

Abstract
BCL11A, the major regulator of HbF(α2γ2) level, represses γ-globin expression through direct promoter binding in adult erythroid cells in a switch to adult-type HbA (α2β2). To uncover how BCL11A initiates repression, we used CRISPR/Cas9, dCas9, dCas9-KRAB, and dCas9-VP64 screens to dissect the γ-globin promoters and identified an activator element near the BCL11A binding site. Using CUT&RUN and base editing, we demonstrate that a proximal CCAAT box is occupied by the activator NF-Y. BCL11A competes with NF-Y binding through steric hindrance to initiate repression. Occupancy of NF-Y is rapidly established upon BCL11A depletion, and precedes γ-globin derepression and LCR-globin loop formation. Our findings reveal that the switch from fetal-to-adult globin gene expression within the >50 kb β-globin gene cluster is initiated by competition between a stage-selective repressor and a ubiquitous activating factor within a remarkably discrete region of the γ-globin promoters.
Introduction
Human hemoglobin expression undergoes two switches during development, embryonic-to-fetal and fetal-to-adult. The second wave initiates in utero and is completed postnatally, when the predominant form switches from fetal hemoglobin (HbF, α2γ2) in fetal liver to adult hemoglobin (HbA, α2β2) in bone marrow. HbF comprises <2% of total hemoglobin in adults. Increasing the level of HbF ameliorates the β-hemoglobinopathies, β-thalassemia and sickle cell disease (SCD).
Sequential expression of β-like globin loci in development - ε-embryonic to γ-fetal to β-adult - is reflected in successive looping between the Locus Control Region1 (LCR) and each globin promoter2–4. Transgenic mouse experiments incorporating individual human γ-globin5, β-globin6, or the β-globin locus lacking the LCR7,8 revealed that developmental specificity resides in and about the globin genes rather than the LCR, which enhances transcription. In human, naturally occurring mutations or deletions in the γ-globin promoters cause continued HbF expression in adults, known as Hereditary Persistence of Fetal Hemoglobin (HPFH). The mutations fall in distinct clusters, consistent with repressor binding at these elements. BCL11A and LRF/ZBTB7A, established HbF repressors9–11, bind directly to the −115 and −200 (relative to γ-globin TSS) HPFH clusters, respectively12,13. We and others found that BCL11A selectively acts at a distal TGACCA motif (−118 to −113), rather than a proximal duplicate in the γ-promoters12,13. Cas9 mediated mutagenesis of the motif impairs BCL11A binding and γ- repression12–14. These findings established local control through cis elements as a primary determinant for hemoglobin switching.
Contributions of distal elements of the β-globin locus in HbF repression are unclear. A boundary element encompassing the HBBP1 gene15, the ncRNA gene BGLT316, and sequences upstream of HBD gene17, have been implicated in repression. Yet, prior work argues against a repressive element upstream of HBD18.
Here we investigate how BCL11A initiates γ-globin gene repression and counteracts transcription activation. Our findings lead to a parsimonious model of hemoglobin switching.
Results
dCas9 placement at BCL11A motif in γ-promoter reduces HbF
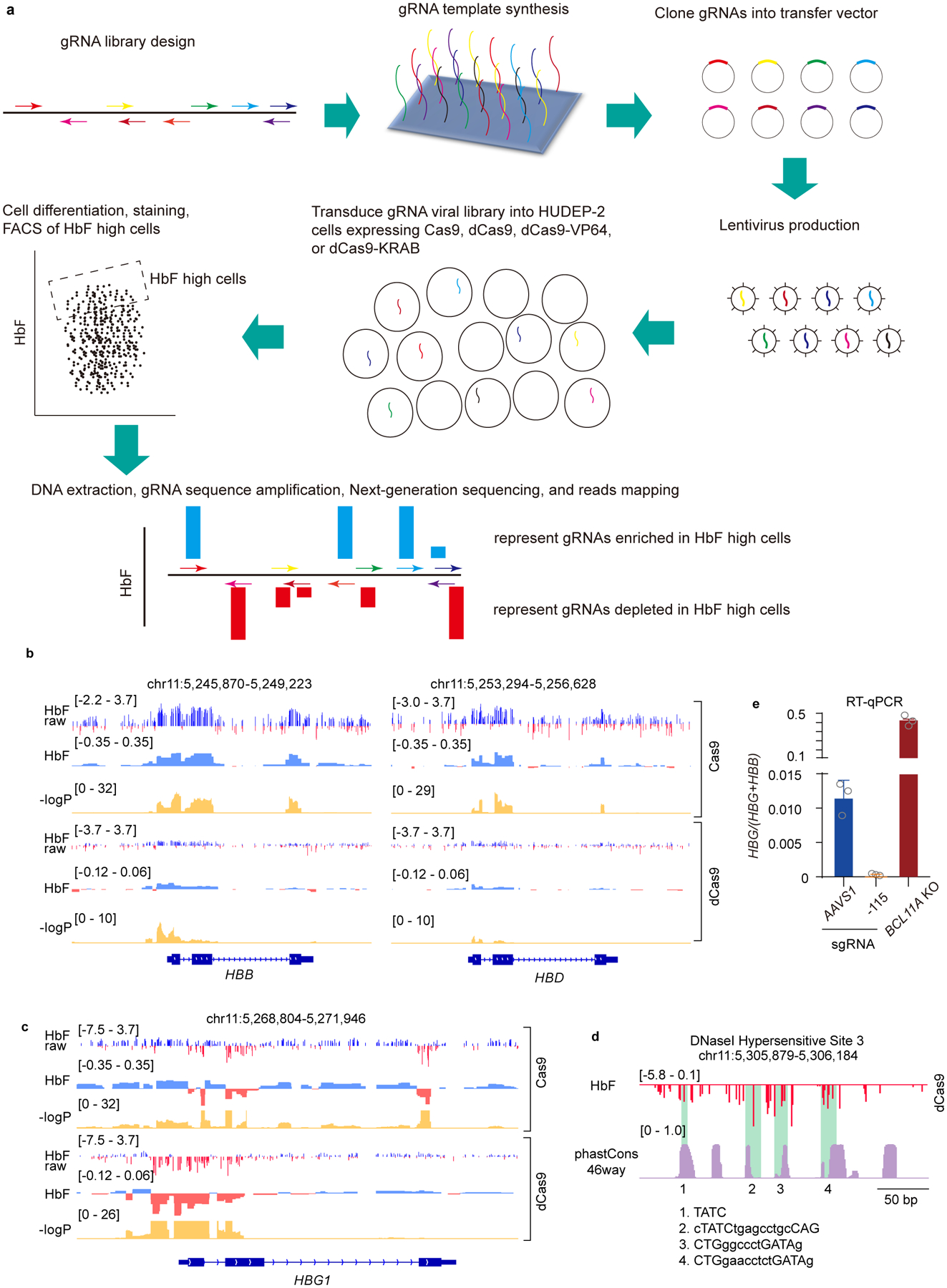
We performed CRISPR/Cas9 dense perturbation throughout the β-globin cluster to identify regulatory elements (see Methods and Extended Data Fig. 1a). We employed adult-type (low HbF) HUDEP-2 cells19 that stably expressed 1) Cas9 to mutate target sequences, 2) inactive Cas9 (dCas9) to bind target sequences but not introduce DNA breaks, or 3) transcription-activating dCas9-VP64 or repressive dCas9-KRAB. Following introduction of pooled, densely spaced gRNAs (9293 gRNAs targeting 106 kb of the cluster, 1 gRNA per ~11 bp), high HbF cells were isolated to assess enrichment or depletion of individual gRNAs20. In principle, enriched gRNAs may target repressive elements, whereas depleted gRNAs may pinpoint activating sequences. We deconvoluted gRNA enrichment scores to identify elements controlling γ-globin expression (Fig. 1a). Based on Cas9-mediated mutagenesis, HbF repressive sequences resided at HBB and HBD coding sequences (Extended Data Fig. 1b), and noncoding sequences proximal to HBG1 (Extended Data Fig. 1c), HBG2 and around HBBP1. We suspect that induction of HbF by targeting these sequences reflects secondary effects of Cas9 editing and subsequent DNA repair. In addition, gRNAs targeting the duplicated HBG1 and HBG2 genes frequently result in 4.9 kb deletions14, which may lead to remodeling of local chromatin or removal of repressive elements. Authentic cis-acting elements may therefore be obscured. Targeting HBG1 and HBG2 coding sequences led to reduced HbF level, as expected. We did not detect distal HbF repressive elements, as proposed to reside between HBD and HBBP121. The effects of dCas9-VP64 (activator) or dCas9-KRAB (repressor) were as expected. Targeting VP64 or KRAB to the body and flanking regions of the γ-globin gene led to induction or reduction of HbF, respectively (Fig. 1a).
Figure 1. dCas9 dense perturbation reveals an activator element at the γ-globin promoters.
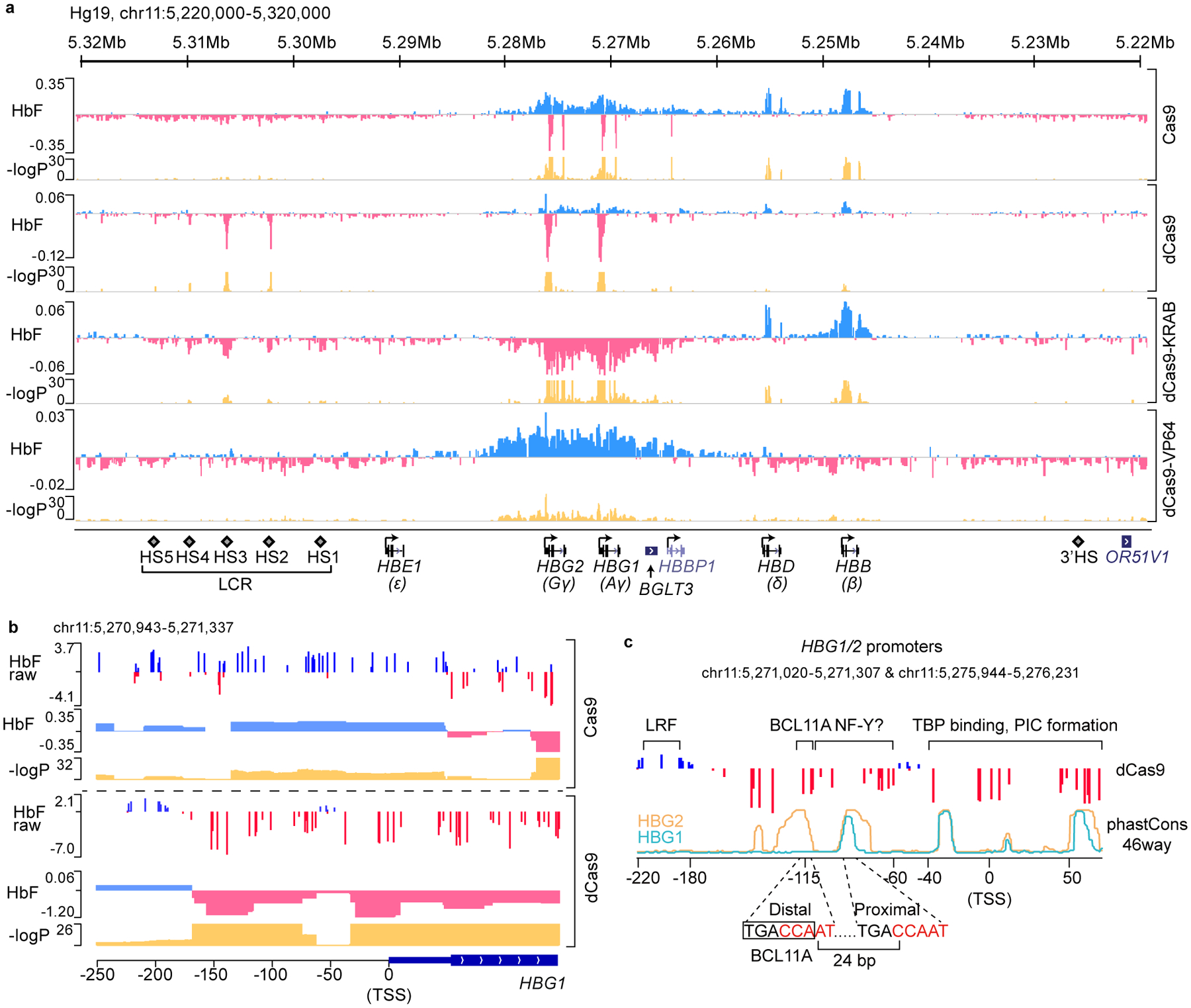
(a) Dense perturbation of genomic sequences around the β-globin gene cluster by pooled gRNA library in Cas9, dCas9, dCas9-KRAB or dCas9-VP64 expressing HUDEP-2 cells. gRNA enrichment in HbF-high cells was used to deconvolute underlying genomic regulatory signal (with “HbF” track displaying beta coefficient). Corresponding p-values are shown on −log10 scale.
(b) A zoomed-in view of the dense perturbation results at the γ-globin (HBG1) promoter and first exon. “HbF raw” scores were calculated as enrichment of reads for each sgRNA in HbF-high compared to the unsorted population at end of erythroid maturation (shown as log2 fold change). “HbF” track depicts the deconvoluted regulatory signal as beta coefficient with associated p-values below. Minor ticks indicate value of zero. The result at the HBG2 promoter was the same as the sequences share 99.3% identity.
(c) Schematic structure of γ-globin promoters. The binding sites of LRF/ZBTB7A, BCL11A, and TBP are indicated. Sequence conservation of HBG1 and HBG2 promoters across 46 vertebrates (phastCons46way) are shown as cyan and orange lines, respectively. Two CCAAT boxes that are potential binding sites of NF-Y are highlighted in red. The distal TGACCA motif through which BCL11A represses γ-globin expression is delineated with a rectangle.
Statistical tests of the beta coefficients were performed empirically through bootstrapping and two-tailed tests. Multiple hypothesis testing was accounted for with the Benjamini-Hochberg (BH) procedure.
