

Abstract
Purpose
The multifunctional profibrotic cytokine TGF-β2 is implicated in the pathophysiology of primary open angle glaucoma (POAG). While the underlying cause of POAG remains unclear, TGF-β2 dependent remodeling of the extracellular matrix (ECM) within the trabecular meshwork (TM) microenvironment is considered an early pathologic consequence associated with impaired aqueous humor (AH) outflow and elevated IOP. Mitochondrial-targeted antioxidants have been recently shown by our group to markedly attenuate TGF-β2 profibrotic responses, strongly implicating oxidative stress as a key facilitator of TGF-β2 signaling in human TM cells. In this study, we determined the mechanism by which oxidative stress facilitates TGF-β2 profibrotic responses in cultured primary human TM cells.
Methods
Semiconfluent cultures of primary or transformed human TM cells were conditioned overnight in serum-free media and subsequently challenged without or with TGF-β2 (5 ng/mL). Relative changes in the mRNA content of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) isoforms, connective tissue growth factor (CTGF), collagen 1α1 and 4α1 isoforms or relative changes in the protein content of Nox4, phospho- and total-Smad2 and -Smad3, collagens I and IV were determined in the absence or presence of GKT137831, a Nox1-Nox4 dual enzyme inhibitor, and quantified by real-time qPCR or by immunoblot, respectively. Relative in situ changes in collagens I and IV and in alpha smooth muscle actin (αSMA) were semiquantified by immunocytochemistry, whereas relative changes in filamentous actin stress fiber formation was semiquantified by phalloidin staining.
Results
Quiescent primary human TM cells cultured in the presence of TGF-β2 exhibited a marked selective increase in endogenous Nox4 mRNA and Nox4 protein expression. Actinomycin D prevented TGF-β2 mediated increases in Nox4 mRNA expression. TM cells reverse transfected with siRNA against Smad3 prevented TGF-β2 mediated increases in Nox4 mRNA expression. Pre-incubating TM cells with GKT137831 attenuated TGF-β2 mediated increases in intracellular reactive oxygen species (ROS), in COL1A1, COL4A1, and CTGF mRNA expression, in Smad3 protein phosphorylation, in collagens I, collagens IV, and αSMA protein expression, and in filamentous actin stress fiber formation.
Conclusions
TGF-β2 promotes oxidative stress in primary human TM cells by selectively increasing expression of NADPH oxidase 4. Dysregulation of redox equilibrium by induction of NADPH oxidase 4 expression appears to be a key early event involved in the pathologic profibrotic responses elicited by TGF-β2 canonical signaling, including ECM remodeling, filamentous actin stress fiber formation, and αSMA expression. Selective inhibition of Nox4 expression/activation, in combination with mitochondrial-targeted antioxidants, represents a novel strategy by which to slow the progression of TGF-β2 elicited profibrotic responses within the TM.
Keywords: nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, TGF-β2, human, trabecular meshwork, glaucoma, fibrosis
Primary open-angle glaucoma (POAG) is the leading cause of irreversible blindness worldwide. Currently, nearly 3 million Americans and approximately 60.5 million people globally are affected by this indolent progressive ocular disorder.1–3 Estimated to affect 111.8 million people by the year 2040, glaucoma remains a major socioeconomic strain on the global economy. Known risk factors for development of POAG include advancing age, ethnic background (i.e. 3–4 times more prevalent among African Americans compared with non-Hispanic White ethnicities), and elevated intraocular pressure (IOP).2,4 Although the loss of retinal ganglion cells (RGCs) and associated degeneration of the optic nerve contributes to the irreversible vision loss, lowering IOP either by pharmacological or surgical intervention remains the first-line therapeutic approach to slow disease progression.5,6 Lowering IOP, however, is not without limitations and include (i) the presence of patient nonresponders, (ii) variable patient compliance with IOP lowering agents, (iii) advancing patient age, and (iv) the indolent and progressive nature of POAG.7
Whereas the underlying cause of POAG remains poorly defined, many laboratories have examined in detail the role transforming growth factor-β2 (TGF-β2) plays in the pathogenesis of POAG. TGF-β2 is a multifunctional cytokine and a member of the TGF-β superfamily of growth factors that is significantly elevated in aqueous humor (AH) of some patients with POAG.8,9 Numerous experimental studies now support a pathologic role for TGF-β2 in the development of elevated IOP associated with POAG.10–13 In healthy eyes, IOP is maintained by a balanced equilibrium between AH production and AH outflow.14,15 Elevated IOP in affected patients with POAG has been largely attributed to a reduction in AH outflow secondary to pathological changes affecting the trabecular meshwork (TM) outflow pathway.16–18 Profibrotic changes elicited by TGF-β2 within the TM have been implicated at increasing IOP and include altered turnover of extracellular matrix (ECM) components, formation of cross-linked actin networks (CLANS), upregulation of alpha smooth muscle actin (αSMA), and aberrant formation of actin stress fibers.10–13 Despite advancements elucidating the mechanistic details by which TGF-β2 confers pathological changes to the TM, the molecular pathways responsible for the initiation and underlying cause of POAG remains to be determined.
Oxidative stress, implicated in the pathology of many neurodegenerative diseases, is now recognized as an early key facilitator to the pathophysiology of POAG.19,20 Supported by numerous studies documenting (i) elevated levels of oxidative stress markers within the AH of affected patients with POAG,21,22 (ii) increased resistance to AH outflow by H2O2 induced TM degeneration,20,23,24 and (iii) altered expression of antioxidant defenses (superoxide dismutase [SOD], catalase, and glutathione pathways) in TM of patients with glaucoma,25,26 the human TM is exquisitely vulnerable to oxidative damage.27 Free radicals and reactive oxygen species (ROS) are known to specifically affect the cellularity of the human TM.28 Selective oxidative damage to TM mitochondria may elicit TM cell dysfunction.29 Increases in ROS production associated with POAG may arise as a consequence of mitochondrial dysfunction.30 Recent in vitro findings from our laboratory demonstrate that TGF-β2 elicits a marked increase in oxidative stress within human TM cells.31 Collectively, the following observations from these and related studies32 can be made: (i) TGF-β alone induces the generation of mitochondrial ROS from complex III of the electron transport chain, (ii) mitochondrial generated ROS are required for TGF-β induced gene expression downstream of Smad3 phosphorylation and nuclear translocation, (iii) TGF-β induced transcription of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 requires mitochondrial generated ROS establishing a feed-forward loop leading to increased intracellular ROS, and (iv) blocking mitochondrial ROS generation with targeted antioxidants (MitoQ or XJB-5-131) markedly attenuates TGF-β induced profibrotic gene expression.31,32
In this study, we investigated the molecular origins of TGF-β2 mediated oxidative stress in human TM cells. TGF-β2 was found to selectively elicit a marked increase in NADPH oxidase 4 mRNA and protein expression. Dysregulation of redox balance by induction of NADPH oxidase 4 expression appears to be a key early upstream event involved in the pathologic profibrotic responses elicited by TGF-β2 signaling. We propose that selective inhibition of NADPH oxidase 4 expression/activation, in combination with mitochondrial-targeted antioxidants, represents a novel strategy by which to restore redox balance to oxidatively stressed TM.
