

Abstract
Therapies employing chimeric antigen receptor T cells (CAR-T cells) targeting tumour-associated antigens (TAAs) can lead to on-target–off-tumour toxicity and to resistance, owing to TAA expression in normal tissues and to TAA expression loss in tumour cells. These drawbacks can be circumvented by CAR-T cells targeting tumour-specific driver gene mutations, such as the four-nucleotide duplication in the oncogene nucleophosmin (NPM1c), which creates a neoepitope presented by the human leukocyte antigen with the A2 serotype (HLA-A2) that has been observed in about 35% of patients with acute myeloid leukaemia (AML). Here, we report a human single-chain variable fragment (scFv), identified via yeast surface display, that specifically binds to the NPM1c epitope–HLA-A2 complex but not to HLA-A2 or to HLA-A2 loaded with control peptides. In vitro and in mice, CAR-T cells with the scFv exhibit potent cytotoxicity against NPM1c+HLA-A2+ leukaemia cells and primary AML blasts, but not NPM1c−HLA-A2+ leukaemia cells or HLA-A2− tumour cells. Therapies using NPM1c CAR-T cells for the treatment of NPM1c+HLA-A2+ AML may limit on-target–off-tumour toxicity and tumour resistance.
Editor's Summary
CAR-T cells specific for a neoantigen derived from the driver oncogene nucleophosmin display potent and specific cytotoxic activity in mouse models of human acute myeloid leukaemia.
The recent Food and Drug Administration approval of CD19-targeted chimeric antigen receptor T cell (CAR-T cell) therapy is a major milestone in the development of genetically modified cell therapies for cancer. Many academic and industry investigators are actively developing approaches to extend this treatment to other haematological malignancies and solid tumours. However, current CARs targeting lineage-restricted or tumour-associated antigens (TAAs) can be accompanied by severe toxicity due to low antigen expression in normal tissues[1, 2]. Furthermore, because TAAs are not required for tumour cell survival, loss of TAA expression is the major cause of development of tumour resistance to CAR-T therapies[2]. Neoantigens are derived from tumour-specific gene mutations, and their formation and expression are restricted to malignant cells[3, 4, 5]. However, the majority of neoantigens are encoded by patient-specific passenger mutations that could be lost due to immune editing, resulting in tumour immune evasion[6]. The neoantigens derived from oncogenic driver gene mutations are less likely to induce immune evasion because cancer cells need to express the driver genes in order to maintain their malignant phenotype[3, 5]. Thus, the neoantigens derived from oncogenic driver gene mutations are ideal cancer-specific targets for immunotherapy.
Acute myeloid leukaemia (AML) is a rapidly progressing haematopoietic malignancy characterized by accumulation of malignant myeloid precursor cells that are arrested in their differentiation in the bone marrow[5, 7]. Current standard therapy for AML still relies on intensive chemotherapy and autologous or allogeneic haematopoietic stem cell transplantation[8, 9]. Although most patients can respond to standard therapy and achieve complete remission, relapses occur in about 50% of patients[10]. Patients with relapsed or refractory AML after intensive chemotherapy or allogeneic haematopoietic stem cell transplantation usually have a very poor prognosis[5], and thus there is a strong need to develop new and effective therapies for these patients.
Genomic analysis of AML has shown a lower mutational load than most other adult cancers, with an average of 13 coding mutations per patient with AML[11, 12, 13]. However, somatic mutations in AML often occur in the same genes[11, 14] and neoantigens derived from these hotspot mutations therefore become attractive targets for tumour-specific immunotherapy[5]. Among the most commonly occurring mutations is a four-nucleotide duplication in a critical driver gene encoding nucleophosmin (NPM1), which occurs in 30–35% of all adult patients with AML[11, 14, 15]. Mutations in NPM1 result in its aberrant cytoplasmic localization, and the mutant protein is referred to as NPM1c. NPM1c produces a leukaemia-specific neoepitope (AIQDLCLAV, abbreviated as AIQ) that is presented by the most common HLA-A*0201 allele (in ~50% of the human population)[16]. It has been shown that NPM1c+ patients with AML with specific CD8 T cell responses to the AIQ neoepitope have a dramatically better overall survival than NPM1c+ patients with AML without a CD8 T cell response to the neoepitope[17]. Recently, a study identified T cell receptors (TCRs) recognizing a NPM1c neoepitope presented by HLA-A2, and showed that TCR-transduced T cells are effective in killing NPM1c+HLA-A2+ AML[5]. Importantly, the mutations in NPM1 are thought to be initiating mutations and present in all leukaemic cells[5]. Therefore, a CAR-T cell therapy that specifically targets the NPM1c neoepitope in complex with HLA-A*0201 (AIQ–HLA-A2) would be a cancer-specific immunotherapy for NPM1c+HLA-A2+ AML with the potential for reduced on-target/off-tumour toxicity and tumour resistance.
In this study, we used yeast surface display to isolate a single-chain variable fragment (scFv) specific for the AIQ–HLA-A2 complex, and developed CAR-T cells that exhibit potent cytotoxicity against NPM1c+HLA-A2+ but not NPM1c−HLA-A2+ leukaemia cells both in vitro and in vivo. Our proof-of-concept study demonstrates that CAR-T cells targeting a neoepitope derived from an oncogenic driver gene mutation are potent and specific.
Results
Isolation of human scFv specific for the AIQ–HLA-A2 complex by yeast surface display
Isolation of human scFv specific for the AIQ–HLA-A2 complex is the basis for developing tumour-specific CAR-T therapy for AML with NPM1c mutation. We used yeast surface display[18] to identify high-affinity scFv specific for the AIQ–HLA-A2 complex. Because yeast surface display allows quantitative discrimination between different scFv variants based on their binding to antigens on the yeast cell surface by flow cytometry, selection of high-affinity scFvs can be achieved during the screening process. Furthermore, because the yeast display library was constructed using the entire repertoire of variable region gene fragments from human splenic B cells, the isolated scFvs are already of human origin and are therefore suitable for therapeutic development. The yeast surface display library used was estimated to have a diversity of 1 × 107 to 1 × 109 different clones[18].
We undertook a series of positive and negative selections to isolate scFvs with specificity to the AIQ–HLA-A2 complex but not to HLA-A2 in the presence or absence of other antigenic peptides, such as the cancer/testis antigen NY-ESO-1 epitope (SLLMWITQC; abbreviated as SLL–HLA-A2) (Fig. 1a,b). Approximately 1 × 1010 yeast cells (at least tenfold of the library diversity) were incubated with biotin-labelled AIQ–HLA-A2 complex, followed by streptavidin-labelled magnetic beads and magnetic-activated cell sorting (MACS) purification (Fig. 1b, round 1). The positively selected yeast cells were expanded approximately tenfold and incubated with biotin-labelled SLL–HLA-A2 complex, followed by streptavidin-labelled magnetic beads and MACS negative selection (Fig. 1b, round 2). The unbound yeast cells were expanded and incubated with biotin-labelled AIQ–HLA-A2 complex, followed by phycoerythrin-labelled antibody specific for mouse immunoglobulin G (IgG) (the Fc portion of the HLA-A2 dimer; Fig. 1a) and fluorescein isothiocyanate (FITC)-labelled anti-c-Myc antibody, which detects the c-Myc epitope at the carboxy terminal of scFv (Fig. 1a). As shown in Fig. 1b (round 3; fluorescence-activated cell sorting (FACS) sorting plot 1), ~3.6% of yeast cells were positive for anti-mouse IgG and c-Myc epitope. The double-positive yeast cells were isolated by FACS (Fig. 1b, round 3).
Fig. 1. Isolation of human scFv specific for the AIQ–HLA-A2 complex by yeast surface display.

a, Schematics of the peptide–HLA-A2 complex (left), scFv displayed on the yeast surface (middle) and binding of the peptide–HLA-A2 complex to scFv on the yeast cell surface (right). HA, haemagglutinin. b, Strategies and steps used to isolate yeast cells displaying scFvs that specifically recognize the AIQ–HLA-A2 complex. The round of selection is indicated on the left. ‘Antigen’ indicates peptide–HLA-A2 complexes or HLA-A2 alone used in positive or negative selection. In the first two rounds of selection, yeast cells were selected by MACS. In the remaining rounds of selection, yeast cells were sorted by flow cytometry (FACS sorting) based on staining with FITC-labelled anti-c-Myc antibody plus phycoerythrin-labelled anti-mouse IgG or APC-labelled streptavidin. The gates for sorted cells are indicated. Sorted yeast cells from rounds 4–9 were expanded, stained with biotin-labelled HLA-A2, GIL–HLA-A2, SLL–HLA-A2 or AIQ–HLA-A2, followed by FITC-labelled anti-c-Myc antibody and APC-labelled streptavidin, and then analysed by flow cytometry (FACS analysis) gating on live cells (DAPI−). The FACS sorting plots are labelled from 1–7 and the FACS analysis plots are labelled from 1–29. c, Yeast cells expressing either YG1 or YG2 clones were stained and analysed as in b. The percentages in b and c indicate the percentages of cells in the gated regions. Diagrams adapted with permission from ref. [19], Springer Nature Ltd.
To further enrich scFv specificity for the AIQ–HLA-A2 complex, the sorted yeast cells were expanded and subjected to a series of positive (with biotin-labelled AIQ–HLA-A2) and negative selection (with biotin-labelled SLL–HLA-A2 or HLA-A2), followed by staining with streptavidin-allophycocyanin (APC) and FITC-labelled anti-c-Myc antibody and cell sorting. To assess the enrichment, the selected yeast cells were analysed for non-specific binding to HLA-A2, GIL–HLA-A2 (HLA-A2 loaded with GILGFVFTL (abbreviated as GIL) peptide from influenza virus M1; ref. [19]) and SLL–HLA-A2, and specific binding to AIQ–HLA-A2 (Fig. 1b; FACS analysis). For example, in round 6 (Fig. 1b; FACS sorting plot 4), yeast cells displaying scFv with high affinity for the AIQ–HLA-A2 complex were purified. However, most of the purified yeast cells bound to both HLA-A2 and AIQ–HLA-A2, but not GIL–HLA-A2 and SLL–HLA-A2 (Fig. 1b; FACS analysis plots 11–14), indicating that most of the selected scFvs can bind to HLA-A2 without peptide. To select yeast cells displaying scFv specific for AIQ–HLA-A2, we stained yeast cells with HLA-A2 and purified HLA-A2-negative yeast cells by cell sorting (Fig. 1b; round 7; FACS sorting plot 5). Around 12% of the selected yeast cells were positive for HLA-A2 and c-Myc (Fig. 1b; FACS analysis plot 16), 22.8% were positive for AIQ–HLA-A2 and c-Myc (Fig. 1b; FACS analysis plot 19) and less than 1% were positive for either GIL–HLA-A2 or SLL–HLA-A2 (Fig. 1b; FACS analysis plots 17 and 18), indicating an enrichment of scFv for AIQ–HLA-A2. With further positive and negative selection (Fig. 1b; rounds 8 and 9), we finally selected yeast cells that were mostly positive for AIQ–HLA-A2 and c-Myc (~60%; Fig. 1b; FACS analysis plot 29) but mostly negative for HLA-A2 (<3%; Fig. 1b; FACS analysis plot 26).
