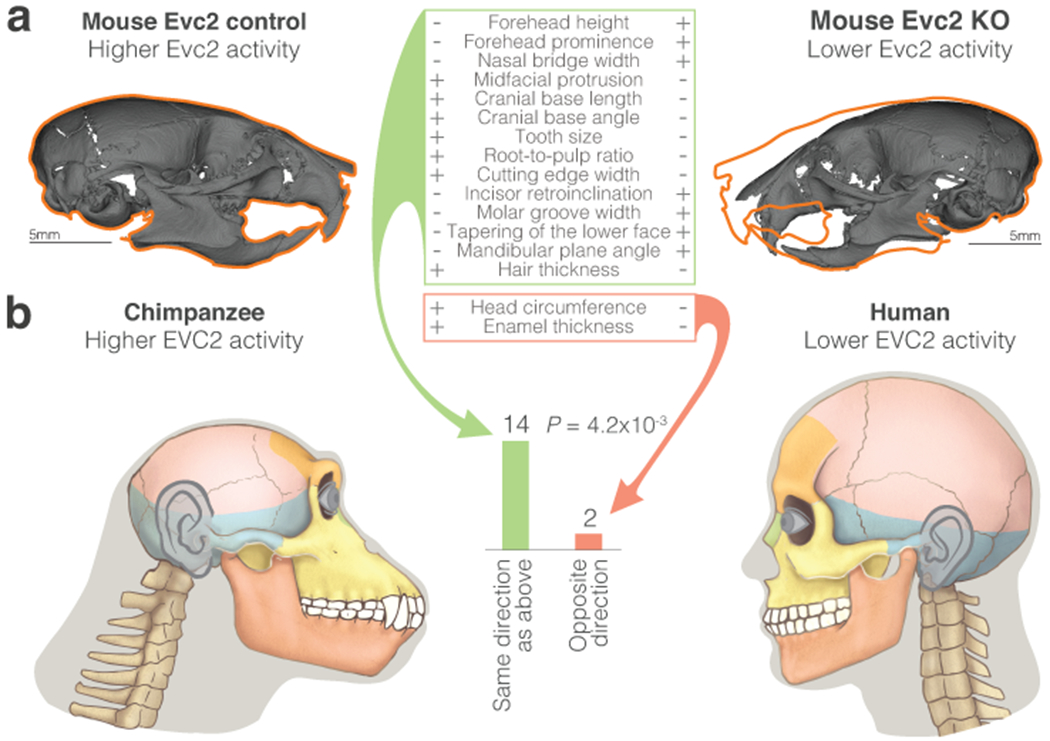
a. Micro-CT models of the control and Evc2 KO mice at day P28. Orange outline shows Evc2 control silhouette. Table shows the directional craniofacial phenotypes that differ between Evc2 KO and control mice, and their respective state in control and KO mice. +/− represent increased/decreased phenotype, respectively. b. The number of phenotypes that show the same directionality between Evc2 KO and control mice as they do between humans and chimpanzees. Two-sided binomial test P-values are shown. Phenotypic differences in Evc2 control vs KO mice resemble phenotypic differences in chimpanzee vs human.
Studies in cattle and mice have shown that the role of EVC2 is conserved in these mammals55,63–68. In humans, homozygous loss-of-function mutations in either EVC2 or EVC cause the Ellis-van Creveld syndrome. Heterozygous truncation of EVC2 (which also inhibit Hh signaling69) lead to the milder, autosomal dominant Weyers Acrofacial Dysostosis syndrome47. The phenotypes of these ciliopathies are mainly skeletal and integumentary, and include (but are not limited to) dental anomalies, retracted midface, high forehead, and nail dysplasia47. SNPs in EVC2 have been associated with milder craniofacial phenotypes70,71. Together, this suggests that the extent of phenotypic change is dependent on the level of EVC2 activity. Next, we examined EVC2 loss-of-function phenotypes in humans. To investigate the link between EVC2 down-regulation and human-chimpanzee divergent phenotypes, we tested whether EVC2 loss-of-function phenotypes resemble craniofacial phenotypes that differ between humans and chimpanzees. For each phenotype in healthy humans vs patients, we examined if it is also divergent between chimpanzees and humans, and whether the direction of divergence matches as well. We found that 25 out of 27 phenotypes (93%) are known to be divergent between humans and chimpanzees. The direction of 23 of the divergent traits (92%) matches human-chimpanzee morphology, compared to 50% expected by chance (P = 1.9x10−5, binomial test, Fig. 6, Supplementary Tables 21–23). Importantly, the key phenotypes that are often used to describe pronounced differences in facial shape between humans and chimpanzees (specifically, midfacial retrusion with a more downward facial trajectory1) are observed in both the current and previous Evc2 KO studies64,65,68, as well as in Ellis-van Creveld patients47,65 (Fig. 6, Extended Data Fig. 5d). Moreover, we found that 24 out of 25 craniofacial phenotypes are human-derived, consistent with the expectation that a gene expression change specific to the human lineage should result in phenotypic changes specific to the human lineage.
Figure 6. Phenotypes driven by reduced levels of functional EVC2 are observed between humans and chimpanzees.
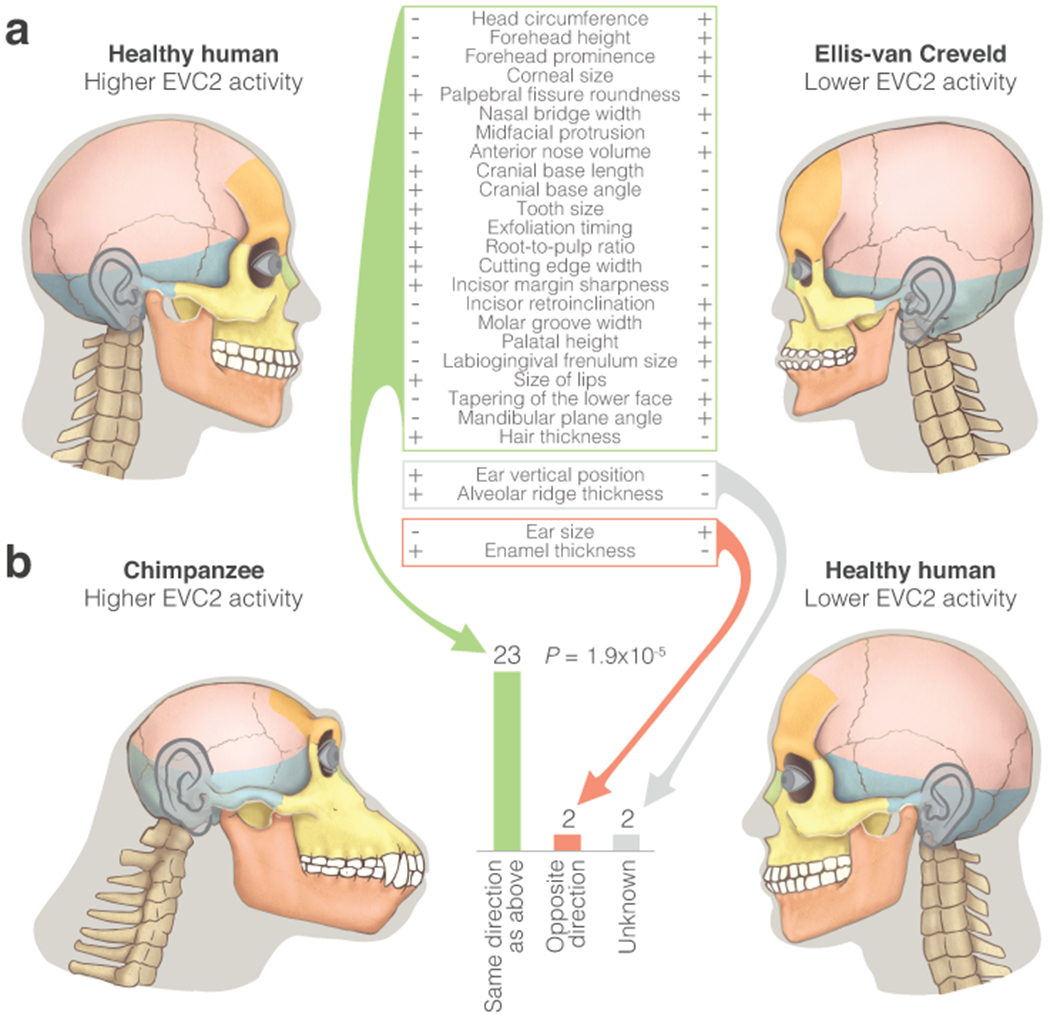
a. Directional craniofacial phenotypes in EVC2 loss-of-function, and their respective state in the syndrome and in healthy individuals. b. The number of phenotypes that show the same directionality between healthy and Ellis-van Creveld syndrome individuals as they do between humans and chimpanzees. Phenotypic differences between healthy and Ellis-van Creveld syndrome individuals resemble phenotypic differences between chimpanzees and humans. Two-sided binomial test P-values are shown. See Supplementary Table 22 for non-craniofacial phenotypes.