Methods
Human Trabecular Meshwork Cell Culture
The use of human material in this study was approved by the Edward Hines Jr. VA Hospital institutional review board. Fresh corneoscleral rims were received in Optisol corneal storage medium from the Illinois Eye Bank (Chicago, IL, USA) at the time of corneal transplant, and primary human TM cell isolates were prepared from collagenase type 1 (4 mg/mL; Worthington Biochemical Corp., Lakewood, NJ, USA), -dispase II (10 mg/mL; Sigma-Aldrich, St. Louis, MO, USA) digested TM tissue, as previously described.33 To objectively validate the identity and stability of isolated TM cells, human primary TM cultures were routinely challenged with dexamethasone and analyzed for expression and release of myocilin, as previously described.31 Consistent with published recommendations,34 cultured primary TM cells from two separate donors (0824 and 2799) were restricted to no more than eight passages and were maintained in either low-glucose (5.6 mM) Dulbecco's Modified Eagles Media (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (ThermoFisher Scientific, Ashville, NC, USA), 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B (ThermoFisher Scientific), or low-glucose (5.6 mM) Minimum Essential Media (MEM) supplemented with 10% fetal bovine serum, 5% adult bovine serum, essential and nonessential amino acids, 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B. SV40-transformed human TM cells (GTM3) were a generous gift from Alcon Laboratories (Fort Worth, TX, USA) and were cultured in low-glucose (5.6 mM) DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 mg/mL streptomycin, and 0.25 µg/mL amphotericin B.
Treatment
Semiconfluent cultures of primary or transformed (GTM3) human TM cells were incubated overnight in serum-free culture media unless noted otherwise. Serum-starved cultures were treated in the absence (vehicle, 2 µM HCl) or presence of recombinant human TGF-β2 (5 ng/mL; R&D Systems, Inc. [Tocris], Minneapolis, MN, USA, or Cell Signaling Technology, Danvers, MA, USA) for 24 hours unless noted otherwise. In some experiments, cells were pretreated (2 hours at 37°C) with SB-431542 (10 µM; Sigma-Aldrich), or with GKT-137831 (20 µM, a selective Nox1-Nox4 dual inhibitor; Cayman Chemicals, Ann Arbor, MI USA). In other experiments, TM cells were reverse transfected with either 20 nM scrambled siRNA (Silencer Select predesigned; ThermoFisher Scientific), with 20 nM siRNA directed against Smad2 (S8397), or Smad3 (S8402) using 0.6% Lipofectamine RNAiMAX according to the manufacturer's protocol (ThermoFisher Scientific) as previously described.35
Real-Time qPCR
Total RNA from TM cells was isolated using TRIzol reagent (ThermoFisher Scientific), quantified (NanoDrop 2000; Thermo Scientific, Wilmington, DE, USA), and 1 µg was reverse transcribed using iScript Supermix (Bio-Rad Laboratories, Hercules, CA, USA). The cDNA sequences were amplified by real-time (iQ SYBR Green Supermix; Bio-Rad Laboratories) PCR using a 96-well CFX Connect PCR detection system with published human specific primer pairs (Table). Human-specific glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primer pairs were used as a reference control. For each sample, the specificity of the real-time reaction product was determined by melt curve analysis. Reaction efficiencies for all primer pairs were typically >90% when cDNA templates were amplified for 40 cycles using a melting temperature of 95°C × 30 seconds, an annealing temperature of 60°C × 45 seconds, and an elongation temperature of 72°C × 30 seconds. The endogenous expression of GAPDH was unaltered by drug treatment (data not shown). Relative fold changes in gene expression in each sample were normalized to expressed levels of GAPDH and analyzed by the 2−ΔΔCt method of Livak.36
Table.
Human Specific Primer Pairs Used in qPCR Experiments
| Gene | 5′-Sense-3′, Forward | 5′-Antisense-3′, Reverse | Product Size, bp |
|---|---|---|---|
| Nox1 | ACA AAT TCC AGT GTG CAG ACC AC | AGA CTG GAA TAT CGG TGA CAG CA | 247 (var. 2) 394 (var. 1, 3) |
| Nox2 | GGG CTG TTC AAT GCT TGT GGC T | ACA TCT TTC TCC TCA TCA TGG TGC | 413 |
| Nox3 | ATG AAC ACC TCT GGG GTC AGC TGA | GGA TCG GAG TCA CTC CCT TCG CTG | 458 |
| Nox4 | CAC CTC TGC CTG TTC ATC TG | GGC TCT GCT TAG ACA CAA TCC | 128 (var. 4, X6) 132 (var. 1, 4B) |
| Nox5 | ATC AAG CGG CCC CCT TTT TTT CAC | CTC ATT GTC ACA CTC CTC GAC AGC | 239 |
| P22 phox | GTG TTT GTG TGC CTG CTG GAG T | CTG GGC GGC TGC TTG ATG GT | 320 |
| COL1A1 | CTA AAG GCG AAC CTG GTG AT | TCC AGG AGC ACC AAC ATT AC | 107 |
| COL4A1 | CGG GCC CTA AAG GAG ATA AAG | GAA CCT GGA AAC CCA GGA AT | 115 |
| CTGF | GGC TTA CCG ACT GGA AGA C | AGG AGG CGT TGT CAT TGG | 143 |
| GAPDH | ACC ACA GTC CAT GCC ATC AC | CCA CCA CCC TGT TGC TGT A | 451 |
Immunoblot Analysis
In separate experiments, treated cells were lysed with either 20 mM Tris buffer (pH 7.5) containing 150 mM NaCl, 1% Triton X-100, and 1 mM EDTA supplemented with a cocktail of protease (Roche Diagnostics Corp., Indianapolis, IN, USA) and phosphatase (Sigma-Aldrich) inhibitors and subsequently clarified by centrifugation (20,000g × 10 minutes at 4°C) for phospho- and total-Smad protein analyses or with 2 times Laemmli sample buffer. To analyze secreted proteins, equal aliquots of conditioned media were collected and concentrated by centrifugal filtration (Amicon ultra centrifugal filters, 10-kDa cutoff; MilliporeSigma, Burlington, MA, USA). Clarified cell lysates and concentrated conditioned media were stored at -80°C until use. Protein concentrations in cell lysates and in culture media concentrates were determined by the BCA method (ThermoFisher Scientific) using BSA as a standard. Proteins (10 µg proteins per lane) in cell lysates or concentrated media were resolved by 4% to 20% SDS-PAGE gel electrophoresis either under reducing/denaturing (for Smads, collagens IV) or reducing (collagen I) conditions and electrotransferred overnight onto 0.2 µm nitrocellulose membranes. Membranes were blocked with 5% Carnation non-fat dried milk (Nestle, Arlington, VA, USA) in 10 mM Tris buffer (pH 7.4) containing 0.1% Tween 20 for 1 hour at 23°C, incubated overnight at 4°C in the presence of a 1:1000 dilution of rabbit monoclonal: (i) anti-Smad2 (clone 86F7), (ii) anti-phospho-Smad2 (clone 138D4; Ser465/467), (iii) anti-Smad3 (clone C67H9), or (iv) anti-phospho-Smad3 (clone C25A9; Ser423/425) primary antibody from Cell Signaling Technology. To quantify relative changes in Nox4 protein content, a 1:2000 dilution of rabbit monoclonal anti-NADPH oxidase 4 (ab133303; Abcam, Cambridge, MA, USA) primary antibody was used, as indicated. Relative changes in collagens were determined using a 1:1000 dilution of sheep polyclonal anti-collagen 1α1 (AF6220-SP; R&D Systems Inc., Minneapolis, MN, USA) or a 1:1000 dilution of rabbit polyclonal anti-collagen IV (ab6586; Abcam) primary antibodies were used as indicated. Immunostained membranes were washed in Tris-Tween buffer and incubated for 1 hour at 23°C with a 1:10,000 dilution of goat anti-rabbit IgG (Jackson ImmunoResearch Labs, Inc, West Grove, PA, USA) or a 1:1000 dilution of goat anti-sheep IgG (R&D Systems, Inc.) horseradish peroxidase-conjugated secondary antibodies, respectively. Immunostained proteins were visualized using Supersignal West Pico chemiluminescent substrate (ThermoFisher Scientific). Relative changes in protein content were quantified by densitometry and normalized to total Smad or total GAPDH content, as indicated.