An unexpected observation in the dCas9 screen caught our attention. Although dCas9 is inactive for DNA breakage, its targeting to chromatin may interfere with binding of endogenous regulators22. The majority of gRNAs in the dCas9 screen were neither enriched nor depleted, suggesting that dCas9 binding at numerous sites did not perturb γ-globin transcription (Fig. 1a). Expression changes were observed when dCas9 was targeted to HS2, HS3 and γ-globin promoters. Within HS2 and HS3, the most depleted gRNAs mapped to composite GATA1-TAL1 motifs (Extended Data Fig. 1d), suggesting that eviction of GATA1 and/or TAL1 at these motifs impairs LCR activity. Within the γ-promoters, however, enriched and depleted gRNAs mapped to discrete regions (Fig. 1b, c). Targeting of dCas9 (or Cas9) to ~ −200 bp , the site at which LRF/ZBTB7A binds9,13, increased HbF, consistent with factor displacement. In contrast, targeting of dCas9 to ~ −115 bp, where BCL11A is normally bound to a TGACCA motif12,13, reduced, rather than increased HbF expression. As expected, targeting of Cas9 with these gRNAs increased HbF expression.
The effect of positioning dCas9 at the BCL11A binding site seemed paradoxical. The further reduction in HbF expression was striking, given the low basal HbF level in HUDEP-2 cells (Fig. 1b and Extended Data Fig. 1e). Moreover, dCas9 targeting to multiple positions from −150 bp to −60 bp led to a similar reduction in HbF. We hypothesized that dCas9 exerted this effect through displacement of an activator.
NF-Y activates γ-globin expression
The cognate BCL11A binding site is duplicated in the γ-promoters and overlaps CCAAT boxes, a conserved activating motif present in ~30% of promoters (Fig. 1c). Moreover, the CCAAT box often co-occurs with other TF motifs with precise spatial positioning, suggesting an architectural role in activation23–25.
NF-Y, a ubiquitous protein complex composed of NFYA, NFYB and NFYC subunits, is a major effector that recognizes CCAAT. NFYA confers sequence specificity whereas NFYB and NFYC form a nucleosome-like structure with their histone fold domains26. NF-Y disrupts nucleosomal structure27, displays pioneer factor activity28, and is essential for recruitment of TATA box binding protein (TBP) and PolII to promoters28–32.
Although NF-Y has been implicated in globin gene transcription33–36, how it acts remained unclear. The paradoxical effect of dCas9 binding at the BCL11A binding site suggested that dCas9 might prevent binding of an activator, such as NF-Y, in either of two ways. NF-Y might rely on the distal CCAAT box for binding, which is prevented by dCas9 occupancy. Alternatively, dCas9 might displace NF-Y binding at the proximal CCAAT box, which lies within the vicinity of, but not adjacent to the functional BCL11A binding site.
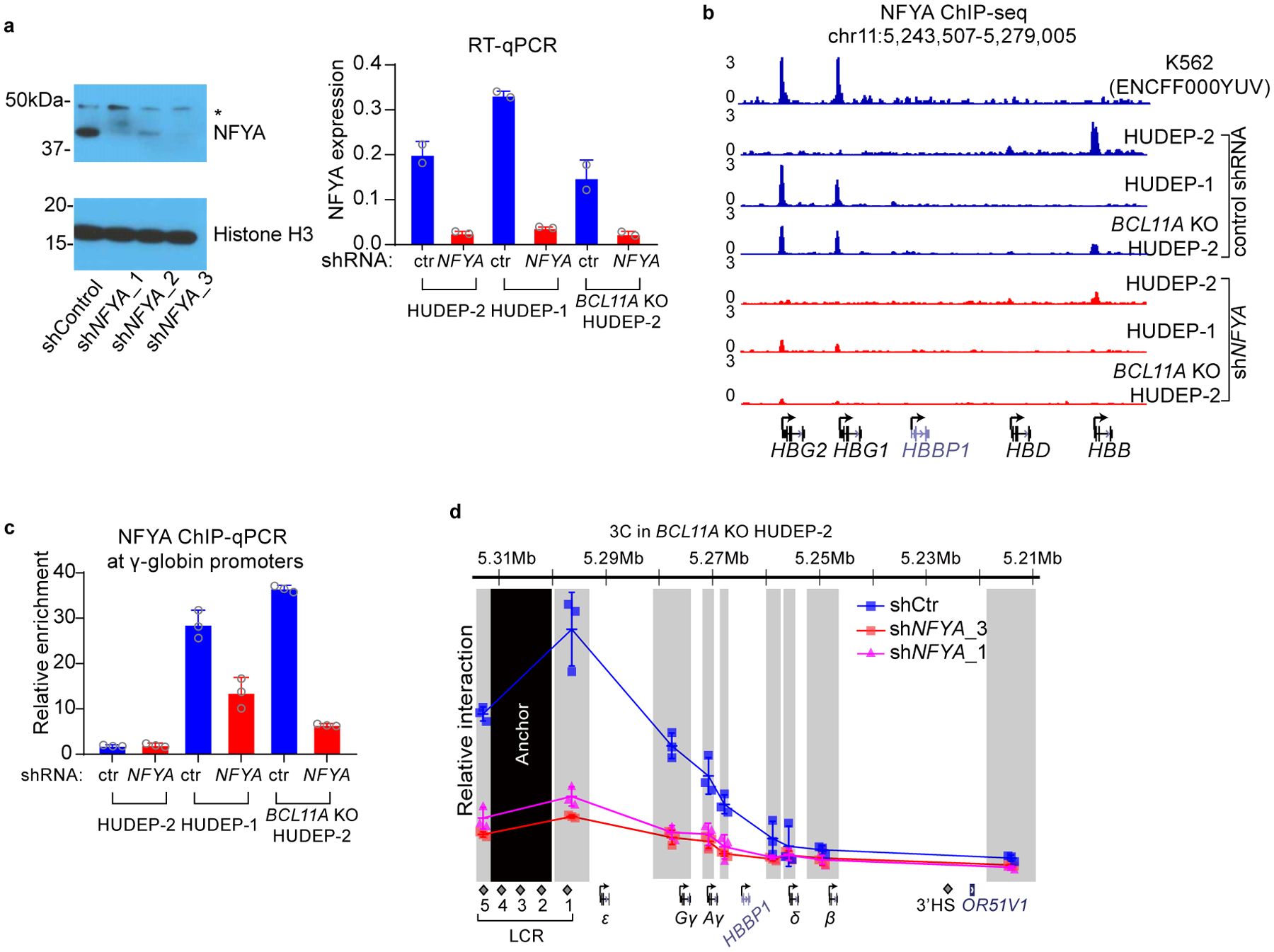
We first explored involvement of NF-Y in γ-globin transcription. shRNA-mediated knockdown of NFYA reduced γ-globin expression in BCL11A KO HUDEP-2 cells, and in HUDEP-1 cells19, where HbF predominates (Fig. 2a and Extended Data Fig. 2a and Source Data). Similar results were obtained in primary human CD34+ cell derived erythroid precursors (see below). ChIP-seq of NFYA revealed occupancy of NF-Y at the γ-globin promoters (Extended Data Fig. 2b–c). These data confirmed that NF-Y regulates γ-globin expression.
Figure 2. NF-Y activates γ-globin through direct binding to the proximal CCAAT.
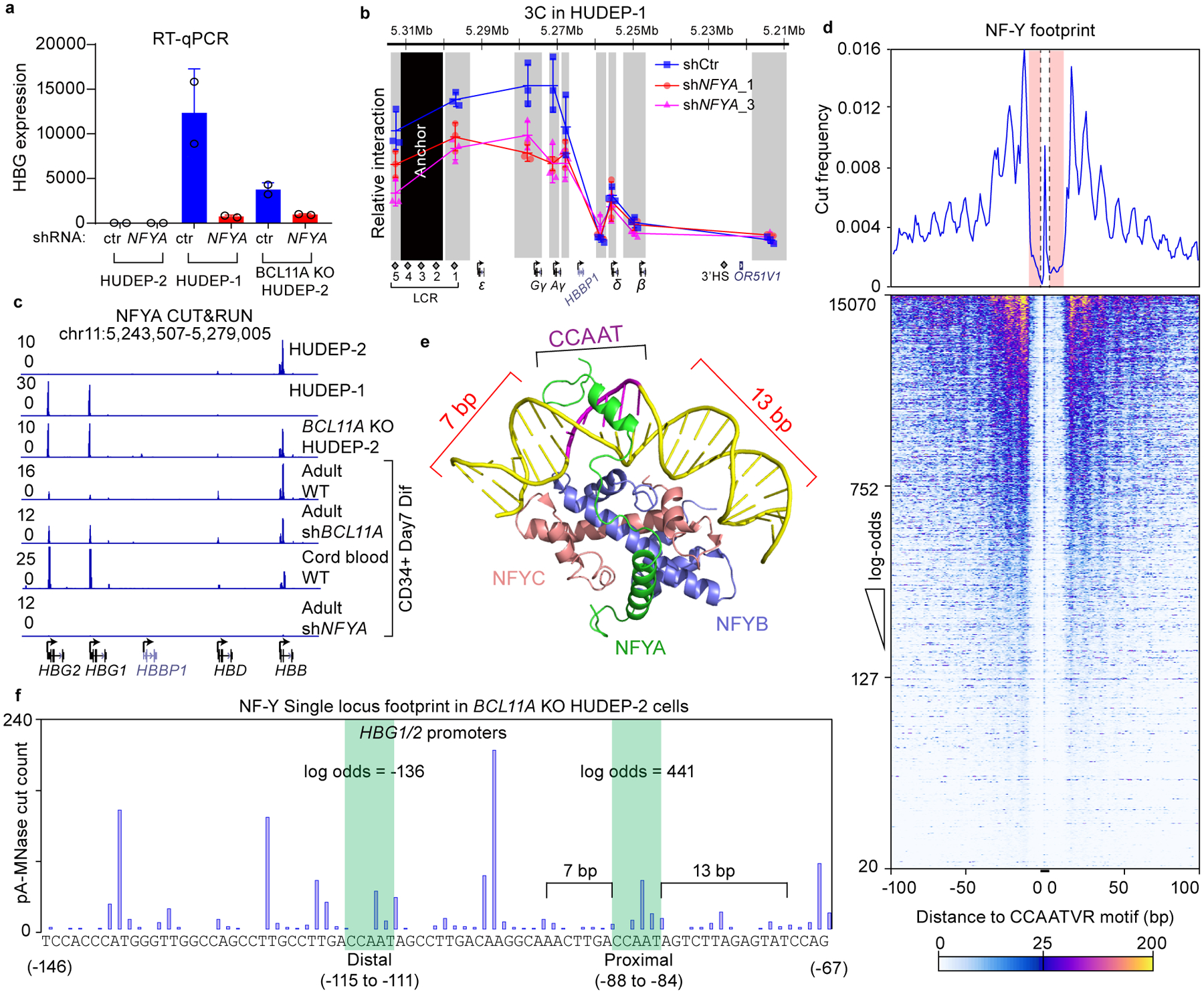
(a) RT-qPCR analysis of γ-globin expression in HUDEP-2, HUDEP-1, BCL11A KO HUDEP-2 cells with and without NFYA knockdown. The result is shown as mean (SD) of two technical replicates and representative of two biological replicates. HPRT1 was used throughout as an endogenous control to normalize between samples.
(b) Chromosome Conformation Capture qPCR in HUDEP-1 cells with or without NFYA knockdown to evaluate LCR-globin interaction. EcoRI fragment encompassing HS2–4 of the LCR was used as anchor point. Each grey box indicates a restriction fragment. The result is shown as mean (SD) of three technical replicates.
(c) NFYA CUT&RUN in HUDEP-2, HUDEP-1, BCL11A KO HUDEP-2 cells and in primary human CD34+ cells derived erythroid cells (four lower tracks are: adult, adult with BCL11A knockdown, fetal, and adult with NFYA knockdown). CUT&RUN tracks in HUDEP cells are representatives of multiple biological replicates. CUT&RUN in CD34+ cells with CRISPR editing yielded similar results (see Figure 4).
(d) Motif footprint analysis of NFYA CUT&RUN. (Upper) Average cut probability of each base surrounding and within CCAATVR motifs was plotted. The core CCAAT motif lies within the dashed lines. Motif flanking regions (7 bp upstream and 11 bp downstream) are shaded red and were protected from nuclease digestion. (Lower) Heat map of NF-Y footprints at all the NF-Y peaks with a log-odds higher than 20. Each row represents one NF-Y binding site, ranked by log-odds value. Color key is shown at the bottom.
(e) Structure of NF-Y/DNA complex adapted from26 and generated with PyMOL. Note that DNA bending is induced by NF-Y binding and flanking sequences are wrapped around NF-Y through histone-fold domains of NFYB and NFYC. NFYA is responsible for motif recognition.
(f) Single locus cut profile at the γ-globin promoters, generated using NFYA CUT&RUN in BCL11A KO HUDEP-2 cells. The proximal CCAAT motif but not the distal one revealed a footprint of NF-Y. The log-odds of NF-Y binding are labeled.
High level expression of individual genes in the β-cluster requires looping of the LCR to the respective gene in a developmental specific manner4. We previously showed that knockout of BCL11A in HUDEP-2 cells shifts LCR interaction from the β- to the γ-globin gene, as revealed by Chromosome Conformation Capture (3C) analysis12,37. Preferential interaction of the LCR with the γ-globin gene in HUDEP-1 and BCL11A KO HUDEP-2 cells was markedly reduced upon NFYA knockdown (Fig. 2b and Extended Data Fig. 2d). Similar results were obtained in CD34+ cell derived erythroid precursors (see below). These results suggest that NF-Y is required for chromosomal looping between the LCR and the γ-globin genes to achieve high-level expression.