In summary, we report EVC2 as the most divergent skeleton-related gene between humans and chimpanzees in iPSCs and CNCCs. This gene is also part of the pathway with the strongest cis-acting down-regulation in humans. The down-regulation of EVC2 is observed across many samples and tissues, at the RNA as well as protein level, is driven mainly by cis changes, and has likely arisen along the human lineage. Inducing EVC2 down-regulation results in diminished Hh signaling output, which in turn is known to affect craniofacial morphology. Indeed, phenotypes driven by EVC2 loss-of-function resemble phenotypes distinguishing humans from chimpanzees. We propose that this process may have contributed to human-specific craniofacial morphology.
Discussion
Various mechanisms are known to generate midfacial retraction in vertebrates. In humans, this retraction is driven predominantly by early cessation of growth in the cartilaginous joints of cranial base bones. This leads to a shortened cranial base, which in turn drives midfacial retraction72. Interestingly, EVC2 plays a key role in the development of these cartilaginous joints61,68. Indeed, Evc2 loss in mouse CNCCs causes early cessation of growth in the cranial base joints, leading to a shortened cranial base and a retracted midface. Likewise, although various Hh signaling disorders show phenotypes that are similar to human-chimpanzee divergent phenotypes, Ellis-van Creveld syndrome exhibits the most similar phenotypes61. Thus, at the phenotypic as well as the mechanistic level, EVC2 loss-of-function shows a striking resemblance to human-specific craniofacial development.
Altered Hh signaling was suggested to play a role in the skeletal diversification of several species, including canids73, cichlids74, and cormorants75. Hh signaling may represent a recurrent target of selection because its dosage-dependent effects allow fine-tuning of morphology. Indeed, the effect of CNCC Hh signaling on facial development was shown to be dosage-dependent, with loss leading to undergrowth and over-activation leading to overgrowth62. Protein sequence divergence may also contribute, and in fact the Hh ligand Sonic Hedgehog went through rapid sequence evolution along the primate lineages leading to humans76.
One of the main motivations of this work was to shed light on genes that could underlie human-specific traits. We used a phenotype directionality prediction approach48 to link regulatory to phenotypic divergence via comparisons to phenotypes in Mendelian disorders48. The use of disease phenotypes as a platform to infer the morphological effects of genes is supported by the observation that genes that underlie disorders tend to underlie morphological variation within humans, as well as between humans and chimpanzees77.
We have also found that genes known to affect the larynx (voice box) are the most enriched for down-regulation in humans. This adds to recent evidence of down-regulation of larynx-affecting genes in humans: we have previously reported that in anatomically modern humans, the most extensive hypermethylation emerged in larynx-affecting genes36. In fact, while less than 2% of genes in the genome are known to affect the larynx, all of the top five hypermethylated genes are larynx-affecting36. Additionally, hypoplasia of the epiglottis (the cartilaginous lid of the larynx) is the phenotype most significantly associated with down-regulated CNCC enhancer marks in humans compared to chimpanzees43. Interestingly, the laryngeal structure and position are particularly divergent in humans. The effect of these anatomical changes on vocalization has been debated for decades, with studies focusing almost exclusively on vocal tract anatomy1,78,79. These new genetic findings now provide an opportunity to begin to elucidate the genetic evolutionary forces that shaped our vocal tract.
We have shown here that a major challenge in genetics – associating divergent gene expression with divergent phenotypes – can be tackled through the use of hybrid cells and loss-of-function phenotypic data. Looking ahead, this strategy could be applied to a wide range of traits and species to uncover genes underlying species divergence.
Online Methods
See accompanying Agoglia et al. work12 for hybrid iPSC generation. In short, cells were labelled with diffusible dyes (Human iPSCs: CellTracker Deep Red, 1.5 μM in DPBS, Thermo Fisher Scientific, C34565, Chimp iPS cells: CellTracker Green CMFDA). Polyethylene glycol 1500 (PEG, Sigma-Aldrich, 10783641001) was used to fuse human and chimpanzee iPSCs, resulting in tetraploid hybrid cells where each nucleus contains the chromosomes of both species. Cells were dissociated, and cells positive for both Deep Red and Green CMFDA dyes and negative for DAPI were sorted. We generated three such tetraploid hybrid iPSC lines from a male-male pair and two additional lines from a female-female pair. PCR and karyotyping confirmed the presence of a full set of human and chimpanzee chromosomes that was stable over dozens of passages12.
Ethics statement
Approval for the derivation of human induced pluripotent stem (iPS) cell lines used in this study was granted by the University of Chicago Institutional Review Board, protocol 11-0524. Human donors in this study consented to the use of their cells (fibroblasts) to generate iPS cells for studies of evolution and cross-species comparisons, and to the generation of other cell types that would be derived from these iPS cells. Donors consented to the deposition of any resulting data from the study onto the Gene Expression Omnibus (GEO). Generation of hybrid iPSCs was approved by the Stanford Stem Cell Research Oversight committee (protocol 534). The experiments described in this manuscript were additionally reviewed by an anonymous reviewer with expertise in ethics.
We note that these tetraploid cells are not approved for use in vivo or for attempting to generate an organism (which biologically is unlikely even possible). We recommend that all future applications of these cells occur in close consultation with bioethicists.
CNCC differentiation
iPSC culture
Human (derived from the H20961 sample, hereinafter, Hu1), chimpanzee (derived from the C3649 sample, hereinafter, Ch1) and human-Chimpanzee hybrid (Hy1_30) induced pluripotent stem cells (iPSCs) lines as well as human embryonic stem cells (hESC) (H9 line) were cultured in in feeder-free, serum-free mTESR-1 medium (StemCell technologies). Pluripotent stem cells (PSCs) were regularly passaged ~1:6 every 5–6 days. For passaging, iPSCs were incubated in ReLeSR (StemCell technologies) for 1 min followed by aspiration, and incubating the culture plates for 6-7 mins at 37°C. mTESR-1 medium was added to the culture plates and plates were gently tapped to detach the cells, which were then re-plated on tissue culture dishes coated with growth-factor-reduced Matrigel (BD Biosciences).