To identify the sequences of scFvs, plasmid DNA was extracted from the selected yeast cell pool and transformed into Escherichia coli. Twenty-five single colonies were randomly picked for plasmid isolation and sequencing. Of these, 24 had the same scFv DNA sequence (referred to as YG1), and the remaining one had a different scFv DNA sequence (referred to as YG2). YG1 and YG2 were then transformed separately into RJY100 competent yeast cells and scFv expression was induced by culturing yeast cells in SGCAA media. Flow cytometry analysis showed that c-Myc-positive YG1-expressing yeast cells were also stained positive for the AIQ–HLA-A2 complex but not HLA-A2 or the GIL–HLA-A2 or SLL–HLA-A2 complexes, while c-Myc-positive YG2-expressing yeast cells were weakly stained for both HLA-A2 and AIQ–HLA-A2 (Fig. 1c). These results show that scFv from YG1 is specific for the AIQ–HLA-A2 complex.
Specific and high-affinity binding of YG1 scFv–Fc to the AIQ–HLA-A2 complex
To further characterize YG1 scFv, we produced an scFv–Fc fusion protein using a switchable yeast display/secretion system (Fig. 2a). The fusion protein was purified using protein A resin and analysed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Under the non-reducing condition, major bands were detected at approximately 60, 140 and 260 kDa, corresponding to the sizes of monomeric, dimeric and tetrameric scFv–Fc protein products (Fig. 2b). Upon reduction, the intensity of the 60 kDa band was increased and the 260 kDa band was gone, consistent with the expected molecular weight of a monomeric scFv–Fc protein product. The persistence of the 140 kDa band was probably due to post-translational modification, such as glycosylation, of the fusion protein[20].
Fig. 2. Specific and high-affinity binding of YG1 scFv–Fc to the AIQ–HLA-A2 complex on AML cells.

a, Schematic of the switchable yeast display/secretion vector for expressing the scFv–Fc fusion protein. In this switchable system, scFv–Fc can be secreted or displayed on the yeast cells depending on whether or not OmeY is added to the culture[20]. b, SDS-PAGE analysis of purified scFv–Fc proteins (lane 1: protein ladder; lane 2: non-reduced scFv–Fc protein (1 μg); lane 3: reduced scFv–Fc protein (1 μg)). Gel was stained using Coomassie Blue. Shown are the representative data from three separate experiments. The image of the full scan (with the boundaries of the crops outlined) is provided in the Supplementary Information. c, Flow cytometry analysis of HLA-A2 expression by OCI-AML3, T2, GMB and PC-3 cells. Red histograms show staining with anti-HLA-A2, whereas cyan histograms show staining with isotype control antibody. Representative data from technical triplicates are shown. d, Flow cytometry analysis of AIQ–HLA-A2 expression by OCI-AML3, T2, GMB and PC-3 cells. Red histograms show staining with YG1 scFv–Fc and anti-haemagglutinin, whereas cyan histograms show staining with BSA followed by anti-haemagglutinin. Representative data from three separate experiments with technical triplicates are shown. e, Kinetic analysis of the interactions between scFv–Fc and AIQ–HLA-A2, SLL–HLA-A2 or HLA-A2 by biolayer interferometry. The streptavidin biosensor tips of the FortéBio Octet RED96 instrument were coated with biotinylated scFv–Fc protein. The tips were dipped in increasing concentrations (indicated at the bottom of binding curve) of AIQ–HLA-A2, SLL–HLA-A2 or HLA-A2 to measure their binding to scFv–Fc (association) and subsequently moved to wells containing buffer to measure the dissociation rate (dissociation). Shown are representative data from three separate experiments.
We used flow cytometry to verify specific binding of the YG1 scFv–Fc protein to the AIQ–HLA-A2 complex in a panel of positive and negative cell lines, including OCI-AML3 (a HLA-A2+ AML cell line with NPM1c mutation)[21], T2 (a HLA-A2+ lymphoblastic cell line with wild-type NPM1)[22], glioblastoma multiforme (GMB; a HLA-A2+ and NPM1c− B cell leukaemia/lymphoma line derived from overexpression of Myc and Bcl2)[23] and PC-3 (a HLA-A2− prostate cancer cell line with wild-type NPM1)[24]. When cells were stained for HLA-A2, OCI-AML3, T2 and GMB cells were positive and PC-3 cells were negative, as expected (Fig. 2c). When the cells were stained with YG1 scFv–Fc fusion protein followed by phycoerythrin-labelled anti-haemagglutinin antibody to detect the haemagglutinin epitope in the fusion protein (Fig. 2a), almost all OCI-AML3 cells were positive but none of the T2, GMB or PC-3 cells were positive (Fig. 2d). Thus, the YG1 scFv is capable of specific binding to the AIQ–HLA-A2 complex on human HLA-A2+ AML cells with the NPM1c mutation.
We measured the affinity of the YG1 scFv–Fc to the AIQ–HLA-A2 complex using biolayer interferometry. The purified YG1 scFv–Fc fusion protein was biotinylated and captured on streptavidin biosensors. Upon addition of AIQ–HLA-A2, the binding amplitudes increased in a concentration-dependent manner (Fig. 2e), whereas the binding amplitudes were not significantly changed upon addition of increasing concentrations of SLL–HLA-A2 or HLA-A2. Based on the association and dissociation kinetics, the association constant (Kon) of YG1 scFv–Fc with AIQ–HLA-A2 was 5.33 ± 0.02 × 104 M−1s−1, the dissociation constant (Koff) was 3.77 ± 0.02 × 10−4s−1 and the equilibrium dissociation constant (Kd) was 7.07 ± 0.08 nM. Together, these results show that the YG1 scFv–Fc fusion protein binds to the AIQ–HLA-A2 complex with high specificity and affinity.
Generation of CAR-T cells specific to the AIQ–HLA-A2 complex
To construct NPM1c CAR, we cloned the YG1 scFv in frame into a CAR backbone containing a CD8α hinge and transmembrane domain, a 4-1BB co-stimulatory domain and a CD3ζ activation domain, followed by self-cleavage P2A and enhanced green fluorescent protein (EGFP) (Fig. 3a,b). As a control, the same structural backbone was used to express CAR specific for CD19 (CD19 CAR). Human CD8+ T cells were purified from donor peripheral blood mononuclear cells, activated with anti-CD3 and anti-CD28 for 4 d and then either not transduced or transduced with lentiviruses expressing NPM1c CAR or CD19 CAR. Four days later, transduced T cells were isolated by sorting for GFP+ cells and expanded for another 10 d. The resulting T cells were analysed for GFP and CAR expression by staining with AF647-labelled anti-human IgG heavy and light chain antibody, which recognizes scFv. As shown in Fig. 3c, T cells transduced with NPM1c CAR-expressing lentiviruses were positive for both GFP and scFv, whereas T cells transduced with CD19 CAR-expressing lentiviruses were positive for GFP but only weakly positive for scFv. The latter is probably because CD19 scFv is humanized from mouse sequences[25]. To determine whether the resulting NPM1c CAR-T cells specifically recognize the AIQ–HLA-A2 complex (Fig. 3b), CAR-T cells were incubated with biotinylated AIQ–HLA-A2, SLL–HLA-A2 or HLA-A2 followed by streptavidin-APC staining. GFP+ NPM1c CAR-T cells were specifically bound to the AIQ–HLA-A2 complex, but not to SLL–HLA-A2 or HLA-A2 (Fig. 3d), while both untransduced T cells and CD19 CAR-T cells did not show binding to any of the three complexes. These results confirm the specificity of NPM1c CAR-T cells to the AIQ–HLA-A2 complex.
Fig. 3. Generation of NPM1c CAR-T cells specific to the AIQ–HLA-A2 complex.

a, Schematic of the CAR vector consisting of scFv (YG1 or CD19), the CD8α extracellular hinge and transmembrane domain (TM), the 4-1BB co-stimulatory domain and the CD3ζ activation domain, followed by self-cleavage P2A and EGFP. b, Schematic of the recognition of NPM1c CAR-T cells by the soluble AIQ–HLA-A2 complex. c, Flow cytometry analysis of CAR expression by untransduced and transduced T cells. Transduced T cells were enriched by sorting for GFP+ cells, expanded and stained with AF647-labelled antibody specific for human IgG heavy and light chains. Untransduced T cells were activated and expanded without sorting. Shown are the GFP versus anti-human IgG staining profiles of live cells (DAPI−). d, NPM1c CAR-T cells recognize the AIQ–HLA-A2 complex. Untransduced and transduced T cells were incubated with biotinylated AIQ–HLA-A2, SLL–HLA-A2 or HLA-A2, followed by streptavidin-APC staining. Shown are the GFP versus streptavidin-APC staining profiles of live (DAPI−) untransduced T cells, NPM1c CAR-T cells and CD19 CAR-T cells. The data in c and d are representative of three separate experiments. The percentages indicate the percentages of cells in the gated regions.
Specific killing of human AML cells by NPM1c CAR-T cells in vitro
To investigate the potency and specificity of NPM1c CAR-T cells, untransduced T cells and NPM1c CAR-T cells were co-cultured with OCI-AML3, GMB and PC-3 tumour cells at different effector-to-target ratios for 24 h. The relative proportions of T cells and tumour cells were quantified by flow cytometry staining for CD8 plus CD33 (for OCI-AML3), CD19 (for GMB) or mCherry (for PC-3). As shown in Fig. 4, NPM1c CAR-T cells killed NPM1c+HLA-A2+ OCI-AML3 cells in a dose-dependent manner, but did not kill NPM1c−HLA-A2+ GMB cells or NPM1c−HLA-A2− PC-3 cells, regardless of the effector-to-target ratio. Consistently, a significantly higher percentage of NPM1c CAR-T cells expressed interferon-γ (IFN-γ) compared with untransduced T cells when co-cultured with OCI-AML3 cells compared with GMB or PC-3 cells (Supplementary Fig. 1a). Similarly, a significantly higher percentage of NPM1c CAR-T cells expressed interleukin-2 (IL-2) compared with untransduced T cells when co-cultured with OCI-AML3 cells, but the percentage was low (<2%; Supplementary Fig. 1b). These results show that NPM1c CAR-T cells can specifically kill NPM1c+HLA-A2+ AML cells in vitro.
Fig. 4. NPM1c CAR-T cells specifically kill HLA-A2+NPM1c+ AML cells in vitro.

