

Graphical abstract
Abbreviations: APR, ancestral protein resurrection; ASR, ancestral sequence reconstruction; CDS, coding sequence; GR, glucocorticoid receptor; GWAS, genome wide association study; InDel, insertion/deletion; MCMC, Markov Chain Monte Carlo; ML, maximum likelihood; MP, maximum parsimony; MR, mineralcorticoid receptor; MSA, multiple sequence alignment; NJ, neighbor-joining; SFS, site frequency spectrum
Keywords: Ancestral sequence reconstruction, Evolution, Genomics, Metabolism, Phylogenetics, Plants
Highlights
-
•
ASR allows to explore the distant past in gene sequence space.
-
•
We provide a primer for non-experts to reconstruct the sequence of an extinct gene.
-
•
ASR, GWAS and fitness landscapes explore molecular evolution at different timescales.
Abstract
Whilst substantial research effort has been placed on understanding the interactions of plant proteins with their molecular partners, relatively few studies in plants - by contrast to work in other organisms - address how these interactions evolve. It is thought that ancestral proteins were more promiscuous than modern proteins and that specificity often evolved following gene duplication and subsequent functional refining. However, ancestral protein resurrection studies have found that some modern proteins have evolved de novo from ancestors lacking those functions. Intriguingly, the new interactions evolved as a consequence of just a few mutations and, as such, acquisition of new functions appears to be neither difficult nor rare, however, only a few of them are incorporated into biological processes before they are lost to subsequent mutations. Here, we detail the approach of ancestral sequence reconstruction (ASR), providing a primer to reconstruct the sequence of an ancestral gene. We will present case studies from a range of different eukaryotes before discussing the few instances where ancestral reconstructions have been used in plants. As ASR is used to dig into the remote evolutionary past, we will also present some alternative genetic approaches to investigate molecular evolution on shorter timescales. We argue that the study of plant secondary metabolism is particularly well suited for ancestral reconstruction studies. Indeed, its ancient evolutionary roots and highly diverse landscape provide an ideal context in which to address the focal issue around the emergence of evolutionary novelties and how this affects the chemical diversification of plant metabolism.
1. Introduction
Over the last decades evolutionary biology and experimental molecular biology have taken quite distinctive trajectories which have mostly developed in isolation from one another. Indeed, the massive expansion of biological research coupled with the increased adoption of chemistry and physics in addressing biological questions likely exacerbated this problem [1]. A consequence of this is that currently few scientists have been trained in both fields. However, fortunately the torrent of information from next-generation sequencing of closely related species [2], [3] means that the techniques, subject matter and philosophies of molecular biology are finally being brought to bear on both classical and modern evolutionary questions [4]. The approaches we present in this review are able to shed new light on both the origin and organization of molecular entities and in particular to reconcile the reductionist approach of molecular biology with the wider view evolutionary genetics can afford into biological systems.
Before attempting to combine molecular biology with evolutionary studies, it is important to acknowledge, as has been eloquently done in several works of Joseph Thornton [1], [4], [5], that they are fundamentally different disciplines. The strength of molecular biology being that it establishes functional links between gene and effect in experiments in which manipulation of single molecular entities (genes, proteins) is carried out under strictly controlled conditions. An example of this approach is that of classical Alanine Scanning, in which site-directed mutagenesis is used to introduce alanine substitutions in a wild-type (i.e., extant) protein. The combinatorial libraries thus obtained can be used to investigate the consequence of alanine insertions on protein structure, stability and function [6]. Whilst setting high standards of evidence-based inference, an unavoidable consequence of these approaches is, however, that they neglect the contribution of evolutionary forces to the biological variation and to the functional diversification we observe today. As such, their inferences are necessarily constrained in a reductionist thinking which does not embrace evolutionary processes [1]. A few examples highlight the problems inherent in this approach. First, in the case of certain functionally defined groups of proteins (such as carbonic anhydrases, alcohol dehydrogenases and serine proteases [7], [8], [9]), which contain members that harbour the same biochemical activity but highly dissimilar overall structures (being evolved independently from distinct ancestral proteins), the functionalist approach cannot link all properties of a protein to its function [10]. A second limitation of the approach is that it assumes that all aspects of proteins have been optimized for function. However, myriad studies have demonstrated that a proteińs sequence, structure, affinity for ligands and many other physical properties drift dramatically across several degrees of freedom in as long as they remain untargeted by purifying selection [11], [12], [13]. Furthermore, they often reflect that constraints imposed by their evolutionary history and those of “tinkering” to produce optimal form [14]. Finally, there are simply too many degrees of freedom in sequence, structure and function to identify the causal links between these phenomena in an abstract manner. As we will demonstrate below, it is, however, possible to evaluate how evolutionary divergence from a common ancestral protein caused structure and function to diverge, and, as such, how the specific and distinctive modern proteins evolved.
By contrast to molecular biologists, evolutionary biologists take a considerably less reductionist approach with biological variation being seen as a favourable outcome rather than an inconvenience. The strength of this approach is that it focuses on real world biological systems in their natural and historical contexts. It is, however, hampered by the fact that statistical associations are not reliable indicators of causality [15]. Thus, despite the fact that several important experimental works in evolutionary biology have been published in the last two decades [16], [17], [18], [19], [20], inferences about historical evolution are seldom as clear cut as those from molecular biology [1]. To address this, Joseph Thornton and others proposed a functional synthesis in which the techniques of evolutionary and phylogenetic analysis are combined with molecular biology, structural biology and biochemistry. In this way, phylogenetic analysis and, by extension, population or quantitative genetics are used to detect mutations whose effect can be then functionally tested, with molecular approaches, and associated to putatively adaptive phenotypes. As such, this “functional synthesis” provides historical insights into identifying the period and indeed the lineage in which a new function emerged, the mutations which arose and the sequence sites bearing putative signals of selection. Moreover, protein structures can help to identify historical amino acids that are likely to have been involved in the evolution of function. Having got so far, molecular biology allows the hypothesis to be tested directly as the production of synthetic genes are resurrected, expressed and functionally characterized [5].
A key theme of this functional synthesis between molecular biology and evolutionary genetics is about how evolutionary novelties emerge. Does this process proceed through quantitative small, gradual steps, or big and sudden jumps? Metabolism - with its protracted evolution and diversification [21], [22] - is a good field to test this hypothesis, and in particular the secondary metabolism of plants, which is characterised by phenomenal diversity. This large phytochemical complexity of plants is partially linked to the fact that gene duplications and even whole genome duplication events are considerably more frequent in Plantae than in the other kingdoms of life [23], [24]. Nevertheless, trying to understand the modalities about how evolutionary novelties emerge - at a molecular level - is a complex task. Analysis of sequence data from extant species is clearly not sufficient, as comparison of present-day data does not provide an estimate of the full spectrum of evolutionary changes that might have occurred starting from the ancestral sequence (e.g., when multiple changes affect the same site during the history of sequence divergence, the number of differences in the extant sequences is always an underestimation of the real number of substitutions which have occurred, an issue known as the “multiple hit problem”). As we will describe below, there are several approaches possible to address this issue. The first, which was initially proposed in the 60s, but has acquired prominence in recent years (at least in animal and microbial systems), is that of ancestral sequence reconstruction (ASR) [25], [26], [27], [28]. Such works - pioneered by the group of Steven Benner - provided, for the first time, historical insights about how protein function and specialization evolved [29], [30] (see also Fig. 1). Essentially, as we will describe in more words below, ASR is based on the reconstruction of extinct protein sequences, followed by their expression in heterologous systems and characterization of their function in comparison with that of modern-day proteins. The approach has been applied to enzymes, DNA binding proteins and receptors, but, surprisingly, has not been taken-up at scale in plants. This is somewhat surprising: as we hope to demonstrate in more words below, resurrecting extinct proteins is a powerful way - and, perhaps, the only possible one, given that intact proteins are rarely preserved intact in fossil or amber specimens - to identify the historical amino acid changes which are responsible for the large diversification of the structure, stability and activity of the proteins existing today. This diversification is particularly evident in plant metabolism: the chemodiversity of specialized metabolites is hardly tractable, estimated between 100,000 and 1 million [31], and is largely generated by the proliferation of relatively few gene families (i.e., terpene synthases, cytochrome P450, glycosyltransferases, acyltransferases, polyketide synthases, see for example [32]) occurring upon the gene and genome duplications events in the plant kingdom [33]. Thus, it is only through ASR that we can identify – and restrict to a discrete interval of evolutionary time – the amino acid changes responsible for the functional divergence of the metabolic enzymes extant today [34], [35]. In fact, classical “horizontal” biochemical approaches, e.g., swap experiments, in which those amino acid residues held responsible for functional shifts are exchanged between extant homologous proteins, usually fail to interconvert protein function ([36], [37], with some notable exceptions, see for example [38], [39]). Extant homologous proteins catalysing different reactions, for example, may differ in several residues, but most often only a minority of these differences are responsible for the functional shift, with the remaining substitutions having at best an ancillary role in the functional divergence. This may make the identification of candidate functional residues for swap experiments particularly difficult. Another reason that accounts for the failure of horizontal swap experiments in interconverting biochemical functions is the pervasive role of intragenic epistasis in modern day sequences. A single or few amino acid replacements could not be sufficient anymore to interconvert function between homologous sequences, as activity in extant protein sequences is heavily nested, and dependent, on the amino acid states at multiple sites [40], [41], [42]. In light of this, the horizontal replacement of one or few amino acids between extant sequences usually yields a nonfunctional product, due to the incompatible interactions of the swapped residues in the amino acid background of the receiving protein [10]. On the other hand, studies based on ASR, by focusing only on the sequence differences between the ancestral and descendant nodes in a phylogeny, drastically reduce the number of amino acid substitutions, making easier the identification of the residues leading to functional divergence. Thus, it is only through the “vertical” comparative approach of ASR, rather than the “horizontal” comparison of extant sequences, which we can effectively distinguish which amino changes result in the functional optimization of a pre-existing activity or in its strict partitioning among paralogs, or, again, in the de novo evolution of a function from an ancestral protein devoid of such activity [43].
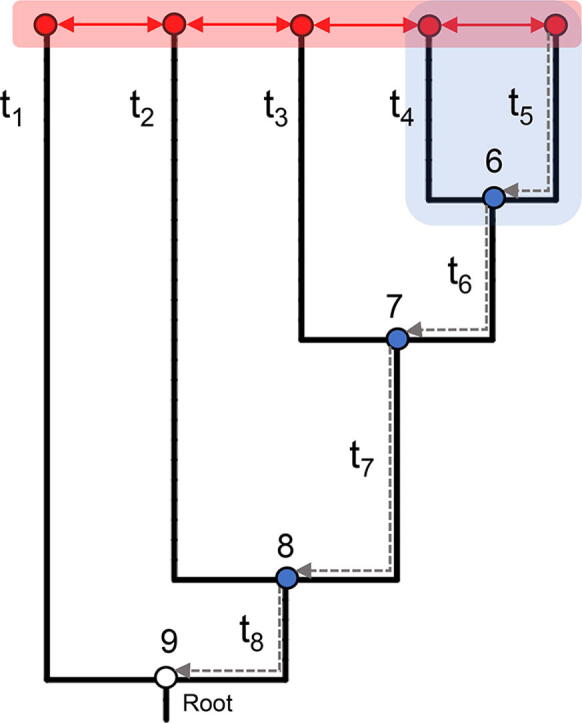
Fig. 1.

Horizontal Vs vertical approach in the analysis of sequence data. The approach followed by ASR operates a shift from the classical, “horizontal” comparison of sequence data of extant species. Starting from a sequence multialignment and a phylogenetic tree (with branch lengths, here represented by t1…t8), the algorithms used by ASR infer the sequences in the ancestral nodes (blue dots). These ancestral sequences can be then aligned to the extant sequences (“vertical” comparison) to identify where and when the historical mutations occurred along the evolutionary trajectories. The ancestral coding sequences can be then expressed in heterologous systems for functional assays. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
That said, validated genome wide association studies (GWAS) represent an additional form of the functional synthesis that is widespread in plants [44], [45] - albeit one that tends to focus on recent evolutionary events. Moreover, fitness landscape studies, such as those championed by Michael Purugganan and co-workers, are additionally complementary approaches which are being carried out in plants and related to ancestral reconstruction studies [46]. We will detail both sets of studies in the section Alternative functional syntheses of evolutionary and molecular biology below. However, before doing so, we will briefly detail the methodology by which ancestral proteins are resurrected, discussing the strengths and limitations of this approach to provide examples of ancestral protein resurrection emanating from all kingdoms of life. We will discuss these examples in the context of protein specialization to finally present the case for a greater adoption of the ancestral sequence resurrection approach, alongside those of GWAS and fitness landscapes, as a potent tool for understanding the evolution of plant metabolism and other processes.
2. How to “resurrect” a dead gene - a primer on the methods
Despite the recent development of sophisticated computational ASR methods, applied especially in the study of protein functional evolution in animals and microbes, the approach of reconstructing ancestral sequences cannot be considered entirely novel: already in the 60s Linus Pauling and Emile Zuckerkandl made the suggestion that it could have been possible, with some approximation, to deduce the ancestral amino acid sequence from a group of homologous sequences [47]. It was not until the 90 s, however, that the first pioneering ASR studies were published by the group of Steven Benner [29], [30]. Since then, ancestral sequence reconstruction approaches have been greatly improved with the adoption of refined methods for orthology prediction, sequence alignment, tree and ancestral sequence inferences [27], [28], [48]and their combination with sequence-based tests of selection [21] and unbiased metabolomics for assessing enzyme specificities [49]. We present in Table 1 a non-exhaustive list of computer programs and database resources for all steps involved in a typical ASR study, starting from the selection of homologous genes to the various algorithms for reconstructing ancestral sequences
Table 1.
