

Abstract
Lipid nanoparticles (LNPs) are the most clinically advanced delivery system for RNA-based drugs but have predominantly been investigated for intravenous and intramuscular administration. Subcutaneous administration opens the possibility of patient self-administration and hence long-term chronic treatment that could enable messenger RNA (mRNA) to be used as a novel modality for protein replacement or regenerative therapies. In this study, we show that subcutaneous administration of mRNA formulated within LNPs can result in measurable plasma exposure of a secreted protein. However, subcutaneous administration of mRNA formulated within LNPs was observed to be associated with dose-limiting inflammatory responses. To overcome this limitation, we investigated the concept of incorporating aliphatic ester prodrugs of anti-inflammatory steroids within LNPs, i.e., functionalized LNPs to suppress the inflammatory response. We show that the effectiveness of this approach depends on the alkyl chain length of the ester prodrug, which determines its retention at the site of administration. An unexpected additional benefit to this approach is the prolongation observed in the duration of protein expression. Our results demonstrate that subcutaneous administration of mRNA formulated in functionalized LNPs is a viable approach to achieving systemic levels of therapeutic proteins, which has the added benefits of being amenable to self-administration when chronic treatment is required.
Keywords: LNP, mRNA, subcutaneous delivery, FGF21, steroid prodrug, tolerability
Graphical abstract
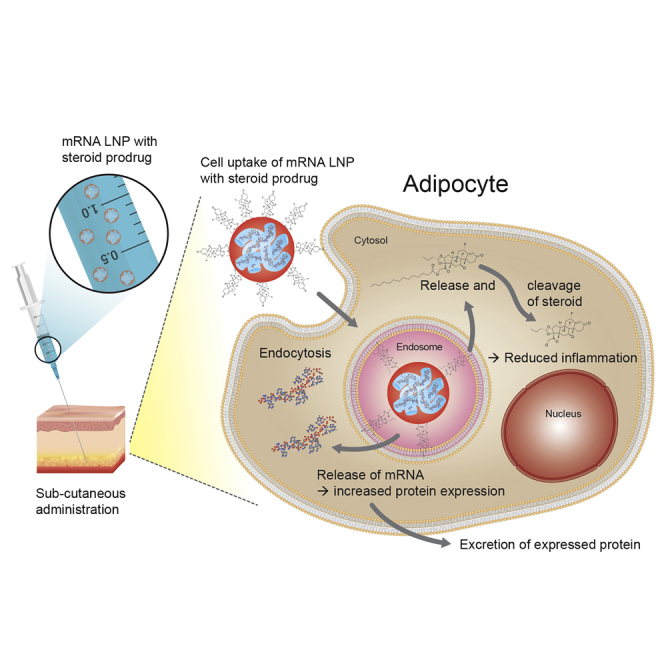
Subcutaneous administration of mRNA formulated within LNPs results in measurable plasma exposure of a secreted protein, albeit with dose-limiting local inflammatory responses. Inclusion of a steroid prodrug in mRNA LNPs resulted in an increased level and duration of protein expression as well as improved tolerability of the LNP following subcutaneous administration.
Introduction
Chemically modified messenger RNA (mRNA) is an emerging class of nucleic acid-based therapeutics that is able to encode for both wild-type and engineered intracellular, transmembrane, and secreted proteins.1 Modified mRNA is chemically engineered to structurally resemble natural, mature, and processed mRNA in the cytoplasm, while not eliciting an immunological response when administered. The endogenous machinery of the transfected cell is utilized for in vivo translation of the message to the corresponding protein that undergoes post-transcriptional modifications and folding prior to secretion.1,2
Substantial investment has been made in the last decades to modify the structural elements of the mRNA (including modifications of the nucleotides and cap structure) to enable increased protein expression and reduced immunogenicity. However, there are still some fundamental challenges with the use of mRNA as therapeutics, including stability, duration of action, in vivo pharmacokinetic/pharmacodynamic (PK/PD), and effective delivery to the target cell type or tissue. In general, the technology has progressed significantly since the first non-clinical studies in the 1990s and has to date been explored for vaccines, protein replacement therapies, and in regenerative medicine applications.1, 2, 3, 4, 5, 6, 7, 8, 9, 10
One of the major obstacles facing the successful development of mRNA-based therapies is the identification of a safe and effective delivery system that can offer protection of the mRNA from endonucleases and exonucleases and effectively deliver the mRNA into the cells in a manner that is acceptable to patients. Broadly speaking, RNA delivery can be mediated by viral and non-viral vectors.2,11,12 Lipid nanoparticles (LNPs), initially developed for in vivo delivery of small interfering RNA (siRNA), have also been investigated for delivery of mRNA and have shown promise as a non-viral delivery system.13, 14, 15, 16
LNPs are multi-component systems that typically consist of an ionizable amino lipid, a phospholipid, cholesterol, and a polyethylene glycol (PEG)-lipid, with all of the components contributing to efficient delivery of the nucleic acid drug cargo and stability of the particle.17 The cationic lipid electrostatically condenses the negatively charged RNA into nanoparticles and the use of ionizable lipids that are positively charged at acidic pH is thought to enhance endosomal escape. The most explored formulations for delivery of siRNA both clinically and non-clinically are predominantly based on cationic lipids such as DLin-MC3-DMA (MC3).11,18 Recently, the therapeutic siRNA Onpattro (patisiran) in MC3 LNPs was awarded breakthrough approval by the US Food and Drug Administration for the treatment of the polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults.19