Quantification of Oxidative Stress
Intracellular ROS were detected using the oxidation-sensitive fluorogenic dyes CellROX green (Life Technologies, Grand Island, NY, USA) or 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA; ThermoFisher Scientific). For quantitative assessment of oxidative stress using CellROX green, TM cells were cultured in 8-well Nunc Lab-Tek II chambered glass slides (ThermoFisher Scientific) to near confluency, serum-starved overnight, and subsequently treated as described above. CellROX green (5 µM) was added to the final 30 minutes of treatment. Dye-loaded cells were rinsed with ice cold PBS and fixed for 20 minutes at 23°C by immersion in 4% buffered (pH 7.4) paraformaldehyde. Fixed cells were rinsed, air dried, and mounted with Fluoroshield containing 4′,6′-diamidino-2 phenylindole (DAPI). Cells were imaged using a Leica TCS SPE confocal microscope (Leica MicroSystems, Buffalo Grove, IL, USA) and LAS-X imaging suite. All cells’ fields were imaged under identical confocal conditions using identical software settings. For quantitative assessment of oxidative stress using carboxy-H2DCFDA, TM cells were cultured in 96-well plates to near confluency, serum-starved overnight, and subsequently incubated with 10 µM carboxy-H2DCFDA at 37°C for 30 minutes, as we have previously described.31 Carboxy-H2DCFDA pre-loaded cells were washed to remove excess dye and then treated as described above. Dichlorofluorescein diacetate fluorescence was quantified using a Labtech FLUOstar Optima luminometer (BMG Labtech, Cary, NC, USA) at 488-nm excitation/520-nm emission wavelengths.
Immunocytochemistry
TM cells were cultured in 8-well Nunc Lab-Tek II chambered glass slides (ThermoFisher Scientific) to near confluency, serum-starved overnight, and subsequently treated as described above. Treated TM cells were rinsed with PBS (pH 7.4) and fixed for 20 minutes at 23°C by immersion in 4% buffered (pH 7.4) paraformaldehyde. Fixed cells were permeabilized for 20 minutes at 23°C with 0.05% Triton X-100 (for ECM proteins) or 0.1% Triton X-100 (for Smad2, Smad3, filamentous actin, and αSMA proteins) in PBS, blocked for 1 hour at 23°C with 5% BSA in PBS, and subsequently incubated overnight at 4°C in the presence of a 1:500 dilution of either sheep polyclonal anti-collagen 1α1 (AF-662-SP; R&D Systems, Minneapolis, MN, USA), rabbit polyclonal anti-collagen IV (ab6586; Abcam), rabbit monoclonal anti-Smad2 (clone 86F7; Cell Signaling Technology) or anti-Smad3 (clone C67H9; Cell Signaling Technology), or rabbit polyclonal anti-αSMA (ab5964, Abcam) primary antibody in 1% BSA, as indicated. Immunostained cells were washed in PBS, incubated for 1 hour at 23°C with a 1:400 dilution of either AlexaFluor 488-conjugated goat anti-sheep IgG or 1:400 dilution of AlexaFluor 594-conjugated goat anti-rabbit IgG secondary antibody (ThermoFisher Scientific), respectively. Filamentous actin stress fibers organization was determined by immunofluorescence with Alexa 488-conjugated phalloidin, as previously described.33 Slides were mounted with Fluoroshield containing 4′,6′- DAPI and imaged using a Leica TCS SPE confocal microscope (Leica MicroSystems) and LAS-X imaging suite. Fluorescent images were Z-stacked at 1-µm steps acquired from below the base of the cells to the top of the cells. Relative fluorescence intensities from images were semi-quantified using Image J (National Institutes of Health, Bethesda, MD, USA) with results expressed as background-corrected integrated fluorescence density.
Statistical Analysis
Unless otherwise specified, results are expressed as the mean ± SD of triplicate cultures, repeated at least two additional times. Statistical significance of parametric data was determined by Student's t-test. Significance between multiple groups was determined by 1-way ANOVA using a Tukey's post hoc analysis. In each case, P < 0.05; was considered statistically significant.
Results
NADPH Oxidase mRNA Expression in Human TM Cells
Quiescent human primary TM cells cultured in serum-free medium constitutively express detectable amounts of all five known mRNA isoforms of NADPH oxidase, albeit at low (Ct >35) endogenous levels, relative to GAPDH expression (Fig. 1A). By comparison, the GAPDH-normalized mRNA content of the obligate membrane accessory protein p22phox was constitutively higher (Ct = 26.0 ± 0.4). Relative levels of the dual oxidases 1 and 2 were not determined in this study. Culturing quiescent cells in the presence of the profibrotic cytokine TGF-β2 had only a marginal effect on Nox1, Nox2, Nox3, Nox5, or on p22phox mRNA expression. In contrast, TGF-β2 elicited a marked (>200-fold) increase in Nox4 mRNA content relative to GAPDH expression (see Fig. 1A) that was detectable within 2 hours of stimulation and cumulative over the 24 hours examined (Fig. 1B). To determine whether TGF-β2 elicits a selective increase in constitutive Nox4 gene expression or in Nox4 mRNA stability, in a separate experiment, we challenged TM cells with TGF-β2 in the absence or presence of actinomycin D, an inhibitor of gene transcription. TGF-β2 reproducibly elicited a time-dependent increase in Nox4 mRNA content (see solid circles, Fig. 1B). However, pretreating TM cells with actinomycin D completely prevented TGF-β2-mediated increases in Nox4 mRNA expression (see solid squares, Fig. 1B). The delayed addition of actinomycin D to TGF-β2 treated (8 hours only) cells (see open diamonds, Fig. 1B) or to cells in the presence of both TGF-β2 and actinomycin D (see semi-solid diamonds, Fig. 1B) resulted in a subsequent decline in TGF-β2-mediated Nox4 mRNA relative content. These data suggest that TGF-β2 signaling elicits a marked selective increase in Nox4 gene expression in human TM cells.
Figure 1.