NF-Y binds the proximal CCAAT box in the γ-globin promoters
We mapped NF-Y binding within the γ-globin promoters by CUT&RUN, as this nuclease-based method maps TF chromatin occupancy at higher resolution than ChIP-seq12,38 (Extended Data Fig. 3a). NFYA CUT&RUN in HUDEP-2 cells mapped 11,900 peaks genome-wide, 6,357 of which overlapped with NFYA ChIP-seq peaks (Extended Data Fig. 3a–b). The large number of CUT&RUN peaks is due to a lack of an immunoprecipitation step, resulting inindirect peaks caused by pA-MNase cutting at proximal regions12,38. Peaks that reflect direct TF binding are identified by presence of a TF footprint (see below). shRNA-mediated NFYA knockdown led to a global reduction of CUT&RUN signals (Extended Data Fig. 3a). de novo motif discovery within CUT&RUN peaks identified CCAATVR as the most highly enriched motif (Extended Data Fig. 3c)39. We observed NF-Y peaks at γ-globin promoters in HUDEP-1 cells and at the β-globin promoter in HUDEP-2 cells, and at both promoters in BCL11A KO cells (Fig. 2c), a pattern consistent with ChIP-seq and expression of the respective genes. Similar results were obtained in CD34+ cells. NF-Y binds strongly at the γ-globin promoters in cord blood CD34+ and BCL11A knockdown adult CD34+ derived erythroid cells, but less strongly in adult CD34+ derived erythroid cells (Fig. 2c).
Prior in vitro EMSA experiments indicated that NF-Y binds either of the two CCAAT motifs of the γ-promoter40,41. However, the profile of the NF-Y CUT&RUN peak at the γ-globin promoters was biased toward the proximal motif (Extended Data Fig. 3d). As reported previously, the BCL11A CUT&RUN peak is biased to the distal TGACCA BCL11A motif12 (Extended Data Fig. 3d). These observations initially raised the possibility that NF-Y might act at the proximal CCAAT site, rather than at the distal BCL11A site.
To determine the CCAAT motifs bound by NF-Y in vivo, we performed digital footprinting using CUT&RUN data. In principle, DNA sequences bound by a TF are protected from nuclease digestion, while flanking regions are cut more frequently. We generated an average NF-Y footprint profile for all CCAATVR motifs using CUT&RUNTools39, and observed a strong but atypical footprint (Fig. 2d). Discrete cleavage was observed within the core motif, yet flanking sequences were protected from digestion. This pattern is compatible with the structure of the NF-Y-DNA complex26. NF-Y binding induces a kink within the CCAAT motif at the adenine residues (Fig. 2e), which are preferred substrates for micrococcal nuclease42. Moreover, the NF-Y-DNA complex forms a nucleosomal structure that protects the upstream 7 bp and downstream 13 bp DNA of the motif26. The computationally derived CUT&RUN footprint faithfully reflects the native conformation of NF-Y bound to DNA.
The high signal-to-noise ratio of CUT&RUN allows single locus footprinting39. We first plotted the single locus cut profile of the promoter of an established NF-Y target gene, CCNB1 (Cyclin B1)43. Duplicated CCAAT motifs, separated by 27 bp reside in the CCNB1 promoter and exhibit footprints similar to that determined for all CCAATVR motifs (Extended Data Fig. 3e). The calculated log-odds of NF-Y binding at the two CCAAT motifs were 809 and 526, values suggesting a high probability of direct binding. Similar results were obtained for duplicated CCAAT motifs in the CDK1 promoter, another NF-Y target (Extended Data Fig. 3e)44. These observations are consistent with a report that CCAAT boxes separated by 27 bp (32 bp end to end) allow synergistic binding of two NF-Y complexes41.
Following validation of NF-Y footprinting by CUT&RUN, we plotted the single locus cut profile at the γ-promoters in BCL11A KO HUDEP-2 cells. In contrast with findings at the CCNB1 and CDK1 promoters, the cut profile revealed a characteristic NF-Y footprint at the proximal, but not distal, CCAAT motif (Fig. 2f). The calculated log-odds of binding at proximal and distal CCAAT boxes were 441 and −136, respectively. Similar results were obtained in HUDEP-1 and CD34+ cells (Extended Data Fig. 3f). These data demonstrated that NF-Y selectively binds the proximal CCAAT motif of the γ-globin promoters in native chromatin.
Base editing of proximal NF-Y motif impairs γ- expression
To correlate NF-Y binding at the proximal CCAAT motif and γ-globin expression, we used base-editing to convert cytidine to thymidine residues45. We employed Target-AID-NG46, which recognizes NG PAM sequences, as no gRNAs with NGG PAM sequence are available at the site. Base editing was conducted in BCL11A KO HUDEP-2 cells (Fig. 2a, c). To achieve adequate expression of the base editor, we engineered a split intein47 ligated Target-AID-NG (Extended Data Fig. 4a, Source Data and Supplementary Note). Separate expression of Cas9NG-Intein-N and Intein-C-AID resulted in high levels of each protein component. Split intein mediated protein ligation in vivo generated full-length Target-AID-NG at a level exceeding that from a vector expressing intact Target-AID-NG (Extended Data Fig. 4a).
The proximal CCAAT NF-Y motif was edited by the split intein Target-AID-NG (Fig. 3a) at a conversion rate of 48% on day 11. In contrast to bulk-edited cells (Fig. 3b), independent, single cell clones with base edits exhibited reduced γ-globin expression (Fig. 3c).
Figure 3. Base editing of the NF-Y motif reduces γ-globin expression.
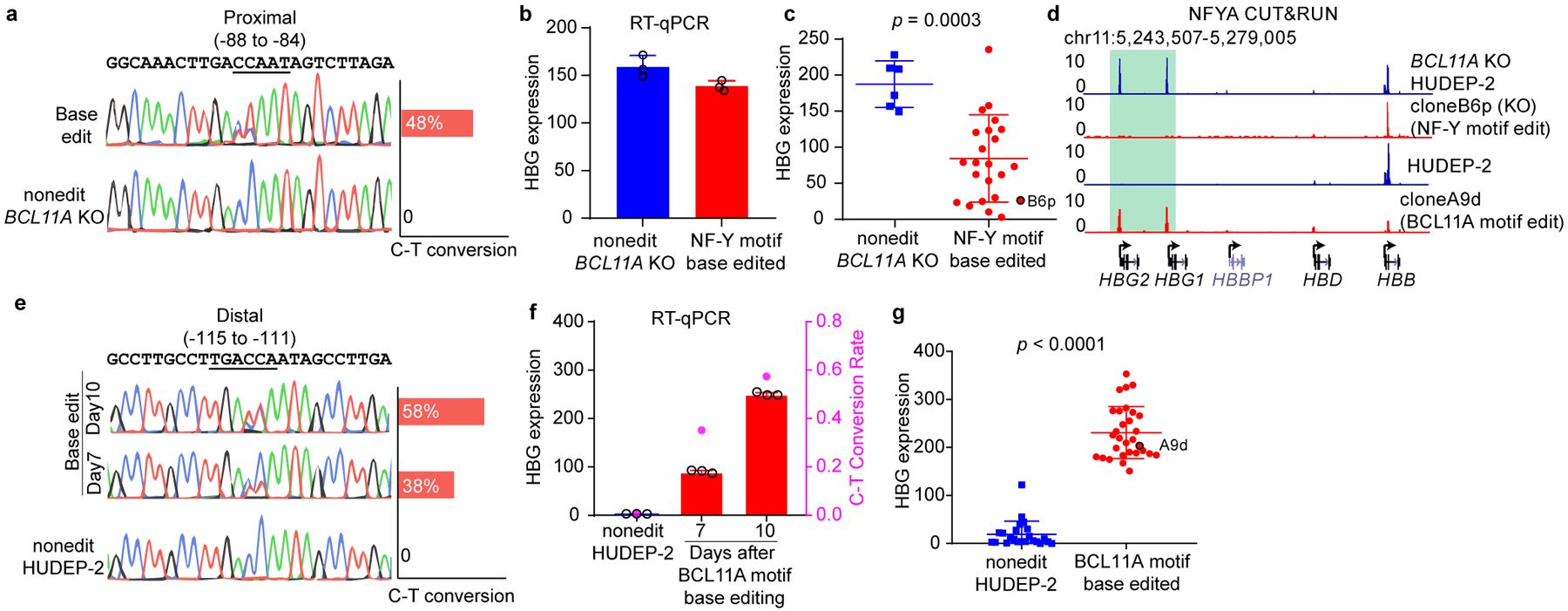
(a) Sanger sequencing confirmed base editing of the NF-Y motif in the γ-globin promoters. Quantification of editing efficiency is shown as bar graph on the right. The editing is performed in BCL11A KO HUDEP-2 cells.
(b) RT-qPCR showing the γ-globin expression level in bulk cells after NF-Y motif editing. The result is shown as mean (SD) of three technical replicates.
(c) RT-qPCR analysis of γ-globin expression in multiple independent clones derived from NF-Y motif base editing. Data is showed as mean (SD) of multiple independent clones. Nonedit: n=6, base edited: n=27. Two-tailed, unpaired t-test, t=4.010, df=32. Clone B6p (circled) was used for CUT&RUN analysis in Figure 3d.
(d) NFYA CUT&RUN in BCL11A KO HUDEP-2, NF-Y motif edit clone B6p (in BCL11A KO background), HUDEP-2, and BCL11A motif edit clone A9d (wild-type background).
(e) Sanger sequencing confirmed base editing of the BCL11A motif in the γ-globin promoters. Quantification of editing frequency is shown as bar graph on the right. The editing is performed in wild-type HUDEP-2 cells.
(f) RT-qPCR showing the γ-globin expression level in bulk cells after BCL11A motif editing. Cells collected at different time points showed different degrees of editing. The result is shown as mean (SD) of three technical replicates. C-T conversion rates are shown as the right y-axis.
(g) RT-qPCR analysis of γ-globin expression in multiple independent clones derived from BCL11A motif base editing. Data is showed as mean (SD) of multiple independent clones. Nonedit: n=23, base edited: n=30. Two-tailed, unpaired t-test, t=17.11, df=51. Clone A9d (circled) was used for CUT&RUN analysis in Figure 3d.
To assess NF-Y occupancy, we performed NF-Y CUT&RUN in 9 independent clones. NF-Y binding was diminished at the γ-promoters in all clones (Fig. 3d and Extended Data Fig. 4b). BCL11A CUT&RUN was not conducted since the clones were generated in a BCL11A null background. Taken together, base editing of the proximal CCAAT motif, the site of NF-Y binding in vivo, impaired NF-Y occupancy and γ-globin gene expression.
Other proteins have been reported to bind CCAAT sequences based on in vitro assays. These include C/EBP (CCAAT Enhancer Binding Protein) family, CDP/CUX1 (CCAAT Displacement Protein) and NFI (Nuclear Factor I) protein families. Subsequent ChIP-seq experiments have determined different binding motifs for each protein family, none of which match the γ-globin CCAAT box. We knocked-out each protein in BCL11A KO HUDEP-2 cells. None revealed contribution to γ-globin activation approximating that of NF-Y (Extended Data Fig. 4c and Source Data). Therefore, we conclude that NF-Y is the predominant factor acting at the proximal CCAAT box of γ-globin promoters.
Base editing of distal BCL11A motif promotes NF-Y binding
We previously reported that BCL11A functions through the distal TGACCA motif, rather than the proximal TGACCA motif that also overlaps a CCAAT sequence12. In contrast, the data presented here indicate that NF-Y acts through the proximal CCAAT and not the distal CCAAT box. Therefore, the functionally relevant BCL11A and NF-Y binding sites are encompassed within 35 bp of the promoter. We next explored how mutation of the BCL11A binding site affects NF-Y occupancy. The unexpected finding in the dCas9 screen (Fig. 1) that positioning dCas9 at the BCL11A motif represses γ-globin led us to ask whether the distal CCAAT also supports activation and is required for NF-Y function. We base edited the distal motif (TGACCAAT to TGATTAAT). As Target-AID-NG failed to edit the distal motif in HUDEP-2 cells, we used a codon-optimized cytidine base editor48 and achieved 38% and 58% C-T conversion on day 7 and day 10, respectively (Fig. 3e). We observed marked derepression of γ-globin expression in bulk edited cells, and the level of derepression correlated with the extent of C-T conversion, supporting the role of the distal motif as a repressor binding site (Fig. 3f and Extended Data Fig. 4d). Edits of a nearby cytidine 10 bp away with a different gRNA failed to perturb γ-globin expression, indicating that the effects are not attributable to base editor binding or base editing per se (Extended Data Fig. 4e). We sorted bulk edited cells based on intracellular HbF levels on day 10 and compared C-T conversion in HbF high and low populations. The distal motif was 87% edited in HbF high cells, but only 7.4% in HbF low cells (Extended Data Fig. 4f). Single clones were isolated and γ-globin expression was assessed by RT-qPCR and flow cytometry. All clones with C-T conversion at the BCL11A motif exhibited increased γ-globin expression (Fig. 3g and Extended Data Fig. 4g), confirming repression through the distal motif. We observed strong NF-Y binding in BCL11A motif edited clones (Fig. 3d and Extended Data Fig. 4b). Single locus footprinting revealed protection of the proximal CCAAT motif, providing additional evidence for NF-Y action at the proximal motif (Extended Data Fig. 4h). Thus, mutation of the distal BCL11A motif was accompanied by NF-Y occupancy at the proximal CCAAT motif and robust γ-globin expression. These results support base editing of the distal BCL11A motif for reactivation of HbF as treatment for sickle cell disease or β-thalassemia49.
BCL11A depletion leads to NF-Y binding and γ- transcription
Knockout and editing experiments provide a static view at the endpoint of a genetic pertubation, and the resulting interpretation may be confounded by secondary effects. To explore the dynamics of NF-Y binding and downstream events upon acute depletion of BCL11A, we knocked-out BCL11A in CD34+ cells undergoing erythroid differentiation by nucleofection of Cas9/sgRNA ribonucleoprotein complex (RNP) (Extended Data Fig. 5a). BCL11A protein decreased to 35% at 32 hrs after RNP delivery (Fig. 4a and Source Data). γ-globin transcripts remained unchanged at 32 hrs, and increased thereafter (Fig. 4b). As RT-qPCR quantifies the total level of old and newly synthesized RNA, it does not directly measure transcriptional dynamics. To compare γ-globin transcription rates at different times, we analyzed nascent transcripts using PRO-seq (Precision nuclear run-on sequencing)50. Two replicates were highly correlated (Extended Data Fig. 5b). We observed promoter pausing of PolII, indicating a successful PRO-seq experiment (Extended Data Fig. 5c). Acute depletion of BCL11A did not lead to global changes in transcription: only 9 and 110 genes were transcribed differentially at 32 or 72 hrs, respectively (Fig. 4c). Consistent with RT-qPCR, γ-globin transcription remained unchanged at 32 hrs, but was increased at 72 hrs in BCL11A KO (Fig. 4c–d, Extended Data Fig. 5d). We chose 32- and 72- hrs for detailed analysis. 32-hr represents a time at which changes at globin promoters are initiated, whereas 72-hr approximates an endpoint.