a,b, NPM1c CAR-T cells were co-cultured with OCI-AML3, GMB and PC-3 tumour cells at the indicated effector-to-target ratios for 24 h. The cell mixtures were stained for CD8 plus CD33, CD19 or mCherry, followed by flow cytometry. The percentages of CAR-T cells were quantified by CD8 staining and the percentages of OCI-AML3, GMB and PC-3 cells were quantified by CD33, CD19 and mCherry, respectively. The percentages of specific lysis of tumour cells were calculated (see Methods for the formula). Shown are examples of CD8 versus CD33, CD19 or mCherry staining profiles (a) and the percentages of specific lysis (b) at different effector-to-target ratios. The percentages of cells in the gated regions are indicated. In a, the plots are representative of three separate experiments. The graphs in b were created using Microsoft Office 2016. The P values (two-sided independent-samples t-test) indicate comparison between NPM1c CAR-T cells and untransduced T cells at the same effector-to-target ratio (n = 3 biologically independent samples; data points and error bars represent means ± s.e.).
To assess the release of a broader range of cytokines by NPM1c CAR-T cells after stimulation with target cells, we used Quantibody Human Cytokine Array to quantitatively measure the secretion of 20 human cytokines and chemokines in supernatants from co-culture of NPM1c CAR-T cells or untransduced T cells, prepared from four healthy donors, with OCI-AML3 target cells. Significantly increased secretion of granulocyte–macrophage colony-stimulating factor (four out of four donors), IL-13 (four out of four donors), macrophage inflammatory protein-1 (four out of four donors), IL-8 (four out of four donors), IFN-γ (three out of four donors), Macrophage inflammatory protein-1α (three out of four donors), RANTES (three out of four donors), IL-2 (two out of four donors) and monocyte chemoattractant protein-1 (two out of four donors) was found in supernatants from NPM1c CAR-T cells compared with untransduced T cells (Supplementary Fig. 2).
To assess whether NPM1c+HLA-A2+ target cells stimulate the proliferation of NPM1c CAR-T cells, NPM1c CAR-T cells or untransduced T cells were co-cultured with OCI-AML3 cells for 5 d, and T cell numbers and Ki-67 expression were quantified by flow cytometry. The absolute number of NPM1c CAR-T cells was significantly higher than that of untransduced T cells, as determined by flow cytometry using precision count beads (Supplementary Fig. 3a). The increased expression of Ki-67 was observed in NPM1c CAR-T cells compared with untransduced T cells, as determined by the mean fluorescence intensity of intracellular Ki-67 staining (Supplementary Fig. 3b). These results indicate the ability of NPM1c CAR-T cells to proliferate in response to NPM1c+HLA-A2+ target cells.
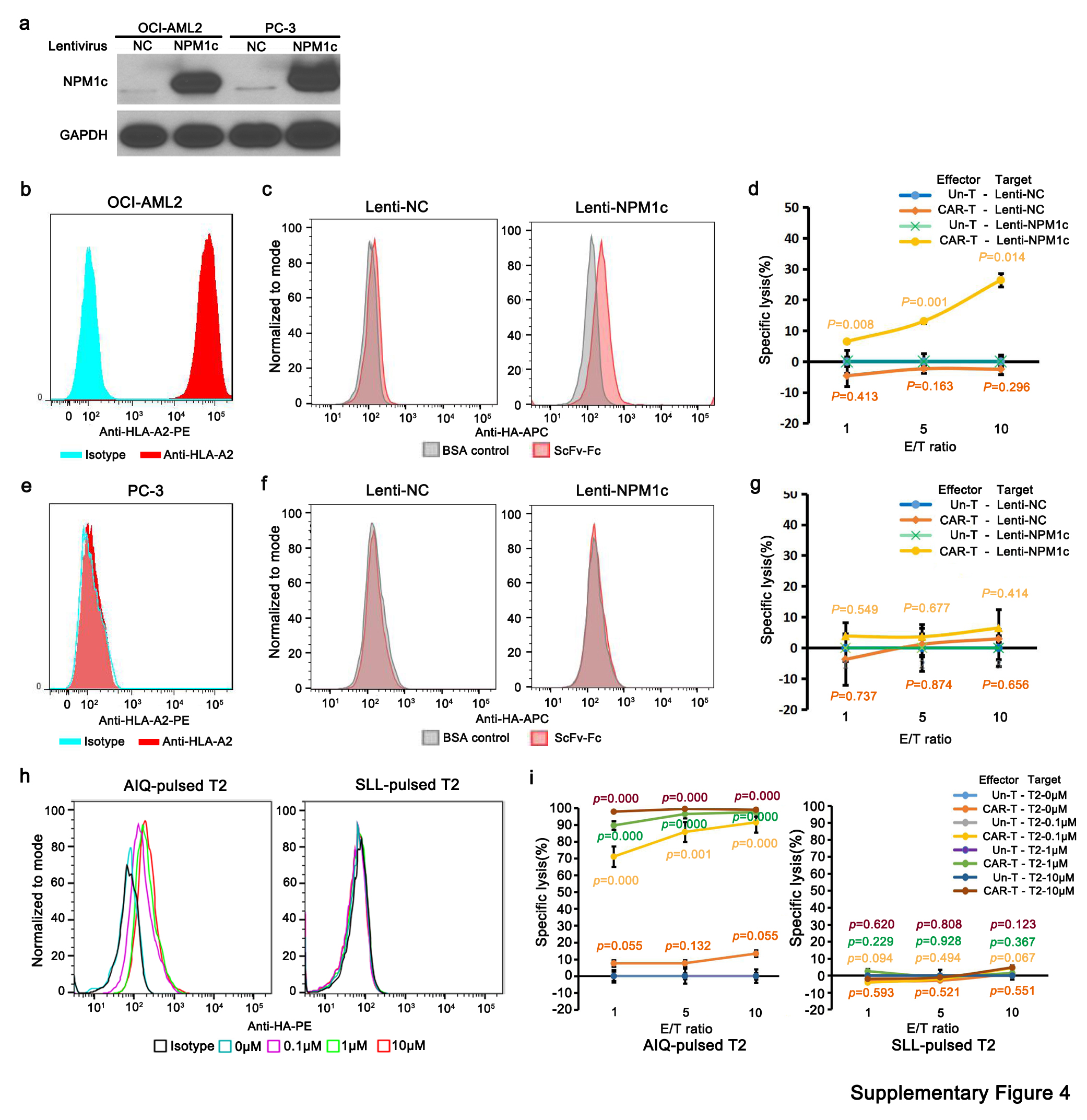
To further verify the specificity of NPM1c CAR-T cells in targeting NPM1c+HLA-A2+ AML cells, OCI-AML2 cells (a HLA-A2+ AML cell line with wild-type NPM1) and PC-3 cells (a HLA-A2− prostate cancer cell line with wild-type NPM1) were transduced with lentivirus to stably express NPM1c (Supplementary Fig. 4a,b,e). Following exogenous NPM1c expression, HLA-A2+ OCI-AML2 cells were stained positive for YG1 scFv–Fc (Supplementary Fig. 4c right) and killed by NPM1c CAR-T cells in an effector-to-target ratio-dependent manner (Supplementary Fig. 4d). However, NPM1c-expressing HLA-A2− PC-3 cells were not stained by YG1 scFv–Fc (Supplementary Fig. 4f; right) and were not killed by NPM1c CAR-T cells at any effector-to-target ratio compared with untransduced T cells (Supplementary Fig. 4g). As PC-3 expresses HLA-A1, HLA-A24, HLA-B13, HLA-B55 and HLA-C01, these results also suggest that the isolated YG1 scFv specifically recognizes the NPM1c epitope (AIQ) presented by HLA-A2, and either the NPM1c peptide is not presented by HLA-A1, HLA-A24, HLA-B13, HLA-B55 and HLA-C01 or the presented epitope in association with these HLA molecules is not recognized by YG1 scFv.
We also used T2 cells loaded with different peptides to demonstrate the antigen specificity of NPM1c CAR-T cells. T2 cells are HLA-A2+ but deficient in transporter associated with antigen processing (TAP). Therefore, many of the HLA-A2 molecules on the T2 cell surface are not loaded with endogenous peptides, but can be loaded with exogenous peptides[26, 27]. T2 cells were pulsed with different concentrations of NPM1c peptide (AIQ) or NY-ESO-1 peptide (SLL) ranging from 0.1 to 1 to 10 μM. T2 cells pulsed with AIQ peptide but not SLL peptide were stained by YG1 scFv–Fc (Supplementary Fig. 4h). Consistently, NPM1c CAR-T killed AIQ-pulsed T2 cells in a peptide concentration-dependent manner but not SLL-pulsed T2 cells (Supplementary Fig. 4i). These results further support the specificity of NPM1c CAR-T cell recognition and the killing of target cells with the NPM1c–HLA-A2 complex on the cell surface.
NPM1c CAR-T therapy reduces leukaemia burden and prolongs survival
To test the antitumour activity of NPM1c CAR-T cells in vivo, NOD-scid IL2rgnu11 (NSG) mice were injected intravenously with luciferase-expressing OCI-AML3 AML tumour cells (1 × 106 per mouse; Fig. 5a). After confirmation of engraftment by bioluminescence imaging (BLI) 4 d post-injection, mice were given a single injection of CAR-T or control untransduced T cells (1 × 107 cells per mouse) intravenously. The leukaemia burden was monitored every 3 d using BLI. Mice treated with NPM1c CAR-T cells showed a significant reduction of leukaemia burden resulting in prolonged survival compared with mice treated with untransduced T cells (Fig. 5b,c). To test the specificity of NPM1c CAR-T cell killing in vivo, NSG mice were injected with luciferase-expressing human HLA-A2+ CD19+ GMB cells[23, 28]. Tumour-bearing mice were given a single dose of NPM1c CAR-T cells, untransduced T cells or CD19 CAR-T cells. Compared with untransduced T cells, NPM1c CAR-T cells did not reduce the lymphoma burden or prolong survival, whereas CD19 CAR-T cells greatly reduced the lymphoma burden and prolonged survival (Fig. 5d,e). These results show that NMP1c CAR-T cells are capable of killing AMLs that are HLA-A2+ and NPM1c+ in vivo, but not HLA-A2+ lymphoma cells without NPM1c mutation, demonstrating killing specificity.
Fig. 5. NPM1c CAR-T cell therapy reduces leukaemia burden and prolongs survival.