List of computer programs and resources for the typical steps of an ASR study. Additional softwares for phylogenetic inference can be found in Joseph Felsenstein’s homepage (https://evolution.genetics.washington.edu/phylip/software.html, with latest updates in 2012) or in [21].
| Name | Description | References |
|---|---|---|
| Orthology prediction (de novo) | ||
| JustOrthologs | fast algorithm for ortholog inference. Avoids BLAST all-vs-all searches comparing instead lengths of CDS and calculates frequencies of dinucleotide occurrences in exon sequences | [50] |
| Orthofinder | inference of orthologs with increased precision (takes into account the gene length bias associated to the BLAST similarity scores). Provides rooted species tree and gene trees for all orthogroups. Maps duplication events along tree branches. | [51], [52] |
| Orthograph | maps coding nucleotide sequences to genes of known orthology (useful for the extension of existing orthogroups) | [53] |
| OrthoMCL | uses the Markov Cluster algorithm (MCL) to group putative orthologs and paralogs in a single orthogroup | [54] |
| Databases of pre-computed orthogroups | ||
| eggnog 5.0 | A database of orthogroups and functional annotations from virus, bacterial and eukaryotic genomes | [55] |
| Genomicus Plants v.41 | a multi-species genome browser allowing to visualize orthology and paralogy relationships | [56], [57] |
| OrthoDB | large catalogs of orthologs (across around 600 eukaryotes and > 3000 bacteria), obtained from best reciprocal hits in Smith-Waterman local sequence alignments | [58], [59] |
| Phylome DB | a website hosting catalogs of precomputed gene phylogenies from multiple genomes (“phylomes”). Provides high-quality orthology and paralogy relationships based on phylogenetic trees. Several plant phylomes available | [60] |
| PLAZA 4.5 | a comparative plant genomics database hosting instances for Eudicots and Monocots. Provides sets of orthologous genes obtained through Markov clustering | [61] |
| Multisequence alignment (MSA) | ||
| BAli-Phy | evolution-based tool for multiple sequence alignment. Incorporates a parametric model of sequence evolution, considering also indels | [62] |
| ClustalΩ (omega) | a fast progressive multialignment employing sequence embedding to reduce the time required to build the guide tree | [63], [64] |
| Expresso | a structure-based sequence alignment tool (protein 3D models from the Protein Data Bank are used as templates to guide the sequence alignment) | [65] |
| Historian | An evolution-based alignment software optimized for assessing indel rates and dN/dS ratios | [66] |
| MAFFT | progressive multialignment, includes iterative refinement methods (for small-scale alignments) and structural methods for RNA | [67] |
| MUSCLE | progressive multialignment based on k-mer counting | [68] |
| PRANK | evolution-based algorithm for alignment of closely-related sequences. Accurate placement of insertions and deletions. | [69] |
| ProbCons | algorithm based on a Markov model progressive alignment in combination with probabilistic sequence conservation information | [70] |
| SATe’-I and SATe’-II | Co-estimation of alignments and phylogenetic trees. Iterative approach using an initial RAxML-computed tree with a MAFFT alignment, followed by further refinements through a divide-and-conquer strategy | [71], [72] |
| T-Coffee | consistency-based multialignment, combining a global pairwise approach (e.g., ClustalW) with a local pairwise alignment (e.g. Lalign) | [73] |
| Alignment curation | ||
| BMGE | Calculates an entropy score for each column in the MSA and compares it with similarity score based on a PAM or BLOSUM matrix. Allows to distinguish, for each aligned character, biological variability from noise | [74] |
| Divvier | Identifies clusters of characters of shared homology, filtering out divergent partitions; alleviates long-branch attraction in trees obtained from filtered MSAs | [75] |
| Gblocks | Eliminates poorly aligned (highly variable) positions from a multialignment. Can be tailored to be more or less stringent according to the value of five different threshold scores | [76] |
| Noisy | Eliminates homoplastic sites from MSAs based on character compatibility | [77] |
| PREQUAL | A pre-alignment filtering tool to remove non-homologous characters in phylogenomic datasets. It uses a probabilistic model to infer homology between amino acids in non-aligned sequences | [78] |
| trimAl | Alignment trimming based on gap, similarity and consistency scores across all columns of a MSAs | [79] |
| Phylogenetic (tree) inference | ||
| BEAST2 | Bayesian analysis of molecular sequences. It uses Markov chain Monte Carlo (MCMC) as a numerical approximation to average over tree space | [80] |
| FastME | Distance-based tree inference (Neighbor-Joining) | [81] |
| FastTree2 | approximate-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequence | [82] |
| IQ-TREE | infers phylogenetic trees by maximum likelihood | [83] |
| MPBoot | Tree reconstruction based on maximum parsimony, suitable for large DNA and protein sequence alignments | [84] |
| MrBayes | Bayesian phylogenetic inference using Markov Chain Monte Carlo methods, with a large selection of evolutionary models for aminoacid and DNA (codon) data | [85], [86] |
| PAML (v4.9j) | a package of several programs for phylogenetic analyses of DNA or protein sequences using ML. Includes the empirical Bayes method for estimation of ancestral sequences using nucleotide, codon or amino acid substitution models [87] | [88] |
| PhyloBayes | A popular bayesian Monte Carlo Markov Chain (MCMC) software for phylogenetic reconstruction and molecular dating. It uses non-parametric methods to characterize sequence evolution | [89], [90], [91] |
| PhyML v3.0 | package for phylogenetic reconstruction using ML from nucleotide or amino acid sequences; several substitution models and tree searching algorithms implemented; introduces the criteria of minimum posterior expected error (MPEE) for ancestral sequence reconstruction [92] | [93], [94] |
| Curation of phylogenetic trees | ||
| Phylo-MCOA | Identifies outlier genes and species in phylogenomic datasets | [95] |
| TreeShrink | Identifies genes leading to long branches | [96] |
| TreSpEx | Identifies artificial signals in phylogenetic reconstructions (paralogy, long-branch attraction) | [97] |
| Ancestral sequence reconstruction (ASR)(ASR methods based on MP and ML are generally implemented as additional functions in tree inference programs, the ones listed below are some additional dedicated resources) | ||
| ANCESCON | ASR software incorporating different substitution rates among sites (“alignment-based rate factors”) with the estimation of phylogenetic trees based on a weighted neighbor-joining method (distance-based, [98]) | [99] |
| FastML | A user-friendly web server for computing ancestral sequences based on ML (includes marginal and joint estimates, with the time required for calculation scaling linearly with the number of sequences, hence it is applicable to very large datasets) | [100], [101], [102] |
| PhyloBot | A web-based tool, designed for non-experts, integrating all common steps for a typical ASR pipeline (sequence alignment, phylogenetic inference, ancestral reconstruction, and prediction of functional effects) | [103] |
| ProtASR/ProtASR2 | prediction of ancestral sequences using a mean-field (MF) substitution model incorporating selection on folding stability | [104], [105] |
| Revenant | a database of resurrected ancestral proteins | [106] |
Studies of ASR are usually focused on reconstructing the evolutionary trajectories of a specific protein family. The objective of a typical ASR experiment is to recreate the sequence of an extinct protein (representing an ancestral node in a phylogenetic tree), assess its functional properties (through expression in heterologous systems and biochemical assays, for example) and compare how these properties differ with respect to extant proteins. By doing so, ASR experiments can answer the focal questions about where (in terms of which part of the sequence) and when (so, along which specific lineage of the gene tree) functional shifts occurred during the evolutionary trajectories leading to protein divergence.
An ideal family which could be suitable for ASR is medium-large in size (in terms of the number of gene family members) and is characterised by both recent and more ancient gene duplications (as these events may lead to functional divergence, ASR can be the ideal approach to understand when functional specialization emerged along ancestral branches). Also, the activity of present-day proteins should be easily measurable, in terms of substrate specificities/reaction products (for enzymes) or other biochemical properties (e.g., DNA binding specificity for transcription factors). Possibly, in order to map the position of some of the amino acid changes along the 3-D protein structure, the crystal structures for some extant enzymes should be also available. However, recent advances in cryo-microscopy are finally on the cusp of delivering the long promised atomic scale resolution and as such may soon represent a more accessible and physiological source of this information [107].
The starting dataset for an ASR study is thus a group of homologous sequences representing an entire gene family. The initial gene (protein) sequences can be collected directly from NCBI or Uniprot, using one or a few query sequences in a similarity search (BLAST); but the identification of gene family members, especially when assaying multiple genomes, can be better done in a phylogenomic framework, using orthology-prediction tools like OrthoMCL [54] or Orthofinder [51], [52]. These tools take the full set of protein sequences encoded in a number of selected genomes and use all-vs-all similarity and clustering algorithms to finally reconnect homolog sequences, from multiple species, into orthogroups.
Once a gene family has been identified as a specific orthogroup, the next step in ASR is to build an accurate multiple sequence alignment (MSA) (although this could be already an output of the orthology prediction program). Several alignment tools exist in this case: i) traditional progressive tools, such as MAFFT and MUSCLE; ii) consistency-based algorithms (e.g. ProbCons and T-Coffee) and the iii) family of evolution-based approaches, like BAli-Phy and PRANK.
Essentially, the progressive tools work by estimating an approximate similarity-based ranking among all sequence pairs, to then build a preliminary tree (the “guidetree”) of all sequences and add progressively the additional sequences to the initial top-scoring aligned pair. These are the faster algorithms in MSA, and are usually employed in the orthology prediction tools. Consistency-based methods tend to be more accurate, albeit significantly slower, as they work by searching the multialignment which maximises the consistency among all previously-computed pairwise alignments. The evolution-based tools include instead an explicit model of sequence evolution (at the level of DNA, amino acid or codon): they thus reconstruct evolutionarily-consistent alignments incorporating not only substitutions but also sequence insertions and deletions. Given that they are considerably more computationally intensive than the progressive or consistency-based methods, they can be used in a later step of the phylogenomic pipeline, for example to calculate an accurate multialignment of the sequences of a specific orthogroup initially obtained from the orthology prediction tool.
The choice of a specific MSA algorithm has important implications for the accuracy of downstream ancestral reconstructions: while most of the alignment algorithms were found to be quite robust to weak perturbances (i.e., in the presence of low tree depths and low frequency of InDels), in cases of more demanding alignments (e.g. higher InDel rate and high number of substitutions/site) the use of different algorithms had quite drastic consequences on the accuracies of the reconstructed ancestral sequences. In general, the MAFFT consistency iterative methods (MAFFT E-INS-i and MAFFT L-INS-I, present in MAFFT version > 7) and the various refinements of PRANK outperformed the progressive-based alignments in terms of reconstruction accuracies [108].
The next step in an ASR study is that of the inference of a correct phylogenetic tree; this is again a critical step before reconstruction of ancestral sequences can be attempted. The tree specifies the genealogical history of the sequences: in a gene tree, internal nodes represent the ancestors and may mark gene duplication events or emergence of a new lineage (Fig. 1). A well supported gene tree, which has been reconciled with the tree of the species under examination, representing a phylogeny which is coherent with the larger tree of life, is of course an essential requirement to prevent erroneous inference of ancestral states [109], [110], [111] (see also [112], [113], [114] for a debated case of ASR based on a controversial, and possibly incongruent, phylogeny of Eukaryotes). Several methods exist for the inference of a phylogenetic trees, and they are broadly classified into distance-based or character-based methods. Given that distance-based methods (e.g., neighbor-joining, minimum evolution) do not model the variation of the single character states (i.e., nucleotides, codons or amino acids in the sequence alignments), but rely solely on the estimation of the distance between sequences, they can be used only for the calculation of the initial sequence tree, but not to reconstruct the ancestral sequence states. We will thus briefly describe only the main approaches for the character-based methods, given that the same principles of these methods also govern the reconstruction of ancestral states, referring the reader to the more extensive excellent reviews on the topic [48], [115], [116].
Character-based methods for phylogenetic inference belong either to: i) maximum parsimony (MP); ii) maximum likelihood (ML) or iii) Bayesian approaches. Parsimony basically calculates the number of character changes over all possible tree topologies and considers as the best estimate of the species (or sequence) phylogeny the tree with the smallest number of changes [117]. As the number of possible topologies cannot be easily explored (there are already 4.95x1038 possible rooted trees for 30 taxa!), current parsimony approaches use some form of fast heuristic search algorithms [84]. Although methods based on MP are generally fast, they consider all character changes as equally probable; in doing so, they do not consider either the transition-transversion bias or the different rates of substitution among sites. The main limitation of MP in phylogenetic inference is thus its lack of an explicit model for sequence evolution. For these reasons, MP has been now almost completely superseded by ML, which effectively incorporates explicit models of sequence evolution. A model of sequence evolution is a description, in terms of probabilities, of how a nucleotide or amino acid sequence changes over time. Many sophisticated substitution models exist [118], with many variables to be estimated (parameters): they all derive however from the simple Jukes-Cantor model (JC, [119]), in which all base changes are equally probable. Under this assumption, the probability of a nucleotide change is only a function of branch lengths. Essentially, the basic principle of likelihood methods is to maximise the probability of observing the data (e.g., a single column in the multi-alignment), optimising the values of the parameters (e.g. those included in the substitution model along with the branch lengths). Since aligned sequence sites are treated as independent variables, the overall likelihood of a tree is the product of the probabilities calculated at each aligned site. Thus, the phylogenetic tree which achieves the highest likelihood is the ML tree and is considered the best representation of the sequence’s evolution.