Building on the experience of LNP delivery of siRNA, several groups have demonstrated effective and tolerable delivery of mRNA using LNPs for transient expression of vaccine antigens as well as of secreted proteins following intravenous (i.v.) or intramuscular (i.m.) administration.4, 5, 6, 7,20, 21, 22 Consequently, a number of mRNA constructs have recently progressed into clinical trials.23 While great progress has been made in achieving efficient and tolerable LNPs for delivery of mRNA for i.v. or i.m. administration, the challenge remains with regard to subcutaneous (s.c.) self-administration of therapeutic mRNA required for chronic/sub-chronic treatment of diseases. Following s.c. administration, LNPs and their mRNA cargo are expected to be largely retained at the site of injection, resulting in high local concentrations. Since LNPs are known to be pro-inflammatory, largely attributed to the ionizable lipid present in the LNPs,14 then it would not be unexpected that s.c. administration of mRNA formulated in LNPs would be associated with dose-limiting inflammatory responses. Previous work has shown that co-administration of dexamethasone with LNP reduces the immune-inflammatory response following i.v. administration,24 and recently Chen et al.25 reported on reduced immune stimulation following systemic administration by incorporating lipophilic dexamethasone prodrugs within LNP-containing nucleic acids. Herein, we report the concept of incorporating hydrophobic prodrugs of anti-inflammatory compounds (AICs) into mRNA-loaded LNPs, i.e., functionalized LNPs to minimize the inflammatory response and maintain protein expression.26 We report, for the first time, the use of functionalized LNPs in vivo that enable tolerable s.c. administration of mRNA encoding for a model secreted protein (human fibroblast growth factor 21 [hFGF21]) to demonstrate the utility of this approach for systemic protein replacement therapies.
Results
Systemic protein exposure and tolerability following i.v. and s.c. administration of mRNA formulated within LNPs
A chemically modified mRNA encoding for the secreted hFGF21 protein (hFGF21 mRNA) was synthesized and formulated within LNPs prepared using DLin-MC3-DMA as the amino, ionizable lipid. The MC3 LNP-formulated hFGF21 mRNA was administered to CD1 mice either i.v. or s.c. at a dose of 0.3 mg/kg mRNA.
Plasma exposure profiles of the secreted hFGF21 protein are reported in Figure 1A. Following s.c. administration, plasma exposure was approximately 20-fold lower compared to i.v. administration, exhibiting a delayed maximal observed plasma concentration (Cmax) at approximately 8 h as compared to a much earlier Cmax for i.v. administration (Table S1; Figure 1B).
Figure 1.

Systemic hFGF21 protein exposure and tolerability following i.v. and s.c. administration of hFGF21 mRNA formulated in MC3 LNPs
Plasma concentration-time profiles (mean ± SE) and exposure of hFGF21 protein, haptoglobin, transaminases, and cytokines in plasma at 24 h post-administration in CD1 mice (n = 5) following i.v. administration of PBS (group 1, green six-pointed star) or s.c. (group 2, orange circle) or i.v. (group 3, turquoise diamond) administration of 0.3 mg/kg hFGF21 mRNA in MC3 lipid nanoparticles. (A) Blood samples were collected at 2, 5, 8, and 24 h after dose administration for plasma hFGF21 protein exposure. (B) Calculated individual and geometric mean (gray x) exposure (AUC<sub>2-24 h</sub>) of FGF21 protein. (C–I) Plasma exposure at 24 h of haptoglobin (C), ALT (D), AST (E) IL-6 (F), IP-10 (F), KC (H), and MCP-1 (I). The PBS group for IL-6 was not included in the statistical evaluation (n.c., not calculated), as three out of five values were below the limit of detection. Statistics for IL-6 and AUC<sub>2–24 h</sub> were therefore evaluated using a Welch two-sample t test. Remaining parameters were evaluated using a one-way ANOVA with unequal variance and adjusted p values. One sample of KC in the PBS groups was a clear outlier and was excluded from the statistical analysis. ∗p < 0.05–0.01, ∗∗p < 0.01–0.001, ∗∗∗p < 0.001. ns, not significant (p > 0.05).
To assess tolerability, plasma levels of the acute phase protein, haptoglobin, and selected inflammatory cytokines/chemokines (interleukin [IL]-6, murine IL-8 homolog [KC], interferon gamma-induced protein 10 [IP-10], and monocyte chemoattractant protein 1 [MCP-1]) were measured at termination of the study (24 h post-dosing) and are reported in Table S1 and Figures 1C–1I. Haptoglobin, IL-6, IP-10, and MCP-1 levels were elevated following i.v. administration when compared with PBS control. However, after s.c. administration, haptoglobin levels were greatly increased, and at the time point measured they were 13-fold higher than after the equivalent i.v. dose. IL-6, IP-10, and MCP-1 were also higher compared with i.v. administration. Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured as biomarkers of liver toxicity; however, they were not elevated above phosphate-buffered saline (PBS) control levels when hFGF21 mRNA LNPs were administered by either i.v. or s.c. administration (Table S1).