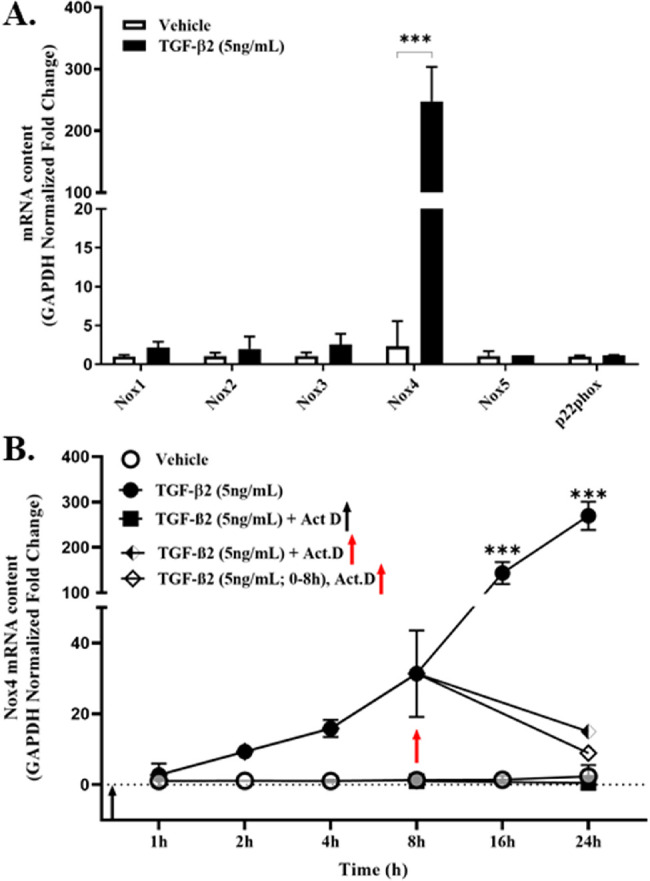
TGF-β2 selectively increases Nox4 mRNA expression in primary human TM cells. Serum-starved confluent cultures of primary TM cells were treated (24 hours) with vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL) and (A) relative fold changes in GAPDH-normalized Nox mRNA and p22phox mRNA isoform expression were quantified by qPCR, as indicated. (B) Time-dependent GAPDH-normalized Nox4 mRNA expression in TM cell cultures assayed in the absence (vehicle) or presence of TGF-β2 (5 ng/mL) treated without (solid circles) or with actinomycin D (1 µM for 1 hour) prior to (solid squares) or at 8 hours post TGF-β2 treatment (open diamonds). The continued presence of TGF-β2 (semi-solid diamonds) in the presence of actinomycin D did not alter Nox4 mRNA degradation. Data shown in A and B are the means ± SD (N = 3–5 replicate wells per condition) from separate experiments. ***P < 0.0001; 1-way ANOVA with Tukey's post hoc analysis.
TGF-β2 Canonical Signaling Mediates Nox4 mRNA Expression
To determine the molecular pathway responsible for TGF-β2 mediated increase in Nox4 gene expression, primary human TM cells were reverse transfected with either scrambled (control) siRNA or with siRNA directed against either Smad2 or Smad3, respectively. Compared to scrambled controls, TM cells expressing attenuated levels of endogenous Smad2 proteins content remained responsive to TGF-β2 mediated signaling (Fig. 2). In marked contrast, reducing the endogenous content of Smad3 proteins significantly attenuated TGF-β2 mediated increase in Nox4 gene expression (see Fig. 2A). Efficiency of siRNA directed knockdown of Smad proteins was confirmed by immunoblot (Fig. 2B). Quiescent TM cells expressed measurable levels of both Smad2 and Smad3 proteins. Compared to scrambled siRNA control cultures, the endogenous content of Smad2 and of Smad3 in transfected TM cells was markedly reduced by their respective siRNAs. It is important to note that knockdown of either Smad2 or Smad3 was selective and did not affect the endogenous content of the reciprocal Smad protein (see Fig. 2B).
Figure 2.
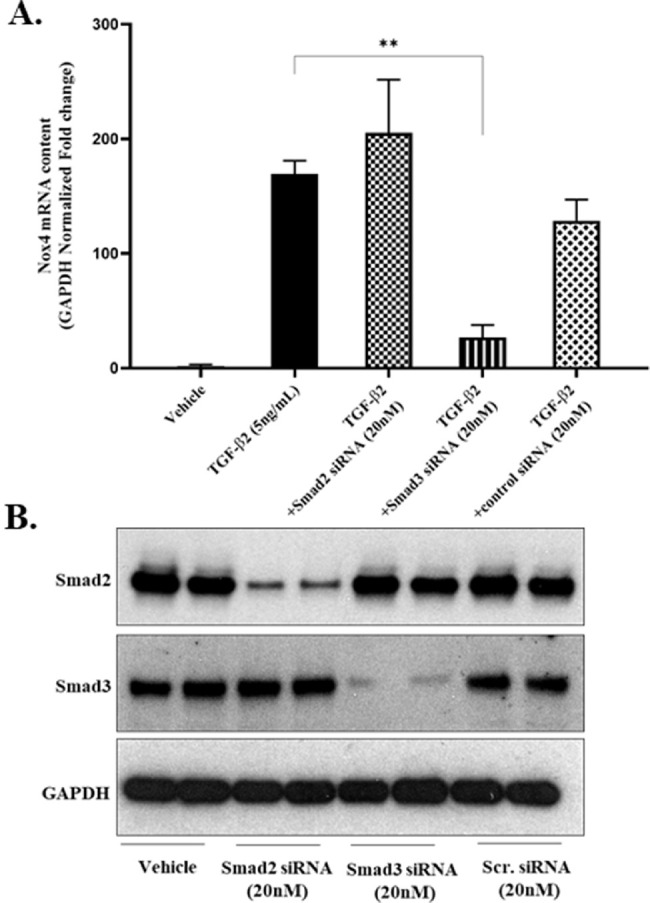
Smad3-knockdown prevents TGF-β2 mediated increases in Nox4 mRNA expression. TM cells were reverse transfected with siRNA (20 nM for 72 hours) against Smad2, Smad3, or scrambled (control), serum starved overnight, and subsequently treated without (vehicle) or with TGF-β2 (5 ng/mL for 24 hours), as indicated. Data shown are the means ± SD (N = 3 replicate wells per condition) from a single experiment representative of two additional experiments. **P < 0.001, 1-way ANOVA with Tukey's post hoc analysis. (A) GAPDH-normalized Nox4 mRNA expression quantified by qPCR. (B) Representative immunoblot of total Smad2 or total Smad3 proteins present in lysates from TM cells reverse transfected with vehicle or siRNA (20 nM) against Smad2 or Smad3 or with control scrambled siRNA (Scr.), as indicated. GAPDH, glyceraldehyde phosphate dehydrogenase protein loading controls.
NADPH Oxidase 4 Protein Expression in Human TM Cells
To determine whether the observed relative increase in TGF-β2 mediated Nox4 mRNA content was of physiologic relevance, we examined by immunoblot the content of Nox4 protein expressed within quiescent and TGF-β2 stimulated primary and transformed (GTM3) TM cells (Fig. 3). Unexpectedly, the endogenous content of Nox4 protein expressed in quiescent primary TM cells was found to be significantly less than that expressed in identically cultured quiescent transformed GTM3 cells (see Fig. 3A). Culturing primary TM cells in the presence of TGF-β2 elicited a significant increase (>50%) in endogenous Nox4 protein expression (Fig. 3B). In contrast, GTM3 cells treated with TGF-β2 exhibited no change in endogenous Nox4 protein expression (Fig. 3C). Consistent with these findings was the observation that GAPDH-normalized Nox4 mRNA content in GTM3 cells was unaltered by TGF-β2 signaling (data not shown).
Figure 3.

Nox4 protein expression in human TM cells. Serum-starved confluent cultures of primary or transformed TM cells were treated in the absence (A) or presence (B, C) of vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL for 24 hours), as indicated. A Immunoblots of Nox4 proteins in lysates from quiescent primary or transformed (GTM3) TM cells, as indicated. B and C Nox4 proteins expressed in lysates from primary B or transformed C TM cells treated without (vehicle) or with TGF-β2, as indicated. GAPDH, glyceraldehyde phosphate dehydrogenase protein loading controls. (Bottom panels) Densitometric analyses of respective Nox4 protein bands. Data shown are the means ± SD (N = 3 replicate wells per condition) of GAPDH-normalized Nox4 protein bands from a single experiment representative of two separate experiments. ns, not significant; *P < 0.05; ***P < 0.0001, Student's t-test.