Figure 4. NF-Y rapidly activates γ-globin after acute depletion of BCL11A.
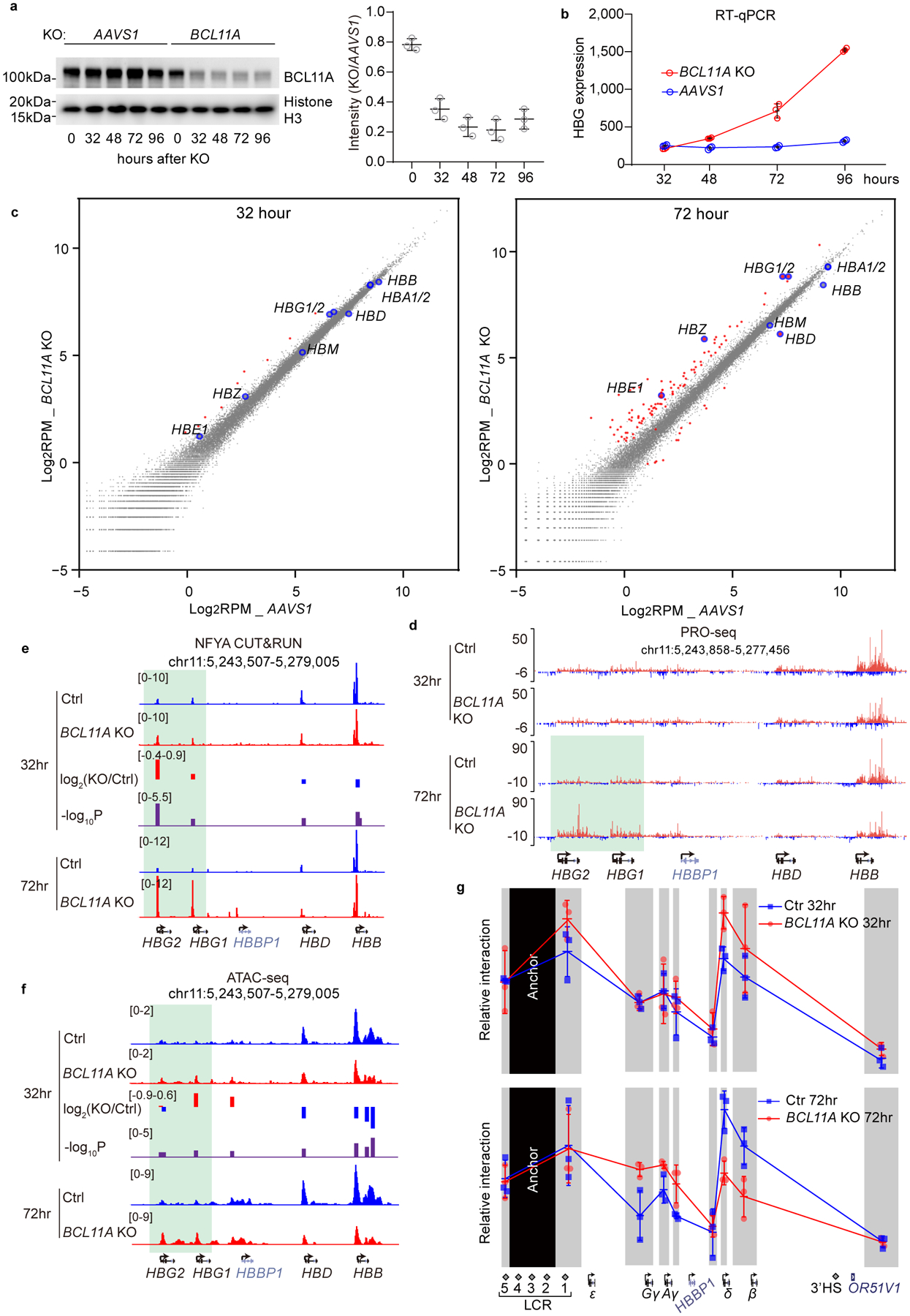
(a) Left, western blot showing the level of BCL11A after CRISPR/Cas9 mediated acute BCL11A KO at different time points in adult primary human CD34+ cells undergoing erythroid differentiation (cropped). Right, quantification of BCL11A depletion of the left experiment using ImageJ. The result is a representative of two biological replicates. The control cells were edited with AAVS1 sgRNA.
(b) RT-qPCR analysis of γ-globin expression level at different time points after acute depletion of BCL11A. Data is showed as mean (SD) of three technical replicates.
(c) Scatter plot of PRO-seq data at 32 hrs (left) and 72 hrs (right) after acute depletion of BCL11A. The x-axis represents log2RPM (reads per million) of each gene in control experiments (AAVS1) and the y-axis represents log2RPM of each gene in BCL11A KO experiments. Each dot represents a gene. Globin genes are enlarged and highlighted in blue circles. Genes that are significantly different between control and KO (log2FC(KO/Ctrl) <−1 or >1) are highlighted in red. The results are shown as means of two biological replicates.
(d) PRO-seq tracks at the β-globin locus. Transcripts of positive and negative strands are shown in different colors. γ-globin remained repressed in BCL11A KO at 32 hrs. Derepression became evident at 72 hrs, as highlighted in green.
(e,f) NFYA CUT&RUN (e) and ATAC-seq (f) in CD34+ cells undergoing erythroid differentiation after 32 or 72 hrs of BCL11A acute depletion. Quantification of KO/Ctrl and the corresponding p-values are reported by MAnorm.
(g) Chromosome Conformation Capture qPCR in CD34+ cells undergoing erythroid differentiation after 32 (upper) or 72 (lower) hrs of BCL11A acute depletion. EcoRI fragment encompassing HS2–4 of the LCR was used as anchor point to evaluate LCR-globin interaction. The result is shown as mean (SD) of three technical replicates and is a representative of two biological replicates.
We compared chromatin accessibility, TF binding, and LCR-globin looping by ATAC-seq, CUT&RUN, and 3C-qPCR, respectively. At 72 hrs, NF-Y and TBP exhibited strong occupancy at the γ-promoters in BCL11A KO (Fig. 4e and Extended Data Fig. 5e). Chromatin accessibility increased (Fig. 4f), and contacts between the LCR and γ-globin genes were more frequent, whereas those with the β-globin gene were reduced (Fig. 4g lower panel). Combined depletion of NF-Y and BCL11A impaired γ-globin expression (Extended Data Fig. 5f–g and Source Data) and reduced LCR-γ-globin interaction at 72 hrs (Extended Data Fig. 5h). These results confirmed opposing roles of BCL11A and NF-Y. Data obtained at 32 hrs after BCL11A KO provided insights into the order of events. At this time, we observed increased NF-Y and TBP binding at the γ-promoters (Fig. 4e and Extended Data Fig. 5e), with a fold increase of 1.9 (p=3.2×10−6) and 1.6 (p=7.9×10−5), respectively, as quantified with MAnorm algorithm51. Chromatin accessibility (Fig. 4f) showed a small but statistically insignificant increase. LCR-γ-globin gene interaction (Fig. 4g upper panel), and γ-globin transcription (Fig. 4b–d) exhibited negligible changes. These data reveal that NF-Y binds rapidly to the γ-globin promoters upon BCL11A depletion to open up local chromatin, which precedes formation of enhancer-promoter contacts and transcriptional activation, consistent with NF-Y pioneer activity25,27,28.
BCL11A displaces NF-Y by steric hindrance
The above findings suggested a dynamic model for how BCL11A initiates γ-globin repression. We posit that binding of BCL11A (or dCas9) at the distal TGACCA sequence constitutes a steric barrier to NF-Y binding at the proximal CCAAT motif.
To test this model, we explored how recruitment of dCas9 at different positions affects γ-globin expression and NF-Y binding. We transduced 8 gRNAs of the dCas9 screen individually into dCas9-expressing cells (Fig. 5a). To detect changes in NF-Y binding upon dCas9 recruitment, we employed BCL11A KO HUDEP-2 cells, rather than wild-type HUDEP-2 cells in which NF-Y binding and γ-globin expression are low. As validation for subsequent experiments, we confirmed that dCas9 recruitment to sequences at −62 to −139 led to reduced γ-globin expression, and dCas9 recruitment to −197 or −208 led to an increase of γ-globin due to displacement of ZBTB7A9 (Fig. 5b). CUT&RUN revealed that NF-Y occupancy was reduced, though partially, upon placement of dCas9 at −102 or −62, which overlaps the NF-Y motif or downstream flanking sequence, respectively, suggesting that dCas9 displaces NF-Y upon recruitment to an NF-Y binding site. These findings are consistent with the modest reduction of γ-globin expression. Recruitment of dCas9 to the distal BCL11A motif (gRNAs-115 and −124) or even further (gRNA-139) perturbs NF-Y binding at the proximal CCAAT motif, and reduces γ-globin expression. Therefore, dCas9 targeted to the BCL11A motif impairs NF-Y binding to its motif 24 bp downstream.
Figure 5. NF-Y binding is affected by steric hindrance at the γ-globin promoters.
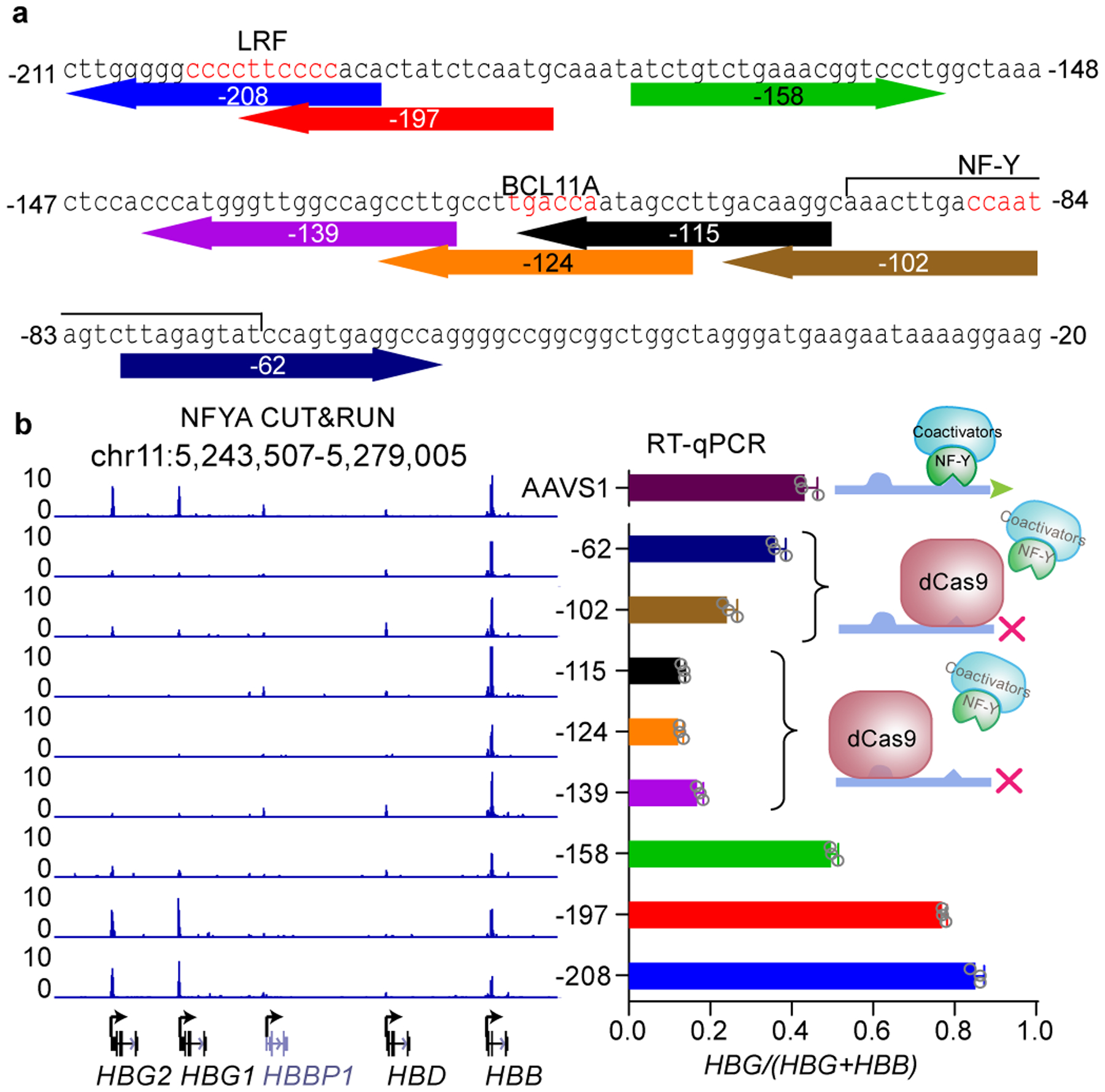
(a) The sequence of γ-globin promoters. The eight sgRNAs used for dCas9 disruption experiment are shown below the sequence and color coded. The numbers (−208, etc) represent the distances between TSS and positions of the 17th nucleotide of each sgRNA. The position of LRF, BCL11A and NF-Y motifs are labeled in red. The flanking sequences of NF-Y motif is also involved in NF-Y binding.
(b) Right, RT-qPCR analysis of the percentage of γ-globin in BCL11A KO HUDEP-2 cells expressing dCas9 with different sgRNAs. Data is showed as mean (SD) of three technical replicates and a representative of two biological replicates. Left, NFYA CUT&RUN in these cells. The cartoon indicates the deduced protein binding at the promoters.
Discussion
Here we reveal that γ-globin repression in adult cells is controlled by competitive binding of BCL11A and NF-Y within the γ-globin promoters. This finding was inspired by a paradoxical observation that dCas9 displacement of BCL11A binding at the γ-globin promoters further represses, rather than activates expression. Protein binding (either BCL11A or dCas9) at the BCL11A motif presents a steric barrier to the activator NF-Y, whose relevant binding motif lies 24 bp downstream. Competitive binding between BCL11A and NF-Y determines the activity of the γ-globin promoter and transcription output.