The use of ML in phylogenetic inference was proposed by Joseph Felsenstein in 1981 [120], when he derived the equations for the likelihood of a given tree (based on the probabilities of character changes) and formulated the iterative method to find the tree topology with branch lengths maximising the likelihood value. The original ML method with its most recent refinements have been, since then, implemented in several softwares (Table 1).
Bayesian inference is the third class of approaches for phylogenetic inference; these methods were introduced in the field of phylogenetics in the mid-1990s, and became, since then, popular alternatives to MP or ML. However, an extensive treatment of these methods is beyond the scope of this review, and we rather refer the reader to excellent prior in-depth descriptions of these topics [116], [121], [122]. Here, suffice to say that Bayesian methods explore the tree space, and yield the optimal phylogeny, combining three quantities: (i) the prior probability, that is, the probability of a tree before the analysis (generally all trees can be equally probable, but in some cases the priors can be weighted according to some a priori knowledge, e.g., on the basis of fossil calibration); (ii) the likelihood (the probability of the observed data given the tree), and (iii) the posterior probability, which is obtained combining the prior probability with the likelihood, resulting in the calculation of the probability of a tree given the data. Although a seemingly simple quantity, the posterior probability needs to be calculated over all possible tree topologies, model parameters and branch lengths; this is achieved computationally using Markov Chain Monte Carlo (MCMC) approaches, which, in simple terms, approximate the calculation of posterior probabilities by random iterative sampling [116]. The main Bayesian programs for phylogenetic analysis are listed in Table 1.
With a sequence multi-alignment and a well-supported phylogenetic tree at hand, the next step is the reconstruction of ancestral sequences. Essentially, the same classes of approaches for inferring a phylogeny, i.e. parsimony, likelihood and Bayesian statistics, also apply in the context of inference of ancestral states.
Historically, parsimony was the first method used for ancestral reconstruction [117]. It is a simple, fast and efficient method based on traversing the tree twice to find the most parsimonious reconstruction of ancestral states (the one which minimizes the number of character changes). An example of the classical Fitch’s algorithm for MP reconstruction of ancestral states is presented in Fig. 2.
Fig. 2.

The Fitch's algorithm of maximum parsimony (MP) to reconstruct ancestral states. To assign ancestral states the tree is traversed twice. The first time the algorithm proceeds from leaves to root, and assigns to each internal node a set of characters based on the intersection of descendant states (or the union of the intersection is empty). In the second step, the algorithm proceeds from the root to the leaves, and assigns to the internal nodes the state which is present both in the ancestral and in the descendant node. When different equally parsimonious reconstructions are possible, multiple solutions exist (see Suppl. Fig. 1).
The same limitations of MP in tree inference, however, also apply in the reconstruction of ancestral states. These limitations stem from the lack of a probabilistic model describing sequence evolution, which is instead the key component of ML and Bayesian methods. In Fitch’s MP algorithm [117], in fact, all changes are equally probable, but this assumption ignores the transition/transversion rate bias, and also the heterogeneity in the rates of evolution at different sequence sites (i.e., the bias at the 3rd codon position). Also, parsimony does not take into account branch lengths, ignoring, for example, the higher frequency of substitutions associated to long branches. Moreover, when multiple equally parsimonious solutions exist, MP cannot distinguish which is the correct one, and thus converges on multiple, ambiguous reconstructions of ancestral states (see Suppl. Fig. 1 for an example of four equally parsimonious solutions in Fitch’s algorithm).
That said, MP can provide an accurate estimation of ancestral sequences only in cases of limited divergence [123] and has thus been used to resurrect ancestral proteins only from the recent past. The first ASR study used MP to resurrect the sequence of a digestive ribonuclease ancestral to the divergence between buffalos and oxen (so, a relatively young ancestral protein, dating back to 10 Mya, [29]), with the same approach being also used to resurrect the older ancestral ribonuclease - an enzyme of 40 Mya - from the base of the Artiodactyl lineage (the order of Mammals including deers, pigs and oxen) [30].
ML and Bayesian methods for reconstruction of ancestral sequences provide a solution to the drawbacks of MP, and have allowed so far to reconstruct sequences from the distant past (dating back to several hundred million years ago; we will present several examples further below). Essentially, the mathematical framework to reconstruct ancestral states is the same as that used in ML-based tree inference. These methods were originally proposed in the 1990s [87], [124], but further developments were introduced later, such as the inclusion of protein structural and folding parameters [102], [104]. Essentially, these methods are based on the calculation of the posterior probabilities of all possible ancestral states given the data. In doing so, they effectively incorporate branch lengths and substitution probabilities, and yield the probabilities not only of the optimal reconstruction, but also of those related to the sub-optimal solutions to allow the further biochemical exploration of alternative sequences [125] (see Fig. 3 for a simple example of a ML-based estimation of ancestral states).
Fig. 3.

Example of a Maximum Likelihood (ML) algorithm for reconstruction of ancestral states. The figure represents a simple case of ancestral reconstruction using ML. We considered only a single site, with two possible character states (H or Q), across a phylogenetic tree with equal branch lengths. The algorithm first traverses the tree from the leaves to the root, and, for each internal node, computes the likelihood of all possible states taking also into account all possible states of the father node(s). In the second step, the algorithm traverses the tree from the root to the leaves assigning the ancestral states which maximise the likelihood. The figure represents the calculation for the subtree composed by the leaf nodes 4 and 5, the internal node 6 and father node 7 (blue rectangle).
In the context of ML-based reconstruction of ancestral sequences, the initial algorithms allowed the calculation of marginal probabilities (i.e., the probability associated to the reconstruction of a single ancestral node, [87], [124]), while further improvements included also the computation of joint likelihoods (i.e., the calculation of the entire set of reconstructions across all ancestral nodes, [100], [126]). Approaches based on marginal probabilities are best indicated if the objective is to reconstruct the ancestral state within a single subtree (e.g., as for the synthesis of the ancestral protein); joint probabilities, on the other hand, could be used to count the number of character changes across the entire tree.
3. Limits of ancestral protein resurrection approaches
The idea of reconstructing extinct sequences, although attractive, is, however, not exempt from limitations. The two main approaches recalled above, MP and ML, have each their own drawbacks. As we have seen, MP might not converge to a unanimous solution (Suppl. Fig. 1), and, in this case, needs to make some assumptions about when character changes preferentially occur in the phylogeny (e.g., at the earliest possible point or along a more recent branch, [127], [128], [129]). In a similar vein, also ML approaches are not exempt from a priori assumptions. The phylogenetic tree is inferred from the data, hence, large ambiguities in the alignment, and thus in the resulting tree topology and branch lengths, have all drastic consequences on the accuracy of reconstructed ancestral sequences [108]. Accurate alignments may be achieved by a comprehensive sampling of extant sequences (e.g., by selecting a specific orthogroup from a large phylogenomic pipeline) and by the use of evolution-based alignment algorithms which can effectively model the insertion of gaps (see Table 1 and [130], [131]).
Although ML methods for ASR are the most commonly used today [28], there are additional limitations which should be considered, especially when the focus of the ASR is the study of biochemical properties related to protein stability (see below). As we have recalled above, ML approaches assume that the multialignment, the tree and the model parameters are known and true a priori, while Bayesian inference incorporates the uncertainty of these parameters in the inference of ancestral sequences (e.g., by sampling over the distribution of all possible tree topologies and model parameters) [132], [133], [134]. Although the incorporation of uncertainty over the tree phylogenies, in a Bayesian framework, does not seem to increase significantly the accuracy of reconstructed sequences [135], a study of simulated protein evolution has pointed to some inherent bias when ML (and MP) are used in ASR. Essentially, these two methods may introduce a systematic bias overestimating protein stability in the reconstructed sequences, independently from the depth at which the ancestral node is located in the phylogeny [136]. This bias stems from the inherent tendency of ML and MP to converge on the most probable (or most parsimonious, in case of MP) ancestral sequence solution across all sites, with the exclusion of all less probable amino acids across sequence sites (some of which can give still suboptimal likelihoods). This results in the maximization not only of the ancestral sequence probability, but also of the thermodynamic stability of the protein. In a simulated protein evolution scenario, in which it is possible to directly compare the ancestral proteins with their true descendants, it was shown that Bayesian inference methods, while yielding only marginally inferior ancestral reconstruction accuracies (introducing suboptimal, slightly detrimental substitutions in the ancestral sequences), outranked ML- and MP-based methods in the estimation of the thermodynamic properties of the ancestral sequences [136], suggesting that caution should be taken in interpreting especially those ML-based ASR studies focusing on the evolution of thermostability [137]. In these cases, the optimal strategy would be of course to functionally test both predictions, from both ML-based and Bayesian statistics, before drawing conclusions on the paths of functional evolution.
A crucial step in ML- and Bayesian-based tree inference is the choice of the probabilistic model for sequence evolution, a selection which brings about assumptions about the amino acid frequencies, exchange tendencies and rate heterogeneity amongst sequence sites. Since many models exist, the usual procedure is to measure the fit of each model to the existing data and select the one yielding the best fit [138]. Although selection of an inaccurate model may produce an unrealistic tree [139], there is currently no consensus about which criteria should be followed to determine the model with the best fit to the data. Most importantly, it has been demonstrated that model selection might not even be strictly necessary in ancestral sequence reconstruction, as the use of different criteria in model selection, or the use of the most parameter-rich model (GTR + I + G), all yielded similar results in terms of tree topologies and ancestral sequences [140].
Perhaps a more critical step for reliable reconstructions is instead to obtain a well supported phylogeny, which needs to be consistent with the larger tree of life and which can effectively reconcile the gene with the species tree under examination. It is thus important that, as a starting point in an ASR study, an orthogroup is selected from a cross-genome orthology inference calculation (see Table 1), as these pipelines generally implement reconciliation of gene trees with the overall species tree [109], [141], [142]. Of course, the resulting reconciled species tree needs to be critically evaluated in light of the systematic relationships among the taxa under examination. No current software is of course a substitute for expert knowledge in evaluating how realistic the gene family tree is in light of the species phylogeny. The recent large-scale phylogenomic investigations across all kingdoms of life (Bacteria and Archaea: [143], [144], Eukaryotes: [145], [146], [147] may provide a guide for biologists to systematically assess the organismal relationships among taxa and provide support for phylogenetic inferences.
A strong phylogenetic support is especially important at the level of the subtrees from which ancestral nodes need to be reconstructed. At this regard, it is also important to rely on the most robust metrics for branch support (e.g., the approximate likelihood- or Bayesian-based measures of branch support or the more traditional bootstrap, [148], [149], [150]) to make sure the target nodes are sufficiently robust to alternative topologies.
Another point that needs to be considered is that the reconstructed (optimal) ancestral sequence represents, in any case, the best guess, and, as such, it should not considered as the true, unequivocal ancestor’s sequence. Ambiguous reconstructions may in fact occur at several sites, where different character states might show almost equal probabilities. A caveat could be in this case not only to consider the optimal ML reconstruction, but also the alternative sequences with lower overall posterior probabilities, and test their biochemical properties to make solid conclusions about the possible paths of protein functional evolution (this approach has been followed, among other studies, in yeast, for resurrecting the ancestral alcohol dehydrogenase [151], and, more recently, on a massive scale, for the half million alternative sequences of the ancestral steroid hormone receptor [125]). When the number of ambiguous sites is extremely large, then synthesis and characterization of all alternative states is impractical: a solution can be in this case to synthesize the worst plausible case (the so called “AltAll”), basically a version including the least probable character states in all ambigous sites. If the AltAll version then shows similar functional properties to the ML ancestral sequence, then it is safe to assume that the functional inference is robust to the uncertainty present in the ambiguous sites [152], [153], [154]. A systematic evaluation of the sequence uncertainty in ASR has been attempted in three protein families characterised by different functions and architectures (guanylate kinases, and the DNA-binding and ligand-binding domains of the steroid hormone receptors); the results showed that similar functions were observed when alternate, lowly probable amino acid states were incorporated into the reconstructed AltAll sequences [155]. Although the results reported in this study can be hardly generalized to all protein families, they suggest that the characterization of the AltAll version may provide a strong support to the conclusions made on the function of the ancestral protein.
4. Evolution of protein specificity
Having covered the basic steps and methods of a typical ASR study, we now turn to examine some exemplary case studies of ASR in vertebrates and other organisms including those that appeared more recently in plants. As recalled above, it was the group of Steven Benner who pioneered studies of ASR in several biological systems [29], [30], [151], but also the lab of Joseph Thornton made, more recently, important contributions to the field. For example, Thornton and his team found that genes for the steroid hormone receptors, previously assumed to be confined to Vertebrates, were also present in the sea slug Aplysia californica [156]. Based on this study, he used the extant genes to climb down the evolutionary tree and deduce the most likely sequence of the common ancestor. He then assembled the gene and inserted it in cells which could authentically produce the ancient protein. Taking this further, Thornton set his own lab up to study when (and where) the differences between the mineralcorticoid (MR) and the glucocorticoid receptor (GR) emerged along their evolutionary history. The MR preferentially binds the steroid hormone aldosterone, which regulates salt and water balance, whilst its closely related glucocorticoid receptor is activated by cortisol and controls stress responses. Intriguingly, although the receptors evolved via a gene duplication event >450 million years ago, aldosterone itself did not arise until many million years later. Resurrecting the ancestral protein surprisingly revealed it to be sensitive to aldosterone, suggesting it had been activated by a similar structure [157]. Building on this work, Thornton’s group next determined the crystal structure of the common ancestor of the GR and MR, revealing that two crucial mutations were responsible for altering the binding pocket of the ancestral receptor such that it preferred binding to cortisol [157]. Attempts to run the evolutionary sequence backwards, however, failed. They instead engineered a hormone irresponsive protein introducing a set of other mutations which had accrued between the ancestral protein and the GR. These additional mutations, whilst playing no role in the receptors' new function, acted as an evolutionary ratchet preventing it from regaining its old function [158]. This finding underlines the value of ASR studies: the contribution of epistasis and especially how it gradually accrues during protein evolution can only be possible through the “vertical” comparison of the reconstructed ancestral and extant sequences [40], [41].