CNCC derivation and culture
The population of cells used in this study are mesenchymal CNCCs which have been delaminated from the neuroepithelial spheres. Three independent CNCC differentiation experiments were performed to generate these cells. In each one, human-chimpanzee hybrid iPSC (Hy1_30), parental human (Hu1), parental chimpanzee (Ch1) iPSC lines, and human embryonic stem cells (hESCs, control) were differentiated into CNCCs, as previously described43,81. Briefly, iPSCs and hESCs were incubated with 2mg/ml collagenase for ~30-50 min leading to detachment of colonies. Detached cells were plated as clusters of 100-200 cells in low-attachment petri dishes and cultured in the presence of CNCC differentiation medium consisting of 1:1 Neurobasal medium/DMEM F-12 medium (ThermoFisher Scientific), 0.5× B-27 supplement with Vitamin A (50× stock, GeminiBio), 0.5× N-2 supplement (100× stock, GeminiBio), 20 ng/ml bFGF (Peprotech), 20 ng/ml EGF (Sigma-Aldrich), 5 μg/ml bovine insulin (Sigma-Aldrich) and 1× Glutamax-I supplement (100× stock, ThermoFisher Scientific). Cells grown in CNCC differentiation medium grew as neural spheres/rosettes. For the first four days of differentiation, spheres were separated from cell debris by gentle centrifugation and re-plated into new petri dishes in fresh CNCC differentiation medium. After four days, the neural spheres were allowed to settle for three days to promote attachment to the culture plate surface. After the neural spheres began to attach to the plate, media was changed daily, and neural crest cells were allowed to migrate out of the neural rosettes for 4-5 days. Afterwards, neuroectodermal spheres were manually picked and removed from the culture dishes leaving behind emigrated neural crest cells, which were dissociated with 1x Accutase and passaged onto fibronectin (7.5μg/ml) (ThermoFisher Scientific) coated plates. The early migratory CNCCs were cultured in the presence of maintenance medium comprising of 1:1 Neurobasal medium/DMEM F-12 medium (Invitrogen), 0.5× B-27 supplement with Vitamin A (50× stock, GeminiBio), 0.5× N-2 supplement (100× stock, GeminiBio), 20 ng/ml bFGF (Peprotech), 20 ng/ml EGF (Sigma-Aldrich), 1 mg/ml bovine serum albumin, serum replacement grade (Gemini Bio-Products # 700-104P) and 1× Glutamax-I supplement (100× stock, ThermoFisher Scientific). The CNCCs were cultured on fibronectin coated dishes, with passaging every three days with 1x Accutase for additional two passages. Afterwards, medium was changed to BMP/ChIR medium by adding 3μM ChIRON 99021 (Selleck, CHIR-99021) and 50pg/ml BMP2 (Peprotech) to the maintenance medium, which increased cell proliferation and decreased migration.
Immunocytochemistry
Immunocytochemistry was performed as described previously82. Briefly, cells were fixed in 4% paraformaldehyde for 10 min RT followed by permeabilization with 0.1% triton X-100 in PBS for 15 mins. Cells were then blocked with blocking buffer (1% BSA/0.01% triton X-100) for 1 hr at RT and incubated with two primary antibodies: goat anti-human PAX3 (1:100; 4°C overnight; Santa Cruz, sc-34916), and mouse anti-human NR2F1 (1:100; 4°C overnight; Perseus Proteomics, PP-H8132-00) diluted in blocking buffer. Subsequently, cells were incubated with anti-mouse or anti-goat Alexa Fluor 488 antibodies (1:400; 1 hour at RT; Invitrogen) diluted in blocking buffer and counter-stained with DAPI nuclear dye (0.5 μg/ml in PBS; 10 min; Sigma). Cells that were incubated with secondary antibodies alone served as negative controls.
CNCC RNA isolation and preparation of RNA-seq libraries
~4x106 CNCCs from each sample in each of the three independent CNCC differentiation experiments were lysed at passage 4 of CNCC differentiation using Trizol reagent (Invitrogen) and total RNA was isolated as per manufacturer’s protocol.
RNA sequencing
RNA quality was assessed using the Agilent Bioanalyzer RNA Pico assay. All samples had an RIN greater than or equal to 8.0. From each sample, 100ng-1ug of total RNA was used for library preparation using the Illumina TruSeq Stranded mRNA kit. Libraries were prepared according to the manufacturer’s instructions. Samples were barcoded with Illumina dual-index adapters. Concentrations of cDNA were measured using a Qubit (HS DNA Assay), then normalized and pooled; the quality of the pooled library was assessed with the Agilent Bioanalyzer HS DNA assay. Libraries were then sequenced on an Illumina HiSeq machine to generate 2x150bp paired-end reads.
Data were deposited in GEO under accession numbers GSE144825 and GSE146481.
Read alignment
Additional human and chimpanzee iPSC83,84 and CNCC43 RNA-seq data were downloaded from GEO under accession number GSE96712, and GSE47626, and from European Nucleotide Archive (ENA) under accession number PRJNA289483. These reads, as well as reads generated in this study were aligned to the human GRCh38 and chimpanzee panTro5 genomes using STAR aligner (v2.6.0)85 with arguments: -outSAMattributes MD NH -outFilterMultimapNmax 1 -sjdbGTFfile -sjdbOverhang 149. Exon-exon junctions from all RNA-seq datasets (both iPSCs and CNCCs, parental and hybrid samples) were used collectively in the final STAR alignment step. Duplicate reads were removed using Picard v2.18.27 with argument DUPLICATE_SCORING_STRATEGY = RANDOM. To minimize potential biases when aligning one species to the genome of another species, we took several measures. First, reads were aligned twice, once to the human GRCh38 genome and once to the chimpanzee panTro5 genome. Only orthologous genes (annotated in both genomes) which show similar values of differential expression across both genomes were kept (see Allele-specific expression and differential expression chapter). Second, we used a modified version of WASP19,86 (https://github.com/TheFraserLab/Hornet) to minimize false signals of allelic imbalance. In this pipeline, only reads that are mapped to the same position after in silico allele swapping are kept, thus ensuring that the variants in themselves do not create biased read mappability. Unless otherwise mentioned, values throughout the manuscript represent GRCh38-aligned values.
Allele-specific expression and differential expression
Single nucleotide variants (SNVs) between the human and chimpanzee genomes were identified by first assembling a list of all variants and indels from a pairwise alignment of GRCh38 and PanTro4. RNA-Seq from Ward et al83, Agoglia et al12, and from this study (for a total of 28 samples) was then used to filter this list. Loci were retained only if: (1) at least 2 reads mapped to the locus when mapping to each genome and (2) greater than 90% of the reads mapped to that locus were assigned to the correct species when mapped to each genome. This resulted in a list of 4 million high-confidence variants to be used for phasing of hybrid RNA-seq reads. UCSC Liftover was used to convert SNV coordinates from PanTro4 to those of PanTro5 when this new genome build became available.
Using the SNV file, reads were assigned to a species only if both paired ends mapped unambiguously to one species, using the 2015.03.24 ASEr package (https://github.com/TheFraserLab/ASEr/) as previously described10. Reads that did not contain variants separating the species were discarded, leaving on average 48% of reads (minimum: 44% for CNCC Ch1_rep1, maximum: 52% for Hy1_25_rep1, Supplementary Tables 1–2). In iPSCs, 13,483 out of 13,809 (98%) of expressed genes (FPKM > 1) had at least 1 SNV. In CNCCs, 14,015 out of 14,785 (95%) genes had at least 1 SNV. Differential expression per gene was computed using DESeq2 [20], using the Likelihood Ratio Test (LRT) and the model ~cond_Cell+cond_Species, where cond_Cell represents the replicates and cond_Species represents the species. This was done for hybrid iPSCs, for hybrid CNCCs, for iPSC parental samples and for CNCC parental samples, with each of these aligned once to the GRCh38 genome and once the panTro5 genome. Differential expression between parental samples was computed using samples from different labs to minimize potential lab-specific effects. Genes with FDR < 0.05 in both genomes, and where the absolute[log2(ASEGRCh38) – log2(ASEpanTro5)] < 1 were considered differentially expressed. The use of additional SNVs extracted from the CNCC data, as well as more junctions being identified in reads from the other sources of iPSC and CNCC RNA-seq slightly increased power to detect differential expression12.
For FPKM, TPM and CPM calculations we used all reads that map to the exons of a gene, regardless of whether they map to human-chimpanzee SNVs. Because FPKM is incompatible with between-sample comparisons, we used FPKM values only for gene expression comparisons within a sample or within the means of samples, and not for differential expression analyses or comparisons of genes between samples.
The contribution of trans and non-genetic factors to the overall differential expression in the parental samples was computed as abs[log2(Parental)] – abs[log2(ASE)]. Cis-contribution was computed as .
Changes observed between alleles within the same hybrid can be attributed to cis-regulatory divergence, with one possible exception: trans-induced epigenetic changes in the parental lines that are stably carried over to the hybrid. We infer their contribution to be small due to several reasons: 1. The epigenetic landscape of the precursor parental cells was shown to have largely been reset during reprogramming to iPSCs and did not explain observed within-species differences80. 2. Such changes are expected to be shared by the human and chimpanzees parents if they are selected for in culture. Indeed, we did not identify an over-representation of these genes87 in our datasets (P = 0.25, one-sided hypergeometric test). Alternatively, if they are stochastic, they are not expected to replicate across samples generated by different labs and at different times, which our algorithm requires for calling differential expression.