We propose a TF competition model for initiation of hemoglobin switching (Fig. 6). In adult cells, BCL11A expression rises with erythroid commitment and peaks during erythroblast maturation, at which time BCL11A binding is detectable at the γ-globin promoters12. BCL11A prevents NF-Y binding through steric hindrance to initiate repression, followed by involvement of corepressors. BCL11A interacts with the NuRD corepressor complex52, which possesses nucleosome remodeling and histone deacetylation activities. NuRD recruitment promotes chromatin compaction and the repressive state of the γ-globin promoters. NuRD components are 100 times more abundant than coactivators in erythroid cells53. This cofactor imbalance ensures the competitive advantage of BCL11A-NuRD. The roles of BCL11A and NuRD can be bypassed by placing dCas9 at the BCL11A motif, which displaces NF-Y. In fetal progenitors, BCL11A protein is expressed at a low level10,54. NF-Y occupies the γ-globin promoters and modifies chromatin through recruitment of cofactors (e.g. P300) to activate γ-globin expression. NF-Y activates γ-globin similarly in erythroid cells harboring mutations at the BCL11A binding motif or BCL11A gene, either in genetically modified cellular models12 or in individuals with HPFH.
Figure 6. A simplified model for hemoglobin switching.
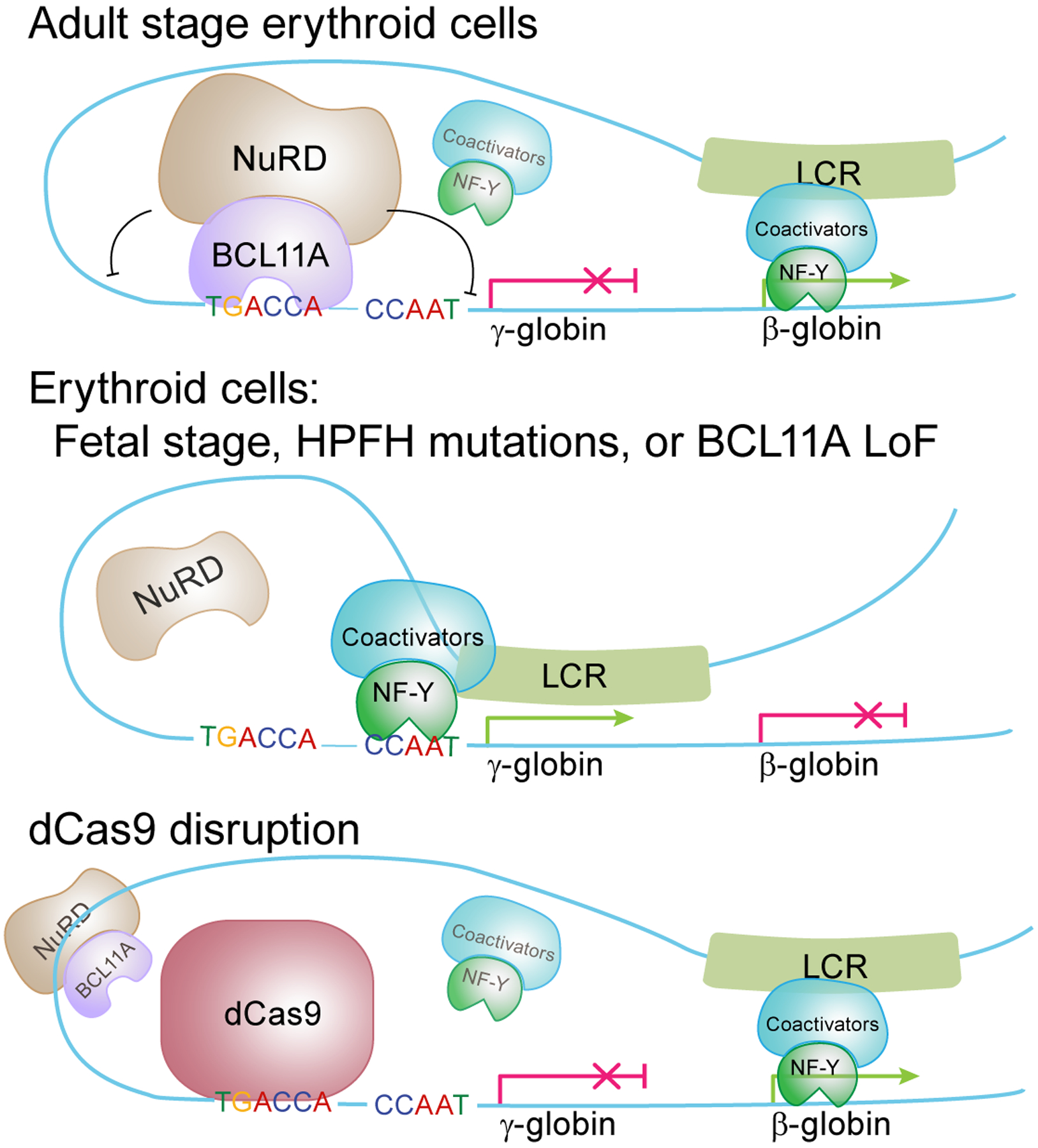
Competitive binding between NF-Y and BCL11A controls hemoglobin switching. In fetal stage erythroid cells, or cells with HPFH mutations or lacking BCL11A, NF-Y binds to the γ-globin promoters and activates expression. In adult stage erythroid cells, BCL11A prevents NF-Y binding and represses γ-globin in concert with NuRD. LRF/ZBTB7A independently recruits NuRD and represses γ-globin through binding to the −200 bp region of the γ-globin promoters (not illustrated in the model). dCas9 binding at the BCL11A motif is sufficient to disrupt NF-Y binding and repress γ-globin expression. When γ-globin is silenced, NF-Y may bind to β-globin and regulate its expression. Other known positive regulators of β-globin including GATA1, KLF1, LDB1, etc are not shown.
In addition to BCL11A-NF-Y competition, coordinated action of other factors likely contributes to γ-globin repression. LRF/ZBTB7A, a second HbF repressor, acts at −200 bp and independently recruits NuRD9,13. We speculate that the concerted action of BCL11A and LRF/ZBTB7A for NuRD recruitment stabilizes the repressed state of the γ-promoters, and loss of either factor increases chromatin accessibility, thereby allowing NF-Y to gain partial competitive advantage. Other repressors suggested to act at the γ-globin promoters (such as TR2/TR4, SOX6, KLF3, COUP-TFII)55 lack supporting evidence to substantiate postulated roles.
The effects upon acute depletion of BCL11A suggest that TF binding precedes and directs LCR-globin looping. Distinct proteins recruited by TFs and the histone marks they deposit may stabilize or restrict looping depending on their biochemical compatibility, as determined by their specific interactions56. This conclusion is consistent with the finding that TF binding is an upstream event of enhancer-promoter looping at the α-globin locus57.
Our work presents a parsimonious model for initiation of hemoglobin switching, and highlights competitive TF binding within 35 bp of the γ-globin promoters in determination of stage specificity for the >50 kb β-globin cluster. Spatial constraints have been previously reported for NF-Y promoter binding. In vitro studies revealed that two NF-Y molecules exhibit synergistic binding to double CCAAT motifs when the distance between motifs is 27 bp (three helical turns). NF-Y may still occupy double CCAAT motifs without synergy when the distance is reduced by a few basepairs, but co-binding was not detected when the distance is reduced to 17 bp40,41. C/EBP and NF-Y binding are mutually exclusive in the albumin gene promoter where a CCAAT motif is juxtaposed to a C/EBP site. Yet, the factors may cobind if the distance between the two motifs is increased by 10 bp58. Studies with synthetic promoters demonstrate that promoter activity is reduced if the distance between cis elements is less than 10–15 bp59. More broadly, instances of competitive protein binding at promoters are observed in lower organisms whose TFs usually lack effector domains60. For example, the E. coli lac repressor sterically restricts RNA polymerase binding. Collectively, TF competition is widely used for modulation of gene activity. In the context of globin gene regulation, the precise architecture of the γ-globin promoters has evolved to provide a platform for dynamic eviction of a ubiquitous activator by a stage-selective repressor as the basis for the fetal-to-adult globin switch.
Online Methods
Culture of Primary cells
Human peripheral blood stem cells (CD34+ cells) (G-CSF mobilized, CD34+ enriched) were purchased from the Center of Excellence in Hematology at the Fred Hutchinson Cancer Research Center. These deidentified samples were exempted from BCH IRB approval. Cells were thawed and recovered to EDM (IMDM (Corning, 15–016-CV) supplemented with 330 μg/mL Holo-Human Transferrin, 10 μg/mL Recombinant Human Insulin, 2 IU/mL Heparin, 5% Inactivated Plasma, 3 IU/mL Erythropoietin, 2 mM L-Glutamine) with three supplements (10−6 M hydrocortisone, 100 ng/mL SCF, 5 ng/mL IL-3) for 7 days to allow erythroid differentiation, then further differentiated in EDM with one supplement (100 ng/mL SCF).
Culture of immortalized Cell lines
Human HEK293T cells (female) were purchased from ATCC, and cultured in DMEM, high glucose (Thermo Fisher Scientific, 11965) with 10% FCS and 2 mM L-Glutamine and passaged every three days.
HUDEP-1 and HUDEP-2 lines (human umbilical cord blood-derived erythroid progenitor, male) were provided by Dr. Yukio Nakamura. HUDEP-2 cells were maintained in expansion medium: StemSpan™ SFEM (STEMCELL Technologies, 09650) with SCF (50 ng/ml), EPO (3 IU/ml), Dexamethasone (10−6 M), Doxycycline (1 μg/ml), and passaged every three days. Erythroid differentiation was carried out by replacing the medium to EDM2 (Erythroid Differentiation Media: IMDM (Corning, 15–016-CV) supplemented with 330 μg/mL Holo-Human Transferrin, 10 μg/mL Recombinant Human Insulin, 2 IU/mL Heparin, 5% Inactivated Plasma, 3 IU/mL Erythropoietin, 2 mM L-Glutamine, 100 ng/mL SCF, 1 μg/mL doxycycline).
Design and synthesis lentiviral sgRNA libraries
We identified every 20-mer sequence upstream of the SpCas9 NGG PAM sequence at the HBB locus from hg19 chr11:5,220,000–5,326,000 (106 kb comprising the HBB gene cluster inclusive of the HS5 to 3’HS1 distal elements). 10,383 spacer sequences were designed (including 100 negative control genome targeting spacers with 95 safe-targeting61 and 5 AAVS1 targeting spacers and 990 positive controls at BCL11A). The sgRNA oligos (synthesized by CustomArray) were cloned using a Gibson Assembly master mix (New England Biolabs) into lentiGuide-Puro (Addgene plasmid #5296362) which had been BsmBI digested, gel purified, and dephosphorylated. Gibson Assembly products were transformed to electrocompetent cells (E. cloni 10G ELITE Electrocompetent Cell, Lucigen,60052). Sufficient colonies were isolated to ensure more than 1000 colonies per spacer sequence. Plasmid libraries were deeply sequenced to confirm representation.
To produce lentivirus, HEK293T cells were cultured with Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Omega Scientific) and 2% penicillin-streptomycin (Life Technologies) in 15 cm tissue culture treated Petri dishes. HEK293T cells were transfected at 80% confluence in 12 ml of media with 13.3 μg psPAX2 (Addgene plasmid #12260, a gift from Didier Trono), 6.7 μg VSV-G (Addgene plasmid #14888, a gift from Tannishtha Reya), and 20 μg of the lentiviral construct plasmid of interest using 180 μg of linear polyethylenimine (Polysciences). Medium was changed 16–24 h after transfection. Lentiviral supernatant was collected at 48 and 72 h post-transfection and subsequently concentrated by ultracentrifugation (24,000 rpm for 2 h at 4°C with Beckman Coulter SW 32 Ti rotor).
Transducution of HUDEP-2 cells with lentiviral library
HUDEP-2 cells were transduced with lentivirus encoding various Cas9 or dCas9 variants (Cas9: LentiCas9-Blast, Addgene plasmid #5296262; dCas9: pGH125_dCas9-Blast, Addgene plasmid #8541763; dCas9-KRAB: pHR-SFFV-dCas9-BFP-KRAB, Addgene plasmid #4691164; dCas9-VP64: pHRdSV40-dCas9–10xGCN4_v4-P2A-BFP, Addgene plasmid #60903 and pHRdSV40-scFv-GCN4-sfGFP-VP64-GB1-NLS, Addgene plasmid #6090465) to produce stably expressing cells. The cells were then transduced with the sgRNA lentivirus library and selected with 1 μg/ml puromycin. Cells were transduced at MOI of 0.3. Cell numbers were maintained throughout the experiment with at least 1000 cells per spacer sequence. After sgRNA transduction, cells were cultured in EDM2 medium for 12 days.
HbF staining and fluorescence activated cell sorting
After differentiation, intracellular staining was performed by fixing cells with 0.05% glutaraldehyde (grade II) (Sigma) for 10 min at room temperature. Cells were centrifuged for 5 min at 600 × g and then resuspended in 0.1% Triton X-100 (Life Technologies) for 5 min at room temperature for permeabilization. Triton X-100 was diluted with phosphate buffered saline (PBS) with 0.1% BSA and then centrifuged at 600 × g for 15 min. Cells were stained with antibodies for HbF (clone HbF-1 with FITC or APC conjugation; Life Technologies, MHFH01) for 20 min in the dark. Cells were washed to remove unbound antibody before FACS. 0.2 μg HbF antibodies were used per 5 million cells. A population of cells with the top 10% HbF expression was sorted by FACS. A total of 12 samples were collected: 3 biological replicates of Cas9-sorted, Cas9-unsorted, dCas9-sorted, and dCas9-unsorted.
Amplification of sgRNA and deep sequencing
After collecting unsorted total and the HbF-high sorted cell populations at the end of erythroid maturation culture, library preparation and deep sequencing were performed. Briefly, genomic DNA was extracted using the Qiagen Blood and Tissue kit. sgRNA integrant PCR to amplify spacer sequences was performed with Herculase II reaction buffer (1X), forward and reverse primers (0.5 μM each), dimethyl sulfoxide (DMSO) (8%), deoxynucleotide triphosphates (dNTPs) (0.25 μM each), Herculase II Fusion DNA Polymerase (0.5 reactions) using the following cycling conditions: 95°C for 2 min; 20 cycles of 95°C for 15 s, 60°C for 20 s, 72°C for 30 s; 72°C for 5 min. Multiple reactions of no more than 200 ng each were used to amplify from 6.6 μg gDNA (~1×106 cell genomes) per pool. Samples were subjected to additional rounds of PCR to add sequencing adaptors and indices before Illumina sequencing.