The results described using MC3 LNPs for the s.c. delivery of mRNA demonstrate the potential of the s.c. route of administration for achieving systemic exposures of protein. However, the observations also clearly highlight the need to improve the tolerability of the mRNA LNP formulation if this is to be a viable path forward for mRNA-based therapeutics that can be self-administered via s.c. injection.
Biodistribution of transfection following i.v. and s.c. administration of mRNA formulated within MC3 LNPs
To better understand protein expression following s.c. compared to i.v. administration of mRNA formulated in LNPs, an in vivo imaging system (IVIS) with luciferase (Luc) mRNA was used to identify where protein was being expressed. The study was conducted using the same dose of mRNA as used for the FGF21 protein systemic exposure study as detailed above (0.3 mg/kg).
Luc protein expression following s.c. administration was predominantly confined to the site of administration, with some expression being observed in the local axial and brachial draining lymph nodes (Figure 2). Protein expression from the liver contributed to less than 1% of total luminescence following s.c. administration of the Luc mRNA formulated in MC3 LNPs (Table 1). This distribution of protein expression did not change with time up to 48 h after administration and hence is assumed to represent where protein expression occurs following s.c. administration of mRNA formulated in LNPs. In contrast, and as expected for MC3 LNPs having this composition,27 most protein expression (>95%) following i.v. administration was observed to occur in the liver (Figures 2B and 2D; Table 1).
Figure 2.

Biodistribution of luciferase (Luc) protein expression following i.v. or s.c. administration of Luc mRNA formulated in MC3 LNPs
(A–G) Representative whole-body and excised tissue IVIS images 8 h following s.c. (A and C) and i.v. (B and D) administration of 0.3 mg/kg Luc mRNA in MC3 lipid nanoparticles and cellular expression as evaluated by immunohistochemistry (brown staining): adipocytes (E), fibroblasts (F), and macrophages (arrowhead) and neutrophils (arrow) (G).
Table 1.
Organ distribution of luminescence following s.c. and i.v. administration of 0.3 mg/kg Luc mRNA in MC3 LNPs (mean ± SEM)
| Route | Time | Injection site/skin (%) | Lymph nodes (%) | Liver (%) | Spleen (%) | Kidneys (%) | Heart (%) | Lungs (%) |
|---|---|---|---|---|---|---|---|---|
| s.c. | 8 h | 99.2 ± 0.5 | 0.71 ± 0.52 | 0.07 ± 0.03 | 0.01 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| 24 h | 98.2 ± 0.8 | 1.72 ± 0.84 | 0.07 ± 0.03 | 0.02 ± 0.01 | 0.00 ± 0.00 | 0.01 ± 0.01 | 0.00 ± 0.00 | |
| 48 h | 98.2 ± 0.9 | 1.39 ± 0.79 | 0.23 ± 0.06 | 0.09 ± 0.02 | 0.01 ± 0.00 | 0.04 ± 0.01 | 0.00 ± 0.00 | |
| i.v. | 8 h | 1.11 ± 0.44 | 0.30 ± 0.07 | 97.6 ± 0.5 | 0.96 ± 0.16 | 0.00 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 |
To further explore s.c. administration of mRNA formulated in LNPs, histological evaluation of the injection site and surrounding tissues was undertaken to identify the cellular distribution of transfection. Luc protein expression (semiquantitative scoring) was predominantly expressed by adipocytes with limited expression in fibroblasts and macrophages (Figures 2E–2G). This cellular distribution of protein expression again did not change with time (up to 48 h, data not shown) and hence is assumed to represent the cellular distribution of where protein expression occurs following s.c. administration of mRNA formulated in LNPs.
Incorporation of an anti-inflammatory steroid within LNPs
Considering that tolerability of the mRNA LNP formulation could be the limiting determinant to the success of s.c. self-administration for systemic protein replacement therapies based on mRNA, it was decided to investigate the concept of incorporating an anti-inflammatory steroid within the LNP. Steroids are potent AICs that interact with numerous pathways involved with inflammatory responses, and hence they have widespread anti-inflammatory effects.28 Two steroids, namely rofleponide and budesonide, were hence investigated for incorporation within LNPs as an approach to improve the tolerability of the mRNA LNP formulation following s.c. administration.
To enable efficient entrapment of the steroid within the LNP, aliphatic ester prodrugs with varying alkyl chain lengths (C5 [rofleponide only], C8 [budesonide only], C14, C16, and C18) were synthesized to increase the lipophilicity/decrease aqueous solubility of the steroids and thereby promote their incorporation within LNPs. For all steroid prodrugs investigated, entrapment within LNPs was greater than 75%.
The ester prodrugs of rofleponide and budesonide require enzymatic cleavage to release the active parent steroid to have an anti-inflammatory effect.29 For this reason, and because of their low aqueous solubility, it was considered that the ester prodrugs would likely need to be located on the surface of the LNP to be accessible for such enzymatic cleavage.