It has been reported that some genes tend to gain methylation in iPSC culture and this methylation is often stable across passages87. As described above, if one species has gained these changes while the other species has not, and if they remain stable post-hybridization, these changes might manifest as cis-regulatory changes. To test this, we examined the 23 genes reported by Weissbein et al.87. and tested how many of them show differential expression in the parental and hybrid CNCCs. We found that 7 out of 23 are differentially expressed (COX7A1, CTSF, CXCL5, MNS1, SLFN12, ZNF471, and ZNF667), which is not higher than expected by chance (P = 0.25, one-sided hypergeometric test).
Aneuploidy
Several measures were taken to detect and control for potential aneuploidies. First, the hybrid cells were karyotyped, revealing a fully tetraploid set of chromosomes across the five hybrid cell samples12. To test whether any aneuploidies arose between karyotyping and sequencing, we tested if the RNA-seq data reveal stretches of chromosomes with a consistent bias towards one species, suggesting these stretches were possibly duplicated or deleted in one of the species. In the iPSCs, this analysis revealed that Hy1_25 and Hy2_9 possibly have an extra chimpanzee copy of chromosome 20. In Hy1_29, we detected a possible loss of the human short arm and gain of the human long arm of chromosome 20 (chromosome 20 aneuploidies are common in pluripotent stem cell culture18). In the rest of the samples we detected no signs of aneuploidy12. As a precaution, we removed chromosome 20 from subsequent iPSC analyses, including from the differential expression we report. We also removed this chromosome from the background list of genes in all iPSC enrichment analyses. We did not observe aneuploidies in the CNCC hybrid samples (Extended Data Fig. 6 and Extended Data Fig. 7). Based on the lack of evidence of a chromosomal bias in the three CNCC samples, we estimate that these samples likely have a balanced number of chimpanzee and human chromosomes. These results are consistent with previous studies showing that human and mouse tetraploid cells tend to retain their tetraploidy in cell culture15–17. Thus, although aneuploidy is a concern in tetraploid (as well as in diploid) cultured cells, we see no evidence of aneuploid CNCC samples. Mitochondrial genes were excluded from the analyses as well, as they show a consistent human-biased expression12. This human-biased mitochondrial expression probably originates in the parental lines, which show significantly higher expression of human mitochondrial genes both in our dataset and in their original publication80. This suggests that the human iPSCs might have had a higher mitochondrial content.
Finally, we did not detect a bias in chromosome X. Despite the chimp-biased expression of XIST in the female iPSC lines, the inactivation of this chromosome appears to be species-independent12.
Overlap of differentially expressed genes with divergent loci
We analyzed 28 datasets reporting genomic divergence between humans and chimpanzees, including sequence divergence23,24,33,34,25–32, transcription factor binding35, DNA methylation36, chromatin accessibility37–40, 3D chromosomal interactions41,42, histone modification marks43, and gene expression83 (Supplementary Table 6). These datasets were divided into two groups: (a) datasets where the pattern of divergence is indicative of the direction of expression change (e.g., a promoter that became hypermethylated along the human lineage is more likely to be associated with decreased rather than increased expression). This group included 8 datasets, divided into Hu>Ch and Ch>Hu marks. (b) datasets where the pattern of divergence is not indicative of changes in gene expression (e.g., sequence insertion). This group included 20 datasets. First, to examine whether differentially expressed genes tend to overlap divergent regions, we tested their overlap with datasets in both groups. For datasets that reported divergent genes (e.g., differentially accessible genes in chimpanzee and human iPSCs, Supplementary Table 7), we examined the fraction of genes in the list that overlap the differentially expressed gene list, and tested the significance of this overlap using a one-sided hypergeometric test. For datasets that report coordinates of loci along the genome, we first took the genes they overlap (either in their gene body or up to 5 kb upstream of the TSS). Genes that do not contain human-chimpanzee variants were removed from all subsequent analyses as these are genes for which we are unable to detect differential expression, and therefore, to minimize bias, should not appear in the list of genes associated with the examined dataset either. Hypergeometric P-values were then FDR-adjusted using the Benjamini-Hochberg procedure.
Such overlap tests are sensitive to genomic composition biases. For example, longer genes are more likely to overlap divergent loci and at the same time, are also more likely to be reported as differentially expressed as they have more RNA reads, which makes them more likely to have sufficient statistical power to detect differential expression. To account for this, we took several measures. First, we ran a randomization test where each locus is assigned new coordinates along the genome, while keeping its original chromosome and length and matching the mean GC content and coding sequence length of the original gene list with the new randomized list. Then, we linked these randomized loci with genes (as described above) and tested the overlap of each randomized list with the list of differentially expressed genes. This was repeated 1,000 times for each dataset and P-values were assigned based on the fraction of iterations where the randomized overlap is higher than the observed overlap. P-values were then FDR-adjusted using the Benjamini-Hochberg procedure. These processes were repeated for each of the two cell types (iPSCs and CNCCs). Second, for the 8 datasets that are potentially informative of the directionality of gene expression changes (group a), we examined if Hu>Ch genes tend to overlap genomic patterns that are indicative of up-regulation in humans compared to chimpanzees, and if Ch>Hu genes tend to overlap genomic patterns that are indicative of up-regulation in chimpanzees compared to humans. While genomic composition may bias to some extent the overall overlap between lists, it is less likely to result by chance in Hu>Ch genes overlapping human up-regulation patterns and Ch>Hu genes overlapping chimpanzee up-regulated patterns. The tests above were conducted for ASE genes as well as parental differentially expressed genes, and for absolute log2(fold-change) thresholds of 0 and 1, and cis-contributions thresholds of 0%, 50%, 75%, 85% and 90%. One-tailed paired t-test was used to examine the overall significance of the overlaps within each of the above runs. To do so, overlap enrichment values within datasets of chimpanzee up-regulation marks were multiplied by −1. Extended Data Fig. 2a shows the most significant result. For other results, see Supplementary Table 7.
Gene ORGANizer enrichment analysis
Body part enrichment analyses were conducted using Gene ORGANizer version 13, which is based on Human Phenotype Ontology47 (HPO) build 115 (23 January, 2017) and DisGeNET88 release from 10 April, 2015. The first part of the analysis was conducted using each of the two lists of significantly differentially expressed genes (Hu>Ch and Ch>Hu genes) in each of the two hybrid cell types (iPSCs and CNCCs) against the Gene ORGANizer46 genomic background using the ORGANize tool with the confident+tentative option. To minimize tissue-specific effects, only expressed genes (FPKM > 1) were used in both the gene list and the background gene list. Analyses were restricted to skeleton-related body parts for iPSCs and head-related phenotypes for CNCCs. The pelvis was analyzed both as an Organ and as a Region. P-values were FDR-adjusted. Body parts which passed the first test (FDR < 0.05) were tested again in a more stringent test (taking only the confident option with both typical and typical+non-typical associations), this time by comparing the Hu>Ch and Ch>Hu genes against one another in each cell type using Fisher’s exact test. By doing so, we further minimized biases that are potentially introduced when looking at a specific cell type where the set of expressed genes is skewed compared to the genomic background. P-values were FDR-adjusted here too. In cases where both the general body part (e.g., jaws) and its more specific sub-parts (e.g., mandible and maxilla) were significantly enriched, we presented in the figure the data for the more specific body parts (Fig. 2a, Supplementary Tables 9–12).
Analyzing gene-trait associations
Gene-phenotype associations were downloaded from the Human Phenotype Ontology47 (HPO) build 1268 (18 Nov, 2019). For CNCC analyses, only craniofacial related phenotypes were used. First, we tested enrichment of specific HPO phenotypes within Hu>Ch and Ch>Hu genes in CNCCs, iPSCs or both, and with log2(fold-change) thresholds of 0, 0.5, and 1. Only phenotypes linked to at least 5 genes were analyzed. Hypergeometric test P-values were then FDR-adjusted using Benjamini-Hochberg procedure (Supplementary Tables 13–15).