Indeed, a recent study demonstrated how a model that statistically identifies epistasis in alignments of present day sequences can illuminate the sequence-function landscape in a manner that will allow the prediction of new mutations [42]. This has clear implications for biotechnology, since features such as high stability, substrate/catalytic promiscuity, conformational flexibility and altered interactive properties have all been postulated [159], [160]. Along these lines, the Thornton group investigated the role of epistasis further. In their 2017 paper, Starr et al. explored the alternative evolutionary histories of transcriptional control of oestrogen response elements compared to steroid response elements [125]. The aim of these series of experiments was to understand the route molecular evolution took by also studying the alternative trajectories which could have been taken but were not. They achieved this by combining ancestral protein resurrection with deep mutational scanning [161], [162] to characterize alternative histories in sequence space of an ancient transcription factor which evolved a novel role via well-characterized mechanisms [163], [164]. They found hundreds of alternative protein sequences that use diverse mechanisms to perform the derived function at least as well as the extant protein. Intriguingly, these alternatives all require permissive substitutions that do not enhance the derived function - but do not all require the same permissive changes. They interpret this to mean that “the outcome of evolution depends on a serial chain of compounding chance events”. Elegant though it is such studies are by no means restricted to the work of Thornton with considerable research into the role of the evolvability of promiscuous protein functions [165] and specifically for glutamine binding [166], esterases and hydoxynitryle lyases [167], beta-lactamases [168], lactate dehydogenases [169], alkaline phosphatases [170], sesquiterpene lactone oxidases [171] and chalcone isomerases [172].
As we have said, parsimony approaches dominated the early ASR studies, with the resurrected proteins being of a relatively young age (10–80 Mya). The later introduction of ML and Bayesian statistics made it possible to resurrect sequence from the distant past, with the most ancient protein resurrected dating back to around 4000 Mya [173]. In any case, the approach of ASR, so far, has been mainly applied to reconstruct ancestral sequences from Vertebrates [174], yeast and bacteria [106]; we will thus focus our attention here on the few recent examples from plants, referring the reader to other excellent reviews for a more comprehensive coverage of ASR studies in Vertebrates and microbes [25], [26], [28].
A recent application of ASR in plants is the study of the ligand-receptor interactions in the self-incompatibility (SI) of Brassicaceae. This is a spectacular example of a diversified allelic series in which numerous highly diverged receptor-ligand combinations are segregating in natural populations [175]. Using in planta ancestral protein reconstruction, the study demonstrated that two allelic variants, segregating as distinct receptor-ligand combinations, diverged through an asymmetrical process whereby one variant has retained the same recognition specificity as their (now extinct) putative ancestor, while the other has functionally diverged and now represents a novel specificity no longer recognized by the ancestor.
Another recent study focused on the ancestral reconstruction of protein interaction networks [176]. The investigation adopted, as a reference timepoint, the gamma genome polyploidization event which occurred at the origin of the core eudicots. To better understand the impact of this whole-genome duplication, the interaction networks of ancestral MADS domain transcription factors were reconstructed from just before and just after the gamma duplication event. The networks were then directly compared to the extant networks of Arabidopsis thaliana and tomato (Solanum lycopersicum). It was found that the gamma duplication expanded the MADS domain interaction network more strongly than subsequent genomic events; it strongly rewired MADS domain interactions allowing for the evolution of new functions, as well as installing robustness through new redundancy. Intriguingly, post gamma, the network evolved from an organization around a single hub to a network organized around multiple hubs with well-connected proteins losing, rather than gaining, novel interactions.
Given the possibility ASR affords in recreating the most probable ancestral sequences, it is not surprising that RuBisCo (Ribulose-1,5-bisphosphate carboxylase-oxygenase), namely the protein catalyzing the first step of CO2 assimilation in plants has been the target of an in-depth evolutionary investigation. ASR allowed the biochemical characterization of a predicted Precambrian variant [177], which was likely present one billion years ago. The findings of this study revealed the divergent evolutionary paths taken by eukaryotic RuBisCos with respect to their bacterial homologs. Eukaryotic enzymes gradually developed improved specificity for CO2, while bacterial homologs had increased rates of carboxylation. This was consistent with the in vivo analysis that showed the preferential association of ancestral RuBisCOs into modern-day carboxysomes, which constitute the cyanobacterial organelles responsible for the CO2-concentrating mechanism.
Perhaps one of the most interesting avenues for the application of ASR, although still relatively unexplored, is that of secondary metabolism of plants. The evolution of metabolic diversity is hardly tractable without the reference points represented by the ancestral enzyme sequences. This is especially true in plant genomes, where the impact of gene duplications led to a phenomenal chemical diversity and enzyme promiscuity. Two papers from the lab of Todd Barkman represent seminal works in which ancestral protein resurrection was used to determine how enzyme function (i.e., substrate specificity) emerged along evolution. In the first of these papers, Huang et al. investigated the role for ancestral functional variation in determining modern-day enzyme specificity in the salicylic acid/benzoic acid/theobromine (SABATH) methyltransferase lineage [35]. In each case, they demonstrated that ancestral non-preferred activities were improved upon in a daughter enzyme after gene duplication, suggesting that these functional shifts were likely coincident with positive selection. In their second study, Huang et al. revealed that the convergent evolution of caffeine in plants [178] was partially the result of the co-option of exapted ancestral enzymatic activities which were maintained for 100 Mya [34]. These exaptations probably became fixed, and rose to prominence, after the initial steps of the caffeine pathway(s) evolved.
That of evolution of plant metabolism is indeed one of the fields where ASR has allowed recently some groundbreaking discoveries. The emergence of a new protein function is generally explained as the partition into the paralogs of activities already present in a generalist ancestral enzyme; in this way, subsequent evolutionary pressures could then act to optimize the individual protein functions in the descendant lineages. This has been the case which probably acted during the evolution of the iridoid synthase (ISY) activity in Nepetinae, a group of plant species part of the larger mint family, Lamiaceae. Iridoids are a class of monoterpenes derived from 8-oxogeranial (8-OG), and generally act as defensive molecules against herbivores. In particular, nepetalactones, a subclass of iridoids derived from 8-OG through the action of iridoid synthase, are volatiles monoterpenes which mimic pheromone activities and induce behavioural responses in cats and other Felids. While phylogenetic reconstruction of ancestral states posits the existence of iridoid biosynthesis at the base of Lamiaceae, the capacity to synthesize these molecules has been lost in most of the Nepetoiadeae (a large subgroup of Lamiaceae), to later reappear only in species of the Nepeta genus. ASR has been used to reconstruct the sequences of the ancestral PRISE, the gene family to which ISY belongs. The characterization of these ancestral forms was in support of an evolutionary model where the ancestral PRISE had only a minor ISY activity which was subsequently optimized, through positive selection acting on one of the paralogs, to become preponderant in extant enzymes of the Nepeta genus [49].
De novo evolution, that is, the emergence of an entirely new function in the descendant which was absent in the ancestral gene, is instead generally considered a rare instance in the emergence of protein function. A recent investigation on the evolution of chalcone isomerase (CHI, an enzyme catalysing an early step in the biosynthesis of plant flavonoids) has instead provided evidence for the de novo evolution of the catalytic function of CHI from a non-enzyme ancestor. Extant CHIs catalyse the enantioselective isomerization of chalconaringenin to (2S)-naringenin, and are phylogenetically related to two protein families devoid of isomerase activity: the CHI-like proteins (CHIL), whose accumulation generally correlates with that of CHIs, and the more distant fatty acid binding proteins (FAPs). Reconstruction of the ancestors of CHIs (ancCHI), CHILs (ancCHIL) and of the ancestor predating the split between CHIs and CHILs (ancCC) showed that none of these possessed isomerase activity. The study also identified the three founder amino acid substitutions which gradually imparted the enzymatic activity to extant CHIs starting from the non-catalytic ancestor (ancCC). These three mutations showed moderate epistasis, as each of them gradually increased isomerase activity irrespective of the order in which they occurred in their non-catalytic ancestor [179].
Partioning and improvement of side activity, as in the case of iridoid synthase or SABATH methyltransferases, or evolution de novo, as for chalcone isomerase, are perhaps only two specific trajectories from a wider spectrum of intermediate possibilities which range from strict partitioning to exclusive evolution de novo. Although it is attractive to classify trajectories of protein functional evolution into the distinct classes of sub- and neofunctionalization of gene paralogs, ASR studies have generally provided evidence for the co-occurrence of different phenomena, yielding a highly complex scenario for the emergence of new protein functions. Some models, and empirical data [180], blur the distinction between neo- and subfunctionalization [37], as the two modalities may co-occur during the evolution of a protein family. Although the simple partition of enzymatic activities from a generalist/promiscuous ancestor seems to be the most common scenario, at least from the ASR studies conducted to date, evolution of an entirely new activity - from an ancestor devoid of such activity - appears to be neither difficult nor rare. Also, when a new function emerged along an evolutionary branch, be it a new catalytic activity or allosteric interaction, very few large-effect amino acid substitutions were sufficient for the functional shift, with the remaining mutations having (at best) an ancillary role in the fine tune and further optimization of the new function [37], [181].
Whilst all of the examples presented above both provide strong testament to the power of ancestral protein reconstruction (as well as telling fascinating biological stories in their own right), they merely represent a minor proportion of the plant-based studies which could be described as fulfilling the requirements of the functional synthesis. Genome-wide association studies (GWAS) and fitness-landscape modelling represent in fact alternative approaches combining, as does ASR (although on a different scale), the power of molecular with evolutionary biology. As these two approaches are currently being far more utilized by the plant community, we shall detail them below.
5. Alternative functional syntheses of evolutionary and molecular biology
As we said earlier, ancestral sequence reconstruction has been seldom attempted in plants, but there are essentially two approaches that represent alternative functional syntheses to that provided by ASR: GWAS and fitness landscape studies. Both are arguably more commonly applied than ancestral protein resurrection, but their pre-eminence and utility is particularly notable in plants.
The basic principle of a GWAS, which was initially developed for use in medical genetics, is that the presence of nucleotide polymorphisms can be associated with the presence of variance in a given trait [182]. Examples of its use in Arabidopsis include studies into the defense metabolite glucosinolate [183], [184], enzyme activities [185] and metabolite levels [186], [187] of primary and secondary metabolism. A detailed evaluation of floral secondary metabolism in a subset of these ecotypes via LC-MS revealed that approximately half of them contained a set of 18 previously unidentified metabolites [188]. Detailed analysis revealed that these compounds are phenylacylated flavonols, and the evaluation of reciprocal Col-0 and C24 introgression lines revealed, respectively, gain- and loss-of-function mutations. Thus, these studies allowed cloning of the responsible gene and subsequent analysis of its evolutionary origin, as well as a functional evaluation of the role of these metabolites in conferring UV-B tolerance [188]. Maintaining this theme, the Arabidopsis accession Pna-10 is a naturally occurring deletion mutant of the enzymes sinapoylglucose:malate sinapoyltransferase and sinapoylglucose:anthocyanin sinapoyltransferase that are responsible for the biosynthesis of sinapoyl malate, which also confers UV-B tolerance [189]. Similarly, a novel amino acid racemase was discovered through the exploration of 440 natural accessions of Arabidopsis and N-malonyl-D-allo-isoleucine was identified, opening up the opportunity to explore the largely untapped metabolism of D -amino acids [190]. Whilst these examples provide interesting insights into how protein function can be assigned from GWAS they represent only a minor subset of what has been achieved to date. Fortunately, much of this has been databased in the AraPheno and AraGWAS Catalog [191], which also includes RNA sequencing and knockout mutation data. Given that spurious associations arising from historical relationships and selection patterns can occur [192], molecular validation of GWAS experiments is needed to provide proof that the associations between genotype and phenotype are indeed reflective of a causal relationship. Unsurprisingly, similar experiments have been employed in crop species with such experiments being instrumental in defining genes, proteins and metabolic pathways underlying glycoalkaloid, flavonoid, terpene and acyl sugar biosynthesis in tomato [193], [194], [195], [196], [197], [198], whilst flavonoid and terpenoid metabolism as well as vitamins have been well studied in the cereal crops maize, rice and wheat [199], [200], [201], [202], [203] and indeed many other species including melon, watermelon (family Cucurbitaceae), sunflower (family Asteraceae) and Rosaceae (for recent reviews see [44], [45], [204]), with many of the genetic polymorphism-trait associations being either cross-validated in alternative populations or confirmed via directed transgenic or gene editing approaches. As for the examples described above for Arabidopsis, many of these examples also include the identification of species-specific genes, enzymes or metabolites and thereby represent a rich source of information regarding the evolution of protein function. Indeed, two recent studies in wheat [205] and tomato [206] explicitly studied the change in the metabolome on the domestication of these species with the latter study taking a multi-omics approach integrating genomics, transcriptomics and metabolomics. Whilst domestication is largely characterized by altered gene expression [207], notable changes in protein coding sequences have additionally been identified, including the loss of efficiency of the cell wall invertase in tomato [208], the origin of six-rowed barley [209], the reduction of grain shattering during rice domestication [210] and the free-threshing trait in wheat [211]. As alluded to above, all these cases are essentially examples of Thornton‘s functional synthesis even if they only cover a mere percentage of the evolutionary timescale achieved by studies of ancestral sequence reconstruction.