The location of the rofleponide-C14 prodrug within the MC3 LNP was therefore investigated by small-angle neutron scattering (SANS) with isotropic contrast variation as previously described.30 In brief, the distribution of the rofleponide-C14 prodrug was elucidated by varying the content of deuterated water (D2O) to match the scattering length densities of different regions of the particle (Figure 3A). Simultaneous fitting of the SANS data (Figure 3B) proposes that a core-shell particle model, where the deuterium-labeled rofleponide-C14 predominantly distributes into the outer shell of the particle, agrees best with the experimental results. In comparison, models that assume more AIC is introduced into the core (“AIC throughout” or “AIC in core”) show progressively poorer fit (Figure 3B). This strongly supports that the rofleponide-C14 prodrug is not homogeneously distributed within the MC3 LNP, but rather it is preferentially located in the outer shell region. Simultaneous fitting of five isotropic contrast datasets estimates that approximately 85% ± 5% of the total mole fraction of the rofleponide-C14 prodrug in the LNP is located at the surface (shell) of the LNP.
Figure 3.

Characterization of steroid prodrug distribution within LNPs
(A) Schematic of the isotopic contrast variation used to highlight the location of AICs in the LNPs with partially deuterated rofleponide-C14 prodrug depicted as red triangles. (B) SANS data (symbols) and best fit (lines) shown as scattering intensity as a function of the scattering vector (q) for mRNA-containing MC3 LNPs with a rofleponide-C14/mRNA 1:1 weight ratio. The data shown are for MC3 LNPs in 15–100 vol % D2O buffer (PBS, pH 7.4). The solid lines show the best fit model, where most AIC molecules are located in the shell. The dotted and broken lines show the models that assume the AIC molecules partitioning in the core or distributed homogenously throughout the particle, respectively. The corresponding models are shown above. The intensity for each dataset contrast is offset by an order of magnitude for clarity.
Effect of incorporating steroid esters with varying alkyl chain lengths within hFGF21 mRNA LNPs on protein expression and tolerability following s.c. administration
The impact of incorporating steroid prodrugs within LNP formulations of hFGF21 mRNA on systemic protein exposure and tolerability following s.c. administration was investigated in CD1 mice. Furthermore, steroid prodrugs having fatty acid esters of various chain lengths were investigated to evaluate whether the activity of the steroid was influenced by the carbon chain length of the ester prodrugs. To investigate the broader applicability of this concept to different LNPs, two steroids, namely rofleponide and budesonide, were respectively incorporated within LNPs formulated using two different amino lipids, that is, either DLin-MC3-DMA or L608.
Incorporation of rofleponide ester prodrugs within MC3 LNPs
Fatty acid ester prodrugs of rofleponide having alkyl chain lengths of C5 (valerate), C14 (myristate), C16 (palmitate), and C18 (stearate) were synthesized and incorporated within hFGF21 MC3 LNPs and compared to LNPs not containing steroid.
Consistent with the previous results, s.c. administration of hFGF21 mRNA formulated in MC3 LNPs without steroid resulted in measurable systemic protein exposures but was again associated with systemic inflammatory responses (Figures 4A–4J; Table 2). Furthermore, edema and focal neutrophilic inflammation at the site of s.c. injection was observed in all mice (Figures 4C and 4L).
Figure 4.

Rofleponide prodrugs formulated in MC3 LNPs improve protein expression and reduce local (edema) as well as systemic inflammatory responses following s.c. administration
Plasma concentration-time profiles (mean ± SE) and exposure of hFGF21 protein, haptoglobin, transaminases, cytokines, and histopathology of the injection site at 24 h post-administration following s.c. administration in CD1 mice (n = 5) of PBS (group 1, green six-pointed star) or hFGF21 mRNA in MC3 lipid nanoparticles (0.3 mg/kg mRNA) containing no steroid (group 2, orange circle) or with rofleponide-C5 (group 3, turquoise diamond), rofleponide-C14 (group 4, magenta triangle), rofleponide-C16 (group 5, blue square), or rofleponide-C18 (group 6, red star) incorporated into the LNP at a prodrug steroid/mRNA weight ratio of 1:1. (A) Blood samples were collected at 2, 5, 8, and 24 h after dose administration for plasma hFGF21 protein exposure. (B and C) Individual and geometric mean (gray x) exposure (AUC2–24 h) of FGF21 protein (B) and incidence of edema at site of injection at 24 h after dosing (C). (D–J) Plasma exposure at 24 h of haptoglobin (D), ALT (E), AST (F), IL-6 (G), IP-10 (H), KC (I), and MCP-1 (J). Statistics evaluated using one-way ANOVA with unequal variance and adjusted p values. ∗p < 0.05–0.01, ∗∗p < 0.01–0.001, ∗∗∗p < 0.001. ns, not significant (p > 0.05). (K–M) Hematoxylin and eosin-stained histological sections of injection site at 24 h after dosing for PBS showing no morphological changes (∗) (K), MC3 LNPs without steroid showing extensive focal acute inflammation (∗) (L), and MC3 LNPs containing rofleponide-C16 showing significant reduction of inflammation (∗) (M). Original magnification, ×5 (K and L) and ×10 (M).
Table 2.