a, Schematic of the experimental process. NSG mice were injected with OCI-AML3 cells (1 × 106) or GMB cells (2 × 106) intravenously (D–4) and imaged for engraftment 4 d later (D0). Mice were then injected intravenously with 1 × 107 NPM1c CAR-T cells, untransduced T cells or CD19 CAR-T cells. The mice were monitored by BLI every 3 d to assess tumour burden and survival. b, Comparison of the OCI-AML3 leukaemia burden by BLI between mice treated with NPM1c CAR-T cells and untransduced T cells at the indicated days (D0–D21) post-T cell injection (n = 5). The scales for imaging are shown to the right. The experiment was repeated twice, with four and five mice per group. c, Comparison of the total flux (luciferase signals from systemic OCI-AML3 leukaemia cells) in the mice (n = 5) from b (left), and Kaplan–Meier survival curves (right; n = 9) of mice treated with either NPM1c CAR-T cells or untransduced T cells. d, Comparison of GMB lymphoma burden by BLI between mice treated with NPM1c CAR-T cells (n = 5), untransduced T cells (n = 5) and CD19 CAR-T cells (n = 3) at the indicated days (D0–D21) post-T cell injection. The scales for imaging are shown to the right. The experiment was repeated twice with five mice for the untransduced T cells and NPM1c CAR-T cells groups and three mice for the CD19 CAR-T cells group. e, Comparison of the total flux (luciferase signals from systemic GMB cells; left) and Kaplan–Meier survival curves (right) of mice treated with either NPM1c CAR-T cells (n = 5), untransduced T cells (n = 5) or CD19 CAR-T cells (n = 3). The graphs for total flux were created using Microsoft Office 2016 and the survival curve graphs were created using SPSS Statistics 22 software. Data points and error bars represent means ± s.e. P values (two-way repeated-measures analysis of variance for total flux and two-sided Mantel–Cox log-rank test for survival comparison) are indicated. Note that in b different scales are used for day 0, days 3–9 and days 12–21, and in d different scales are used for days 0–12 and days 15–21, for better comparison among the mice at different days post-treatment.
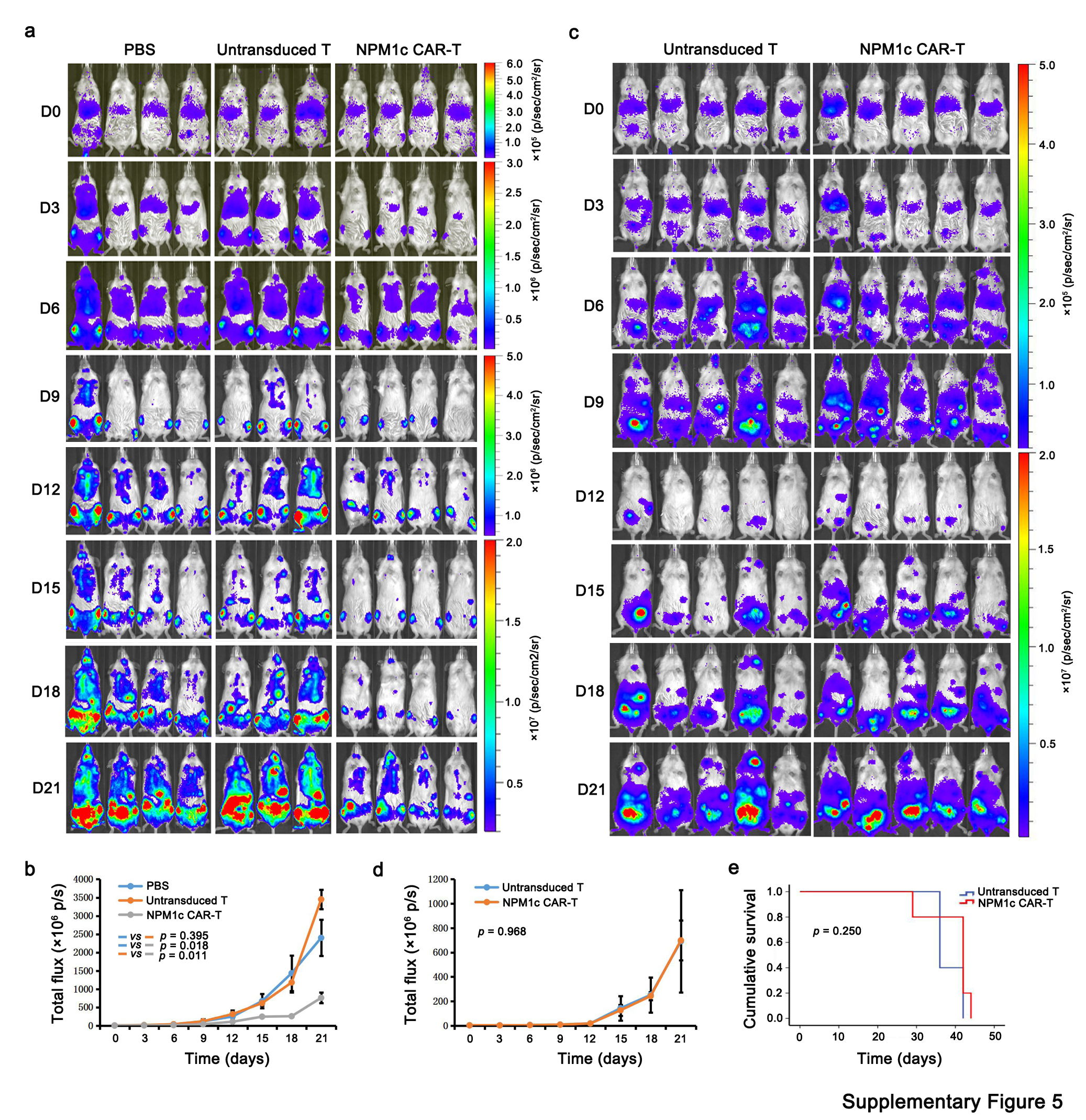
OCI-AML2 cells have a similar background to OCI-AML3 cells but express wild-type NPM1 protein[5]. As a negative control for NPM1c CAR-T cell specificity, NSG mice were injected with luciferase-expressing OCI-AML2 cells (1 × 106 per mouse), followed by a single injection of NPM1c CAR-T or control untransduced T cells (1 × 107 cells per mouse) intravenously 4 d later. As shown in Supplementary Fig. 5a,b, NPM1c CAR-T cells effectively reduced the OCI-AML3 leukaemia burden compared with phosphate-buffered saline (PBS) control or untransduced T cells. However, NPM1c CAR-T cells did not reduce the OCI-AML2 leukaemia burden in NSG mice, nor did they prolong the survival of the treated mice compared with untransduced T cells (Supplemental Fig. 5c-e). These results further support that NPM1c CAR-T cells specifically kill NPM1c-positive AML cells but not NPM1c-negative AML cells.
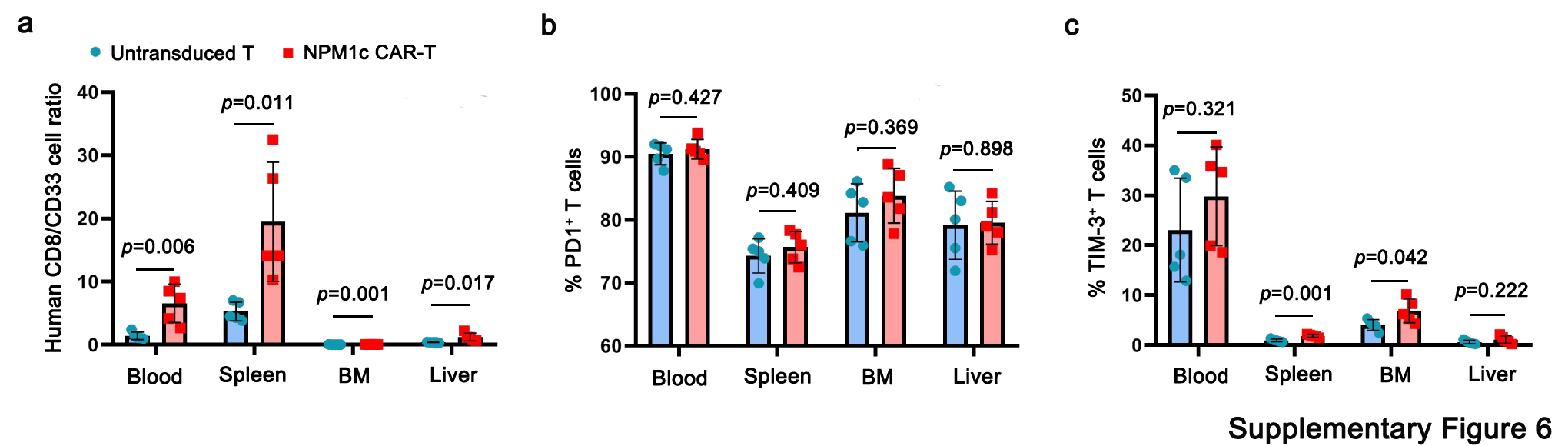
To examine the anti-leukaemia activity of NPM1c CAR-T cells, we analysed CAR-T cells and leukaemia cells in different tissues by flow cytometry 18 d post CAR-T cell transfer. As shown in Fig. 6a,b, the leukaemia burden was similar on the day of T cell transfer (day 4 after OCI-AML3 injection), but by 18 d post-T cell transfer, the leukaemia burden was significantly lower in mice that received NPM1c CAR-T cells compared with mice that received untransduced T cells. On day 18, blood, spleen, bone marrow and liver were harvested and single-cell suspensions were prepared and stained for mouse CD45 (mCD45), human CD45 (hCD45), hCD8, hCD33, human programmed cell death protein-1 (PD-1) and human T cell immunoglobulin mucin-3 (Tim-3). As shown in Fig. 6c, hCD45+ cells consisted of hCD8+ T cells and hCD33+ leukaemic cells. The numbers and percentages of hCD33+ leukaemic cells were significantly lower in all four tissues in mice treated with NPM1c CAR-T cells than those treated with untransduced T cells (Fig. 6d,e). Consistently, there were higher percentages of hCD8+ T cells in all four tissues in mice treated with NPM1c CAR-T cells than those treated with untransduced T cells. Furthermore, the ratio of hCD8+ T cells over hCD33+ leukaemic cells was significantly higher in all tissues in mice treated with NPM1c CAR-T cells than mice given untransduced T cells (Supplementary Fig. 6a). Human CD8+ T cells were also analysed for PD-1 and Tim-3 expression (Fig. 6c). Although the percentages of hCD8+ T cells that expressed PD-1 varied significantly among blood, spleen, bone marrow and liver, there was no significant difference in the same tissue between mice given untransduced T cells or NPM1c CAR-T cells (Supplementary Fig. 6b). Interestingly, the percentages of T cells that expressed Tim-3 were significantly higher in the spleen and bone marrow of mice given NPM1c CAR-T cells, but these percentages were low (<5%; Supplementary Fig. 6c). These results are consistent with BLI analysis of leukaemic burden.
Fig. 6. NPM1c CAR-T cells reduce the leukaemia burden in blood, spleen, bone marrow and liver.