Next, we analyzed the link between divergent expression and divergent phenotypes. To link HPO phenotypes to divergent traits between humans and chimpanzees we re-annotated the chimpanzee divergent trait dataset from Gokhman et al.48 to include 1,774 additional phenotypes from HPO build 1268, following the lines previously described48 (Supplementary Tables 13–15). For each group of genes analyzed, we first tested which of the HPO phenotypes associated with them are known to be divergent between humans and chimpanzees. Then, we assigned a predicted direction of phenotypic change for each HPO phenotype linked to each gene; as most HPO phenotypes are the result of partial or complete loss-of-function47,89, we conjectured that down-regulation of a gene might result in a similar direction of phenotypic change (but not necessarily the same extent). Therefore, the species where the gene is down-regulated was linked to the HPO phenotype (Fig. 2b). Next, we computed the fraction of traits matching the phenotypic directionality between humans and chimpanzees out of all divergent traits. If a gene was differentially expressed in both cell types, its CNCC log2(fold-change) values were used. HPO phenotypes with contradicting directions of phenotypic change between the species (e.g., Aplasia/hypoplasia of the humerus, HP:0006507), unknown direction of divergence (e.g., Decreased osteoclast count, HP:0030328), ambiguous definition (e.g., Shuffling gait, HP:0002362), or non-directional phenotypes (e.g., Abnormal facial shape, HP:0001999) were discarded. The pipeline was applied repeatedly on increasingly higher log2(fold-change) thresholds on ASE genes and on differentially expressed genes in the parental samples with various cis-contribution minimum thresholds (0%, 50%, 75%, 85% and 90%).
P-values were calculated using a randomization test, where each gene was randomly assigned a direction of expression change (i.e., Hu>Ch or Ch>Hu) while keeping its absolute log2(fold-change) value. We then repeated the process above and computed the fraction of correct predictions per trait. Next, we computed the area under curve (AUC), which represents the overall prediction accuracy, and the linear regression slope, which represents the improvement in prediction accuracy with increasing log2(fold-change) thresholds. These two values were then compared to the observed AUC and slope in the real data. P-values were generated by repeating the test 10,000 times.
Additional RNA-seq data
Six human and ten chimpanzee fibroblast RNA-seq samples80,90 were downloaded from SRA and GEO under accession numbers: SRP102410 and GSE61343, respectively. Five gorilla and five human iPSC RNA-seq samples57 were downloaded from GEO under accession number GSE50781.
See Supplementary Information for EVC2 and Hedgehog signaling experiments.
Statistics
The overlap analyses of differentially expressed genes with divergent regulation loci were done using a one-sided hypergeometric test. Randomization tests for overlap with 28 previously published data sets were done by keeping the original chromosome and length of each locus, and matching the mean GC content and coding sequence length of the original gene list with the new randomized list. This was repeated 1,000 times for each dataset. P-values were assigned based on the fraction of iterations where the randomized overlap is higher than the observed overlap. P-values were then FDR-adjusted using the Benjamini-Hochberg procedure. Additionally, to test the overall overlap of these datasets (n = 16) with differentially expressed genes, we used a one-tailed paired t-test. P-values were then FDR-adjusted using Benjamini-Hochberg procedure.
Enrichment tests (HPO, Gene ORGANizer, and Gene Ontology) were done using a one-sided hypergeometric test, and P-values were then FDR-adjusted using the Benjamini-Hochberg procedure. KEGG pathway sign test was done using a binomial test with p = 0.5 and n = number of genes per pathway. P-values were FDR-adjusted using the Benjamini-Hochberg procedure.
For the phenotype directionality prediction, we used a one-sided randomization test, where each gene was randomly assigned a direction of expression change (i.e., Hu>Ch or Ch>Hu) while keeping its absolute log2(fold-change) value. We then repeated the process above and computed the fraction of correct predictions per phenotype. Next, we computed the area under curve (AUC), which represents the overall prediction accuracy, and the linear regression slope, which represents the improvement in prediction accuracy with increasing log2(fold-change) thresholds. These two values were then compared to the observed AUC and slope in the real data. P-values were generated by repeating the test 10,000 times.
Evc2 mouse KO vs wildtype phenotypic comparison was done using a two-tailed paired t-test (n = 5 in each group). Differential expression in the EVC2 reporter assay was tested using a one-tailed t-test in two independent experiments of quadruplet measurements (n = 8). EVC2 phenotype resemblance tests in mouse KO vs wildtype compared to human vs chimpanzee, and in Ellis-van Creveld patients vs healthy individuals compared to human vs chimpanzee were done using binomial tests, where a success was defined as a match in the phenotypic directions between the two pairs, p = 0.5. We note that this assumes that traits are independent of one another (i.e., knowing the directionality of one trait difference does not provide information about the directionalities of other traits), though overlapping phenotypes were merged as previously described48, and the results would remain significant even if several traits were not independent.
Data availability
Data were deposited in GEO under accession number GSE144825 and GSE146481.
Code availability
Code used in this study is available at https://github.com/TheFraserLab/ASEr, https://github.com/TheFraserLab/Agoglia_HumanChimpanzee2020, and https://github.com/TheFraserLab/Hornet/tree/master
Extended Data
Extended Data Fig. 1. Identifying human-chimpanzee expression changes using hybrid cells.
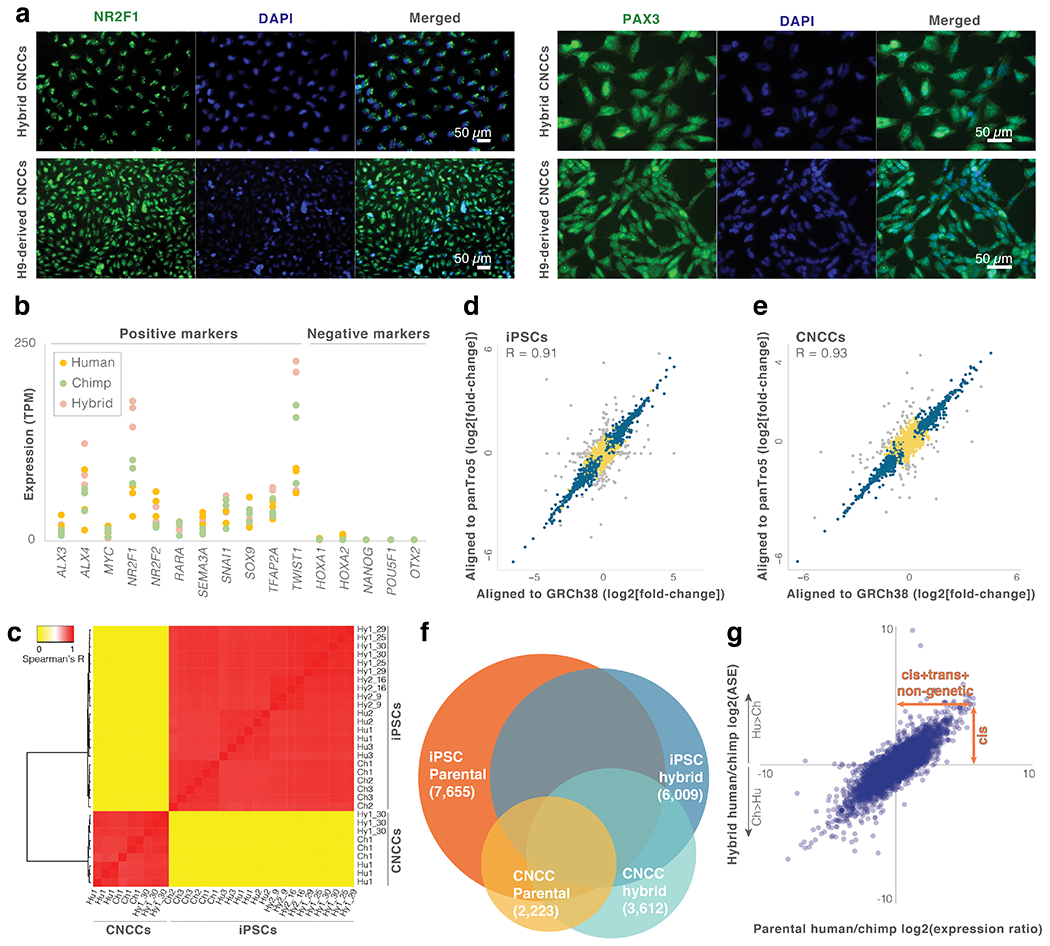
a. Immunostaining for CNCC markers NR2F1 and PAX3 was performed to confirm CNCC differentiation. b. Expression levels of positive and negative markers in the parental and hybrid CNCCs. c. Heatmap and dendrogram of total gene expression across iPSC and CNCC samples. d,e. Fold-change per gene for hybrid iPSCs and hybrid CNCCs when aligned to the human (GRCh38) vs chimpanzee (panTro5) genomes. Grey points are genes where the absolute difference in log2(fold-change) when aligned to the human vs. chimpanzee genome is greater than 1 (i.e., genes with potential alignment bias that were excluded from the analysis). Genes with no observable alignment bias are marked with blue (significant ASE: q-value < 0.05) or yellow (non-significant ASE). f. Venn diagram of genes with significant human-chimpanzee expression changes in parental and hybrid samples. g. Parental vs hybrid iPSC expression changes. See Fig. 1d legend.