Plasma hFGF21 protein exposure in mice after s.c. administration of 0.3 mg/kg hFGF21 mRNA in MC3 LNPs (MC3) and plasma chemistry measured at termination, 24 h after dosing
| Parameter | PBS control | MC3 | MC3 + rofleponide-C5 | MC3 + rofleponide-C14 | MC3 + rofleponide-C16 | MC3 + rofleponide-C18 |
|---|---|---|---|---|---|---|
| AUC2–24 h (nmol·h/L) | N/A | 0.29 ± 0.05 | 0.41 ± 0.08 | 1.1 ± 0.35 | 0.65 ± 0.14 | 0.70 ± 0.08 |
| Haptoglobin (μg/mL) | 84 ± 32 | 1,680 ± 110 | 607 ± 67 | 286 ± 53 | 429 ± 68 | 318 ± 22 |
| IL-6 (pg/mL) | 50 ± 24 | 301 ± 58 | 316 ± 79 | 55 ± 12 | 51 ± 43 | 142 ± 49 |
| KC (pg/mL) | 163 ± 37 | 395 ± 42 | 427 ± 71 | 78 ± 18 | 157 ± 100 | 403 ± 150 |
| IP-10 (pg/mL) | 102 ± 17 | 700 ± 62 | 742 ± 83 | 260 ± 53 | 203 ± 85 | 389 ± 112 |
| MCP-1 (pg/mL) | 69 ± 11 | 488 ± 53 | 383 ± 44 | 53 ± 12 | 98 ± 62 | 316 ± 170 |
| ALT (U/L) | 90 ± 9 | 104 ± 9 | 123 ± 22 | 114 ± 7 | 120 ± 10 | 143 ± 30 |
| AST (U/L) | 37 ± 2 | 34 ± 1 | 35 ± 2 | 51 ± 4 | 51 ± 4 | 66 ± 9 |
Values are mean ± SEM. AUC2–24 h, area under the plasma drug concentration-time curve during the time interval 2–24 h after dosing; N/A, not applicable; IL-6, interleukin-6; KC, murine IL-8 homolog, IP-10, interferon gamma-induced protein 10; MCP-1, monocyte chemoattractant protein 1; ALT, alanine transaminase; AST, aspartate transaminase.
Inclusion of rofleponide prodrugs within hFGF21 mRNA MC3 LNPs reduced both local (edema) as well as systemic inflammatory responses (Figure 4; Table 2). Interestingly, the reduction in inflammatory response seemed to be more pronounced for the longer carbon chain ester prodrugs (C14, C16, and C18) as compared to the C5 ester prodrug, where fewer or no mice were observed to have edema at the site of administration. Plasma haptoglobin and cytokines were lower when rofleponide prodrugs were incorporated within MC3 LNPs, with levels approaching those observed following administration of a PBS, particularly for the longer chain length prodrugs. Histological evaluation of the injection site also showed significantly reduced neutrophilic inflammation when the longer chain length rofleponide prodrugs were incorporated into the hFGF21 mRNA MC3 LNPs (Figure 4M).
An unexpected, additional benefit of incorporating rofleponide prodrugs within hFGF21 mRNA MC3 LNPs was prolonged protein expression and elevated plasma protein exposures. For example, systemic exposure of hFGF21 (AUC2–24 h [area under the plasma drug concentration-time curve during the time interval 2–24 h after dosing]) was increased 3.8- and 2.3-fold, respectively, when rofleponide-C14 or budesonide-C16 was incorporated into the MC3 LNP (Figures 4A and 4B; Table 2).
Incorporation of budesonide ester prodrugs within L608 LNPs
To investigate whether the effects observed when incorporating steroid prodrugs within LNPs were generally applicable, ester prodrugs of a second steroid, namely budesonide, were incorporated within an alternative LNP prepared using L608 as the ionizable amino lipid. For this, fatty acid ester prodrugs of budesonide having alkyl chain of C8 (caprylate), C14 (myristate), C16 (palmitate), and C18:1 (oleate) were synthesized, incorporated within hFGF21 mRNA L608 LNPs, and compared to L608 LNPs not containing steroid.
Similar to MC3 LNPs, hFGF21 mRNA formulated in L608 LNPs resulted in measurable plasma exposures of hFGF21 protein following s.c. administration (although plasma levels in three out of five mice were below the limit of quantification of the assay at the 24-h termination time point). Furthermore, s.c. administration of the L608 LNPs not containing steroid was again associated with both local and systemic inflammatory responses (Figure 5).
Figure 5.

Budesonide prodrugs formulated in L608 LNPs improve protein expression and reduce local (edema) as well as systemic inflammatory responses following s.c. administration
Plasma concentration-time profiles of hFGF21 protein (mean ± SE); plasma haptoglobin, edema, and histopathology of the injection site at 24 h post-administration following s.c. administration in CD1 mice (n = 5) of hFGF21 mRNA formulated in L608 LNPs (0.3 mg/kg mRNA) containing no steroid (group 1, orange circle) or with budesonide-C8 (group 2, turquoise diamond), budesonide-C14 (group 3, magenta triangle), budesonide-C16 (group 4, blue square), or budesonide-18:1 (group 5, red star) incorporated into the LNP at an equivalent parent steroid/mRNA weight ratio of 2:3. (A) Blood samples were collected at 4, 7, and 24 h after dose administration for plasma hFGF21 protein exposure. (B) Individual and geometric mean (gray x) haptoglobin concentrations in plasma 24 h after dosing. (C) Incidence of edema at site of injection at 24 h after dosing. (D and E) Histological sections of injection site at 24 h after dosing for L608 LNPs without steroid showing severe acute inflammation (∗) (D) and L608 LNPs containing budesonide-C16 showing significant reduction of the inflammation (∗) (E). Original magnification, ×5. Statistics were evaluated using a one-way ANOVA with unequal variance and adjusted p values. ∗p < 0.05–0.01, ∗∗p < 0.01–0.001, ∗∗∗p < 0.001. ns, not significant (p > 0.05).