Data analysis for Cas9 and dCas9 screens
In total 8,639 sgRNAs (including 977 positive control and 100 negative control guides) were retained after filtering by MIT Specificity Score >= 5. For each sgRNA, the read counts were calculated from raw fastq files with custom codes. Quality control meta data was shown in Supplementary Table 1. DESeq221 (run on R 4.0.1) was then used to compare the sgRNA counts between sorted and unsorted samples expressing Cas9 and dCas9 variants. The log2 fold change values for HBB locus were generated by DESeq2. gRNA positions in the bedGraph files were displayed at the default cleavage position 17–18 for Cas9 and the center of the guide for dCas9. Analysis of the dense perturbation data was also performed with CRISPR-SURF66. First, the sgRNA counts were used as input into CRISPR-SURF Count with the following parameters: -nuclease cas9, -pert indel (for Cas9 samples) or -pert crispri (for dCas9 samples). With the resulting output, CRISPR-SURF Deconvolution was applied with the following parameters: -pert cas9, -sim_n 1000, and the perturbation range parameter -range 7 (for Cas9) or -range 20 (for dCas9). The CRISPR-SURF deconvolution analysis provides p-values and statistical power for the CRISPR dense perturbation data.
Validation of dCas9 disruption at the γ-globin promoters
Plasmids that express 8 gRNAs shown in Figure 5a were individually constructed, followed by lentivirus packaging with the protocols described above except using a small scale. BCL11A KO HUDEP-2 cells expressing dCas9 were transduced with each virus and selected using puromycin. After five days of erythroid differentiation, cells were collected for gene expression analysis and CUT&RUN. gRNA sequences are listed in Supplementary Table 2.
RT-qPCR
Total RNA was extracted from 1 million freshly collected cells or frozen pellets using Qiagen RNeasy Mini Kit. Synthesis of cDNA was carried out using iScript™ cDNA Synthesis Kit (Bio-rad). The cDNA was diluted 5 times and 1 μL was used for each qPCR reaction. qPCR was performed with iQ SYBR Green Supermix (Bio-rad) using Bio-rad CFX Real-Time PCR Detection system. HPRT1 was used as endogenous control to normalized between samples. All the RT-qPCR primers are listed in Supplementary Table 2. The data were analyzed using 2−ΔΔCT method and visualized using GraphPad. Three technical replicates, at least two biological replicates were performed for each experiment.
shRNA knockdown
The vectors of shRNAs were purchased from Sigma-Aldrich. Entrance numbers were listed in Supplementary Table 3. Lentivirus encoding shRNAs were packaged using HEK293T cells. 100 μL culture supernatant containing virus particles were added to 1 mL HUDEP or CD34+ cell suspension. The next day, fresh media supplemented with puromycin was added to select transduced cells. Knockdown efficiency was validated through RT-qPCR or immunoblot.
CUT&RUN
Briefly, 1 million cells were collected and frozen in 10% DMSO in FBS, as necessary. Frozen cells were thawed, pelleted, and washed once with PBS, and processed according to the following steps: 1) addition of 0.5 mL of NE buffer (20 mM HEPES-KOH, pH 7.9, 10 mM KCl, 0.5 mM Spermidine, 0.1% Triton X-100, 20% Glycerol and 1x protease inhibitor cocktails from Sigma) for nuclei isolation. Nuclei were pelleted at 600 × g for 3 min, resuspended with 0.4 mL of NE buffer. 2) 25 μL BioMagPlus Concanavalin A beads slurry was washed twice and resuspended in 200 μL Binding Buffer (20 mM HEPES pH 7.5, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2), and added to the nuclei suspension. The mixture was thoroughly mixed on a rocker for 10 min to allow binding of nuclei to the beads. 3) The nuclei were pelleted with a magnet stand and blocked with Wash Buffer (20 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 0.1% BSA and 1x protease inhibitor cocktails from Sigma) supplemented with 2 mM EDTA. After washing once with Wash Buffer, nuclei were resuspended in 200 μL Wash buffer containing 2 μg of antibody, and incubated overnight to allow antibody binding. 4) The next day, wash the nuclei twice on a magnet stand with wash buffer and incubate with 1:1000 pA-MN in 200 μL Wash Buffer. After 1 hr, wash twice to remove unbound pA-MN. 5) Resuspend the nuclei in 150 μL Wash Buffer, chill on a 0°C metal block that sits in water-ice mixture. Add 3 μL 100 mM CaCl2 to activate pA-MN, and incubate at 0°C for 60 min. 6) The reaction was stopped by adding 150 μL 2XSTOP buffer (200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 μg/mL RNase A and 40 μg/mL glycogen). The protein-DNA complex was released by centrifugation and then digested by proteinase K at 50°C overnight. DNA was extracted by ethanol precipitation, followed by Qubit fluorometer and bioanalyzer quality control.
Antibodies used for CUT&RUN: BCL11A, ab191401, Abcam; NFYA, sc-17753, Santa Cruz Biotechnology; TBP, ab220788, Abcam.
CUT&RUN library preparation
Library preparation of CUT&RUN was performed using NEB Ultra II library preparation kit with important modifications. Briefly, less than 30 ng DNA was used as input. End prep was performed at 20°C for 30 min and then 50°C for 60min (instead of 65°C for 30 min in the manual). The reduced temperature prevents melting of short DNA fragments. 5 pmol adaptor was added and ligated to end prep products at 20°C for 15 min. USER enzyme was then used to cleave the uracil in the loop. 1.7x volume of AMPure beads were used to purify the ligation product. The use of 1.7x beads is essential for recovery of short fragments. To amplify the library, the ligation product was mixed with 2x Ultra II Q5 mix, universal primer and index primers. PCR was carried out as follows: 98°C 30 s, 12 cycles of 98°C 10s and 65°C 10s, and final extension 65°C 5 min. The PCR product was subjected to double size selection with 0.8x and then 1.2x AMPure beads. The purified library was quantified with Qubit and Tapestation. Libraries were pooled at similar molar amount and sequenced using Nextseq500 platform. Paired-end sequencing, read length 42 bp × 2, 6 bp index was performed.
For detailed protocol please see: dx.doi.org/10.17504/protocols.io.wvgfe3w
CUT&RUN data processing
Raw data was processed using FastQC and CUT&RUNTools39. The global settings were: fastq_sequence_length=42, organism_build=hg19, num_bp_from_summit=100, num_peaks=5000, total_peaks=15000, motif_scanning_pval=0.001, num_motifs=15. Parameters of individual software called by CUT&RUNTools, including Bowtie267, MACS268, Trimmomatic69, Picard (http://broadinstitute.github.io/picard/), Samtools70, MEME71, Bedtools72, Bedops73, and CENTIPEDE74 were kept unchanged.
CUT&RUNTools performs digital footprinting as an integrated step. CENTIPEDE74 was employed to calculate log-odds, which equals log(p/(1-p)) where p is probability of binding based on the strength of footprints reflected by the cut profile. Average footprint profile, log-odds of all motif instances were retrieved in fimo.result folder. Single locus cut profiles were retrieved by running get_cuts_single_locus.sh script by specifying the coordinates of the regions of interest.
The signal to noise ratio of CUT&RUN is high. Different noise levels in parallel experiments can have a significant impact on data normalization. To eliminate such effect, we normalized the data based on the fraction of reads in peaks instead of total sequencing depth. First, intersecting peaks between each pair of control and treat expriments were collected using bedtools72, and the percentage of reads that reside in these peaks was caculated. A scaling factor, calculated as percentage_in_peaks_control divided by percentage_in_peak_treat, was used to scale up/down the signals in treatment sample using bamCoverage tool from deeptools package75 with the following parameters: --scaleFactor $scale –normalizeUsing CPM.
Statistical significance of differential TF binding were quantified with MAnorm51. 500 bp was used as the window size (“-w”).
Base editing and single-cell cloning
To base edit the NF-Y motif, the three components of split intein Target-AID-NG were sequentially expressed in BCL11A KO HUDEP-2 cells by lentivirus transduction. Cas9NG-Intein-N expressing cells were purified using FACS and Intein-C-AID cells were purified by blasticidin selection. The expression of each component and the ligated full-length split intein Target-AID-NG were confirmed by immunoblot using α-Flag M2 antibody (F1804, Sigma-Aldrich). Each gRNA was introduced to cells through lentivirus transduction.
To base edit the BCL11A motif, we used a codon optimized base editor FNLS48. Base editor and gRNA were introduced to HUDEP-2 cells through lentivirus transduction. After base editing, cells were collected and lysed for genomic DNA extraction. The promoter sequence of γ-globin was amplified and subjected to Sanger sequencing. Sequencing traces were visualized with Snapgene, and the C-T conversion rate is quantified with TIDER76.
To isolate single-cell clones, 30 cells were seeded in a 96 well plate and left unperturbed for 7–10 days. Wells with more than one colonies were excluded. Single clones were expanded and genotyped with the above-described method. Clones with at least 50% C-T conversion were selected, and clones that were not edited were kept as controls. In addition, clones that had small indels or large deletions in the γ-globin promoters were discarded, due to potential indirect effects caused by double-strand DNA break and subsequent repair.
The gRNA sequences for base editing and the primers for amplifying the γ-globin promoters were listed in Supplementary Table 2.
Flow cytometry analysis of single clones
Base edited clones were stained for intracellular HbF using the same protocol as above mentioned except that this was done in 96 well plates. After staining the cells were analyzed in BD Accuri™ C6 flow cytometer, and the results were analyzed using FlowJo. For each clone, the percentage of cells with high HbF was quantified.
CRISPR/Cas9 mediated knock out in CD34+ or HUDEP-2 cells
Cas9/sgRNA Ribonucleoprotein complexes were assembled as follows. For each knockout experiment, 500 pmol Cas9 protein (IDT) and 1000 pmol sgRNA (Synthego) were mixed and incubated for 15 min at room temperature. Five million CD34+ cells on day 5 of erythroid differentiation were collected and washed once with PBS. Cells were resuspended with 100 μL solution P3 for primary cells (Lonza). 5 μL of electroporation enhancer (IDT) and preassembled RNP were then added to the cell suspension, and nucleofection was performed in a 4D-Nucleofector X unit (Lonza) using program EO-100. Cells were centrifuged, and transferred to 10 mL of EDM II for growth under differentiation conditions. For acute depletion of BCL11A, the time of nucleofection was set as day 0. Cells were collected at successive times for analyses. To assess the genome editing efficiency, cells were lysed to extract genomic DNA. Primers spanning the edited sites were used to amplify the genomic region. The PCR products were subject to Sanger sequencing, and editing efficiency was evaluated by TIDE77. The sequences of gRNAs and genotyping primers are listed in Supplementary Table 2. Western blot was used to assess BCL11A levels before and after nucleofection, and the band intensities were quantified using ImageJ. Briefly, the pixel densities of target bands and blank regions were measured. The pixel numbers of blank regions represented background and were subtracted from the band. Three independent measurements were performed for each band, and BCL11A signals were normalized using Histone H3.
For CRSIPR/Cas9 KO in HUDEP-2 cells, the procedure was the same as that described above except using a smaller volume. 50,000 cells were resuspended with 20 μL solution P3, and combined with RNP complex assembled with 100 pmol Cas9 and 150 pmol sgRNAs. After nucleofection, cells were centrifuged, and differentiated for 3 days as described above. Cells were collected after differentiation for western blot or RT-qPCR analysis. The gRNA sequences used are listed in Supplementary Table 2. Antibodies used: C/EBPB, NBP1–46179, NovusBio; C/EBPG, sc-517003, Santa Cruz Biotechnology; CDP, sc-514008, Santa Cruz Biotechnology; NFIA, HPA008884, Sigma-Aldrich; NFIC, A303–123A-T, Bethyl Laboratories; Histone H3, ab24834, Abcam. All antibodies were used at a concentration of 0.5 μg/mL.
Statistics
Statistical analysis were performed using GraphPad Prism 8.0. Details for Student’s t-test were indicated in the figure legends. Sample sizes were indicated either in the legend (n) or by the numbers of individual data points in the figures. For CRISRP-SURF, statistical tests of the beta coefficients were performed empirically through bootstrapping and two-tailed tests. Multiple hypothesis testing was accounted for with the Benjamini-Hochberg (BH) procedure.
Additional methods in Supplementary Note:
Methods for Chromosome Conformation Capture (3C), Construction of split Target-AID-NG46,47,78, Chromatin Immunoprecipitation sequencing and qPCR, ChIP-seq data analysis, ATAC-seq and data analysis79, PRO-seq80, PRO-seq data processing can be found in Supplementary Note.
Data availability
All raw and processed CRISPR screen, CUT&RUN, ChIP-seq, PRO-seq and ATAC-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE150530 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150530). All unprocessed western blot gels for Figure 4a, Extended Data Figures 2a, 4a, c, 5f can be found in Source Data.
Code availability
We have made use of publically available softwares for processing high throughput sequencing raw data. For single-locus CUT&RUN footprinting, the code can be found at https://bitbucket.org/qzhudfci/cutruntools/src/master/. Code for deconvolution of CRISPR screen data can be found at https://github.com/pinellolab. Custom codes used in this study can be found at https://github.com/yao-qiuming/Nan_NG2020.
Extended Data
Extended Data Fig. 1. Dense perturbation of the β-globin locus.
(a) Flow chart of the dense perturbation experiment design.
(b) Zoomed in view of the dense perturbation results at HBB and HBD genes. gRNAs that target the exons are enriched in Cas9 experiment. HbF raw score is enrichment of individual gRNAs in HbF-high compared to unsorted population at end of erythroid maturation, plotted as log2 fold change. HbF score shows deconvoluted underlying genomic regulatory signal with corresponding p-values shown on −log10 scale.
(c) Zoomed in view of the dense perturbation results at HBG1 gene. Not that gRNAs that target the exons are depleted in Cas9 experiment.