NADPH Oxidase Activity Mediates TGF-β2 Induced Oxidative Stress
As we have previously reported,31 TGF-β2 promotes marked oxidative stress in cultured human TM cells as quantified using two distinct fluorogenic dyes (Fig. 4). Pre-incubating TM cells with GKT-137831, a selective Nox1-Nox4 dual inhibitor, significantly prevented TGF-β2 mediated increases in intracellular ROS (see Figs. 4B, 4C). To determine whether endogenous NADPH oxidase enzymatic activity affects TGF-β2 canonical signaling, relative changes in Smad2 and Smad3 phosphorylation were quantified by immunoblot. Compared to vehicle controls, primary human TM cells cultured in the presence of TGF-β2 exhibited significant increases in the phosphorylation of Smad2 (Fig. 5A) and Smad3 proteins (Fig. 5B). Pretreating TM cells with the Alk5 inhibitor SB-431542 prevented TGF-β2 mediated increases in Smad phosphorylation. By comparison, pretreating TM cells with GKT-137831 significantly attenuated TGF-β2 mediated increases in Smad3, but not Smad2, phosphorylation (see Figs. 5A, 5B). To determine whether NADPH oxidase activity affects nuclear translocation of Smad proteins, TGF-β2 treated TM cells were immunostained in situ in the absence or presence of GKT-137831. Relative to vehicle treated control cultures, incubating TM cells with TGF-β2 elicited a marked increase in nuclear staining over diffuse cytosolic background staining for total Smad2 (Figs. 5C, 5D) and total Smad3 proteins (Figs. 5C, 5E). In contrast, TM cells pretreated with either SB-431542 or with GKT-137831 exhibited nuclear staining of total Smad2 and total Smad3 proteins that was statistically indistinguishable from vehicle-treated control cultures (see Fig. 5).
Figure 4.

A dual inhibitor of Nox1-Nox4 enzyme activity prevents TGF-β2 mediated oxidative stress. Serum-starved confluent cultures of primary TM cells were pre-treated (2 hours) with SB-431542 (10 µM) or GKT137831 (20 µM) and incubated with vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL) for an additional 22 hours, as indicated. (A) Representative confocal images of CellROX Green fluorescence in treated cells. Blue, DAPI-stained nuclei. Scale Bar, 100 µm. (B) Quantitative comparison of CellROX Green fluorescence. Data shown are the means ± SD (N = 4 separate wells per condition, 2 fields per well, 15 cells per field). (C) Quantitative comparison of DCFDA fluorescence. Data shown are the means ± SD (N = 8 replicate wells per condition) from a single experiment representative of two separate experiments. **P < 0.001; ***P < 0.0001, 1-way ANOVA with Tukey's post hoc analysis.
Figure 5.

A dual inhibitor of Nox1-Nox4 enzyme activity attenuates Smad3-dependent TGF-β2 canonical signaling. Serum-starved confluent cultures of primary TM cells were pretreated (2 hours) with SB-431542 (10 µM) or GKT137831 (20 µM) and incubated with vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL) for an additional 22 hours, as indicated. (A, B) Immunoblots and respective densitometric analyses of Smad2 or Smad3 phospho- and total-proteins in lysates from primary TM cells treated without (vehicle) or with TGF-β2, as indicated. Densitometric data shown are the means ± SD (N = 3 separate experiments) of normalized phospho-Smad protein bands. (C) Representative confocal immunofluorescent images of total Smad2 or Smad3 proteins expressed in treated cells. Blue, DAPI-stained nuclei. Scale Bar, 100 µm. (D, E) Quantitative comparison of D Smad2 or E Smad3 immunofluorescence. Data shown are the means ± SD (N = 4–6 separate wells per condition, 2 fields per well, 15 cells per field). *P < 0.05; **P < 0.001; ***P < 0.0001, 1-way ANOVA with Tukey's post hoc analysis.
NADPH Oxidase Activity Mediates TGF-β2 Induced Increases in Collagen Expression
To determine whether the activity of NADPH oxidases affects TGF-β2 mediated increases in synthesis and release of extracellular matrix constituents, cultured primary and transformed human TM cells were pre-incubated without or with GKT-137831 and subsequently incubated in the presence of TGF-β2. As we and others have previously reported,31 primary TM or transformed GTM3 cells incubated in the presence of TGF-β2 exhibit marked GAPDH-normalized increases in COL1A1 (Figs. 6A, 6D) and COL4A1 (Figs. 6B, 6E) mRNA expression. By comparison, pre-incubating TM cells with GKT-137831 attenuated TGF-β2 mediated changes in collagen isoform mRNA expression (see Fig. 6). In contrast, pre-incubating TM cells with SB-431542 completely prevented these TGF-β2 mediated responses. TGF-β2 mediated GAPDH-normalized changes in CTGF mRNA content was minimally affected by pre-incubation with GKT-137831 (Fig. 6C). To determine whether NADPH oxidase activity affects TGF-β2 mediated increases in collagen isoform protein expression and release, cultured primary human TM cells were pre-incubated without or with GKT-137831 and subsequently incubated in the presence of TGF-β2. Consistent with our previous observations,31 human primary TM cell cultures incubated in the presence of TGF-β2 exhibit markedly significant increases in the expression and release of collagens I and collagens IV proteins when analyzed by immunoblot (data not shown) or in situ by immunocytochemistry (Fig. 7). In contrast, TM cells pre-incubated with either SB-431542 or with GKT-137831 showed significantly blunted collagen isoform responses to TGF-β2 (see Figs. 7B, 7C).
Figure 6.

A dual inhibitor of Nox1-Nox4 enzyme activity attenuates TGF-β2 mediated collagen mRNA expression. Serum-starved confluent cultures of (A–C) primary or (D–F) transformed TM cells were pretreated (2 hours) with SB-431542 (10 µM) or GKT137831 (20 µM) and incubated with vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL) for an additional 22 hours, as indicated. A and D GAPDH-normalized COL1α1 mRNA B and E GAPDH-normalized COL4α1 mRNA (C and F) GAPDH-normalized CTGF mRNA expression. Data shown are the means ± SD (N = 3 replicate wells per condition) representative of two separate experiments A to C or from a single experiment (D to F). *P < 0.05; **P < 0.001; ***P < 0.0001, 1-way ANOVA with Tukey's post hoc analysis.
Figure 7.

A dual inhibitor of Nox1-Nox4 enzyme activity attenuates TGF-β2 mediated collagen protein expression. Serum-starved confluent cultures of primary TM cells were pretreated (2 hours) with SB-431542 (10 µM) or GKT137831 (20 µM) and incubated with vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL) for an additional 22 hours, as indicated. (A) Representative confocal immunofluorescent images of collagens I (green) or collagens IV (red) proteins expressed in treated cells. Blue, DAPI-stained nuclei. Scale Bar, 100 µm. (B, C) Quantitative comparison of B collagen I or C collagen IV immunofluorescence. Data shown are the means ± SD (N = 4–8 separate wells per condition, 2 fields per well, 15 cells per field). **P < 0.001; ***P < 0.0001, 1-way ANOVA with Tukey's post hoc analysis.