Similar to the incorporation of rofleponide prodrugs within MC3 LNPs, inclusion of budesonide prodrugs within L608 LNPs was able to maintain plasma protein levels of hFGF21 during the 24-h duration of the study (Figure 5A). Incorporation of the shorter chain length prodrug budesonide-C8 within L608 LNPs failed to protect from local inflammatory responses, with edema being observed in four of the five mice treated (similar to the incorporation of the shorter chain length prodrug rofleponide-C5 within MC3 LNPs). Better local tolerability was observed upon incorporating the longer chain length esters of budesonide-C16 and budesonide-C18:1 where edema was only observed in one or two mice (Figure 5C). Interestingly, and in contrast to the observation for MC3 LNPs with rofleponide esters, incorporation of the C14 prodrug of budesonide did not improve local tolerability, with all mice showing edema at the site of injection. The improved local tolerability upon incorporation of budesonide prodrugs having longer alkyl chain lengths into L608 LNPs was confirmed by histological evaluation of the injection site, where less cell infiltrates, fibrin buildup, and cell necrosis were observed (exemplified by histological section of hFGF21 mRNA L608 LNPs with and without B-C16, Figures 5D and 5E).
In terms of systemic inflammatory responses, there was a trend of decreasing plasma haptoglobin with increasing the ester prodrug chain length of budesonide, with levels for the C16 and C18:1 being significantly lower compared with the L608 LNP not containing steroid (Figure 5B). Incorporation of budesonide prodrugs, however, had little effect on cytokine levels, which were overall close to baseline and not particularly elevated for hFGF21 mRNA L608 LNPs (data not shown).
Effect of steroid ester prodrug on duration of protein expression and comparison to parent steroid
Investigations to identify the most suitable prodrug ester of the two steroids indicated that incorporation of the steroid prodrugs within MC3 or L608 mRNA LNPs resulted in prolonged protein expression, at least during the 24-h period investigated in these studies (Figures 4A and 5A). To further investigate this phenomenon, follow-up studies were conducted to extend the duration of pharmacokinetic sampling to 72 h.
Due to the limited amount of blood that could be sampled, as well as the plasma volumes required for various analyses, three parallel groups of mice were used in these studies, wherein one group was sampled during the period 0–24 h, a second group during the period 24–48 h, and a third group during the period 48–72 h. Separate studies were carried out comparing either the steroid ester prodrug (rofleponide-C16 or budesonide-C16) or parent steroid, all formulated in MC3 LNPs to MC3 LNPs not containing steroid.
Incorporation of both rofleponide-C16 and budesonide-C16 within the mRNA MC3 LNP formulation at an equivalent parent steroid/mRNA weight ratio of 2:3 resulted in prolonged protein expression and plasma exposures compared to an MC3 LNP formulation containing no steroid (AUC2–58 h being 2.6- and 3-fold greater for rofleponide-C16 and budesonide-C16, respectively; Figure 6A). Incorporation of the parent steroid within the mRNA MC3 LNP formulation at the same parent steroid/mRNA weight ratio also yielded some benefits in terms of plasma exposures compared to an MC3 LNP formulation containing no steroid, with AUC2–58 h being 1.2- and 1.9-fold greater for rofleponide and budesonide, respectively (Figure 6D).
Figure 6.

Incorporation of steroid prodrugs (C16) within MC3 LNPs prolongs duration of protein expression and improves tolerability compared to parent steroid following s.c. administration
Plasma concentration-time profiles of hFGF21 protein (mean ± SE, n = 4–12); plasma haptoglobin and IL-6 levels at 24 h post-administration following s.c. administration in CD1 mice of hFGF21 mRNA formulated in MC3 LNPs (0.3 mg/kg mRNA) containing no steroid (green diamond), rofleponide-C16 (turquoise circle), budesonide-C16 (magenta triangle), rofleponide parent co-administered (blue circle), or budesonide parent co-administrated (red triangle) at an equivalent parent steroid/mRNA weight ratio of 2:3. (A and D) hFGF21 protein concentration in plasma was determined in blood samples collected up to 72 h after dose administration. (B, C, E, and F) Plasma concentration of haptoglobin (B and E) and IL-6 (C and F) were determined in blood samples collected at termination (24, 48, or 72 h after dosing).