(d) Zoomed in view of the dCas9 dense perturbation result at HS3 of the LCR aligned to PhastCons46way scores. The four regions highlighted in green contain GATA1 or GATA1-TAL1 composite motifs (CTG[N8–9]GATA), with the sequences shown below.
(e) RT-qPCR showing that dCas9/sgRNA binding at −115 of γ-globin promoters reduced γ-globin expression in HUDEP-2 cells. Note that the γ-globin is only expressed at a basal level in cells expressing AAVS1 control sgRNA. The result is shown as mean (SD) of three technical replicates.
Statistical tests of the beta coefficients were performed empirically through bootstrapping and two-tailed tests. Multiple hypothesis testing was accounted for with the Benjamini-Hochberg (BH) procedure.
Extended Data Fig. 2. NFYA binds to γ-globin promoters and is required for LCR-γ-globin interaction.
(a) Left, western blot gel showing validation of NFYA knockdown efficiency (cropped). All three shRNAs tested showed efficient depletion of NFYA. Right, validation of NFYA knockdown efficiency at mRNA level using RT-qPCR. shRNA3 exhibited efficient knockdown of NFYA mRNA in all three cells tested and was used thereafter. The result is shown as mean (SD) of two technical replicates.
(b) ChIP-seq tracks of NFYA in HUDEP-2, HUDEP-1, BCL11A KO HUDEP-2 cells with or without NFYA knockdown.
(c) ChIP-qPCR validation of NFYA binding at the γ-globin promoters in HUDEP-1 and BCL11A KO HUDEP-2 cells. No strong binding was detected in HUDEP-2 cells which does not express γ-globin. The result is shown as mean (SD) of three technical replicates.
(d) Chromosome Conformation Capture qPCR in BCL11A KO HUDEP-2 cells with or without NFYA knockdown. EcoRI fragment encompassing HS2–4 of the LCR was used as anchor point to evaluate LCR-globin interaction. The result is shown as mean (SD) of three technical replicates.
Extended Data Fig. 3. NF-Y binds to the proximal CCAAT in the γ-globin promoters.
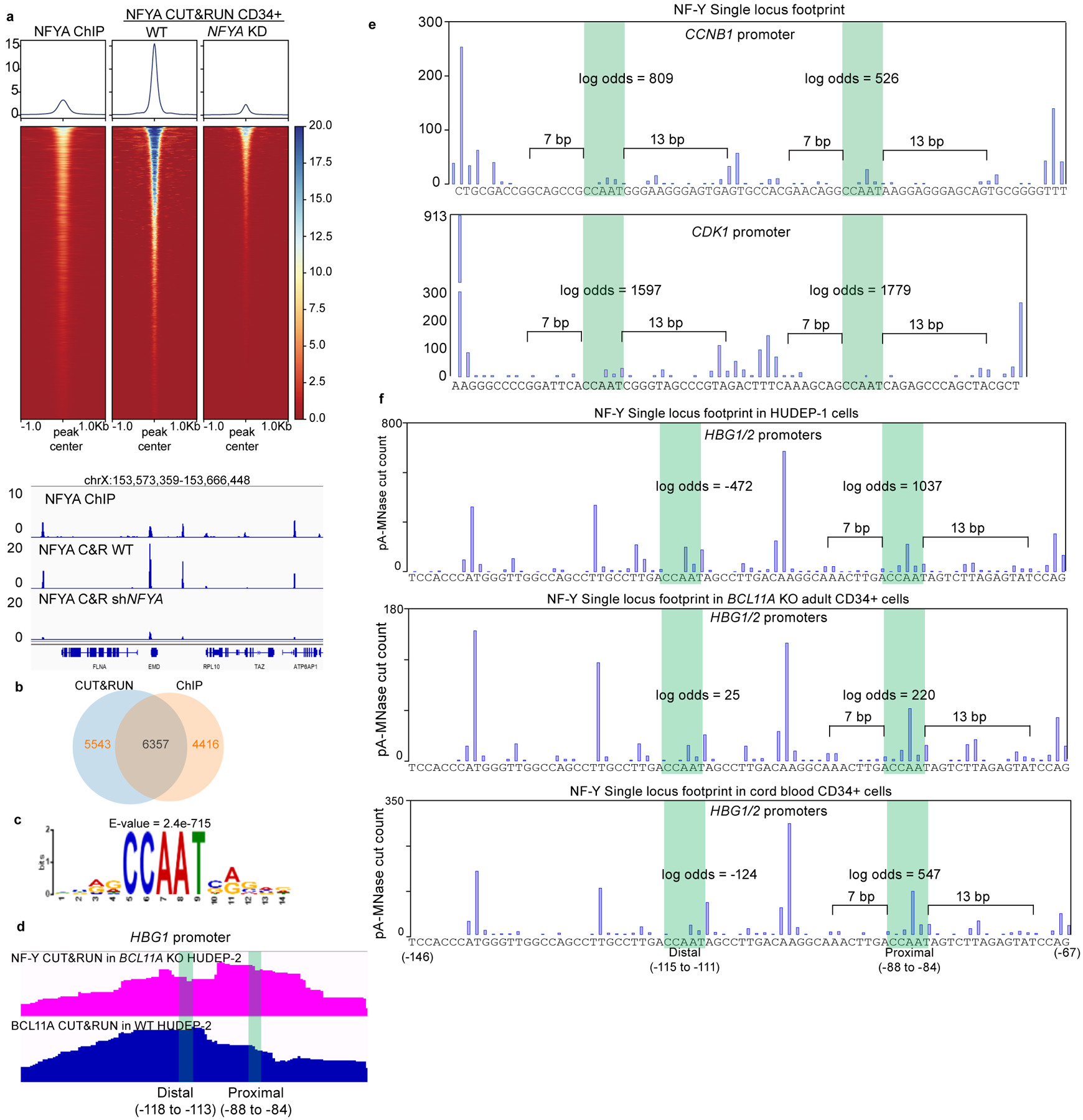
(a) Upper panel, heatmap comparison of NFYA ChIP-seq in HUDEP-2 cells, NFYA CUT&RUN in primary human CD34+ derived erythroid cells with or without NFYA knockdown. Lower panel, comparing the signal of the above three experiments at a representative genomic region.
(b) Venn diagram showing the overlap between NFYA CUT&RUN and ChIP-seq peaks.
(c) Motif analysis from 5000 random peaks of NFYA CUT&RUN identifies CCAAT as the highest ranked motif. E-value is reported by MEME.
(d) Zoomed in view of BCL11A CUT&RUN in HUDEP-2 and NFYA CUT&RUN in BCL11A KO HUDEP-2 cells at the -globin promoters. Distal (−118 to −113) indicates the distal TGACCA motif that BCL11A binds, and proximal (−88 to −84) indicates the proximal CCAAT motif.
(e) Single locus footprint of NF-Y at the CCNB1 promoter (upper) and CDK1 promoter (lower). Both CCAAT motifs show strong NF-Y footprints in the two promoters.
(f) Single locus footprint of NF-Y at the γ-globin promoters in HUDEP-1 (upper), BCL11A KO adult CD34+ derived erythroid cells (middle) and cord blood CD34+ derived erythroid cells. Only the proximal motif shows NF-Y footprint.
Extended Data Fig. 4. Base editing of the BCL11A and NF-Y motif.
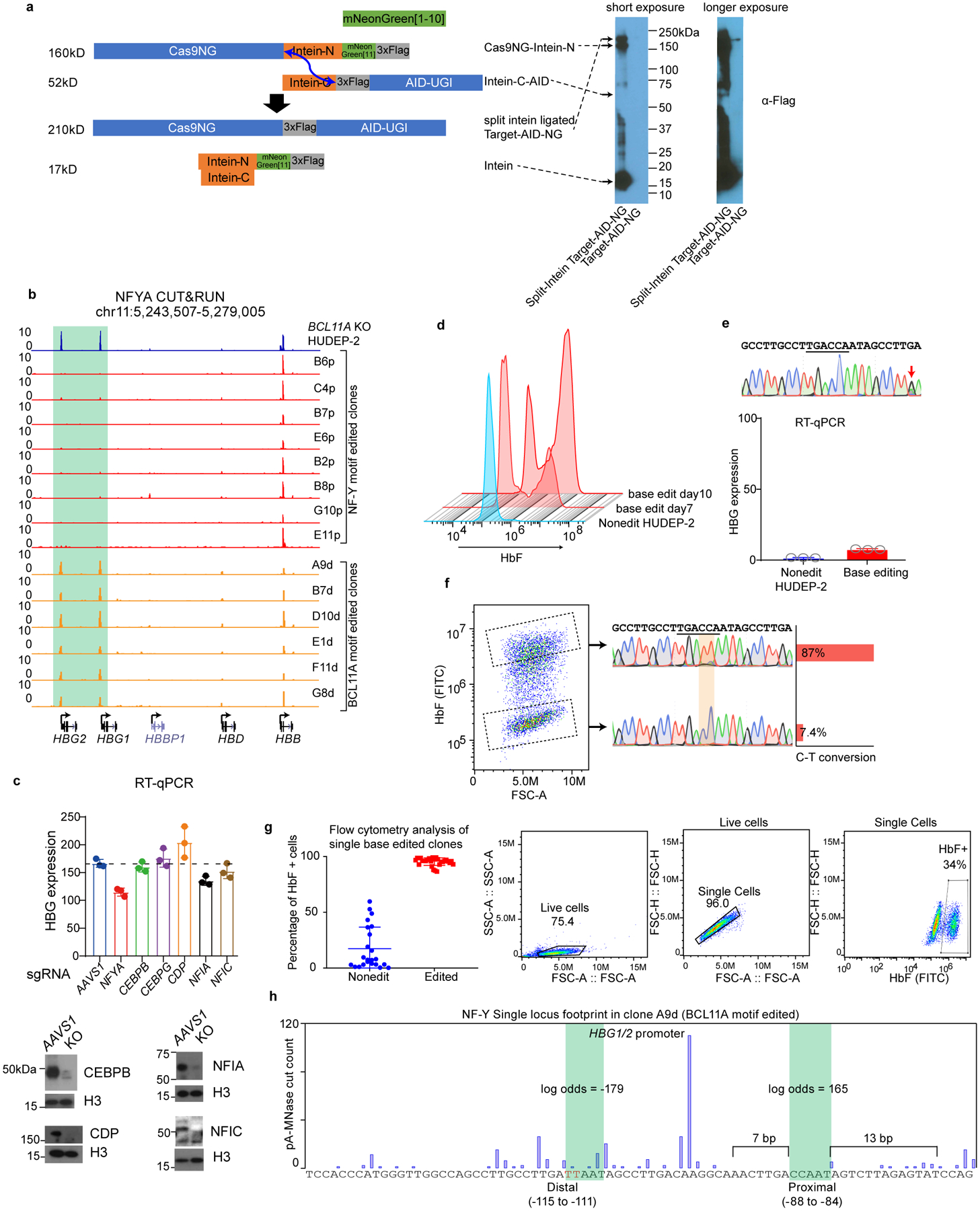
(a) Left, split-intein mediated ligation of Cas9NG-Intein-N and Intein-C-AID, producing full-length Target-AID-NG. Blue arrow indicates the ligation sites. Right, immunoblot validating the expression of each component and the ligation products. The ligation is incomplete, but the level of ligated product is much higher than the original vector (cropped).
(b) NFYA binding at the γ-promoters diminished in all the NF-Y motif-edited clones (red), and increased in all the BCL11A motif-edited clones (orange), as revealed by NFYA CUT&RUN. NF-Y motif editing was carried out in BCL11A KO HUDEP-2 cells while BCL11A motif editing was carried out in wild-type HUDEP-2 cells.
(c) Upper, RT-qPCR analysis of γ-globin expression after acute depletion of C/EBPβ, C/EBPγ, CDP, NFIA and NFIC. Lower, immunoblot validating protein depletion (cropped). BCL11A KO HUDEP-2 cells were differentiated for 3 days after nucleofection. The result is shown as mean (SD) of three technical replicates.
(d) Flow cytometry analysis of HbF levels for BCL11A base-edited clones at day 7 and 10. Longer editing resulted in higher base editing rate (Figure 3e) and higher percentage of HbF positive cells.
(e) A control base editing experiment in which a nucleotide 9 bp away from the BCL11A motif was edited. Sanger sequencing confirmed C-T conversion.
(f) Left, FACS of BCL11A motif base-edited bulk cells into high and low HbF populations. The C-T conversion rate of BCL11A motif in each population was measured by Sanger sequencing and quantified with TIDER. HbF high cells show 87% conversion and HbF low cells show only 7.4% conversion.
(g) Left, flow cytometry analysis of HbF level in individual clones derived from BCL11A motif base editing. Data is showed as mean (SD) of multiple independent clones. Nonedit: n=23, base edited: n=30. Right, gating strategy.
(h) Single locus footprint of NF-Y at the γ-promoters in clone A9d, a BCL11A motif-edited clone.
Extended Data Fig. 5. Acute depletion of BCL11A leads to rapid binding of NF-Y.