The second approach, which is increasingly being applied in plants, is that of developing fitness consequence maps. These maps, from an evolutionary perspective, are able to predict which mutations are beneficial in terms of improving fitness-related traits, whilst from a molecular biological one provide information regarding which area of a genome impacts on cellular function [212]. Following such strategies, fitness effect predictions can be made based on theories of population genetics, quantifying either the proportion of sites under selection or the strength of selection acting on collections of sites in the genome. For this purpose, four approaches are generally taken. First, constraint-based models that use phylogenetic and homology-based inference can be employed to identify sites with low rates of substitution across a phylogeny [213], [214]. However, in some cases this approach might not be applicable. For example, in plant genomes, transcription factor binding sites experience a more rapid evolutionary turnover [215], limiting the use of the constraint-based approaches. The second class of approaches is that of site frequency spectra (SFS), which use histograms of allele frequencies to estimate the magnitude of fitness effects in pre-defined sites in comparison to a neutral class. In doing so, this approach distinguishes sites subject to selection, allowing estimation of fitness effects to be made [216]. Frequency spectra are, by nature, limited by the prior site definition, but have nevertheless proven effective in defining the accumulation and distributions of deleterious mutations in maize [217] as well as in sunflower and other crops from the Asteraceae family [218]. Thirdly, comparative population genomic methods use intra-specific diversity and between-species divergence rates to estimate the proportion of sites subject to selection [219] or the magnitude and orientation of the fitness effect [220]. Finally, effect class methods are used to predict the impact of a mutation on fitness/function by considering properties unique to each effect class, thereby returning a score indicating the likelihood that a given mutation will impact function [221]. That said, most powerful are methods that combine population genomics and divergence data, such as the fitCons method which is based on Natural Selection from Interspersed Genomically Coherent Elements (INSIGHT; [222]). In the rest of this section, we will discuss some examples from five seminal papers utilizing fitness consequence models - two in yeast, one in the model plant Arabidopsis thaliana and two in the important crop plant rice. In the first of these Hietpas et al. [161] exploited deep sequencing to experimentally determine the fitness of all possible individual point mutations for a nine amino acid region of the chaperone Hsp90. The results from this experimental analysis were consistent with the neutral theory, that is, with the majority of amino acid substitutions found to be deleterious and with the remaining mutations being essentially neutral or nearly neutral and governed by genetic drift. More recently, this work was expanded to encompass a large and complete multiallelic intragenic fitness landscape of 640 systematically engineered mutations in the protein. Intriguingly, they report local ruggedness in the fitness-landscape topography as well as the existence of epistatic hotspot mutations which combine to render predictability inherently difficult in the absence of mutation-specific information [223]. The study in Arabidopsis is even more epic. In their work Exposito-Alonso et al. grew 517 accessions in Spain and Germany and exposed them both to high and low precipitation [224]. In this experiment, hot-dry conditions resulted in the death of 63% of the lines; a significant proportion of this climate-driven natural selection was predictable from signatures of local adaptation. This study thus both provided a powerful functional evolutionary study but also, as the authors themselves state, the predictions generated there could represent a first step in the design of conservation strategies to catalyse evolutionary rescue of species. Finally, two papers on rice from the group of Michael Purugganan merit discussion. The first of these studies [46] used the INSIGHT approach to infer fitness-consequence scores from nine functional genomics and epigenomic datasets. These scores were then integrated with genome-wide polymorphism and divergence data from 1477 rice accessions and 11 reference genomes. This massive study concluded that approximately 9% of the genome would have fitness consequences if mutated in more than half of the bases. It also demonstrated that 2% of the genome showed evidence of weak negative selection, predominantly located in candidate regulator loci such as enhancer elements. In their second investigation [225], gene expression in rice was analyzed in order to estimate the type and strength of selection on the levels of 15 000 transcripts. The results indicated that variation in expression appears neutral or under very weak stabilizing selection in wet paddy conditions but that selection was much stronger under conditions of drought. It furthermore showed that drought selected for early flowering and a higher expression of a MADS transcription factor known to regulate this trait. These five examples are all linked by the fact that they associate changes in fitness directly to their molecular bases, and in contrast to ancestral protein resurrection, they often do so without experimentally accessing protein function directly. Of all the alternative functional syntheses we presented here, the fitness landscape maps represent perhaps powerful companion approaches to ASR, in that they allow to further increase the resolution of “vertical” comparisons (e.g., ancestral Vs extant sequences) with the dimension of comparative population genomics of large collection of sequences from extant species. These methods all rely essentially on the comparison between intra- and inter-specific divergence data (of extant species) to compute the proportion of genomic sites subject to selection (either in the coding part of the genome, as when applied on large-scale gene expression data [225], or in the non-coding regions [226]). In doing so, fitness landscape maps could be used to further validate the sites under selection obtained from the classical branch-site tests in ASR ([34], [35], [49], [227], [228]), which calculate the codons under positive selection along the lineages of the phylogenetic tree, and verify the persistence of these selective constraints, using comparative genomic methods [212], in the recent diversification characterizing the populations of extant species. This combined approach would significantly reinforce the conclusions of an ASR study extending the validity of its functional inferences to the natural diversity present in modern species.
6. Summary and outlook
Since its initial inception, the strategy of resurrecting genes has allowed exciting discoveries into the mechanisms of molecular evolution. The approach of ASR adds in fact a further dimension to the comparison of extant sequences, as it allows to trace the historical genesis - and impact - of amino acid substitutions on protein function. In doing so, ASR, especially when combined with deep mutational scanning [229], has the power to explore prevalence and implications of intragenic epistasis during the course of evolutionary history, an outcome that is not normally accessible from the horizontal analysis of extant sequences. The alternative functional approaches highlighted here (GWAS and fitness landscape studies) represent complementary approaches to investigate evolutionary phenomena at different scales. In the plant field, genome-wide association studies, especially when applied on large genotype collections including wild relatives and domesticated forms, may allow to identify genomic regions targeted by domestication processes or subjected to local adaptation. In doing so, GWAS may provide, with respect to ASR, correlative (and, if the results are then validated) also functional insights on very recent evolutionary processes (plant domestication is considered to have occurred in the last 10–12000 years, [230]). Fitness landscape studies, and in particular those based on comparative population genomics, have been crucial in detecting, for example, regions targeted by positive selection.
We think an interesting next step in the development of ancestral reconstruction approaches could be the analysis of non-coding sequences. Protein (or, more generally, coding sequences) have been the primary focus of ASR so far given the massive development of probabilistic models to describe their sequence evolution. We now know, however, that transcriptional regulation and a significant degree of selection constraints lie in the non-coding portion of a genome [46], [226], and there is now an increasing interest to explore the molecular evolution trajectories taken by, for example, promoters and transcription factor binding sites. Indeed, one of the first studies of ancestral reconstruction was targeted at a LINE-1 sequence, a transcriptional inactive retrotransposon dispersed in the mouse genome which is not capable to transpose anymore. Once the sequence of LINE-1 was reconstructed, by parsimony, and transfected into mouse cells, it showed instead promoter activity [231]. With the availability of alignment tools and sequence evolution models specifically designed for non-coding sequences [232], [233], [234], the scenario of reconstructing ancestral promoters, transposons, and cis-regulatory sequences opens up exciting possibilities to explore, in combination with protein resurrection, the evolution of molecular trajectories adopted by gene regulatory networks.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
A.R.F. acknowledges the financial support of the EU Horizon 2020 Research and Innovation Programme, project PlantaSYST (SGA-CSA No. 739582 under FPA No. 664620). A.R.F. is supported by the Max-Planck Society. The authors sincerely thank the anonymous reviewers for critically reading the manuscript and suggesting substantial improvements on the earlier draft.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.03.008.
Contributor Information
Federico Scossa, Email: scossa@mpimp-golm.mpg.de.
Alisdair R. Fernie, Email: fernie@mpimp-golm.mpg.de.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Dean A.M., Thornton J.W. Mechanistic approaches to the study of evolution: the functional synthesis. Nat Rev Genet. 2007;8:675–688. doi: 10.1038/nrg2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tautz D., Ellegren H., Weigel D. Next generation molecular ecology. Mol Ecol. 2010;19(Suppl 1):1–3. doi: 10.1111/j.1365-294X.2009.04489.x. [DOI] [PubMed] [Google Scholar]
- 3.Exposito-Alonso M., Drost H.G., Burbano H.A., Weigel D. The Earth BioGenome project: opportunities and challenges for plant genomics and conservation. Plant J. 2020;102:222–229. doi: 10.1111/tpj.14631. [DOI] [PubMed] [Google Scholar]
- 4.Harms M.J., Thornton J.W. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet. 2013;14:559–571. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thornton J.W. Resurrecting ancient genes: experimental analysis of extinct molecules. Nat Rev Genet. 2004;5:366–375. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- 6.Morrison K.L., Weiss G.A. Combinatorial alanine-scanning. Curr Opin Chem Biol. 2001;5:302–307. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 7.Liljas A., Laurberg M. A wheel invented three times. The molecular structures of the three carbonic anhydrases. EMBO Rep. 2000;1:16–17. doi: 10.1093/embo-reports/kvd016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ekici O.D., Paetzel M., Dalbey R.E. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elleuche S., Fodor K., von der Heyde A., Klippel B., Wilmanns M. Group III alcohol dehydrogenase from Pectobacterium atrosepticum: insights into enzymatic activity and organization of the metal ion-containing region. Appl Microbiol Biotechnol. 2014;98:4041–4051. doi: 10.1007/s00253-013-5374-z. [DOI] [PubMed] [Google Scholar]
- 10.Hochberg G.K.A., Thornton J.W. Reconstructing ancient proteins to understand the causes of structure and function. Annu Rev Biophys. 2017;46:247–269. doi: 10.1146/annurev-biophys-070816-033631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bloom J.D., Romero P.A., Lu Z., Arnold F.H. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biology Direct. 2007;2:17. doi: 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodsell D.S., Olson A.J. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 13.Hart K.M., Harms M.J., Schmidt B.H., Elya C., Thornton J.W. Thermodynamic system drift in protein evolution. PLoS Biol. 2014;12 doi: 10.1371/journal.pbio.1001994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacob F. Evolution and tinkering. Science. 1977;196:1161–1166. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- 15.Ness R.O., Sachs K., Vitek O. From Correlation to Causality: Statistical Approaches to Learning Regulatory Relationships in Large-Scale Biomolecular Investigations. J Proteome Res. 2016;15:683–690. doi: 10.1021/acs.jproteome.5b00911. [DOI] [PubMed] [Google Scholar]
- 16.Elena S.F., Lenski R.E. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- 17.Rainey P.B., Rainey K. Evolution of cooperation and conflict in experimental bacterial populations. Nature. 2003;425:72–74. doi: 10.1038/nature01906. [DOI] [PubMed] [Google Scholar]
- 18.Denver D.R., Morris K., Streelman J.T., Kim S.K., Lynch M. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat Genet. 2005;37:544–548. doi: 10.1038/ng1554. [DOI] [PubMed] [Google Scholar]
- 19.Harrison G.F., Sanz J., Boulais J., Mina M.J., Grenier J.C. Natural selection contributed to immunological differences between hunter-gatherers and agriculturalists. Nat Ecol Evol. 2019;3:1253–1264. doi: 10.1038/s41559-019-0947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L., Ren Y., Yang T., Li G., Chen J. Rapid evolution of protein diversity by de novo origination in Oryza. Nat Ecol Evol. 2019;3:679–690. doi: 10.1038/s41559-019-0822-5. [DOI] [PubMed] [Google Scholar]
- 21.Scossa F., Fernie A.R. The evolution of metabolism: How to test evolutionary hypotheses at the genomic level. Comput Struct Biotechnol J. 2020;18:482–500. doi: 10.1016/j.csbj.2020.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scossa F., Roda F., Tohge T., Georgiev M.I., Fernie A.R. The Hot and the Colorful: Understanding the Metabolism, Genetics and Evolution of Consumer Preferred Metabolic Traits in Pepper and Related Species. Crit Rev Plant Sci. 2019;38:339–381. [Google Scholar]
- 23.Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–298. [Google Scholar]
- 24.Fox D.T., Soltis D.E., Soltis P.S., Ashman T.L., Van de Peer Y. Polyploidy: a biological force from cells to ecosystems. Trends Cell Biol. 2020;30:688–694. doi: 10.1016/j.tcb.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merkl R., Sterner R. Ancestral protein reconstruction: techniques and applications. Biol Chem. 2016;397:1–21. doi: 10.1515/hsz-2015-0158. [DOI] [PubMed] [Google Scholar]
- 26.Gumulya Y., Gillam E.M. Exploring the past and the future of protein evolution with ancestral sequence reconstruction: the 'retro' approach to protein engineering. Biochem J. 2017;474:1–19. doi: 10.1042/BCJ20160507. [DOI] [PubMed] [Google Scholar]