Compared to inclusion of parent steroid within the mRNA MC3 LNP formulation, the steroid ester prodrugs also offered benefits in terms of tolerability. Plasma haptoglobin levels (Figures 6B and 6E) were markedly lower upon incorporation of the C16 prodrugs compared to the parent steroid, as were levels of inflammatory cytokines levels, as exemplified by IL-6 (Figures 6C and 6F).
Effect of steroid prodrug/mRNA weight ratio on protein expression and inflammation
hFGF21 mRNA MC3 LNPs were formulated to incorporate rofleponide-C16 at steroid prodrug/mRNA ratios of 1:1, 1:10, or 1:30 (w/w), respectively, approximating 8, 0.8, and 0.3 mol% of the lipidic LNP components. As most evident in the edema scores (Figure 7C), there was an inverse correlation between the dose of prodrug and the degree of local inflammation. Decreasing the amount of rofleponide-C16 in the LNPs, however, had little effect on prolongation of protein expression (at least when the ratio was 10:1 or less) or systemic haptoglobin levels (Figures 7A and 7B).
Figure 7.

Effect of steroid prodrug (rofleponide-C16)/mRNA weight ratio formulated within MC3 LNPs on hFGF21 protein expression and inflammation following s.c. administration
Plasma concentration-time profiles (mean ± SE) of hFGF21 protein; haptoglobin and edema at 24 h post-administration following s.c. administration in CD1 mice (n = 5) of PBS (group 1, green six-pointed star) or hFGF21 mRNA formulated in MC3 LNPs (0.3 mg/kg mRNA) containing no steroid (group 2, orange circle) or rofleponide-C16 at a prodrug steroid/mRNA ratio of 1:1 (group 3, turquoise diamond), 1:10 (group 4, magenta triangle), or 1:30 (group 5, blue square). (A) hFGF21 protein concentration in plasma was determined in blood samples collected up to 24 h after dose administration. (B) Individual and geometric mean (gray x) haptoglobin concentrations in plasma 24 h after dosing. (C) Incidence of edema at site of injection at 24 h after dosing. Statistics were evaluated using a one-way ANOVA with unequal variance and adjusted p values. ∗p < 0.05–0.01, ∗∗p < 0.01–0.001, ∗∗∗p < 0.001. ns, not significant (p > 0.05).
Steroid pharmacokinetics
To better understand why incorporation of the steroid prodrugs into LNPs both improves the tolerability and prolongs protein expression compared to inclusion of the parent steroid within mRNA LNP formulations, the systemic pharmacokinetics of the parent steroid and steroid prodrugs following s.c. administration of the respective hFGF21 mRNA MC3 LNPs were evaluated and compared with the pharmacokinetic properties of the respective parent steroid following i.v. administration.
The plasma concentration-time profiles of rofleponide and budesonide following i.v. administration, as well as following s.c. administration of rofleponide and budesonide or their respective prodrugs formulated in hFGF21 mRNA MC3 LNPs, are shown in Figure 8. The corresponding calculated pharmacokinetic parameters are reported in Table 3.
Figure 8.

Pharmacokinetics of parent steroid in CD1 mice following i.v. or s.c. administration of parent drug or C5 prodrug, C14 prodrug , or C16 prodrug
Parent steroid was formulated in 0.6% ethanol in PBS solution; steroid prodrugs were formulated in hFGF21 mRNA MC3 LNPs. (A and B) Blood was collected up to 24 h after dose administration for quantification of rofleponide (A) or budesonide (B) in plasma; line represents group mean, and markers show individual values (n = 4). A filled amber diamond indicates i.v. administration, a filled magenta triangle indicates s.c. administration, and an open red circle, open green five-pointed star, and open blue square indicate administration of C5 prodrug, C14 prodrug, and C16 prodrug, respectively.
Table 3.
Steroid pharmacokinetic parameters based on composite mean plasma concentration-time profiles
| Parameter | Rofleponide parent | Rofleponide-C5 | Rofleponide-C14 | Rofleponide-C16 | Budesonide parent | Budesonide-C16 | ||
|---|---|---|---|---|---|---|---|---|
| Route | i.v. | s.c. | s.c. | s.c. | s.c. | i.v. | s.c. | s.c. |
| Dose (mg/kg) | 0.15 | 0.2 | 0.47 | 0.48 | 0.46 | 0.2 | 0.2 | 0.47 |
| F (%) | – | 48 | 70 | 89 | 83 | – | 59 | 62 |
| CL (mL/min/kg) | 74.5 | – | – | – | – | 121 | – | – |
| AUC (nmol·h/L) | 72 | 46 | 134 | 142 | 121 | 65 | 38 | 60 |
| Cmax (nmol/L) | 267 | 74 | 182 | 30 | 13 | 240 | 73 | 7 |
| tmax (h) | 0.03 | 0.5 | 0.5 | 1.5 | 2.5 | 0.03 | 0.17 | 1.5 |
| t½ (h) | 0.33 | 0.34 | 1.8 | 3.3 | 7.9 | 0.73 | 1.0 | 7.0 |
Pharmacokinetic parameters were calculated using non-compartmental analysis. F, bioavailability; CL, clearance; AUC, area under the plasma concentration-time curve; Cmax, maximal observed plasma concentration; tmax, time of Cmax; t½, half-life.