NADPH Oxidase Activity Mediates TGF-β2 Induced Increases in Filamentous Actin Stress Fiber Formation and Alpha Smooth Muscle Action Expression
The effect of NADPH oxidase activity on TGF-β2 mediated increases in filamentous actin stress fiber formation and on αSMA protein expression was determine in situ by phalloidin fluorescence and by immunocytochemistry, respectfully. Human primary TM cell cultures incubated in the presence of TGF-β2 exhibit markedly significant increases in filamentous actin formation and in the protein expression of αSMA (Fig. 8). In contrast, TM cells pre-incubated with either SB-431542 or with GKT-137831 were unable to organize filamentous stress fibers (see Figs. 8A, 8B) nor did they express changes in αSMA protein expression (see Figs. 8A, 8C) in response to a TGF-β2 challenge.
Figure 8.

A dual inhibitor of Nox1-Nox4 enzyme activity attenuates TGF-β2 mediated F-actin stress fiber formation and αSMA expression. Serum-starved confluent cultures of primary TM cells were pretreated (2 hours) with SB-431542 (10 µM) or GKT137831 (20 µM) and incubated with vehicle (0.1% DMSO) or TGF-β2 (5 ng/mL) for an additional 22 hours, as indicated. (A) Representative confocal fluorescent images of phalloidin stained F-actin stress fibers (green) or immunostained αSMA IV (red) proteins expressed in treated cells. Blue, DAPI-stained nuclei. Scale Bar, 100 µm. (B, C) Quantitative comparison of B phalloidin fluorescence or C αSMA immunofluorescence (N = 4–8 separate wells per condition, 2 fields per well, 15 cells per field). ***P < 0.0001, 1-way ANOVA with Tukey's post hoc analysis.
Discussion
Recently, we reported that cells isolated from healthy human TM tissue exhibit enhanced oxidative stress when challenged with TGF-β2, a profibrotic cytokine strongly implicated in the pathogenesis of POAG.31 In this study, we investigated the molecular origins responsible for TGF-β2 enhanced production of ROS in human TM cells. Quiescent human TM cells cultured in the presence of TGF-β2 exhibited a selective increase in endogenous NADPH oxidase 4 (Nox4) mRNA and protein expression. Other NADPH oxidase isoforms (1, 2, 3, and 5) were endogenously expressed at low GAPDH-normalized amounts and minimally responsive to a TGF-β2 challenge. The TGF-β2 mediated increase in Nox4 expression was prevented by actinomycin D, an inhibitor of gene transcription. The siRNA targeted against Smad3, but not Smad2, similarly prevented TGF-β2 mediated increases in Nox4 expression. Pre-incubating TM cells with GKT137831, a dual Nox1-Nox4 inhibitor, attenuated TGF-β2 mediated increases in intracellular ROS, in COL1A1, COL4A1, and CTGF mRNA expression, in Smad3 protein phosphorylation, in collagens I, collagens IV, and αSMA protein expression and in filamentous actin stress fiber formation. These findings support dysregulation of redox equilibrium by induction of Nox4 expression as a key early upstream event involved in the downstream profibrotic responses elicited by TGF-β2 canonical signaling, including ECM remodeling, filamentous actin stress fiber formation, and αSMA expression.
Oxidative stress is a well-established age-associated facilitator of many chronic neurodegenerative disorders, including glaucomatous optic neuropathy.19,20 Implicated in the early pathophysiologic stages of POAG,37 oxidative stress refers to a metabolic state in which a normal healthy cellular production of ROS overwhelms the ability of endogenous phase II enzymatic antioxidant defense pathways to maintain redox equilibrium within the affected cell.38 Despite continuing controversy as to the overall pathologic significance that changes in IOP contribute toward the development of POAG, elevated IOP remains the most recognized risk factor for this indolent progressive disorder.39–41 Morphologic alterations deep within the TM, including remodeling of the ECM, are considered early pathologic hallmarks that are directly associated with increased AH outflow resistance and elevated IOP.31 Although several advancements have elucidated the role TM cells play at maintaining a healthy balanced IOP, the cellular consequences responsible for the initiation, and hence the cause, of age-associated TM cell failure in POAG remains unclear.
Growing evidence obtained from clinical and experimental studies strongly implicate oxidative stress in the degeneration of retinal ganglion cells42 and its association with open-angle glaucoma.37,42 The anterior chamber, along with the TM itself, has also proven to be quite vulnerable to oxidative stress-induced damage.20,27,43 Under physiologic conditions, redox equilibrium within metabolically active cells, such as the TM, is maintained by a host of nonenzymatic (e.g. glutathione, ascorbic acid, α-tocopherols, and metallothionein) and enzymatic (e.g. superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, and heme oxygenase I) molecular antioxidant defenses. In patients with open-angle glaucoma, however, total antioxidant status was reduced while levels of phase II antioxidant enzymes present in AH were elevated compared to controls.37 The observed elevation in phase II antioxidant expression within AH of affected patients may best be reconciled as an Nrf2-mediated compensatory response to an imbalance in redox equilibrium.44
Although several external sources of ROS (e.g. ultraviolet A irradiation) that can potentially damage the TM have been described,20 the majority of oxygen and nitrogen free radicals that are produced under physiologic conditions are short-lived byproducts of normal cellular metabolism.45 Although mitochondria consumes over 90% of cellular oxygen, up to 5% of this oxygen can be converted into harmful superoxide free radicals that are prematurely produced by leakage of electrons at complex I and complex III of the electron transport chain. Subcellular organelles known to produce physiologic amounts of intracellular ROS also include peroxisomes (xanthine oxidase),46 endoplasmic reticulum (cytochrome P450 mono-oxygenases), and several cytosolic and plasma membranes associated oxidases. Normally short-lived and protective, age-associated dysfunction of mitochondrial electron transport and proton pump activity often leads to excessive production of ROS resulting in harmful redox disequilibrium.47 Oxidative stress associated mitochondrial dysfunction in patients with POAG is well-documented.48–50 These findings collectively support mitochondrial dysfunction as a potential source of generated ROS and early facilitator of TM cell failure in POAG. What remains unclear is the molecular initiator of age-associated redox disequilibrium within the TM.
Previous studies have shown that AH of some patients with POAG contain elevated levels of the profibrotic cytokine TGF-β2, strongly implicating this cytokine in the pathogenesis of this disorder.8,29,51 Our own preliminary studies utilizing AH samples from patients at Edward Hines Jr. VA Hospital also show a trend toward elevated levels of biologically active TGF-β2 in Veterans with POAG (0.69 ± 0.49 pg/µL; N = 5) compared to age-matched control subjects (0.28 ± 0.10 pg/µL; N = 7). Previous studies from our laboratory have demonstrated that human TM cells constitutively express and secrete biologically active TGF-β2, highlighting the TM as a viable source of active TGF-β2.52 More recently, we reported that mitochondrial-targeted antioxidants attenuate TGF-β2 mediated oxidative stress associated changes in Smad-dependent transcriptional activity, including marked reductions in CTGF and collagen isoform gene and protein expression.31 These findings further implicate TGF-β2 as a putative initiator of oxidative stress induced TM cell failure.
It is important to note that oxidative stress in the form of ROS has been shown to perpetuate TGF-β signaling in fibroblasts, establishing a vicious feed-forward profibrotic cycle.53,54 ROS generated by this “redox-fibrotic” cycle is reported to be essential for TGF-β mediated profibrotic gene expression in lung fibroblasts.32 Furthermore, in this same study Jain et al. showed that disrupting TGF-β dependent mitochondrial ROS generation attenuated Nox4 expression in lung fibroblasts.32 Although the role of TGF-β mediated Nox4 dependent oxidative stress in POAG remains unclear, preliminary studies of Nox4 expression in human TM cells have been reported.55,56 Here, we show that physiologic concentrations of TGF-β2 elicit a marked isoform-selective increase in NADPH oxidase 4 mRNA and protein expression in cultured primary human TM cells. In contrast, TGF-β2 failed to elicit any significant change in Nox isoform mRNA expression in transformed (GTM3) human TM cells (data not shown). However, constitutive relative expression of Nox4 protein was found to be markedly elevated in quiescent GTM3 cells when compared with primary TM cells. What is not clear from these data is whether enhanced constitutive expression of Nox4 protein seen in quiescent GTM3 cells is a consequence of cellular transformation or uniquely characteristic of a previously unrecognized glaucomatous phenotype. A more thorough examination of this serendipitous observation is warranted.