Extended Data Fig. 2. Differentially expressed genes are associated with divergent chromatin and phenotypes.
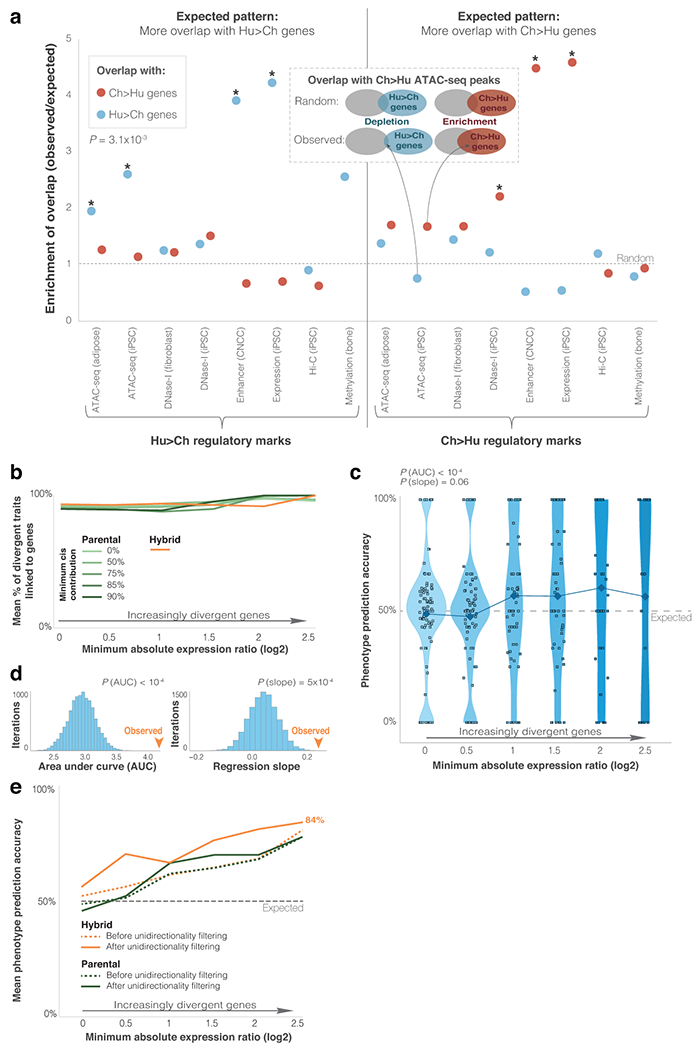
a. Overlap of ASE genes in CNCCs with loci showing divergent regulatory marks. Each of the datasets was examined twice: (1) against Ch>Hu genes (red), and (2) against Hu>Ch genes (blue). In 14 out of 16 datasets, expression differences reflect regulatory differences, i.e., Hu>Ch regulatory marks show more overlap with Hu>Ch genes than with Ch>Hu genes, and vice versa. P-value shows one-tailed paired t-test for overall overlap (see Methods). Asterisks mark significant randomization test overlap (FDR < 0.05). See Supplementary Table 8. b. Mean fraction of divergent phenotypes for groups of genes with increasingly higher fold-change thresholds. c. Violin plots showing phenotype assignment accuracy in groups of genes with increasingly more divergent differential expression in parental cells. Randomization test P-values are shown for overall accuracy compared to random (PAUC), and accuracy increase compared to random (Pslope), as shown in d. See Fig. 2c legend. d. Randomization output for the phenotype assignment pipeline. Genes associated with each phenotype were randomly assigned a direction of expression change, while keeping their absolute fold-change. Randomization test P-values are shown for overall accuracy compared to random (PAUC), and accuracy increase compared to random (Pslope). e. Phenotype assignment accuracy before and after applying unidirectionality filtering, for ASE and parental differential expression with cis-contribution ≥ 90%. See Fig. 2d legend. In the unidirectionality filter, only phenotypes where all genes point in the same phenotypic direction (i.e., complete agreement) are analyzed48.
Extended Data Fig. 3. EVC2 down-regulation in humans.
a. The down:up ratio of Hh signaling genes across increasingly more stringent FDR and fold-change thresholds. b. Differential expression along all of the exons of EVC2 in a CNCC hybrid (Hy1_30_rep1), showing that the majority of reads come from the chimpanzee alleles. Introns are not shown to scale. c. Violin plots of EVC2 expression across iPSC and CNCC non-hybrid samples from various sources, showing consistent EVC2 down-regulation in humans compared to chimpanzees. Diamonds show mean expression levels. DESeq2 FDR-adjusted P-values are presented for cell type. The observation that the human-chimpanzee ratios are similar to the ones observed within the hybrid cells suggests that the majority of differential expression is driven by cis changes. d. EVC2 expression across nine additional tissues for which both human and chimpanzee data are available56, showing that EVC2 down-regulation is not restricted to iPSCs and CNCCs. Dashed line shows mean expression. One-sided t-test P-values are shown. e. Gorilla vs human EVC2 expression. One-sided t-test P-values are shown. f. Western blot of EVC2 protein levels in human and chimpanzee DPSCs. The samples derive from the same experiment and blots were processed in parallel. For gel source data, see Source Data.
Extended Data Fig. 4. Differentially regulated regions in EVC2.

ATAC-seq read pileup along EVC2 and for the three loci showing species-biased peaks within EVC2. Arrows mark peaks. b,c. NR2F1 and TFAP2A ChIP-seq read pileup for loci <10 kb away from the ATAC-seq peaks. d. MUSCLE103 sequence alignment of rhesus, gorilla, chimp and human sequences. Regions with a high proportion of mismatches are colored in red. e. Reporter assay comparing relative firefly/Renilla luciferase activity for chimpanzee and human EVC2 sequences following transient transfection in human DPSCs. Empty vector (pGL4.11b) was used as negative control. Box plots show mean (center), 2nd and 3rd quartiles (box boundaries), and minima and maxima (whiskers). One-tailed t-test P-values in two independent experiments of quadruplet measurements (n = 8) are shown.
Extended Data Fig. 5. Reduced levels of EVC2 result in reduced Hedgehog signaling output and affect craniofacial phenotypes.
a. Western blot of Gli1 protein levels (a measure of Hh signaling output induced by Shh) at different Evc2 and Hh signaling input levels. EvcC2 was introduced at various levels into Evc2−/− mouse NIH/3T3 fibroblasts through retroviral infection. Cells with higher levels of Evc2 show higher Hh signaling output. p38 served as positive control. Pearson’s R and P-value are shown for 40nM SHH. The samples derive from the same experiment and blots were processed in parallel. For gel source data, see Source Data. b. Micro-CT radiographic images of the palate bone, enamel (extra bright) and roots of the first mandibular molar in Evc2 control and Evc2 KO mice at P28. c. Diagram of the mandible indicating the landmarks for the parameters measured. d. Mean skull and mandible measurements from Evc2 control and Evc2 KO mice at P28. (n = 5 for each group, FDR-adjusted two-tailed t-test P-values are shown). Whiskers show one standard deviation in each direction. Landmarks used are shown in the titles.