a,b, NSG mice were injected with OCI-AML3 AML cells and then either untransduced T cells or NPM1c CAR-T cells, as in Fig. 5a. Mice (n = 5) were imaged on the day of T cell injection (day 0) and 18 d later. Shown are BLI images (a) and total flux (b). c, Representative flow cytometry plots showing the gating strategy and expression profiles. Blood, spleen, bone marrow and liver were harvested on day 18 and single-cell suspensions were prepared and stained for mCD45, hCD45, CD8, CD33, PD-1 and Tim-3, followed by flow cytometry. Shown are representative staining profiles and gating strategies, including: mCD45 versus hCD45 gating on live cells (DAPI−); hCD33 versus hCD8 gating on hCD45+ cells; human PD-1 versus hCD8 gating on hCD8+ cells; and human Tim-3 versus hCD8 gating on hCD8+ cells. The numbers indicate the percentages of cells in the gated region. d, Comparison of total numbers of hCD33+ leukaemic cells and hCD8+ T cells in different tissues between mice given NPM1c CAR-T cells and those given untransduced T cells. e, Comparison of percentages of hCD33+ leukaemic cells and hCD8+ T cells among hCD45+ cells in different tissues between mice given NPM1c CAR-T cells and those given untransduced T cells. The blue bars in b, d and e represent treatment with untransduced T cells, whereas the pink bars represent treatment with CAR-T cells. The graphs were created using GraphPad Prism version 8.00 software. Bars heights and error bars represent means ± s.e. P values (two-sided independent-samples t-test) are indicated in b, d and e (n = 5).
We further analysed the leukaemia burden in the bone marrow from surviving mice 30 d after injection with NPM1c CAR-T cells, untransduced T cells or PBS (these mice are shown in Supplementary Fig. 5a). OCI-AML3 leukaemia cells were almost completely absent in the bone marrow of three of the four NPM1c CAR-T-treated mice (Supplementary Fig. 7), whereas a large population of human T cells was detected in the bone marrow. In contrast, a large number of leukaemia cells were detected in the bone marrow from one surviving mouse (the other mice died within 30 d) treated with untransduced T cells and two surviving mice (the other mice died within 30 d) treated with PBS (Supplementary Fig. 7). Therefore, NPM1c CAR-T cells appear to be capable of expanding and killing OCI-AML3 cells in bone marrow and significantly controlling disease progression.
NMP1c CAR-T cells effectively kill primary human AML blasts in vitro and in vivo
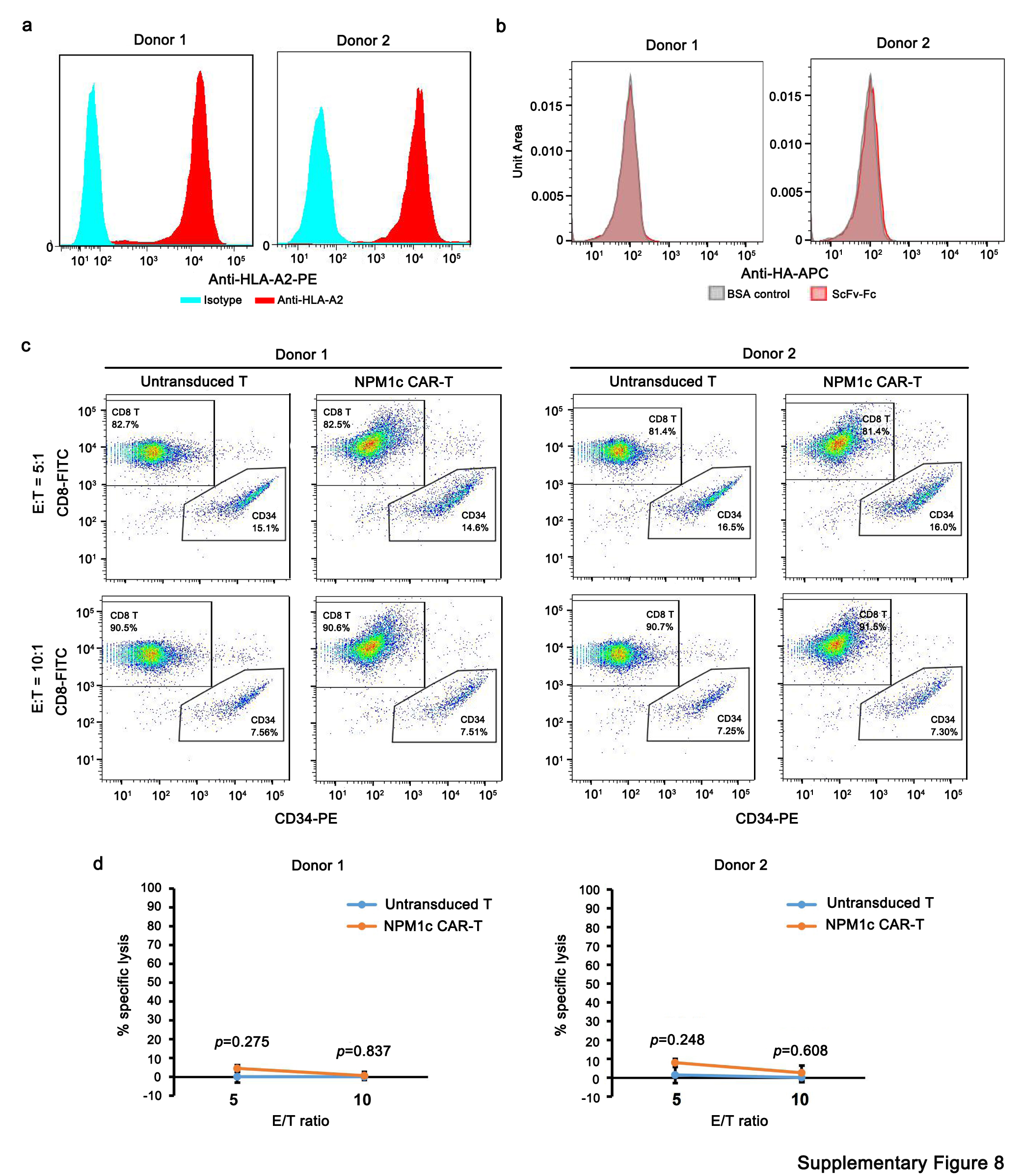
Next, we tested whether NPM1c CAR-T cells can kill NPM1c+HLA-A2+ primary AML blasts. NPM1c+HLA-A2+ primary AML blasts from three different donors were incubated with NPM1c CAR-T cells at different effector-to-target ratios and the number of AML blasts was quantified 24 h later by flow cytometry with precision count beads. As shown in Fig. 7a, all three primary AML samples were effectively killed by NPM1c CAR-T cells in an effector-to-target ratio-dependent manner, although killing activities were variable between samples. In contrast, human HLA-A2+CD34+ haematopoietic stem/progenitor cells from two different donors were not stained by YG1 scFv–Fc (Supplementary Figs. 8a and 4b) and were not killed by NPM1c CAR-T cells compared with untransduced T cells (Supplementary Figs. 8c and 4d).
Fig. 7. NMP1c CAR-T cells effectively kill primary human AML blasts in vitro and in vivo.

a, NPM1c CAR-T cells kill NPM1c+HLA-A2+ primary AML blasts from three donors in vitro. NPM1c CAR-T cells or untransduced T cells were incubated with AML blasts at the indicated ratios for 24 h. The absolute numbers of AML blasts were quantified by staining for CD8 and CD33, followed by flow cytometry with precision count beads. The percentages of specific lysis of tumour cells at different effector-to-target ratios were calculated (see Supplementary Information). The graphs were created using Microsoft Office 2016 (n = 3 biological replicates. b, NPM1c CAR-T cell treatment reduces the leukaemia burden in primary HLA-A2+NPM1c+ AML xenografts. NSGS mice were engrafted with human AML blasts. Two weeks later, when AML blasts were detectable in the blood, mice were given NPM1c CAR-T cells or untransduced T cells. At the indicated days after T cell transfer, mice were bled and mononuclear cells were stained for mCD45, hCD45 and hCD8. Shown are representative hCD45 versus mCD45 staining profiles gating on hCD8− live cells. AML blasts were hCD45+hCD8−. The numbers indicate the percentages of cells in the gated regions. c, Comparison of the percentages of hCD45+CD8− AML blasts in the peripheral blood between mice given NPM1c CAR-T cells and those given untransduced T cells before T cell injection (day 0) and 9 and 18 d post-T cell injection (n = 5). The blue bars represent treatment with untransduced T cells, whereas the pink bars represent treatment with NPM1c CAR-T cells. The graphs were created using GraphPad Prism version 8.00 software. In a and c, bar heights and error bars represent means ± s.e. and P values (two-sided independent-samples t-test) are indicated.
To assess the efficacy of NPM1c CAR-T therapy in patient-derived xenografts, the triple transgenic NSG-SGM3 (NSGS) mice expressing human IL3, GM-CSF and SCF were injected with HLA-A2+NPM1c+ human primary AML blasts. To improve the engraftment efficiency of human primary AML, NSGS mice were hydrodynamically injected with 100 μg DNA plasmids encoding human IL-3 and granulocyte–macrophage colony-stimulating factor 24 h before primary AML injection, as previously described[29]. The AML burden was quantified in the peripheral blood by tail vein bleeding and flow cytometry assaying for hCD45+CD8− cells. AML blasts were detected in the blood 2 weeks after injection (Fig. 7b) and the mice were injected with NPM1c CAR-T cells or untransduced T cells. The level of AML blasts in the blood was monitored every 9 d. As shown in Fig. 7b,c, the level of AML blasts was reduced in mice treated with NPM1c CAR-T cells compared with untransduced T cells at both 9 and 18 d after T cell injection. The difference became significant by day 18. These results show that NPM1c CAR-T cells were also effective in killing primary HLA-A2+NPM1c+ AML blasts in a patient-derived xenograft model.
Discussion
Since the approval of CD19-directed CAR-T cell therapy[30], hundreds of clinical trials have evaluated CAR-T cells targeting different antigens for different tumours[31]. However, almost all of these CAR-T cells target TAAs that are also expressed in healthy tissues (albeit at low levels), which usually results in significant on-target/off-tumour toxicity[32]. For example, CAR-T therapies targeting CD123 or CD33 for the treatment of AML have shown promising antitumour responses in preclinical models, but also resulted in severe myeloablation, due to low CD123 and CD33 expression on normal haematopoietic stem/progenitor cells[33, 34]. Currently, most CARs are designed to bind to antigens on the surface of target cells. However, most proteins from mutated genes are expressed inside the cell, making them unavailable as targets for conventional CARs[35]. Our identified scFv specifically recognizes the AIQ–HLA-A2 complex that is present only on HLA-A2+ AML cells with the intracellular mutant NPM1c protein. NPM1c is an essential driver gene mutation for malignant transformation early in leukaemogenesis and present in all leukaemic cells[5]. Therefore, NPM1c CAR-T cells should be able to specifically target all leukaemia cells, regardless of the heterogeneity of the tumour cell population, and thus reduce the development of tumour resistance. Importantly, different from previous CAR-T cells against AML, NPM1c CAR-T cells did not react to CD34+ normal haematopoietic stem/progenitor cells. Similarly, due to the absence of NPM1c expression in healthy tissues, NPM1c CAR-T cells are expected to mediate antitumour immunity with no or minimal on-target/off-tumour toxicity.