In agreement with our previous observations,31 TGF-β2 elicited a measurable increase in oxidative stress in primary TM cells. Preincubating TM cells with GKT137831, a dual Nox1-Nox4 inhibitor, however, completely prevented TGF-β2 mediated ROS production. These findings support Nox4 as a major molecular source of TGF-β2 mediated oxidative stress in TM cells. Functional significance of this observation was further demonstrated by the ability of GKT137831 to attenuate TGF-β2 mediated increases in Smad3 phosphorylation, in Col1α1, Col4α1, and CTGF mRNA expression, in collagens I and IV protein expression, and in filamentous actin stress fiber formation and αSMA protein expression.
NADPH oxidases are the only known enzyme family with the sole function to produce ROS. In addition to being the most widely expressed isoform, Nox4 is unique among the five distinct NADPH oxidases (Nox 1–5) and two known duoxidases (Duox 1 and 2) in that (i) it is constitutively expressed, (ii) generates H2O2 rather than superoxide as its primary ROS, (iii) requires only p22phox as a binding partner, and (iv) interacts with DNA-directed polymerase delta-interacting protein 2 (PolDip2).57 Exactly how TGF-β2 signaling elicits selective increases in Nox4 isoform mRNA expression in TM cells remains unclear, but may involve activation of bromodomain-containing protein 4, an epigenetic “reader” of acetylated lysine groups on histones.58 In addition, the Nox4 promoter has been shown to contain an AP1/Smad binding element.59 Inhibiting Nox4 enzyme activity with GKT137831 does, however, attenuate TGF-β2 mediated increases in Smad3 phosphorylation, suggesting that Nox4-generated H2O2 regulates Smad3-dependent TGF-β2 canonical signaling in TM cells. This thesis is not without precedence, as TGF-β1 induced increases in H2O2 have been previously shown to regulate TGF-β1 mediated gene expression in lung fibroblasts.60 Further studies are warranted to elucidate the novel role of TGF-β2 mediated H2O2 signaling cascade in human TM cells.
In conclusion, we show in this study that TGF-β2 promotes oxidative stress in primary human TM cells by selective expression of NADPH oxidase 4. Dysregulation of redox balance by induction of NADPH oxidase 4 expression appears to be a key early event involved in the pathologic profibrotic responses elicited by TGF-β2 canonical signaling, including ECM remodeling, filamentous actin stress fiber formation, and αSMA expression. Selective inhibition of Nox4 expression/activation, in combination with mitochondrial-targeted antioxidants, represents a novel strategy by which to slow the progression of TGF-β2 elicited profibrotic responses within the TM.
Acknowledgments
The authors thank Charles Bouchard for assistance with procuring corneoscleral rims for preparation of primary human TM cells, and Joo Hong and ZhenGuo Wang for technical assistance. Supported, in part, by grants from the Department of Veterans Affairs (EBS: 1I21RX001593 and 1I01BX003938), Glaucoma Research Foundation Shaffer Grant, the Illinois Society for the Prevention of Blindness, the Richard A. Perritt, Charitable Foundation.
Disclosure: V.R. Rao, None; E.B. Stubbs Jr, None
References
- 1. Kapetanakis VV, Chan MPY, Foster PJ, Cook DG, Owen CG, Rudnicka AR. Global variations and time trends in the prevalence of primary open angle glaucoma (POAG): a systematic review and meta-analysis. Br J Ophthalmol. 2016; 100: 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006; 90: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tham Y-C, Li X, Wong TY, Quigley HA, Aung T, Cheng C-Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014; 121: 2081–2090. [DOI] [PubMed] [Google Scholar]
- 4. McMonnies CW. Glaucoma history and risk factors. J Optom. 2017; 10: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beidoe G, Mousa SA. Current primary open-angle glaucoma treatments and future directions. Clin Ophthalmol (Auckland, NZ). 2012; 6: 1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Izzotti A, Di Marco B, De Flora S, Sacca S. [Open angle glaucoma: epidemiology, pathogenesis and prevention]. Recenti Prog Med. 2006; 97: 37–45. [PubMed] [Google Scholar]
- 7. Kalouda P, Keskini C, Anastasopoulos E, Topouzis F. Achievements and limits of current medical therapy of glaucoma. Glaucoma Surgery. Unionville, CT: Karger S Publishers; 2017: 1–14. [DOI] [PubMed] [Google Scholar]
- 8. Inatani M, Tanihara H, Katsuta H, Honjo M, Kido N, Honda Y. Transforming growth factor-beta 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch Clin Exp Ophthalmol. 2001; 239: 109–113. [DOI] [PubMed] [Google Scholar]
- 9. Picht G, Welge-Luessen U, Grehn F, Lutjen-Drecoll E. Transforming growth factor beta 2 levels in the aqueous humor in different types of glaucoma and the relation to filtering bleb development. Graefes Arch Clin Exp Ophthalmol. 2001; 239: 199–207. [DOI] [PubMed] [Google Scholar]
- 10. Fleenor DL, Shepard AR, Hellberg PE, Jacobson N, Pang IH, Clark AF. TGFbeta2-induced changes in human trabecular meshwork: implications for intraocular pressure. Invest Ophthalmol Vis Sci. 2006; 47: 226–234. [DOI] [PubMed] [Google Scholar]
- 11. Fuchshofer R, Birke M, Welge-Lussen U, Kook D, Lutjen-Drecoll E. Transforming growth factor-beta 2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Invest Ophthalmol Vis Sci. 2005; 46: 568–578. [DOI] [PubMed] [Google Scholar]
- 12. Fuchshofer R, Tamm ER. The role of TGF-beta in the pathogenesis of primary open-angle glaucoma. Cell Tissue Res. 2012; 347: 279–290. [DOI] [PubMed] [Google Scholar]
- 13. Zode GS, Sethi A, Brun-Zinkernagel AM, Chang IF, Clark AF, Wordinger RJ. Transforming growth factor-beta2 increases extracellular matrix proteins in optic nerve head cells via activation of the Smad signaling pathway. Mol Vis. 2011; 17: 1745–1758. [PMC free article] [PubMed] [Google Scholar]
- 14. Goel M, Picciani RG, Lee RK, Bhattacharya SK. Aqueous humor dynamics: a review. Open Ophthalmol J. 2010; 4: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Llobet A, Gasull X, Gual A. Understanding trabecular meshwork physiology: a key to the control of intraocular pressure? Physiology. 2003; 18: 205–209. [DOI] [PubMed] [Google Scholar]
- 16. Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp Eye Res. 2008; 86: 543–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tan JC, Peters DM, Kaufman PL. Recent developments in understanding the pathophysiology of elevated intraocular pressure. Curr Opin Ophthalmol. 2006; 17: 168–174. [DOI] [PubMed] [Google Scholar]
- 18. Tamm ER. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res. 2009; 88: 648–655. [DOI] [PubMed] [Google Scholar]
- 19. Izzotti A, Bagnis A, Sacca SC. The role of oxidative stress in glaucoma. Mutat Res. 2006; 612: 105–114. [DOI] [PubMed] [Google Scholar]
- 20. Zhao J, Wang S, Zhong W, Yang B, Sun L, Zheng Y. Oxidative stress in the trabecular meshwork (Review). Int J Mol Med. 2016; 38: 995–1002. [DOI] [PubMed] [Google Scholar]