Extended Data Fig. 6. No aneuploidies observed in CNCC hybrid samples.
Figure shows ASE (top), sliding window ASE median over 20 genes (middle), and Wilcoxon rank sum test P-values for each sliding window against the entire genome (bottom). Dashed line shows mean for ASE and sliding window ASE, and shows Bonferroni P-value cutoff for the Wilcoxon rank sum test. An example of data is presented for autosomal chromosomes 1 and 20, and for chromosome X from the CNCC Hy1_30_rep1 sample. No significant deviations were detected in any of the CNCC hybrid samples. See Agoglia et al. for iPSC aneuploidy analyses12.
Extended Data Fig. 7. No chromosomal duplications or losses observed in the CNCC hybrid samples.
Density plots of percentage of human-aligned reads per gene per chromosome for each of the CNCC hybrid samples. Vertical dashed lines show mean per sample.
Supplementary Material
Acknowledgements
S. Bar, L. Carmel, and members of the Fraser, Petrov and Pritchard labs for critical comments, and the Gilad lab (Chicago University) and You lab (University of Pennsylvania) for sharing data and cells. DG was funded by the Human Frontier, Rothschild and Zuckerman fellowships. HBF is supported by NIH grant 2R01GM097171-05A1. The cells used in this study were derived from the iPSCs generated by Gallego Romero et al.80, whose study was supported by the National Institutes of Health, Office of Research Infrastructure Programs/OD [P51OD011132].
Footnotes
Competing Interests Statement
The authors declare no competing interests.
References for main text
- 1.Aiello L & Dean C An Introduction to Human Evolutionary Anatomy. (Elsevier, 2002). [Google Scholar]
- 2.King MC & Wilson AC Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (1975). [DOI] [PubMed] [Google Scholar]
- 3.Enard D, Messer PW & Petrov DA Genome-wide signals of positive selection in human evolution. Genome Res. (2014). doi: 10.1101/gr.164822.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraser HB Gene expression drives local adaptation in humans. Genome Res. 23, 1089–1096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wittkopp PJ & Kalay G Cis-regulatory elements: Molecular mechanisms and evolutionary processes underlying divergence. Nature Reviews Genetics (2012). doi: 10.1038/nrg3095 [DOI] [PubMed] [Google Scholar]
- 6.Tirosh I, Reikhav S, Levy AA & Barkai N A yeast hybrid provides insight into the evolution of gene expression regulation. Science (80-. ). (2009). doi: 10.1126/science.1169766 [DOI] [PubMed] [Google Scholar]
- 7.Wittkopp PJ, Haerum BK & Clark AG Evolutionary changes in cis and trans gene regulation. Nature (2004). doi: 10.1038/nature02698 [DOI] [PubMed] [Google Scholar]
- 8.Pastinen T Genome-wide allele-specific analysis: Insights into regulatory variation. Nature Reviews Genetics (2010). doi: 10.1038/nrg2815 [DOI] [PubMed] [Google Scholar]
- 9.Fraser HB Genome-wide approaches to the study of adaptive gene expression evolution. BioEssays (2011). doi: 10.1002/bies.201000094 [DOI] [PubMed] [Google Scholar]
- 10.Combs PA et al. Tissue-Specific cis-Regulatory Divergence Implicates eloF in Inhibiting Interspecies Mating in Drosophila. Curr. Biol. (2018). doi: 10.1016/j.cub.2018.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Soloway PD & Clark AG Paternally biased X inactivation in mouse neonatal brain. Genome Biol. (2010). doi: 10.1186/gb-2010-11-7-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agoglia A et al. Generation of human-chimpanzee hybrid stem cell-derived organoids to investigate cis-regulatory evolution of the cerebral cortex. co-submitted (2020).
- 13.Sommer. Neural crest-derived stem cells. StemBook (2010). doi: 10.3824/stembook.1.51.1 [DOI] [Google Scholar]
- 14.Øvrebø JI & Edgar BA Polyploidy in tissue homeostasis and regeneration. Development (Cambridge) (2018). doi: 10.1242/dev.156034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin D-H et al. Characterization of Tetraploid Somatic Cell Nuclear Transfer-Derived Human Embryonic Stem Cells. Dev. Reprod. 21, 425–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowan CA, Atienza J, Melton DA & Eggan K Nuclear Reprogramming of Somatic Cells After Fusion with Human Embryonic Stem Cells. Science (80-. ). 309, 1369 LP–1373 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Broughton KM et al. Cardiac interstitial tetraploid cells can escape replicative senescence in rodents but not large mammals. Commun. Biol. (2019). doi: 10.1038/s42003-019-0453-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Initiative ISC et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van De Geijn B, Mcvicker G, Gilad Y & Pritchard JK WASP: Allele-specific software for robust molecular quantitative trait locus discovery. Nature Methods (2015). doi: 10.1038/nmeth.3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014). doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Li YI & Pritchard JK Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell (2019). doi: 10.1016/j.cell.2019.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wittkopp PJ, Haerum BK & Clark AG Regulatory changes underlying expression differences within and between Drosophila species. Nat. Genet. (2008). doi: 10.1038/ng.77 [DOI] [PubMed] [Google Scholar]
- 23.Peyrégne S, Boyle MJ, Dannemann M & Prüfer K Detecting ancient positive selection in humans using extended lineage sorting. Genome Res. 27, 1563–1572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Racimo F, Kuhlwilm M & Slatkin M A test for ancient selective sweeps and an application to candidate sites in modern humans. Mol. Biol. Evol. (2014). doi: 10.1093/molbev/msu255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kronenberg ZN et al. High-resolution comparative analysis of great ape genomes. Science (80-. ). (2018). doi: 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prüfer K et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science (80-. ). 358, 655–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prabhakar S, Noonan JP, Pääbo S & Rubin EM Accelerated evolution of conserved noncoding sequences in humans. Science (80-. ). (2006). doi: 10.1126/science.1130738 [DOI] [PubMed] [Google Scholar]
- 28.Lindblad-Toh K et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kostka D, Holloway AK & Pollard KS Developmental loci harbor clusters of accelerated regions that evolved independently in ape lineages. Mol. Biol. Evol. (2018). doi: 10.1093/molbev/msy109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLean CY et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature (2011). doi: 10.1038/nature09774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gittelman RM et al. Comprehensive identification and analysis of human accelerated regulatory DNA. Genome Res. (2015). doi: 10.1101/gr.192591.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marnetto D, Molineris I, Grassi E & Provero P Genome-wide identification and characterization of fixed human-specific regulatory regions. Am. J. Hum. Genet. (2014). doi: 10.1016/j.ajhg.2014.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature (2016). doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gayà-Vidal M & Albà MM Uncovering adaptive evolution in the human lineage. BMC Genomics (2014). doi: 10.1186/1471-2164-15-599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glinsky GV Transposable elements and DNA methylation create in embryonic stem cells human-specific regulatory sequences associated with distal enhancers and noncoding RNAs. Genome Biol. Evol. (2015). doi: 10.1093/gbe/evv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gokhman D et al. Differential DNA methylation of vocal and facial anatomy genes in modern humans. Nat. Commun. 11, 1189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata Y et al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. (2012). doi: 10.1371/journal.pgen.1002789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swain-Lenz D et al. Comparative Analyses of Chromatin Landscape in White Adipose Tissue Suggest Humans May Have Less Beigeing Potential than Other Primates. Genome Biol. Evol. (2019). doi: 10.1093/gbe/evz134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edsall LE et al. Evaluating chromatin accessibility differences across multiple primate species using a joint modelling approach. bioRxiv (2019). doi: 10.1101/617951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romero IG, Gopalakrishnan S & Gilad Y Widespread conservation of chromatin accessibility patterns and transcription factor binding in human and chimpanzee induced pluripotent stem cells. bioRxiv 466631 (2018). doi: 10.1101/466631 [DOI] [Google Scholar]