- 27.Joy J.B., Liang R.H., McCloskey R.M., Nguyen T., Poon A.F. Ancestral Reconstruction. PLoS Comput Biol. 2016;12 doi: 10.1371/journal.pcbi.1004763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selberg A.G.A., Gaucher E.A., Liberles D.A. Ancestral sequence reconstruction: from chemical paleogenetics to maximum likelihood algorithms and beyond. J Mol Evol. 2021 doi: 10.1007/s00239-021-09993-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stackhouse J., Presnell S.R., McGeehan G.M., Nambiar K.P., Benner S.A. The ribonuclease from an extinct bovid ruminant. FEBS Lett. 1990;262:104–106. doi: 10.1016/0014-5793(90)80164-e. [DOI] [PubMed] [Google Scholar]
- 30.Jermann T.M., Opitz J.G., Stackhouse J., Benner S.A. Reconstructing the evolutionary history of the artiodactyl ribonuclease superfamily. Nature. 1995;374:57–59. doi: 10.1038/374057a0. [DOI] [PubMed] [Google Scholar]
- 31.Fang C., Fernie A.R., Luo J. Exploring the diversity of plant metabolism. Trends Plant Sci. 2019;24:83–98. doi: 10.1016/j.tplants.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Naake T., Maeda H.A., Proost S., Tohge T., Fernie A.R. Kingdom-wide analysis of the evolution of the plant type III polyketide synthase superfamily. Plant Physiol. 2020 doi: 10.1093/plphys/kiaa086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiao X., Li Q., Yin H., Qi K., Li L. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019;20:38. doi: 10.1186/s13059-019-1650-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang R., O'Donnell A.J., Barboline J.J., Barkman T.J. Convergent evolution of caffeine in plants by co-option of exapted ancestral enzymes. Proc Natl Acad Sci U S A. 2016;113:10613–10618. doi: 10.1073/pnas.1602575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang R., Hippauf F., Rohrbeck D., Haustein M., Wenke K. Enzyme functional evolution through improved catalysis of ancestrally nonpreferred substrates. Proc Natl Acad Sci U S A. 2012;109:2966–2971. doi: 10.1073/pnas.1019605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerlt J.A., Babbitt P.C. Enzyme (re)design: lessons from natural evolution and computation. Curr Opin Chem Biol. 2009;13:10–18. doi: 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siddiq M.A., Hochberg G.K., Thornton J.W. Evolution of protein specificity: insights from ancestral protein reconstruction. Curr Opin Struct Biol. 2017;47:113–122. doi: 10.1016/j.sbi.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanzawa Y., Money T., Bradley D. A single amino acid converts a repressor to an activator of flowering. Proc Natl Acad Sci U S A. 2005;102:7748–7753. doi: 10.1073/pnas.0500932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wulff B.B., Thomas C.M., Smoker M., Grant M., Jones J.D. Domain swapping and gene shuffling identify sequences required for induction of an Avr-dependent hypersensitive response by the tomato Cf-4 and Cf-9 proteins. Plant Cell. 2001;13:255–272. doi: 10.1105/tpc.13.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Starr T.N., Thornton J.W. Epistasis in protein evolution. Protein Sci. 2016;25:1204–1218. doi: 10.1002/pro.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starr T.N., Thornton J.W. Exploring protein sequence-function landscapes. Nat Biotechnol. 2017;35:125–126. doi: 10.1038/nbt.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hopf T.A., Ingraham J.B., Poelwijk F.J., Scharfe C.P., Springer M. Mutation effects predicted from sequence co-variation. Nat Biotechnol. 2017;35:128–135. doi: 10.1038/nbt.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohno S. Springer; Berlin, Heidelberg: 1970. Evolution by Gene Duplication. [Google Scholar]
- 44.Fang C., Luo J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 2019;97:91–100. doi: 10.1111/tpj.14097. [DOI] [PubMed] [Google Scholar]
- 45.Liu H.J., Yan J. Crop genome-wide association study: a harvest of biological relevance. Plant J. 2019;97:8–18. doi: 10.1111/tpj.14139. [DOI] [PubMed] [Google Scholar]
- 46.Joly-Lopez Z., Platts A.E., Gulko B., Choi J.Y., Groen S.C. An inferred fitness consequence map of the rice genome. Nat Plants. 2020;6:119–130. doi: 10.1038/s41477-019-0589-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pauling L., Zuckerkandl E. Chemical paleogenetics molecular restoration studies of extinct forms of life. Acta Chem Scand. 1963;17:9-+. [Google Scholar]
- 48.Kapli P., Yang Z., Telford M.J. Phylogenetic tree building in the genomic age. Nat Rev Genet. 2020;21:428–444. doi: 10.1038/s41576-020-0233-0. [DOI] [PubMed] [Google Scholar]
- 49.Lichman B.R., Godden G.T., Hamilton J.P., Palmer L., Kamileen M.O. The evolutionary origins of the cat attractant nepetalactone in catnip. Sci Adv. 2020;6:eaba0721. doi: 10.1126/sciadv.aba0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller J.B., Pickett B.D., Ridge P.G. JustOrthologs: a fast, accurate and user-friendly ortholog identification algorithm. Bioinformatics. 2019;35:546–552. doi: 10.1093/bioinformatics/bty669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emms D.M., Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emms D.M., Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petersen M., Meusemann K., Donath A., Dowling D., Liu S. Orthograph: a versatile tool for mapping coding nucleotide sequences to clusters of orthologous genes. BMC Bioinf. 2017;18:111. doi: 10.1186/s12859-017-1529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L., Stoeckert C.J., Jr., Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huerta-Cepas J., Szklarczyk D., Heller D., Hernandez-Plaza A., Forslund S.K. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Louis A., Muffato M., Roest Crollius H. Genomicus: five genome browsers for comparative genomics in eukaryota. Nucleic Acids Res. 2013;41:D700–705. doi: 10.1093/nar/gks1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nguyen N.T.T., Vincens P., Roest Crollius H., Louis A. Genomicus 2018: karyotype evolutionary trees and on-the-fly synteny computing. Nucleic Acids Res. 2018;46:D816–D822. doi: 10.1093/nar/gkx1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kriventseva E.V., Kuznetsov D., Tegenfeldt F., Manni M., Dias R. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019;47:D807–D811. doi: 10.1093/nar/gky1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zdobnov E.M., Kuznetsov D., Tegenfeldt F., Manni M., Berkeley M. OrthoDB in 2020: evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2020 doi: 10.1093/nar/gkaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huerta-Cepas J., Capella-Gutierrez S., Pryszcz L.P., Marcet-Houben M., Gabaldon T. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014;42:D897–D902. doi: 10.1093/nar/gkt1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Bel M., Diels T., Vancaester E., Kreft L., Botzki A. PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018;46:D1190–D1196. doi: 10.1093/nar/gkx1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suchard M.A., Redelings B.D. BAli-Phy: simultaneous Bayesian inference of alignment and phylogeny. Bioinformatics. 2006;22:2047–2048. doi: 10.1093/bioinformatics/btl175. [DOI] [PubMed] [Google Scholar]
- 63.Blackshields G, Sievers F, Shi WF, Wilm A, Higgins DG. Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms for Molecular Biology 5; 2010. [DOI] [PMC free article] [PubMed]
- 64.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7 doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Armougom F., Moretti S., Poirot O., Audic S., Dumas P. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006;34:W604–608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holmes I.H. Historian: accurate reconstruction of ancestral sequences and evolutionary rates. Bioinformatics. 2017;33:1227–1229. doi: 10.1093/bioinformatics/btw791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katoh K., Standley D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loytynoja A. Phylogeny-aware alignment with PRANK. Methods Mol Biol. 2014;1079:155–170. doi: 10.1007/978-1-62703-646-7_10. [DOI] [PubMed] [Google Scholar]
- 70.Do C.B., Mahabhashyam M.S.P., Brudno M., Batzoglou S. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu K., Raghavan S., Nelesen S., Linder C.R., Warnow T. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science. 2009;324:1561–1564. doi: 10.1126/science.1171243. [DOI] [PubMed] [Google Scholar]
- 72.Liu K., Warnow T.J., Holder M.T., Nelesen S.M., Yu J.Y. SATe-II: Very Fast and Accurate Simultaneous Estimation of Multiple Sequence Alignments and Phylogenetic Trees. Syst Biol. 2012;61:90–106. doi: 10.1093/sysbio/syr095. [DOI] [PubMed] [Google Scholar]
- 73.Notredame C., Higgins D.G., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 74.Criscuolo A., Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ali R.H., Bogusz M., Whelan S. Identifying clusters of high confidence homologies in multiple sequence alignments. Mol Biol Evol. 2019;36:2340–2351. doi: 10.1093/molbev/msz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 77.Dress A.W., Flamm C., Fritzsch G., Grunewald S., Kruspe M. Noisy: identification of problematic columns in multiple sequence alignments. Algorithms Mol Biol. 2008;3:7. doi: 10.1186/1748-7188-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Whelan S., Irisarri I., Burki F. PREQUAL: detecting non-homologous characters in sets of unaligned homologous sequences. Bioinformatics. 2018;34:3929–3930. doi: 10.1093/bioinformatics/bty448. [DOI] [PubMed] [Google Scholar]
- 79.Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bouckaert R., Vaughan T.G., Barido-Sottani J., Duchene S., Fourment M. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2019;15 doi: 10.1371/journal.pcbi.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lefort V., Desper R., Gascuel O. FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol. 2015;32:2798–2800. doi: 10.1093/molbev/msv150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Price M.N., Dehal P.S., Arkin A.P. FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE. 2010;5 doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nguyen L.T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoang D.T., Vinh L.S., Flouri T., Stamatakis A., von Haeseler A. MPBoot: fast phylogenetic maximum parsimony tree inference and bootstrap approximation. BMC Evol Biol. 2018;18:11. doi: 10.1186/s12862-018-1131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huelsenbeck J.P., Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 87.Koshi J.M., Goldstein R.A. Probabilistic reconstruction of ancestral protein sequences. J Mol Evol. 1996;42:313–320. doi: 10.1007/BF02198858. [DOI] [PubMed] [Google Scholar]
- 88.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 89.Lartillot N (2020) PhyloBayes: Bayesian Phylogenetics Using Site-heterogeneous Models. In: Scornavacca C, Delsuc F, Galtier N, editors. Phylogenetics in the Genomic Era: No commercial publisher | Authors open access book. pp. 1.5:1--1.5:16.
- 90.Lartillot N., Lepage T., Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- 91.Lartillot N., Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- 92.Oliva A., Pulicani S., Lefort V., Brehelin L., Gascuel O. Accounting for ambiguity in ancestral sequence reconstruction. Bioinformatics. 2019;35:4290–4297. doi: 10.1093/bioinformatics/btz249. [DOI] [PubMed] [Google Scholar]
- 93.Guindon S., Delsuc F., Dufayard J.F., Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 94.Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 95.de Vienne D.M., Ollier S., Aguileta G. Phylo-MCOA: a fast and efficient method to detect outlier genes and species in phylogenomics using multiple co-inertia analysis. Mol Biol Evol. 2012;29:1587–1598. doi: 10.1093/molbev/msr317. [DOI] [PubMed] [Google Scholar]
- 96.Mai U., Mirarab S. TreeShrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genomics. 2018;19:272. doi: 10.1186/s12864-018-4620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Struck T.H. TreSpEx-detection of misleading signal in phylogenetic reconstructions based on tree information. Evol Bioinform Online. 2014;10:51–67. doi: 10.4137/EBO.S14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bruno W.J., Socci N.D., Halpern A.L. Weighted neighbor joining: a likelihood-based approach to distance-based phylogeny reconstruction. Mol Biol Evol. 2000;17:189–197. doi: 10.1093/oxfordjournals.molbev.a026231. [DOI] [PubMed] [Google Scholar]
- 99.Cai W., Pei J., Grishin N.V. Reconstruction of ancestral protein sequences and its applications. BMC Evol Biol. 2004;4:33. doi: 10.1186/1471-2148-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pupko T., Pe'er I., Shamir R., Graur D. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol Biol Evol. 2000;17:890–896. doi: 10.1093/oxfordjournals.molbev.a026369. [DOI] [PubMed] [Google Scholar]
- 101.Ashkenazy H., Penn O., Doron-Faigenboim A., Cohen O., Cannarozzi G. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 2012;40:W580–584. doi: 10.1093/nar/gks498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moshe A., Pupko T. Ancestral sequence reconstruction: accounting for structural information by averaging over replacement matrices. Bioinformatics. 2019;35:2562–2568. doi: 10.1093/bioinformatics/bty1031. [DOI] [PubMed] [Google Scholar]
- 103.Hanson-Smith V., Johnson A. PhyloBot: a web portal for automated phylogenetics, ancestral sequence reconstruction, and exploration of mutational trajectories. PLoS Comput Biol. 2016;12 doi: 10.1371/journal.pcbi.1004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arenas M., Weber C.C., Liberles D.A., Bastolla U. ProtASR: an evolutionary framework for ancestral protein reconstruction with selection on folding stability. Syst Biol. 2017;66:1054–1064. doi: 10.1093/sysbio/syw121. [DOI] [PubMed] [Google Scholar]
- 105.Arenas M., Bastolla U. ProtASR2: ancestral reconstruction of protein sequences accounting for folding stability. Methods Ecol Evol. 2020;11:248–257. [Google Scholar]
- 106.Carletti MS, Monzon AM, Garcia-Rios E, Benitez G, Hirsh L, et al. (2020) Revenant: a database of resurrected proteins. Database (Oxford) 2020. [DOI] [PMC free article] [PubMed]
- 107.Nakane T., Kotecha A., Sente A., McMullan G., Masiulis S. Single-particle cryo-EM at atomic resolution. Nature. 2020;587:152–156. doi: 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vialle R.A., Tamuri A.U., Goldman N. Alignment modulates ancestral sequence reconstruction accuracy. Mol Biol Evol. 2018;35:1783–1797. doi: 10.1093/molbev/msy055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Groussin M., Hobbs J.K., Szollosi G.J., Gribaldo S., Arcus V.L. Toward more accurate ancestral protein genotype-phenotype reconstructions with the use of species tree-aware gene trees. Mol Biol Evol. 2015;32:13–22. doi: 10.1093/molbev/msu305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Szollosi G.J., Tannier E., Daubin V., Boussau B. The inference of gene trees with species trees. Syst Biol. 2015;64:e42–62. doi: 10.1093/sysbio/syu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nichols R. Gene trees and species trees are not the same. Trends Ecol Evol. 2001;16:358–364. doi: 10.1016/s0169-5347(01)02203-0. [DOI] [PubMed] [Google Scholar]
- 112.Hadzipasic A., Wilson C., Nguyen V., Kern N., Kim C. Ancient origins of allosteric activation in a Ser-Thr kinase. Science. 2020;367:912–917. doi: 10.1126/science.aay9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Park Y., Patton J.E.J., Hochberg G.K.A., Thornton J.W. Comment on “Ancient origins of allosteric activation in a Ser-Thr kinase”. Science. 2020;370 doi: 10.1126/science.abc8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wilson C., Kern D. Response to Comment on “Ancient origins of allosteric activation in a Ser-Thr kinase”. Science. 2020;370 doi: 10.1126/science.abd0364. [DOI] [PubMed] [Google Scholar]
- 115.Scornavacca C, Delsuc F, Galtier N (2020) Phylogenetics in the Genomic Era: No commercial publisher | Authors open access book. p.p. 1-568 p.