- 21. Ferreira SM, Lerner SF, Brunzini R, Evelson PA, Llesuy SF. Oxidative stress markers in aqueous humor of glaucoma patients. Am J Ophthalmol. 2004; 137: 62–69. [DOI] [PubMed] [Google Scholar]
- 22. Goyal A, Srivastava A, Sihota R, Kaur J. Evaluation of oxidative stress markers in aqueous humor of primary open angle glaucoma and primary angle closure glaucoma patients. Curr Eye Res. 2014; 39: 823–829. [DOI] [PubMed] [Google Scholar]
- 23. Ammar DA, Hamweyah KM, Kahook MY. Antioxidants protect trabecular meshwork cells from hydrogen peroxide-induced cell death. Transl Vis Sci Technol. 2012; 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yan DB, Trope GE, Ethier CR, Menon IA, Wakeham A. Effects of hydrogen peroxide-induced oxidative damage on outflow facility and washout in pig eyes. Invest Ophthalmol Vis Sci. 1991; 32: 2515–2520. [PubMed] [Google Scholar]
- 25. Ghanem AA, Arafa LF, El-Baz A. Oxidative stress markers in patients with primary open-angle glaucoma. Curr Eye Res. 2010; 35: 295–301. [DOI] [PubMed] [Google Scholar]
- 26. Majsterek I, Malinowska K, Stanczyk M, et al.. Evaluation of oxidative stress markers in pathogenesis of primary open-angle glaucoma. Exp Mol Pathol. 2011; 90: 231–237. [DOI] [PubMed] [Google Scholar]
- 27. Izzotti A, Sacca SC, Longobardi M, Cartiglia C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Invest Ophthalmol Vis Sci. 2009; 50: 5251–5258. [DOI] [PubMed] [Google Scholar]
- 28. Sacca SC, Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008; 173: 385–407. [DOI] [PubMed] [Google Scholar]
- 29. Tripathi RC, Li J, Chan WF, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp Eye Res. 1994; 59: 723–727. [DOI] [PubMed] [Google Scholar]
- 30. Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. 2013; 13: 12–15. [DOI] [PubMed] [Google Scholar]
- 31. Rao VR, Lautz JD, Kaja S, Foecking EM, Lukacs E, Stubbs EB Jr. Mitochondrial-targeted antioxidants attenuate TGF-beta2 signaling in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2019; 60: 3613–3624. [DOI] [PubMed] [Google Scholar]
- 32. Jain M, Rivera S, Monclus EA, et al.. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biolog Chem. 2013; 288: 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stubbs EB Jr, Von Zee CL. Prenylation of Rho G-proteins: a novel mechanism regulating gene expression and protein stability in human trabecular meshwork cells. Molec Neurobiol. 2012; 46: 28–40. [DOI] [PubMed] [Google Scholar]
- 34. Keller KE, Bhattacharya SK, Borras T, et al.. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp Eye Res. 2018; 171: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langert KA, Pervan CL, Stubbs EB Jr. Novel role of Cdc42 and RalA GTPases in TNF-alpha mediated secretion of CCL2. Small GTPases. 2014; 5: e29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 37. Tang B, Li S, Cao W, Sun X. The association of oxidative stress status with open-angle glaucoma and exfoliation glaucoma: a systematic review and meta-analysis. J Ophthalmol. 2019; 2019: 1803619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim HJ, Nel AE. The role of phase II antioxidant enzymes in protecting memory T cells from spontaneous apoptosis in young and old mice. J Immunol. 2005; 175: 2948–2959. [DOI] [PubMed] [Google Scholar]
- 39. Gordon MO, Beiser JA, Brandt JD, et al.. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002; 120: 714–720; discussion 829–730. [DOI] [PubMed] [Google Scholar]
- 40. Kass MA, Heuer DK, Higginbotham EJ, et al.. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002; 120: 701–713; discussion 829–730. [DOI] [PubMed] [Google Scholar]
- 41. Leske MC. The epidemiology of open-angle glaucoma: a review. Am J Epidemiol. 1983; 118: 166–191. [DOI] [PubMed] [Google Scholar]
- 42. Liu XF, Zhou DD, Xie T, et al.. The Nrf2 signaling in retinal ganglion cells under oxidative stress in ocular neurodegenerative diseases. Int J Biol Sci. 2018; 14: 1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sacca SC, Pascotto A, Camicione P, Capris P, Izzotti A. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch Ophthalmol. 2005; 123: 458–463. [DOI] [PubMed] [Google Scholar]
- 44. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013; 53: 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005; 37: 2478–2503. [DOI] [PubMed] [Google Scholar]
- 46. Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta. 2012; 1822: 1363–1373. [DOI] [PubMed] [Google Scholar]
- 47. Sastre J, Pallardo FV, Vina J. Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life. 2000; 49: 427–435. [DOI] [PubMed] [Google Scholar]
- 48. Abu-Amero KK, Morales J, Bosley TM. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2006; 47: 2533–2541. [DOI] [PubMed] [Google Scholar]
- 49. Pilz YL, Bass SJ, Sherman J. A review of mitochondrial optic neuropathies: from inherited to acquired forms. J Optom. 2017; 10: 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schrier SA, Falk MJ. Mitochondrial disorders and the eye. Curr Opin Ophthalmol. 2011; 22: 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamamoto N, Itonaga K, Marunouchi T, Majima K. Concentration of transforming growth factor beta2 in aqueous humor. Ophthalmic Res. 2005; 37: 29–33. [DOI] [PubMed] [Google Scholar]
- 52. Pervan CL, Lautz JD, Blitzer AL, Langert KA, Stubbs EB Jr. Rho GTPase signaling promotes constitutive expression and release of TGF-beta2 by human trabecular meshwork cells. Exp Eye Res. 2016; 146: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015; 6: 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R, Kietzmann T. Redox-fibrosis: Impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox Biol. 2015; 6: 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rao PV, Stubbs EB Jr. TGF-β2 selectively increases NADPH oxidase type 4 (NOX4) expression in human trabecular meshwork (TM) cells. Invest Ophthalmol Vis Sci. 2020; 61: 3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. RK G M I, OB CJ. TGF-β induces NOX4 and fibrotic genes in trabecular meshwork cells: role in glaucoma. Invest Ophthalmol Vis Sci. 2019; 60: 3800. [Google Scholar]
- 57. Altenhofer S, Kleikers PW, Radermacher KA, et al.. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012; 69: 2327–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sanders YY, Lyv X, Zhou QJ, et al.. Brd4-p300 inhibition downregulates Nox4 and accelerates lung fibrosis resolution in aged mice. JCI Insight. 2020; 5(14): e137127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bai G, Hock TD, Logsdon N, Zhou Y, Thannickal VJ. A far-upstream AP-1/Smad binding box regulates human NOX4 promoter activation by transforming growth factor-beta. Gene. 2014; 540: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Junn E, Lee KN, Ju HR, et al.. Requirement of hydrogen peroxide generation in TGF-beta 1 signal transduction in human lung fibroblast cells: involvement of hydrogen peroxide and Ca2+ in TGF-beta 1-induced IL-6 expression. J Immunol. 2000; 165: 2190–2197. [DOI] [PubMed] [Google Scholar]