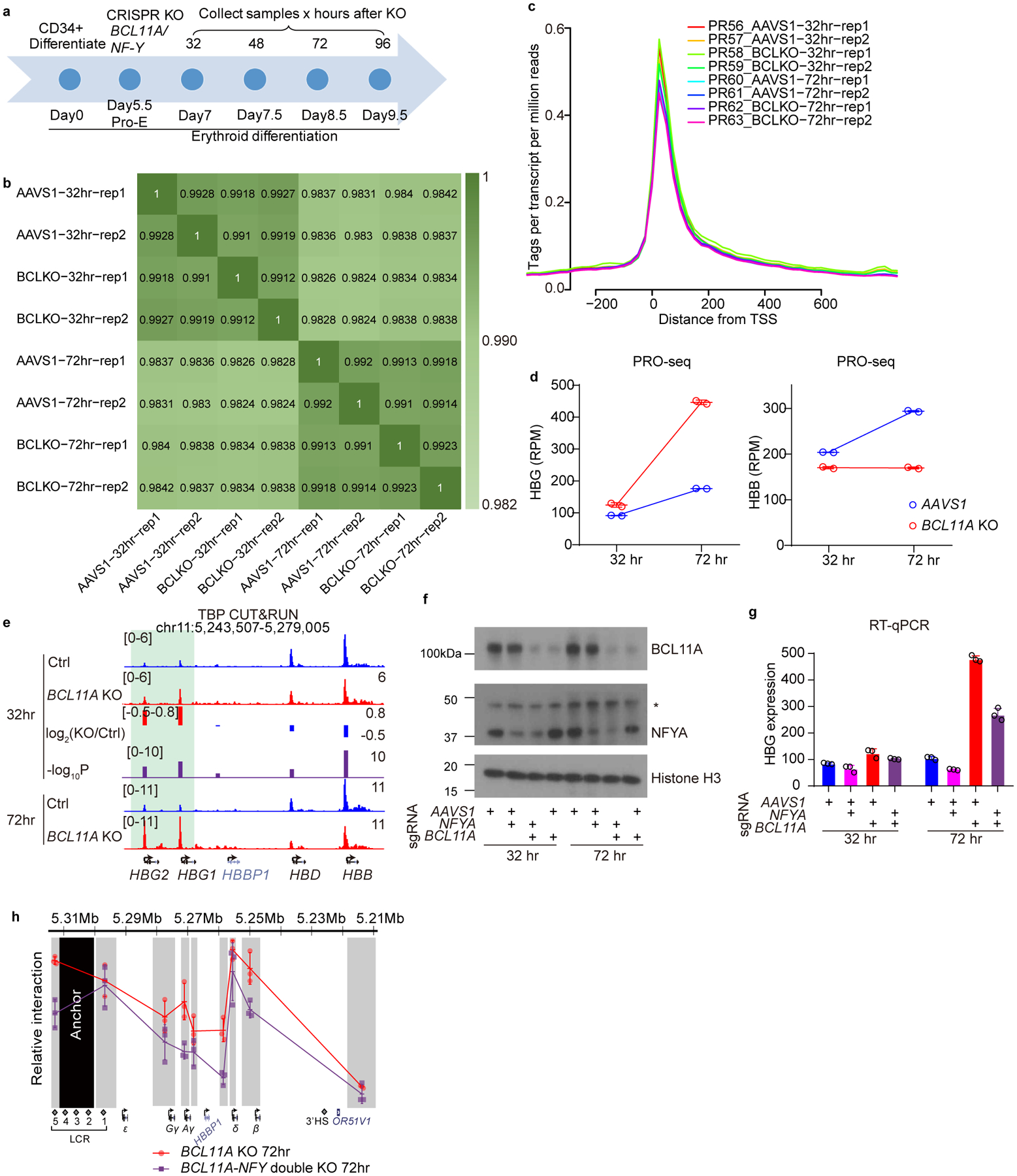
(a) Schematic diagram of primary human CD34+ differentiation and acute depletion of BCL11A using CRISPR/Cas9.
(b) Pairwise correlation of PRO-seq experiments. All the experiments in each time point showed high degree of correlation, indicating very minor transcriptional fluctuation upon BCL11A depletion.
(c) Average PRO-seq signal at −200 to +600 bp relative to TSS exhibited promoter pausing of PolII.
(d) Quantification of PRO-seq reads on HBG1/2 and HBB genes after 32 or 72 hrs of BCL11A acute depletion. The y-axis shows Reads Per Million (RPM) for HBG1+HBG2 or HBB. The result is shown as mean (SD) of two biologically independent samples (independent cell cultures and CRISPR KO).
(e) CUT&RUN of TBP in CD34+ cells undergoing erythroid differentiation after 32 or 72 hrs of BCL11A acute depletion. The result shown is representative of two biological replicates. Quantification of KO/Ctrl and the corresponding p-values are reported by MAnorm.
(f) Western blot for BCL11A and NFYA in adult primary human CD34+ derived erythroid cells upon KO of NFYA, BCL11A or both (cropped).
(g) RT-qPCR analysis of γ-globin expression in adult primary human CD34+ derived erythroid cells upon KO of NFYA, BCL11A or both. Knockout of NFYA after 72 hours decreases γ-globin expression. The result is shown as mean (SD) of three technical replicates.
(h) Chromosome Conformation Capture qPCR in adult primary human CD34+ derived erythroid cells, comparing BCL11A KO and BCL11A/NF-Y double KO. EcoRI fragment encompassing HS2–4 of the LCR was used as anchor point to evaluate LCR-globin interaction. The result is shown as mean (SD) of three technical replicates.
Supplementary Material
Acknowledgment
We thank S. Henikoff at the Fred Hutchinson Cancer Research Center for pA-MNase, T. Muir at Princeton University Department of Chemistry for split intein cDNA, and B. Huang at UCSF Department of Pharmaceutical Chemistry for split mNG2 cDNA. We thank Y. Nakamura at Cell Engineering Division in RIKEN BioResource Center for HUDEP-1 and HUDEP-2 cell lines. We thank Z. Herbert, M. Berkeley and A. Caruso at the Molecular Biology Core Facilities for high throughput DNA sequencing, S. Goldman at the Nascent Transcriptomics Core for generating PRO-seq libraries and performing analyses, and all members at the Hematologic Neoplasia Flow Cytometry core for sorting cells. We also thank M. Cole, M. Canver and C. Smith for critical input and experimental contributions, A. Bowker for technical assistance, and members of the Orkin, Bauer, and Yuan laboratories for input. D.E.B. was supported by the Burroughs Wellcome Fund and NHLBI (DP2HL137300 and P01HL032262). S.H.O. is an Investigator of the Howard Hughes Medical Institute, and supported by NHLBI R01HL032259 and P01HL032262, and by the Burroughs Wellcome Fund. G.C.Y. was supported by NHGRI R01HG009663. L.P. was supported by NHGRI Genomic Innovator Award R35HG010717. N.L. was supported by NIDDK K99DK120925.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hay D et al. Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet 48, 895–903 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter D, Chakalova L, Osborne CS, Dai Y & Fraser P Long-range chromatin regulatory interactions in vivo. Nat. Genet 32, 623–626 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Tolhuis B et al. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol. Cell 10, 1453–65 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Palstra RJ et al. The β-globin nuclear compartment in development and erythroid differentiation. Nature Genetics vol. 35 190–194 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Chada K, Magram J & Costantini F An embryonic pattern of expression of a human fetal globin gene in transgenic mice. Nature 319, 685–689 (1986). [DOI] [PubMed] [Google Scholar]
- 6.Magram J, Chada K & Costantini F Developmental regulation of a cloned adult β-globin gene in transgenic mice. Nature 315, 338–340 (1985). [DOI] [PubMed] [Google Scholar]
- 7.Starck J et al. Developmental regulation of human gamma- and beta-globin genes in the absence of the locus control region. Blood 84, 1656–1665 (1994). [PubMed] [Google Scholar]
- 8.Bender MA, Bulger M, Close J & Groudine M β-globin Gene switching and DNase I sensitivity of the endogenous β-globin locus in mice do not require the locus control region. Mol. Cell 5, 387–393 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Masuda T et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science (80-. ). 351, 285–289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sankaran VG et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science (80-. ). 322, 1839–1842 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Sankaran VG et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature 460, 1093–1097 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martyn GE et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet 50, 498–503 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Traxler EA et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med 22, 987–990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang P et al. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 31, 1704–1713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivaldi MS et al. Fetal g-globin genes are regulated by the BGLT3 long noncoding RNA locus. Blood 132, 1963–1973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sankaran VG et al. A Functional Element Necessary for Fetal Hemoglobin Silencing. N. Engl. J. Med 365, 807–814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaensler KML et al. Sequences in the Aγ-δ intergenic region are not required for stage-specific regulation of the human β-globin gene locus. Proc. Natl. Acad. Sci. U. S. A 100, 3374–3379 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurita R et al. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8, e59890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shariati SA et al. Reversible Disruption of Specific Transcription Factor-DNA Interactions Using CRISPR/Cas9. Mol. Cell 74, 622–633.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani R The molecular biology of the CCAAT-binding factor NF-Y. Gene 239, 15–27 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Dolfini D, Zambelli F, Pedrazzoli M, Mantovani R & Pavesi G A high definition look at the NF-Y regulome reveals genome-wide associations with selected transcription factors. Nucleic Acids Res. 44, 4684–4702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleming JD et al. NF-Y coassociates with FOS at promoters, enhancers, repetitive elements, and inactive chromatin regions, and is stereo-positioned with growth-controlling transcription factors. Genome Res. 23, 1195–1209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nardini M et al. Sequence-Specific Transcription Factor NF-Y Displays Histone-like DNA Binding and H2B-like Ubiquitination. Cell 152, 132–143 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Coustry F, Hu Q, de Crombrugghe B & Maity SN CBF/NF-Y functions both in nucleosomal disruption and transcription activation of the chromatin-assembled topoisomerase IIalpha promoter. Transcription activation by CBF/NF-Y in chromatin is dependent on the promoter structure. J. Biol. Chem 276, 40621–30 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Oldfield AJ et al. Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol. Cell 55, 708–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellorini M et al. CCAAT binding NF-Y-TBP interactions: NF-YB and NF-YC require short domains adjacent to their histone fold motifs for association with TBP basic residues. Nucleic Acids Res. 25, 2174–2181 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frontini M et al. NF-Y recruitment of TFIID, multiple interactions with histone fold TAFIIs. J. Biol. Chem 277, 5841–5848 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Kabe Y et al. NF-Y Is Essential for the Recruitment of RNA Polymerase II and Inducible Transcription of Several CCAAT Box-Containing Genes. Mol. Cell. Biol 25, 512–522 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oldfield AJ et al. NF-Y controls fidelity of transcription initiation at gene promoters through maintenance of the nucleosome-depleted region. Nat. Commun 10, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu X et al. NF-Y Recruits Both Transcription Activator and Repressor to Modulate Tissue- and Developmental Stage-Specific Expression of Human γ-Globin Gene. PLoS One 7, e47175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan Z, Stamatoyannopoulos G & Li Q Role of NF-Y in in vivo regulation of the gamma-globin gene. Mol. Cell. Biol 21, 3083–95 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang X, Han H, Stamatoyannopoulos G & Li Q Developmentally Specific Role of the CCAAT Box in Regulation of Human γ-Globin Gene Expression. J. Biol. Chem 279, 5444–5449 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martyn GE, Quinlan KGR & Crossley M The regulation of human globin promoters by CCAAT box elements and the recruitment of NF-Y. Biochim. Biophys. Acta - Gene Regul. Mech 1860, 525–536 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Xu J et al. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 24, 783–789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Q, Liu N, Orkin SH & Yuan GC CUT and RUNTools: A flexible pipeline for CUT and RUN processing and footprint analysis. Genome Biol. 20, 192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liberati C, Ronchi A, Lievens P, Ottolenghi S & Mantovani R NF-Y organizes the γ-globin CCAAT boxes region. J. Biol. Chem 273, 16880–16889 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Liberati C, di Silvio A, Ottolenghi S & Mantovani R NF-Y binding to twin CCAAT boxes: role of Q-rich domains and histone fold helices. J. Mol. Biol 285, 1441–1455 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Luo D et al. MNase, as a probe to study the sequence-dependent site exposures in the +1 nucleosomes of yeast. Nucleic Acids Res. 46, 7124–7137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Q, Lu J-F, Luo R, Sen S & Maity SN Inhibition of CBF/NF-Y mediated transcription activation arrests cells at G2/M phase and suppresses expression of genes activated at G2/M phase of the cell cycle. Nucleic Acids Res. 34, 6272–85 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kao CY, Tanimoto A, Arima N, Sasaguri Y & Padmanabhan R Transactivation of the human cdc2 promoter by adenovirus E1A. E1A induces the expression and assembly of a heteromeric complex consisting of the CCAAT box binding factor, CBF/NF-Y, and a 110-kDa DNA-binding protein. J. Biol. Chem 274, 23043–23051 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishimasu H et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevens AJ et al. A promiscuous split intein with expanded protein engineering applications. Proc. Natl. Acad. Sci. U. S. A 114, 8538–8543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zafra MP et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol (2018) doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L et al. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Research vol. 30 276–278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahat DB et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shao Z, Zhang Y, Yuan GC, Orkin SH & Waxman DJ MAnorm: A robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 13, R16 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu J et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci 110, 6518–6523 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gillespie MA et al. Absolute Quantification of Transcription Factors Reveals Principles of Gene Regulation in Erythropoiesis. Mol. Cell (2020) doi: 10.1016/j.molcel.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basak A et al. Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat. Genet 52, 138–145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilber A, Nienhuis AW & Persons DA Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood 117, 3945–3953 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Arensbergen J, van Steensel B & Bussemaker HJ In search of the determinants of enhancer-promoter interaction specificity. Trends Cell Biol. 24, 695–702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oudelaar AM et al. Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nat. Commun 11, 2722 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milos PM & Zaret KS A ubiquitous factor is required for C/EBP-related proteins to form stable transcription complexes on an albumin promoter segment in vitro. Genes Dev. 6, 991–1004 (1992). [DOI] [PubMed] [Google Scholar]
- 59.Stracke R, Thiedig K & Kuhlmann M What Have We Learned About Synthetic Promoter Construction? Plant Synthetic Promoters vol. 1482 (2016). [Google Scholar]
- 60.Rojo F Repression of transcription initiation in bacteria. Journal of Bacteriology vol. 181 2987–2991 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgens DW et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods vol. 11 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS & Vale RD A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu JY et al. CRISPR-SURF: discovering regulatory elements by deconvolution of CRISPR tiling screen data. Nature Methods vol. 15 992–993 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolger AM, Lohse M & Usadel B Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Machanick P & Bailey TL MEME-ChIP: Motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quinlan AR BEDTools: The Swiss-Army tool for genome feature analysis. Curr. Protoc. Bioinforma 2014, 11.12.1–11.12.34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neph S et al. BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pique-Regi R et al. Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 21, 447–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brinkman EK et al. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 46, e58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168–e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feng S et al. Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Elrod ND et al. The Integrator Complex Attenuates Promoter-Proximal Transcription at Protein-Coding Genes. Mol. Cell 76, 738–752.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed CRISPR screen, CUT&RUN, ChIP-seq, PRO-seq and ATAC-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE150530 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150530). All unprocessed western blot gels for Figure 4a, Extended Data Figures 2a, 4a, c, 5f can be found in Source Data.