- 116.Nascimento F.F., Reis M.D., Yang Z. A biologist's guide to Bayesian phylogenetic analysis. Nat Ecol Evol. 2017;1:1446–1454. doi: 10.1038/s41559-017-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fitch W.M. Toward defining the course of evolution: minimum change for a specific tree topology. Syst Zool. 1971;20:406–416. [Google Scholar]
- 118.Pupko T, Mayrose I (2020) A gentle Introduction to Probabilistic Evolutionary Models. In: Scornavacca C, Delsuc F, Galtier N, editors. Phylogenetics in the Genomic Era: No commercial publisher | Authors open access book. pp. 1.1:1--1.1:21.
- 119.Jukes TH, Cantor CR (1969) CHAPTER 24 - Evolution of Protein Molecules. In: Munro HN, editor. Mammalian Protein Metabolism: Academic Press. pp. 21-132.
- 120.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 121.Huelsenbeck J.P., Ronquist F., Nielsen R., Bollback J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- 122.Lartillot N (2020) The Bayesian Approach to Molecular Phylogeny. In: Scornavacca C, Delsuc F, Galtier N, editors. Phylogenetics in the Genomic Era: No commercial publisher | Authors open access book. pp. 1.4:1--1.4:17.
- 123.Zhang J., Nei M. Accuracies of ancestral amino acid sequences inferred by the parsimony, likelihood, and distance methods. J Mol Evol. 1997;44(Suppl 1):S139–146. doi: 10.1007/pl00000067. [DOI] [PubMed] [Google Scholar]
- 124.Yang Z., Kumar S., Nei M. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics. 1995;141:1641–1650. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Starr T.N., Picton L.K., Thornton J.W. Alternative evolutionary histories in the sequence space of an ancient protein. Nature. 2017;549:409–413. doi: 10.1038/nature23902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pupko T, Doron-Faigenboim A, Liberles DA, Cannarozzi GM (2007) Probabilistic models and their impact on the accuracy of reconstructed ancestral protein sequences. In: Liberles DA, editor. Ancestral Sequence Reconstruction: Oxford University Press.
- 127.Herbst L., Li H., Steel M. Quantifying the accuracy of ancestral state prediction in a phylogenetic tree under maximum parsimony. J Math Biol. 2019;78:1953–1979. doi: 10.1007/s00285-019-01330-x. [DOI] [PubMed] [Google Scholar]
- 128.Swofford D.L., Maddison W.P. Reconstructing ancestral character states under Wagner parsimony. Math Biosci. 1987;87:199–229. [Google Scholar]
- 129.Agnarsson I., Miller J.A. Is ACCTRAN better than DELTRAN? Cladistics. 2008;24:1032–1038. doi: 10.1111/j.1096-0031.2008.00229.x. [DOI] [PubMed] [Google Scholar]
- 130.Aadland K., Kolaczkowski B. Alignment-integrated reconstruction of ancestral sequences improves accuracy. Genome Biol Evol. 2020;12:1549–1565. doi: 10.1093/gbe/evaa164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maiolo M., Zhang X., Gil M., Anisimova M. Progressive multiple sequence alignment with indel evolution. BMC Bioinf. 2018;19:331. doi: 10.1186/s12859-018-2357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huelsenbeck J.P., Bollback J.P. Empirical and hierarchical Bayesian estimation of ancestral states. Syst Biol. 2001;50:351–366. [PubMed] [Google Scholar]
- 133.Pagel M., Meade A., Barker D. Bayesian estimation of ancestral character states on phylogenies. Syst Biol. 2004;53:673–684. doi: 10.1080/10635150490522232. [DOI] [PubMed] [Google Scholar]
- 134.Hall B.G. Simple and accurate estimation of ancestral protein sequences. Proc Natl Acad Sci U S A. 2006;103:5431–5436. doi: 10.1073/pnas.0508991103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hanson-Smith V., Kolaczkowski B., Thornton J.W. Robustness of ancestral sequence reconstruction to phylogenetic uncertainty. Mol Biol Evol. 2010;27:1988–1999. doi: 10.1093/molbev/msq081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Williams P.D., Pollock D.D., Blackburne B.P., Goldstein R.A. Assessing the accuracy of ancestral protein reconstruction methods. PLoS Comput Biol. 2006;2 doi: 10.1371/journal.pcbi.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gaucher E.A., Thomson J.M., Burgan M.F., Benner S.A. Inferring the palaeoenvironment of ancient bacteria on the basis of resurrected proteins. Nature. 2003;425:285–288. doi: 10.1038/nature01977. [DOI] [PubMed] [Google Scholar]
- 138.Darriba D., Taboada G.L., Doallo R., Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bruno W.J., Halpern A.L. Topological bias and inconsistency of maximum likelihood using wrong models. Mol Biol Evol. 1999;16:564–566. doi: 10.1093/oxfordjournals.molbev.a026137. [DOI] [PubMed] [Google Scholar]
- 140.Abadi S., Azouri D., Pupko T., Mayrose I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat Commun. 2019;10:934. doi: 10.1038/s41467-019-08822-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Emms D.M., Kelly S. STRIDE: species tree root inference from gene duplication events. Mol Biol Evol. 2017;34:3267–3278. doi: 10.1093/molbev/msx259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Emms DM, Kelly S (2018) STAG: Species Tree Inference from All Genes. bioRxiv: 267914.
- 143.Hug L.A., Baker B.J., Anantharaman K., Brown C.T., Probst A.J. A new view of the tree of life. Nat Microbiol. 2016;1:16048. doi: 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- 144.Zhu Q., Mai U., Pfeiffer W., Janssen S., Asnicar F. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat Commun. 2019;10:5477. doi: 10.1038/s41467-019-13443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Burki F., Roger A.J., Brown M.W., Simpson A.G.B. The new tree of eukaryotes. Trends Ecol Evol. 2020;35:43–55. doi: 10.1016/j.tree.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 146.Wong G.K., Soltis D.E., Leebens-Mack J., Wickett N.J., Barker M.S. Sequencing and analyzing the transcriptomes of a thousand species across the tree of life for green plants. Annu Rev Plant Biol. 2020;71:741–765. doi: 10.1146/annurev-arplant-042916-041040. [DOI] [PubMed] [Google Scholar]
- 147.Laumer C.E., Fernandez R., Lemer S., Combosch D., Kocots K.M. Proceedings of the Royal Society B-Biological Sciences 286. 2019. Revisiting metazoan phylogeny with genomic sampling of all phyla. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chatzou M., Floden E.W., Di Tommaso P., Gascuel O., Notredame C. Generalized bootstrap supports for phylogenetic analyses of protein sequences incorporating alignment uncertainty. Syst Biol. 2018;67:997–1009. doi: 10.1093/sysbio/syx096. [DOI] [PubMed] [Google Scholar]
- 149.Anisimova M., Gil M., Dufayard J.F., Dessimoz C., Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol. 2011;60:685–699. doi: 10.1093/sysbio/syr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Soltis P.S., Soltis D.E. Applying the bootstrap in phylogeny reconstruction. Statistical Sci. 2003;18:256–267. [Google Scholar]
- 151.Thomson J.M., Gaucher E.A., Burgan M.F., De Kee D.W., Li T. Resurrecting ancestral alcohol dehydrogenases from yeast. Nat Genet. 2005;37:630–635. doi: 10.1038/ng1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Akanuma S., Yokobori S., Nakajima Y., Bessho M., Yamagishi A. Robustness of predictions of extremely thermally stable proteins in ancient organisms. Evolution. 2015;69:2954–2962. doi: 10.1111/evo.12779. [DOI] [PubMed] [Google Scholar]
- 153.Anderson D.P., Whitney D.S., Hanson-Smith V., Woznica A., Campodonico-Burnett W. Evolution of an ancient protein function involved in organized multicellularity in animals. Elife. 2016;5 doi: 10.7554/eLife.10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gaucher E.A., Govindarajan S., Ganesh O.K. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature. 2008;451:704–707. doi: 10.1038/nature06510. [DOI] [PubMed] [Google Scholar]
- 155.Eick G.N., Bridgham J.T., Anderson D.P., Harms M.J., Thornton J.W. Robustness of reconstructed ancestral protein functions to statistical uncertainty. Mol Biol Evol. 2017;34:247–261. doi: 10.1093/molbev/msw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Thornton J.W., Need E., Crews D. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science. 2003;301:1714–1717. doi: 10.1126/science.1086185. [DOI] [PubMed] [Google Scholar]
- 157.Bridgham J.T., Carroll S.M., Thornton J.W. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312:97–101. doi: 10.1126/science.1123348. [DOI] [PubMed] [Google Scholar]
- 158.Bridgham J.T., Ortlund E.A., Thornton J.W. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–519. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zakas P.M., Brown H.C., Knight K., Meeks S.L., Spencer H.T. Enhancing the pharmaceutical properties of protein drugs by ancestral sequence reconstruction. Nat Biotechnol. 2017;35:35–37. doi: 10.1038/nbt.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Risso V.A., Sanchez-Ruiz J.M., Ozkan S.B. Biotechnological and protein-engineering implications of ancestral protein resurrection. Curr Opin Struct Biol. 2018;51:106–115. doi: 10.1016/j.sbi.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 161.Hietpas R.T., Jensen J.D., Bolon D.N. Experimental illumination of a fitness landscape. Proc Natl Acad Sci U S A. 2011;108:7896–7901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sarkisyan K.S., Bolotin D.A., Meer M.V., Usmanova D.R., Mishin A.S. Local fitness landscape of the green fluorescent protein. Nature. 2016;533:397–401. doi: 10.1038/nature17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Anderson D.W., McKeown A.N., Thornton J.W. Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. Elife. 2015;4 doi: 10.7554/eLife.07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McKeown A.N., Bridgham J.T., Anderson D.W., Murphy M.N., Ortlund E.A. Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell. 2014;159:58–68. doi: 10.1016/j.cell.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Aharoni A., Gaidukov L., Khersonsky O., Mc Q.G.S., Roodveldt C. The 'evolvability' of promiscuous protein functions. Nat Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 166.Clifton B.E., Jackson C.J. Ancestral protein reconstruction yields insights into adaptive evolution of binding specificity in solute-binding proteins. Cell Chem Biol. 2016;23:236–245. doi: 10.1016/j.chembiol.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 167.Devamani T., Rauwerdink A.M., Lunzer M., Jones B.J., Mooney J.L. Catalytic promiscuity of ancestral esterases and hydroxynitrile lyases. J Am Chem Soc. 2016;138:1046–1056. doi: 10.1021/jacs.5b12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Risso V.A., Gavira J.A., Mejia-Carmona D.F., Gaucher E.A., Sanchez-Ruiz J.M. Hyperstability and substrate promiscuity in laboratory resurrections of Precambrian beta-lactamases. J Am Chem Soc. 2013;135:2899–2902. doi: 10.1021/ja311630a. [DOI] [PubMed] [Google Scholar]
- 169.Boucher J.I., Jacobowitz J.R., Beckett B.C., Classen S., Theobald D.L. An atomic-resolution view of neofunctionalization in the evolution of apicomplexan lactate dehydrogenases. Elife. 2014;3 doi: 10.7554/eLife.02304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.van Loo B., Bayer C.D., Fischer G., Jonas S., Valkov E. Balancing specificity and promiscuity in enzyme evolution: multidimensional activity transitions in the alkaline phosphatase superfamily. J Am Chem Soc. 2019;141:370–387. doi: 10.1021/jacs.8b10290. [DOI] [PubMed] [Google Scholar]
- 171.Nguyen T.D., Kwon M., Kim S.U., Fischer C., Ro D.K. Catalytic plasticity of germacrene a oxidase underlies sesquiterpene lactone diversification. Plant Physiol. 2019;181:945–960. doi: 10.1104/pp.19.00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Waki T., Mameda R., Nakano T., Yamada S., Terashita M. A conserved strategy of chalcone isomerase-like protein to rectify promiscuous chalcone synthase specificity. Nat Commun. 2020;11:870. doi: 10.1038/s41467-020-14558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Perez-Jimenez R., Ingles-Prieto A., Zhao Z.M., Sanchez-Romero I., Alegre-Cebollada J. Single-molecule paleoenzymology probes the chemistry of resurrected enzymes. Nat Struct Mol Biol. 2011;18:592–596. doi: 10.1038/nsmb.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chang B.S., Jonsson K., Kazmi M.A., Donoghue M.J., Sakmar T.P. Recreating a functional ancestral archosaur visual pigment. Mol Biol Evol. 2002;19:1483–1489. doi: 10.1093/oxfordjournals.molbev.a004211. [DOI] [PubMed] [Google Scholar]