Yeast surface display is a powerful approach for isolating and engineering antibodies to increase their affinity, specificity and stability[18]. Unlike other display technologies, such as ribosome and phage display, it enables quantitative screening through the use of FACS[18]. The antigen-binding signal is normalized for expression, eliminating artefacts due to host expression bias and allowing for fine discrimination between different scFv variants. Furthermore, because the display library is derived from fully human antibody sequences, the isolated scFvs are expected to have reduced immunogenicity and to be suitable for therapeutic development for human use. In our study, we isolated an scFv that specifically binds to the AIQ–HLA-A2 complex with high affinity (~7 nM). Its specificity was further confirmed in binding assays using recombinant scFv–Fc to different peptide–HLA-A2 complexes displayed on the cell surface, and using AIQ–HLA-A2 binding to CAR-T cells. Interestingly, we observed that the yeast cell population that bound to the AIQ–HLA-A2 complex but not control peptide–HLA-A2 complexes also bound to HLA-A2 without loaded peptide during the screening process. We were able to enrich yeast cells that only bind to the AIQ–HLA-A2 complex but not HLA-A2 through negative selection with HLA-A2. These results suggest that the isolated scFv specifically forms contacts with AIQ peptide as well as HLA-A2.
Our engineered CAR-T cells showed an efficient and specific in vitro anti-leukaemia activity against human NPM1c+HLA-A2+ AML cells, and exhibited increased IFN-γ expression when co-cultured with OCI-AML3 cells. However, only a small fraction of CAR-T cells (<2%) expressed IL-2 when co-cultured with OCI-AML3 target cells. The results from Quantibody Human Cytokine Array further verified the low level of IL-2 production by NPM1c CAR-T cells co-cultured with OCI-AML3 cells, which may be due to the low density of NPM1c peptide–HLA-A2 complex present on the surface of OCI-AML3 cells. Supporting this possibility, a study has shown that the target antigen density that is required to induce T cell cytokine production is much higher than that required to stimulate CAR-mediated lysis: CD20-specific CAR-T cells could lyse target cells with the lowest density of CD20 (~200 molecules per cell), but cytokine production required a higher density of CD20 (~5,000 molecules per cell)[36]. Recently, another study investigated the antigen density required to activate CAR-T cells using Nalm6 cells and found that CAR-T cells could lyse target cells with the lowest CD20 expression and induce IFN-γ production with moderate antigen density, but a significantly higher threshold of CD20 density was required for IL-2 production compared with IFN-γ production[37]. Although we do not know the density of AIQ–HLA-A2 on OCI-AML3 cells, the density of peptide–major histocompatibility complex class I (MHC I) complex on the cancer cells usually ranges from 100 to a few thousand molecules[38], which is sufficient to trigger the cytolytic activity of CAR-T cells, but may not be enough to trigger CAR-T cells to produce cytokines[39]. In this respect, the risk of cytokine release syndrome may potentially be lower using CAR-T cells targeting peptide–MHC complexes than highly expressed TAAs.
NPM1c CAR-T cells appear to be less potent than CD19 CAR-T cells in eliminating respective target tumour cells in NSG mice. We showed that the cytolytic activity of the NPM1c CAR-T cells was proportional to the level of NPM1c–HLA-A2 complex on the surface of AIQ-pulsed T2 target cells, suggesting that the lower cytolytic activity of NPM1c CAR-T cells towards OCI-AML3 than CD19 CAR-T cells towards B cell leukaemic cells is probably due to a lower level of cognate peptide–MHC complexes on OCI-AML3 cells than CD19 on B cell leukaemic cells. Besides antigen density, other factors such as CAR affinity could also influence the anti-leukaemia activity of CAR-T cells. In general, the affinity of scFv-based CAR to its target is in the nM range (Kd = 10−6–10−9 M), which is much higher than the affinity of TCR to peptide–MHC complex. Higher CAR affinity could activate CAR-T cells more potently, leading to higher cytolytic activity. However, high affinity of CAR-T may prevent serial killing, promote T cell exhaustion and even cause activation-induced T cell death[39]. The affinity of our scFv for the AIQ–HLA-A2 complex was about 7 nM, which is in the range of the affinities of scFv-based CARs.
TCR-T cell therapies have been developed against peptide–HLA complexes on tumour cells and have shown efficacy in preclinical experiments[40]. Intracellular proteins such as PRAME (preferentially expressed antigen in melanoma) and WT1 (Wilms tumour 1 antigen) are effective antigens for targeting AML by TCR-T cell therapy, but low antigen expression on some normal tissue cells leads to a risk for toxicity[41, 42, 43]. A recent report has shown that T cells transduced with TCRs specific for a different neoepitope derived from NPM1c in complex with HLA-A2 were effective in killing AML cells[5]. A drawback of TCR-T cell therapy is the risk of TCR chain mispairing between exogenous and endogenous TCR-α and -β chains, resulting in potential altered specificity and reduced efficacy[5, 44, 45]. Another major challenge for TCRs is their low binding affinity to tumour antigens, although several methods have been proposed to enhance the affinity of TCRs to antigens[40]. The affinity of our scFv for the AIQ–HLA-A2 complex was about 7 nM, which is ten- to 100-fold higher than the affinity of a typical TCR for its cognate peptide–MHC complex.
Both our CAR-T cell therapy and the TCR-T cell therapy described by the other study[5] showed specific recognition of NPM1c peptide presented by HLA-A2 and specific lysis of NPM1c+HLA-A2+ AML in vitro and in vivo. In our study, we measured the affinity of our scFv–Fc to NPM1c epitope/HLA-A2 complex, with a Kd of ~7 nM. In the study on TCRs, the authors did not measure the affinity of their TCRs for NPM1c epitope/HLA-A2 complex. Based on previous studies[46], the affinity of TCRs for peptide–MHC complex is about 100- to 1,000-fold lower compared with antibody–antigen interactions. The potential difference in the affinity of our scFv versus TCR for NPM1c epitope/HLA-A2 could have significant implications for their utility. Because of the low affinity of TCRs for peptide/MHC complexes, the isolated TCR is pretty much limited to TCR-T cell application, where co-receptor CD8 may facilitate the interactions between TCRs and peptide–MHC complexes. In contrast, our scFv could have wider applications. As we showed, the scFv can be engineered into CAR for CAR-T cell application. We expect that scFv can be engineered into CAR for CAR-natural killer cell application. Similarly, the isolated scFv can also be engineered into bi-specific antibodies for clinical applications. In addition, our scFv–Fc can be used as a diagnostic tool to identify NPM1c/HLA-A2 complexes on AML cells by flow cytometry.
Methods
Preparation of peptide–HLA-A2 complexes
The CD8 T cell epitope peptide (AIQDLCLAV) from mutant NPM1c and control peptides from cancer/testis antigen NY-ESO-1 (SLLMWITQC; abbreviated as SLL) and influenza virus M1 protein (GILGFVFTL; abbreviated as GIL) were synthesized and purified by GenScript. Recombinant human HLA-A2:Ig fusion protein (DimerX I) was obtained from BD Biosciences. Peptide–HLA-A2 complexes were prepared according to the manufacturer’s protocol. Briefly, peptide was mixed with HLA-A2:Ig fusion protein at a molar ratio of 640:1, and the mixture was incubated at 37 °C overnight to allow for spontaneous complex assembly. The peptide-loaded HLA-A2:Ig fusion protein, referred to as peptide–HLA-A2, was stored at 4 °C for up to 1 week.
Yeast media formulations
SDCAA media contained 20 g dextrose, 6.7 g Difco yeast nitrogen base, 5 g Bacto casamino acids, 10.2 g Na2HPO4·7H2O and 8.56 g NaH2PO4·H2O, dissolved in deionized H2O to a volume of 1 l and filter sterilized using 0.2-μm filter units.
SGCAA madia contained 20 g galactose, 6.7 g Difco yeast nitrogen base, 5 g Bacto casamino acids, 10.2 g Na2HPO4·7H2O and 8.56 g NaH2PO4·H2O, dissolved in deionized H2O to a volume of 1 l and filter sterilized using 0.2-μm filter units.
SDCAA plates were generated from 10.2 g Na2HPO4·7H2O, 8.56 g NaH2PO4·H2O, 182 g sorbitol and 15 g Bacto agar, dissolved in deionized H2O to a volume of 900 ml and autoclaved. In a separate beaker, 20 g dextrose, 6.7 g Difco yeast nitrogen base and 5 g bacto casamino acids were dissolved in deionized H2O to a volume of 100 ml and sterilized by filtration. The autoclaved mixture was cooled with stirring until below 50 °C, combined with the filter-sterilized solution, mixed and poured into plates.
SG-2×SCAA media contained SG (6.7 g Difco yeast nitrogen base, 20 g galactose, 10.2 g Na2HPO4·7H2O and 8.56 g NaH2PO4·H2O), 190 mg Arg, 400 mg Asp, 1,260 mg Glu, 130 mg Gly, 140 mg His, 290 mg Ile, 400 mg Leu, 440 mg Lys, 108 mg Met, 200 mg Phe, 220 mg Thr, 52 mg Tyr, 380 mg Val and 1 g bovine serum albumin (BSA), dissolved in deionized H2O to a volume of 1 l and sterilized by filtration.
Cell line culture
OCI-AML3, T2, 293T and PC-3 cells were purchased from the American Type Culture Collection (ATCC). OCI-AML2 cells were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ). OCI-AML3 and OCI-AML2 cells were cultured in RPMI-1640 medium (Gibco) supplemented with 10% foetal bovine serum (FBS; Life Technologies and VWR) and 2 mM l-glutamine (Thermo Fisher Scientific). GMB cells were generated by engrafting human haematopoietic stem cells transduced with the oncogenes c-Myc and Bcl2 into immunodeficient mice as previously described[23, 28]. The RJY100 yeast strain was constructed using standard homologous recombination approaches and has been described in detail previously[20]. GMB cells were cultured in DMEM medium (Gibco) supplemented with 110 mg l−1 pyruvate sodium, 1 × Non-Essential Amino Acids, 1× 2-mercaptoethanol and 10% FBS. T2 cells (174× CEM.T2; ATCC; CRL1992) were cultured in IMDM medium (Gibco) supplemented with 20% FBS. PC-3 cells were cultured with F-12K medium (ATCC) supplemented with 10% FBS. All media was supplemented with 1% vol/vol penicillin-streptomycin solution (Life Technologies).