- 41.Glinsky GV Mechanistically distinct pathways of divergent regulatory DNA creation contribute to evolution of human-specific genomic regulatory networks driving phenotypic divergence of homo sapiens. Genome Biol. Evol. (2016). doi: 10.1093/gbe/evw185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eres IE, Luo K, Hsiao CJ, Blake LE & Gilad Y Reorganization of 3D genome structure may contribute to gene regulatory evolution in primates. PLoS Genet. (2019). doi: 10.1371/journal.pgen.1008278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prescott SL et al. Enhancer Divergence and cis-Regulatory Evolution in the Human and Chimp Neural Crest. Cell 163, 68–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reilly SK & Noonan JP Evolution of Gene Regulation in Humans. Annu. Rev. Genomics Hum. Genet. (2016). doi: 10.1146/annurev-genom-090314-045935 [DOI] [PubMed] [Google Scholar]
- 45.Cotney J et al. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154, 185–196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gokhman D et al. Gene ORGANizer: Linking genes to the organs they affect. Nucleic Acids Res. 45, W138–W145 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Köhler S et al. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 42, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gokhman D et al. Reconstructing Denisovan Anatomy Using DNA Methylation Maps. Cell 179, 180–192.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Orr HA Testing natural selection vs. genetic drift in phenotypic evolution using quantitative trait locus data. Genetics (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanehisa M, Sato Y, Kawashima M, Furumichi M & Tanabe M KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xavier GM et al. Hedgehog receptor function during craniofacial development. Developmental Biology (2016). doi: 10.1016/j.ydbio.2016.02.009 [DOI] [PubMed] [Google Scholar]
- 52.Ramsbottom SA & Pownall ME Regulation of hedgehog signalling inside and outside the cell. Journal of Developmental Biology (2016). doi: 10.3390/jdb4030023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang SH, Hsiao CJ, Khan Z & Pritchard JK Post-translational buffering leads to convergent protein expression levels between primates. Genome Biol. (2018). doi: 10.1186/s13059-018-1451-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dorn KV, Hughes CE & Rohatgi R A Smoothened-Evc2 Complex Transduces the Hedgehog Signal at Primary Cilia. Dev. Cell (2012). doi: 10.1016/j.devcel.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H et al. Elevated Fibroblast Growth Factor Signaling Is Critical for the Pathogenesis of the Dwarfism in Evc2/Limbin Mutant Mice. PLoS Genet. (2016). doi: 10.1371/journal.pgen.1006510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pipes L et al. The non-human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Res. (2013). doi: 10.1093/nar/gks1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wunderlich S et al. Primate iPS cells as tools for evolutionary analyses. Stem Cell Res. (2014). doi: 10.1016/j.scr.2014.02.001 [DOI] [PubMed] [Google Scholar]
- 58.Briscoe J & Small S Morphogen rules: Design principles of gradient-mediated embryo patterning. Development (Cambridge) (2015). doi: 10.1242/dev.129452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young NM, Chong HJ, Hu D, Hallgrímsson B & Marcucio RS Quantitative analyses link modulation of sonic hedgehog signaling to continuous variation in facial growth and shape. Development (2010). doi: 10.1242/dev.052340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu D & Helms JA The role of Sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development (1999). [DOI] [PubMed] [Google Scholar]
- 61.Pan A, Chang L, Nguyen A & James AW A review of hedgehog signaling in cranial bone development. Front. Physiol. (2013). doi: 10.3389/fphys.2013.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jeong J, Mao J, Tenzen T, Kottmann AH & McMahon AP Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. (2004). doi: 10.1101/gad.1190304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang H et al. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis (2015). doi: 10.1002/dvg.22879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Badri MK et al. Expression of Evc2 in craniofacial tissues and craniofacial bone defects in Evc2 knockout mouse. Arch. Oral Biol. (2016). doi: 10.1016/j.archoralbio.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Badri MK et al. Ellis Van Creveld2 is Required for Postnatal Craniofacial Bone Development. Anat. Rec. (2016). doi: 10.1002/ar.23353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takeda H et al. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc. Natl. Acad. Sci. U. S. A (2002). doi: 10.1073/pnas.152337899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caparrós-Martín JA et al. The ciliary EVC/EVC2 complex interacts with smo and controls hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. (2013). doi: 10.1093/hmg/dds409 [DOI] [PubMed] [Google Scholar]
- 68.Kulkarni AK et al. A Ciliary Protein EVC2/LIMBIN Plays a Critical Role in the Skull Base for Mid-Facial Development. Front. Physiol. 9, 1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pusapati GV et al. EFCAB7 and IQCE Regulate Hedgehog Signaling by Tethering the EVC-EVC2 Complex to the Base of Primary Cilia. Dev. Cell (2014). doi: 10.1016/j.devcel.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li X et al. Genome-wide linkage study suggests a susceptibility locus for isolated bilateral microtia on 4p15.32–4p16.2. PLoS One (2014). doi: 10.1371/journal.pone.0101152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Claes P et al. Modeling 3D Facial Shape from DNA. PLoS Genet. 10, e1004224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lieberman DE & McCarthy RC The ontogeny of cranial base angulation in humans and chimpanzees and its implications for reconstructing pharyngeal dimensions. J. Hum. Evol. (1999). doi: 10.1006/jhev.1998.0287 [DOI] [PubMed] [Google Scholar]
- 73.Pilot M et al. Diversifying selection between pure-breed and free-breeding dogs inferred from genome-wide SNP analysis. G3 Genes, Genomes, Genet. (2016). doi: 10.1534/g3.116.029678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu Y & Albertson RC Hedgehog signaling mediates adaptive variation in a dynamic functional system in the cichlid feeding apparatus. Proc. Natl. Acad. Sci. U. S. A. (2014). doi: 10.1073/pnas.1323154111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burga A et al. A genetic signature of the evolution of loss of flight in the Galapagos cormorant. Science (80-. ). (2017). doi: 10.1126/science.aal3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dorus S et al. Sonic Hedgehog, a key development gene, experienced intensified molecular evolution in primates. Hum. Mol. Genet. (2006). doi: 10.1093/hmg/ddl123 [DOI] [PubMed] [Google Scholar]
- 77.Claes P et al. Genome-wide mapping of global-to-local genetic effects on human facial shape. Nat. Genet. 50, 414–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lieberman P The Evolution of Human Speech: Its Anatomical and Neural Bases. Curr. Anthropol. 48, 39–66 (2007). [Google Scholar]
- 79.Boë L-J et al. Which way to the dawn of speech?: Reanalyzing half a century of debates and data in light of speech science. Sci. Adv. 5, eaaw3916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 80.Romero IG et al. A panel of induced pluripotent stem cells from chimpanzees: A resource for comparative functional genomics. Elife (2015). doi: 10.7554/eLife.07103.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rada-Iglesias A et al. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell (2012). doi: 10.1016/j.stem.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bajpai VK et al. Reprogramming Postnatal Human Epidermal Keratinocytes Toward Functional Neural Crest Fates. Stem Cells (2017). doi: 10.1002/stem.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ward MC et al. Silencing of transposable elements may not be a major driver of regulatory evolution in primate iPSCs. Elife (2018). doi: 10.7554/eLife.33084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marchetto MCN et al. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature (2013). doi: 10.1038/nature12686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dobin A et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics (2013). doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tehranchi A et al. Fine-mapping cis-regulatory variants in diverse human populations. Elife (2019). doi: 10.7554/elife.39595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weissbein U, Plotnik O, Vershkov D & Benvenisty N Culture-induced recurrent epigenetic aberrations in human pluripotent stem cells. PLoS Genet. (2017). doi: 10.1371/journal.pgen.1006979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Piñero J et al. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamosh A, Scott AF, Amberger JS, Bocchini CA & McKusick VA Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pizzollo J et al. Comparative serum challenges show divergent patterns of gene expression and open chromatin in human and chimpanzee. Genome Biol. Evol. (2018). doi: 10.1093/gbe/evy041 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were deposited in GEO under accession number GSE144825 and GSE146481.