- 175.Chantreau M., Poux C., Lensink M.F., Brysbaert G., Vekemans X. Asymmetrical diversification of the receptor-ligand interaction controlling self-incompatibility in Arabidopsis. Elife. 2019;8 doi: 10.7554/eLife.50253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang Z., Coenen H., Ruelens P., Hazarika R.R., Al Hindi T. Resurrected protein interaction networks reveal the innovation potential of ancient whole-genome duplication. Plant Cell. 2018;30:2741–2760. doi: 10.1105/tpc.18.00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shih P.M., Occhialini A., Cameron J.C., Andralojc P.J., Parry M.A. Biochemical characterization of predicted Precambrian RuBisCO. Nat Commun. 2016;7:10382. doi: 10.1038/ncomms10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Scossa F., Benina M., Alseekh S., Zhang Y., Fernie A.R. The integration of metabolomics and next-generation sequencing data to elucidate the pathways of natural product metabolism in medicinal plants. Planta Med. 2018;84:855–873. doi: 10.1055/a-0630-1899. [DOI] [PubMed] [Google Scholar]
- 179.Kaltenbach M., Burke J.R., Dindo M., Pabis A., Munsberg F.S. Evolution of chalcone isomerase from a noncatalytic ancestor. Nat Chem Biol. 2018;14:548–555. doi: 10.1038/s41589-018-0042-3. [DOI] [PubMed] [Google Scholar]
- 180.Voordeckers K., Brown C.A., Vanneste K., van der Zande E., Voet A. Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLoS Biol. 2012;10 doi: 10.1371/journal.pbio.1001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gamiz-Arco G., Gutierrez-Rus L.I., Risso V.A., Ibarra-Molero B., Hoshino Y. Heme-binding enables allosteric modulation in an ancient TIM-barrel glycosidase. Nat Commun. 2021;12:380. doi: 10.1038/s41467-020-20630-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zeng P., Zhao Y., Qian C., Zhang L., Zhang R. Statistical analysis for genome-wide association study. J Biomed Res. 2015;29:285–297. doi: 10.7555/JBR.29.20140007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kliebenstein D.J., Kroymann J., Brown P., Figuth A., Pedersen D. Genetic control of natural variation in Arabidopsis glucosinolate accumulation. Plant Physiol. 2001;126:811–825. doi: 10.1104/pp.126.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chan E.K.F., Rowe H.C., Kliebenstein D.J. Understanding the evolution of defense metabolites in arabidopsis thaliana using genome-wide association mapping. Genetics. 2010;185:991. doi: 10.1534/genetics.109.108522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sulpice R., Pyl E.T., Ishihara H., Trenkamp S., Steinfath M. Starch as a major integrator in the regulation of plant growth. Proc Natl Acad Sci U S A. 2009;106:10348–10353. doi: 10.1073/pnas.0903478106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Wu S., Alseekh S., Cuadros-Inostroza Á., Fusari C.M., Mutwil M. Combined use of genome-wide association data and correlation networks unravels key regulators of primary metabolism in arabidopsis thaliana. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu S., Tohge T., Cuadros-Inostroza Á., Tong H., Tenenboim H. Mapping the arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol Plant. 2018;11:118–134. doi: 10.1016/j.molp.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 188.Tohge T., Wendenburg R., Ishihara H., Nakabayashi R., Watanabe M. Characterization of a recently evolved flavonol-phenylacyltransferase gene provides signatures of natural light selection in Brassicaceae. Nat Commun. 2016;7:12399. doi: 10.1038/ncomms12399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Li X., Bergelson J., Chapple C. The ARABIDOPSIS accession Pna-10 is a naturally occurring sng1 deletion mutant. Mol Plant. 2010;3:91–100. doi: 10.1093/mp/ssp090. [DOI] [PubMed] [Google Scholar]
- 190.Strauch R.C., Svedin E., Dilkes B., Chapple C., Li X. Discovery of a novel amino acid racemase through exploration of natural variation in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2015;112:11726–11731. doi: 10.1073/pnas.1503272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Togninalli M., Seren U., Freudenthal J.A., Monroe J.G., Meng D. AraPheno and the AraGWAS Catalog 2020: a major database update including RNA-Seq and knockout mutation data for Arabidopsis thaliana. Nucleic Acids Res. 2020;48:D1063–D1068. doi: 10.1093/nar/gkz925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Larsson S.J., Lipka A.E., Buckler E.S. Lessons from Dwarf8 on the strengths and weaknesses of structured association mapping. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Itkin M., Heinig U., Tzfadia O., Bhide A.J., Shinde B. Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science. 2013;341:175–179. doi: 10.1126/science.1240230. [DOI] [PubMed] [Google Scholar]
- 194.Kang J.H., Gonzales-Vigil E., Matsuba Y., Pichersky E., Barry C.S. Determination of residues responsible for substrate and product specificity of Solanum habrochaites short-chain cis-prenyltransferases. Plant Physiol. 2014;164:80–91. doi: 10.1104/pp.113.230466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kim J., Matsuba Y., Ning J., Schilmiller A.L., Hammar D. Analysis of natural and induced variation in tomato glandular trichome flavonoids identifies a gene not present in the reference genome. Plant Cell. 2014;26:3272–3285. doi: 10.1105/tpc.114.129460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Matsuba Y., Nguyen T.T., Wiegert K., Falara V., Gonzales-Vigil E. Evolution of a complex locus for terpene biosynthesis in solanum. Plant Cell. 2013;25:2022–2036. doi: 10.1105/tpc.113.111013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Schilmiller A.L., Moghe G.D., Fan P., Ghosh B., Ning J. Functionally divergent alleles and duplicated Loci encoding an acyltransferase contribute to acylsugar metabolite diversity in Solanum trichomes. Plant Cell. 2015;27:1002–1017. doi: 10.1105/tpc.15.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schwahn K., de Souza L.P., Fernie A.R., Tohge T. Metabolomics-assisted refinement of the pathways of steroidal glycoalkaloid biosynthesis in the tomato clade. J Integr Plant Biol. 2014;56:864–875. doi: 10.1111/jipb.12274. [DOI] [PubMed] [Google Scholar]
- 199.Chen J., Hu X., Shi T., Yin H., Sun D. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol J. 2020;18:1722–1735. doi: 10.1111/pbi.13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wen W., Li K., Alseekh S., Omranian N., Zhao L. Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. Plant Cell. 2015;27:1839–1856. doi: 10.1105/tpc.15.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wen W., Li D., Li X., Gao Y., Li W. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun. 2014;5:3438. doi: 10.1038/ncomms4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chen W., Gao Y., Xie W., Gong L., Lu K. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. 2014;46:714–721. doi: 10.1038/ng.3007. [DOI] [PubMed] [Google Scholar]
- 203.Chen W., Wang W., Peng M., Gong L., Gao Y. Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat Commun. 2016;7:12767. doi: 10.1038/ncomms12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fernie A.R., Gutierrez-Marcos J. From genome to phenome: genome-wide association studies and other approaches that bridge the genotype to phenotype gap. Plant J. 2019;97:5–7. doi: 10.1111/tpj.14219. [DOI] [PubMed] [Google Scholar]
- 205.Beleggia R., Rau D., Laido G., Platani C., Nigro F. Evolutionary metabolomics reveals domestication-associated changes in tetraploid wheat kernels. Mol Biol Evol. 2016;33:1740–1753. doi: 10.1093/molbev/msw050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhu G., Wang S., Huang Z., Zhang S., Liao Q. Rewiring of the fruit metabolome in tomato breeding. Cell. 2018;172(249–261) doi: 10.1016/j.cell.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 207.Fernie A.R., Yan J. De novo domestication: an alternative route toward new crops for the future. Mol Plant. 2019;12:615–631. doi: 10.1016/j.molp.2019.03.016. [DOI] [PubMed] [Google Scholar]
- 208.Fridman E., Carrari F., Liu Y.S., Fernie A.R., Zamir D. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science. 2004;305:1786–1789. doi: 10.1126/science.1101666. [DOI] [PubMed] [Google Scholar]
- 209.Komatsuda T., Pourkheirandish M., He C., Azhaguvel P., Kanamori H. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc Natl Acad Sci U S A. 2007;104:1424–1429. doi: 10.1073/pnas.0608580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Li C., Zhou A., Sang T. Rice domestication by reducing shattering. Science. 2006;311:1936–1939. doi: 10.1126/science.1123604. [DOI] [PubMed] [Google Scholar]
- 211.Simons K.J., Fellers J.P., Trick H.N., Zhang Z., Tai Y.S. Molecular characterization of the major wheat domestication gene Q. Genetics. 2006;172:547–555. doi: 10.1534/genetics.105.044727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Joly-Lopez Z., Flowers J.M., Purugganan M.D. Developing maps of fitness consequences for plant genomes. Curr Opin Plant Biol. 2016;30:101–107. doi: 10.1016/j.pbi.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 213.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Burgess D.G., Xu J., Freeling M. Advances in understanding cis regulation of the plant gene with an emphasis on comparative genomics. Curr Opin Plant Biol. 2015;27:141–147. doi: 10.1016/j.pbi.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 216.Eyre-Walker A., Keightley P.D. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8:610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 217.Mezmouk S., Ross-Ibarra J. The pattern and distribution of deleterious mutations in maize. G3 (Bethesda) 2014;4:163–171. doi: 10.1534/g3.113.008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Renaut S., Rieseberg L.H. The accumulation of deleterious mutations as a consequence of domestication and improvement in sunflowers and other compositae crops. Mol Biol Evol. 2015;32:2273–2283. doi: 10.1093/molbev/msv106. [DOI] [PubMed] [Google Scholar]
- 219.Gulko B., Hubisz M.J., Gronau I., Siepel A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat Genet. 2015;47:276–283. doi: 10.1038/ng.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sawyer S.A., Hartl D.L. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–1176. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Katsonis P., Koire A., Wilson S.J., Hsu T.K., Lua R.C. Single nucleotide variations: biological impact and theoretical interpretation. Protein Sci. 2014;23:1650–1666. doi: 10.1002/pro.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gronau I., Arbiza L., Mohammed J., Siepel A. Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol Biol Evol. 2013;30:1159–1171. doi: 10.1093/molbev/mst019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Flynn J.M., Rossouw A., Cote-Hammarlof P., Fragata I., Mavor D. Comprehensive fitness maps of Hsp90 show widespread environmental dependence. Elife. 2020;9 doi: 10.7554/eLife.53810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Exposito-Alonso M., Genomes Field Experiment T., Burbano H.A., Bossdorf O., Nielsen R. Natural selection on the Arabidopsis thaliana genome in present and future climates. Nature. 2019;573:126–129. doi: 10.1038/s41586-019-1520-9. [DOI] [PubMed] [Google Scholar]
- 225.Groen S.C., Calic I., Joly-Lopez Z., Platts A.E., Choi J.Y. The strength and pattern of natural selection on gene expression in rice. Nature. 2020;578:572–576. doi: 10.1038/s41586-020-1997-2. [DOI] [PubMed] [Google Scholar]
- 226.Haudry A., Platts A.E., Vello E., Hoen D.R., Leclercq M. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat Genet. 2013;45:891–898. doi: 10.1038/ng.2684. [DOI] [PubMed] [Google Scholar]
- 227.Yang Z., Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 228.Yang Z., dos Reis M. Statistical properties of the branch-site test of positive selection. Mol Biol Evol. 2011;28:1217–1228. doi: 10.1093/molbev/msq303. [DOI] [PubMed] [Google Scholar]
- 229.Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Meyer R.S., DuVal A.E., Jensen H.R. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 2012;196:29–48. doi: 10.1111/j.1469-8137.2012.04253.x. [DOI] [PubMed] [Google Scholar]
- 231.Adey N.B., Tollefsbol T.O., Sparks A.B., Edgell M.H., Hutchison C.A., 3rd Molecular resurrection of an extinct ancestral promoter for mouse L1. Proc Natl Acad Sci U S A. 1994;91:1569–1573. doi: 10.1073/pnas.91.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.He X., Ling X., Sinha S. Alignment and prediction of cis-regulatory modules based on a probabilistic model of evolution. PLoS Comput Biol. 2009;5 doi: 10.1371/journal.pcbi.1000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Keightley P.D., Johnson T. MCALIGN: stochastic alignment of noncoding DNA sequences based on an evolutionary model of sequence evolution. Genome Res. 2004;14:442–450. doi: 10.1101/gr.1571904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Duque T., Samee M.A., Kazemian M., Pham H.N., Brodsky M.H. Simulations of enhancer evolution provide mechanistic insights into gene regulation. Mol Biol Evol. 2014;31:184–200. doi: 10.1093/molbev/mst170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.