Isolation of human scFv specific for the AIQ–HLA-A2 complex
Human scFv that recognizes the AIQ–HLA-A2 complex was isolated from a non-immune human scFv library displayed on a yeast surface using MACS followed by selection using FACS (FACSAria II), as previously described[18]. To enrich scFv specific for the AIQ–HLA-A2 complex, the SLL–HLA-A2 complex and HLA-A2 protein were used for negative selection. AIQ–HLA-A2, SLL–HLA-A2 and HLA-A2 were biotinylated before each round of selection. The selected yeast pool from each round was propagated in SDCAA media overnight and then induced to express scFv in SGCAA media. The numbers of induced yeast cells used for the subsequent round of selection were about tenfold the number of selected yeast cells from the previous round of selection. The strategy for isolation of scFv recognizing the AIQ–HLA-A2 complex is shown in Fig. 1a,b. After two rounds of MACS selection including positive selection with AIQ–HLA-A2 followed by negative selection with SLL–HLA-A2, the selected yeast pool was subject to multiple rounds of FACS selection (Fig. 1b; FACS sorting). During each round of FACS selection, the selected yeast cell pool was assayed by flow cytometry for binding to HLA-A2, GIL–HLA-A2, SLL–HLA-A2 and AIQ–HLA-A2 (Fig. 1b; FACS analysis).
Isolation of plasmid DNA from the selected yeast cells and sequencing of scFvs
Plasmid DNA from the selected population of yeast cells was isolated using a Zymoprep kit (Zymo Research) according to the manufacturer’s instructions. The number of yeast cells used for plasmid isolation was at least tenfold higher than the expected population diversity, so as to yield display plasmids from each of the different yeast clones in the selected population[18]. Alpha-Select Gold E. coli competent cells (Bioline) were transformed with 5 μl plasmid DNA, plated onto LB ampicillin plates and incubated overnight at 37 °C. Twenty-five single colonies were randomly picked, inoculated in LB ampicillin media and grown overnight at 37 °C, with shaking at 200 r.p.m. Plasmid DNA was then prepared from these 25 cultures by Miniprep (Qiagen), and scFv was sequenced using the following primers: forward (5′-GTCAGTAATTGCGGTTCTCACC-3′) and reverse (5′-GTACAGTGGGAACAAAGTCG-3′).
Characterization of scFv clones
The RJY100 competent yeast cells were prepared using a Frozen-EZ Yeast Transformation II kit (Zymo Research). The display plasmid for a single scFv clone of interest was transformed into competent yeast cells according to the manufacturer’s protocol. Single clones were obtained by plating the transformed yeast cells on an SDCAA plate and incubating at 30 °C for more than 2 d until they formed visible colonies. Three to five clones were randomly selected and inoculated separately in a 5-ml SDCAA culture and grown at 30 °C, with shaking at 250 r.p.m. overnight. To induce scFv expression on the yeast surface, 5 × 107 yeast cells from SDCAA culture were inoculated into 5-ml SGCAA media and induced at 20 °C, with shaking at 250 r.p.m. for at least 20 h. The resulting yeast cells were incubated with biotin-labelled HLA-A2, GIL–HLA-A2, SLL–HLA-A2 or AIQ–HLA-A2, followed by staining with streptavidin-APC (BioLegend) and FITC-labelled anti-c-Myc antibody (Abcam) on ice for 20 min in the dark. Stained yeast cells were analysed by flow cytometry (LSR II HTS-1), collecting at least 20,000 events per sample.
Expression of soluble scFv–Fc protein
Soluble scFv–Fc protein was produced using a switchable yeast display/secretion system[20]. ScFv DNA was synthesized (Integrated DNA Technologies) and the Fc fragment was amplified by PCR using Q5 High-Fidelity 2× Master Mix (New England Biolabs). The PCR primers were 5′-CCGGGGTAGAACCTAAAAGTTCCG-3′ (forward) and 5′-TTTGTTCTGCACGCGTGGATC-3′ (reverse). The switchable yeast display/secretion vector backbone pCHA-FcSup-TAG was doubly digested with the enzymes NheI and BamHI. After gel purification, the vector backbone, scFv DNA and Fc DNA fragments were assembled using Gibson Assembly Master Mix (New England Biolabs) according to the manufacturer’s protocol. A total of 2 μl of the assembly reaction mixture was transformed into α-Select Gold E. coli competent cells (Bioline). The transformed cells were shaken (200 r.p.m.) at 37 °C for 60 min and then plated on an LB ampicillin plate. Colonies were picked and grown in liquid LB media containing ampicillin for plasmid extraction. The resulting plasmids were sequenced and the sequencing primers were 5′-GGGTAATTAATCAGCGAAGCGATG-3′ (forward) and 5′-GTACAGTGGGAACAAAGTCG-3′ (reverse). The correct plasmid was transformed into the RJY100 competent yeast cells. The transformed yeast cells were grown to saturation at 30 °C in SD-SCAA media, with shaking at 250 r.p.m. overnight. The saturated cultures were pelleted and resuspended to an OD600 of 1.0 in 200 ml SG-2×SCAA induction media. The resuspended cultures were then grown for 4 d at 20 °C with shaking at 250 r.p.m. The yeast cells were pelleted at 10,000g for 15 min and the supernatant was filtered using a 0.2-μM filter (Pall Corporation). The pH of filtrate was adjusted to pH 7.4 with 10×PBS pH 7.4 (Gibco), to a final concentration of 1×. scFv–Fc was purified by passing the filtrate twice through a pre-equilibrated protein A column containing 1 ml resin (GenScript) according to the manufacturer’s protocol. The flow-through media was collected for measurement of the binding efficiency to the resin by SDS-PAGE. Resin with bound scFV–Fc was washed three times using 10 ml Binding/Wash Buffer. scFv–Fc was eluted from the resin using 10 ml Elution Buffer. The eluate containing scFv–Fc was immediately neutralized to pH 7.4 with Neutralization Buffer (1/10 volume of total eluate). The neutralized eluate was concentrated and buffer exchanged into 1× PBS using centrifugal filtration units (Millipore; 30 kDa molecular weight cut-off). scFv–Fc was quantified by A280 measurement on a Nanodrop spectrophotometer (Thermo Fisher Scientific) and the purity was assessed by SDS-PAGE using 5% stacking gel and 6% separating gel. Specific binding of the purified scFv–Fc protein to the AIQ–HLA-A2 complex on the surface of target cells was assessed by flow cytometry using 5 nM protein to incubate with 5 × 105 target cells in 200 μl PBS buffer for 30 min at room temperature, followed by staining with phycoerythrin-or APC-labelled anti-haemagglutinin tag antibody.
scFv–Fc affinity determination by biolayer interferometry
BLI experiments were performed using the Octet RED96 instrument (FortéBio) at 25 °C. The same buffer used for all of the steps comprised binding buffer with 0.05% Tween 20 and 0.1% BSA. The scFv–Fc fusion proteins were biotinylated using EZ-Link NHS-PEG4-Biotin (Thermo Fisher Scientific). Streptavidin-coated biosensors (Streptavidin Dip and Read Biosensors; FortéBio) were loaded with a 200-μl volume of 20 μg ml−1 biotinylated scFv–Fc protein until all sensors (except for the reference) reached a capture threshold of 0.5 nm. After a 60-s rinse and baseline steps in buffer alone, the sensors were exposed to a 1:2 dilution series (0, 2.5, 5, 10, 20, 40 and 80 nM) of the antigen (AIQ–HLA-A2 complex, SLL–HLA-A2 complex or HLA-A2). The association with antigen was monitored for 1,000 s and dissociation was carried out for 1,500 s in buffer alone. Data analysis was performed using FortéBio Data Analysis 8.2 (FortéBio). The dataset was fit to a 1:1 binding model to determine Kon, Koff and Kd.
CAR vector design
The sequence of CAR, consisting of the YG1 or CD19 scFv, the CD8α leader sequence, an extracellular hinge domain and transmembrane domain, the 4-1BB co-stimulatory domain and the CD3ζ activation domain, was custom synthesized by Integrated DNA Technologies. The second fragment consisting of self-cleavage P2A followed by EGFP (P2A–EGFP) was synthesized the same way. The pHIV vector (plasmid 21373) was doubly digested with the enzymes XbaI and ClaI. After gel purification of the vector backbone, the pHIV backbone, CAR fragment and P2A–EGFP fragment were assembled based on their overlap region at the 5′ and 3′ terminals using HiFi DNA Assembly Master Mix (New England Biolabs) according to the manufacturer’s protocol. The resulting plasmids were sequenced using the following sequencing primers: 5′-GTTAGGCCAGCTTGGCACTTGATGT-3′ (forward) and 5′-AGGCACAATCAGCATTGGTAGCTG-3′ (reverse). The plasmid with the correct sequence was named pHIV-CAR-GFP.
Generation of CAR-expressing primary human T cells
Lentivirus was generated by transfecting 293T cells with pHIV-CAR-GFP, pCMV-VSVG, pCMV-Δ8.9 and pAdv plasmids. Culture supernatants were collected at 48 and 72 h and lentivirus particles were pelleted by ultracentrifugation at 25,000 r.p.m. and 4 °C for 2 h. Lentivirus particles were suspended in 100 μl serum-free DMEM media and frozen at −80 °C. Human CD8+ T cells were isolated from HLA-A2+ donor peripheral blood mononuclear cells using an EasySep Human CD8+ T Cell Isolation Kit (STEMCELL Technologies). The purity of the isolated T cells (>95%) was quantified using FACS with CD3 and CD8 stainings. The CD8+ T cells were activated and expanded using T Cell TransAct (Miltenyi Biotec) and cultured in TexMACS Medium (Miltenyi Biotec) supplemented with 50 IU ml−1 IL-2 (Miltenyi Biotec), 3% FBS and 1% penicillin-streptomycin (Life Technologies). T cells were transduced with lentivirus (multiplicity of infection = 10) 4 d after TransAct stimulation. GFP+ CAR-T cells were purified by FACS, and CAR expression on T cells was determined using Alexa Fluor 647 AffiniPure F(ab’)2Fragment Goat Anti-Human IgG (H + L) (Jackson ImmunoResearch). CAR-T cells were expanded in TexMACS Medium (Miltenyi Biotec) supplemented with 10 ng ml−1 recombinant human IL-7 (PeproTech), 5 ng ml−1 recombinant human IL-15 (PeproTech), 3% FBS and 1% vol/vol penicillin-streptomycin solution (Life Technologies) for 10 d before use.
Cytotoxicity of CAR-T cells in vitro
To assess the ability of CAR-T cells to kill target cells, CD8+ CAR-T cells were incubated with target cells at the indicated effector-to-target ratios. Immediately after mixing the cells (0 h) and 24 h after incubation, the cells were washed in PBS without serum and stained using Live/Dead Fixable Dead Cell Stain kits (Invitrogen) according to the manufacturer’s protocol. Cells were then stained with FITC-anti-human CD8, PE-anti-human CD33 or PE-anti-human CD19 antibody on ice for 30 min in the dark. Cells were washed with FACS buffer and analysed by flow cytometry (LSR II HTS-1). Specific lysis of each sample was calculated using the following formula: lysis (%) = (1 – ((percentage of target cells at 24 h/percentage of target cells at 0 h) in the CAR-T cell group/(percentage of target cells at 24 h/percentage of target cells at 0 h) in the untransduced T cell group)) × 100.
Intracellular cytokine staining
CAR-transduced or untransduced human CD8+ T cells were co-cultured with target cells (1:1 ratio; 2 × 106 cells per ml), in 96-well round-bottom plates, in RPMI-1640 containing 10% FBS without any cytokines, in the presence of the protein transport inhibitor monensin (BD Biosciences) and brefeldin A (BioLegend) at 37 °C and 5% CO2 for 12 h. Cells were washed and stained with Live/Dead Fixable Aqua Dead Cell Stain, followed by surface staining for CD3, then fixed, permeabilized and intracellularly stained with anti-IFN-γ-APC-Cy7 (BioLegend) and anti-IL-2-APC (BioLegend). The cells were analysed by flow cytometry (LSRFortessa HTS-2) and live CD3+ lymphocytes were gated for further analysis.
CAR-T cell killing of target cells in vivo
NOD-scid IL2rgnu11 (NSG) mice were purchased from The Jackson Laboratory and housed in specific pathogen-free vivarium at the Massachusetts Institute of Technology. All animal work described in this Article complied with local animal ethical and welfare standards. All experiments with mice were conducted under protocol number 031902022 approved by the Institutional Animal Care and Use Committee at the Massachusetts Institute of Technology. Briefly, luciferase-expressing OCI-AML3 cells (1× 106), GMB cells (2 × 106) or OCI-AML2 cells (1 × 106) were injected in 200 μl PBS into NSG mice by tail vein injection. After 4 d, 1 × 107 CAR-T cells (sorted based on GFP expression) or activated but untransduced human T cells were injected into the tumour-bearing mice. BLI was performed every 3 d using a Xenogen IVIS-200 Spectrum camera.
To analyse T cells and tumour cells by flow cytometry, blood, spleen, bone marrow and liver were harvested 18 d post-T cell injection. About 200 μl blood was collected by heart puncture into a microcentrifuge tube containing 1 ml PBS with 4 mM EDTA. Cells were pelleted by centrifugation at 1,500 r.p.m. for 5 min, resuspended with 1 ml ACK Lysis Buffer (Lonza Bioscience) by gently pipetting the mixture up and down, and kept at room temperature for 5 min. The cells were centrifuged at 1,500 r.p.m. for 5 min and then resuspended in FACS buffer. Before organ harvesting, liver was perfused with 5 ml PBS through the superior mesenteric vein. Spleen and liver were mechanically disrupted by pressing them through a 70-μm strainer using a syringe plunger. The disrupted liver was digested with 2 mg ml−1 collagenase D (Sigma–Aldrich) at 37 °C for 30 min. Bone marrow cells were collected from the bilateral femurs by flushing with 5 ml cold PBS. Single-cell suspensions were prepared and lysed of red blood cells. Cells were washed with FACS buffer, centrifuged at 1,500 r.p.m. and 4 °C for 5 min, resuspended with FACS buffer and counted with trypan blue staining.
For flow cytometry analysis, 5 million cells were aliquoted and mixed with 20 μl human serum to block the Fc receptor for 5 min. Then, cells were stained with the following conjugated antibodies on ice for 30 min in the dark: anti-mCD45.1-BUV737 (BD Biosciences), anti-hCD45-APC-Cy7 (BioLegend), anti-hCD8-PE (BioLegend), anti-CD33-APC (BioLegend), anti-hPD1-PE-Cy7 (BioLegend) and anti-Tim-3-BV711 (BioLegend). The cells were washed with FACS buffer and resuspended in FACS buffer containing 4′,6-diamidino-2-phenylindole (DAPI). Flow cytometry analysis was performed using an LSRFortessa HTS-2 flow cytometer (BD Biosciences), collecting 100,000 events per sample.
Statistical analysis
Data are expressed as means ± s.e. from at least three independent experiments unless otherwise stated. A two-tailed t-test was used to compare two independent groups. Tumour growth in vivo was compared using two-way repeated-measures analysis of variance. The Kaplan–Meier method was used to analyse survival patterns in tumour-bearing mice and statistical differences were evaluated according to the Mantel–Cox log-rank test. A P value of <0.05 was considered statistically significant after any adjustment. All of the statistical analyses were performed using SPSS Statistics 22 software. Unless otherwise stated, graphs were created using GraphPad Prism version 8.00 for Windows (GraphPad Software; www.graphpad.com).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We are grateful to K. D. Wittrup and A. W. Tisdale for the yeast display library and yeast display/secretion system. We thank B. Turner and S. A. Nordeen for assistance with the Octet biolayer interferometry experiment, V. Spanoudaki for assisting with mouse BLI, and the Koch Institute Flow Cytometry Core for assistance. This work was supported in part by National Institutes of Health grants AI69208, CA197605 and NS104315, the Ivan R. Cottrell Professorship and Research Fund, Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, National Institutes of Health Pre-Doctoral Training Grant T32GM007287 and a gift from C2Y Therapeutics.
Footnotes
Supplementary information
is available for this paper at https://doi.org/10.1038/s41551-020-00625-5.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Competing interests The authors have filed a provisional patent application on the identified scFv and its applications.
Data availability
The data that support the main findings of this study are available within the paper and its Supplementary Information. Raw data generated for this study are available in Figshare with the identifier 10.6084/m9.figshare.12922520 (https://doi.org/10.6084/m9.figshare.12922520) (ref. [47]).
References
- 1.Coulie PG, Van den Eynde BJ, van der Bruggen P & Boon T Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Srivastava S & Riddell SR Chimeric antigen receptor T cell therapy challenges to bench-to-bedside efficacy. J. Immunol 200, 459–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blankenstein T, Leisegang M, Uckert W & Schreiber H Targeting cancer-specific mutations by T cell receptor gene therapy. Curr. Opin. Immunol 33, 112–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Van der Lee DI et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Invest 129, 774–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verdegaal EM et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536, 91–95 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Thomas D & Majeti R Biology and relevance of human acute myeloid leukemia stem cells. Blood 129, 1577–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dombret H & Gardin C An update of current treatments for adult acute myeloid leukemia. Blood 127, 53–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dohner H, Weisdorf DJ & Bloomfield CD Acute myeloid leukemia. N. Engl. J. Med 373, 1136–1152 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Ossenkoppele GJ, Janssen JJ & van de Loosdrecht AA Risk factors for relapse after allogeneic transplantation in acute myeloid leukemia. Haematologica 101, 20–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ley TJ et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 368, 2059–2074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandoth C et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papaemmanuil E et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med 374, 2209–2221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falini B et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med 352, 254–266 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Greiner J et al. Mutated regions of nucleophosmin 1 elicit both CD4+ and CD8+ T-cell responses in patients with acute myeloid leukemia. Blood 120, 1282–1289 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Greiner J et al. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut). Blood 122, 1087–1088 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Chao G et al. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc 1, 755–768 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Choo JA, Liu J, Toh X, Grotenbreg GM & Ren EC The immunodominant influenza A virus M158–66 cytotoxic T lymphocyte epitope exhibits degenerate class I major histocompatibility complex restriction in humans. J. Virol 88, 10613–10623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Deventer JA, Kelly RL, Rajan S, Wittrup KD & Sidhu SS A switchable yeast display/secretion system. Protein Eng. Des. Sel 28, 317–325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quentmeier H et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 19, 1760–1767 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Lorente E, Garcia R & Lopez D Allele-dependent processing pathways generate the endogenous human leukocyte antigen (HLA) class I peptide repertoire in transporters associated with antigen processing (TAP)-deficient cells. J. Biol. Chem 286, 38054–38059 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leskov I et al. Rapid generation of human B-cell lymphomas via combined expression of myc and Bcl2 and their use as a preclinical model for biological therapies. Oncogene 32, 1066–1072 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsueda S et al. Identification of prostate-specific G-protein coupled receptor as a tumor antigen recognized by CD8+ T cells for cancer immunotherapy. PLoS ONE 7, e45756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jennifer B, Carl HJ, Andreas L, Marcela M & John S Treatment of cancer using humanized anti-CD19 chimeric antigen receptor. US patent 20140271635A1[P] (2014).
- 26.Hosken NA & Bevan MJ Defective presentation of endogenous antigen by a cell line expressing class I molecules. Science 248, 367–370 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Bossi G et al. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2, e26840 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pallasch CP et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 156, 590–602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Q, Khoury M & Chen J Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc. Natl Acad. Sci. USA 106, 21783–21788 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah NN & Fry TJ Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol 16, 372–385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmikangas P, Kinsella N & Chamberlain P Chimeric antigen receptor T-cells (CAR T-cells) for cancer immunotherapy—moving target for industry? Pharm. Res 35, 152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brudno JN & Kochenderfer JN Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. 34, 45–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill S et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 123, 2343–2354 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kenderian SS et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 29, 1637–1647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhlen M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Watanabe K et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol 194, 911–920 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Walker AJ et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubrovsky L et al. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology 5, e1049803 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe K, Kuramitsu S, Posey AJ & June CH Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front. Immunol 9, 2486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J & Wang L The emerging world of TCR-T cell trials against cancer: a systematic review. Technol. Cancer Res. Treat 18, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morris EC & Stauss HJ Optimizing T-cell receptor gene therapy for hematologic malignancies. Blood 127, 3305–3311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amir AL et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin. Cancer Res 17, 5615–5625 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Provasi E et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med 18, 807–815 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bendle GM et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med 16, 565–570 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Van Loenen MM et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl Acad. Sci. USA 107, 10972–10977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsui K et al. Low affinity interaction of peptide-MHC complexes with T cell receptors. Science 254, 1788–1791 (1991). [DOI] [PubMed] [Google Scholar]
- 47.Xie G et al. Dataset for CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Figshare 10.6084/m9.figshare.12922520 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the main findings of this study are available within the paper and its Supplementary Information. Raw data generated for this study are available in Figshare with the identifier 10.6084/m9.figshare.12922520 (https://doi.org/10.6084/m9.figshare.12922520) (ref. [47]).