As can be seen, both rofleponide and budesonide were rapidly eliminated from plasma following i.v. administration, exhibiting a half-life of about 20 and 40 min, respectively. Following s.c. administration of the parent rofleponide and budesonide included in the mRNA MC3 LNP formulation, systemic absorption of the steroid was rapid, with peak plasma concentrations being observed at 30 and 10 min for rofleponide and budesonide, respectively. Elimination half-lives for both steroids were similar to those observed following i.v. administration, resulting in similar plasma profiles for s.c. and i.v. administration of the parent steroid.
In contrast, appearance of the parent steroid in plasma is delayed and more sustained when administered as the ester prodrug incorporated within the hFGF21 mRNA MC3 LNPs (Figure 8). As shown for rofleponide (Figure 8A), a clear relationship is observed between the pharmacokinetic properties and the length of the alkyl chain of the prodrug. Increasing alkyl chain length results in a delayed time of Cmax (tmax), reduced Cmax, and a prolonged half-life (Table 3). Pharmacokinetic parameters were also observed to be similar between the C16 ester prodrug of rofleponide and budesonide, exhibiting a similar delayed tmax and elimination half-lives. It would thus seem that the rate of hydrolysis of the steroid prodrugs is inversely proportional to the alkyl chain length of the steroid prodrug, which in turn determines its retention at the site of administration.
This was further confirmed by incubating the steroid prodrugs in vitro with human adipocytes. Conversion of the steroid prodrugs in vitro was observed to be in the same rank order as observed for the appearance of parent steroid in plasma, with the longer chain length prodrugs having a slower rate of conversion (Figure S1; Table S3).
Discussion
In the current study, we have developed a viable and tolerable formulation for effective murine delivery of mRNA encoding hFGF21 via the s.c. route of administration that results in sustained increases in plasma hFGF21 levels. s.c. administered hFGF21 mRNA formulated in MC3 LNP alone resulted in measurable systemic plasma concentrations of the secreted hFGF21 protein over a duration of 24 h, although the observed plasma exposures were much lower compared to i.v. administration of the same dose of mRNA formulated in LNPs (Figure 1A; Table S1). Furthermore, s.c. administration of hFGF21 mRNA formulated in MC3 LNP was associated with dose-limiting inflammatory responses, which appeared to be exaggerated when compared with i.v. administration of the same dose (Figures 1C–1I; Table S1). The dose-limiting inflammatory responses observed following s.c. administration are likely a result of the high local concentration of the mRNA LNPs at the site of administration, which are known to be pro-inflammatory and attributed to the ionizable amino lipid present in the LNPs.14 The inflammatory response following s.c. administration of such LNPs manifests itself in the form of localized inflammation and edema at the injection site, as well as systemic responses including elevated levels of haptoglobin (an acute phase marker of an inflammatory response)31 as well as various cytokines/chemokines (Figure 4; Table 2).
Investigation of the biodistribution of mRNA LNPs following s.c. and i.v. administration using Luc mRNA (expressing the non-secreted Luc protein) showed that following s.c. administration, protein expression was predominantly confined to the site of administration (Figure 2; Table 1). In contrast, following i.v. administration of mRNA in MC3 LNPs, most protein expression (>95%) was observed to occur in the liver, consistent with previous observations for these types of LNPs.27 This likely explains the almost 20-fold lower hFGF21 protein systemic exposure observed following s.c. administration compared to i.v. administration. Following s.c. administration, mRNA LNPs are largely confined to the injection site, with minimal drainage occurring to the systemic circulation, as indicated by the very low liver expression of Luc protein. As such, the number of cells transfected in the vicinity of the injection site is likely to be much lower following s.c. compared to i.v. administration, where the entire cell population of the liver is able to take up mRNA LNPs and express protein. Immunohistochemical evaluation of the s.c. injection site showed that the predominant cell type transfected was adipocytes. This may not be surprising, as the type of LNP used in the current investigations, incorporating an ionizable lipid such as MC3, is known to be internalized by cells (such as hepatocytes) via the low-density lipoprotein (LDL) receptor following adsorption of apolipoprotein E onto the surface of LNPs.32 Adipocytes are known to also express LDL receptors,33 which may explain why adipocytes are efficient translators of mRNA following s.c. administration when using ionizable LNPs as a delivery vector.34
Prodrugs of rofleponide and budesonide with various alkyl chain lengths were synthesized to enable incorporation into the LNP in an attempt to control the inflammatory responses and enable delivery of mRNA via s.c. administration. The resulting LNP formulations were well tolerated at the doses investigated, particularly when incorporating prodrugs having longer alkyl chain lengths (Figures 4 and 5; Table 2). Interestingly, incorporation of steroid within the LNPs altered the dynamics of protein production when compared with LNP formulations that did not include steroid. Systemic protein exposures were increased up to 3-fold when the steroid prodrugs were incorporated into the LNPs, and this was accompanied by a prolongation in hFGF21 protein exposure of up to 72 h (Figure 6). Importantly, these finding were not limited to MC3 LNPs and were repeated with L608 LNPs (Figure 5), demonstrating potential for broad application of this strategy across LNP delivery systems. These results demonstrate that s.c. administration of mRNA can be a viable route of administration for clinical application of mRNA-protein replacement and regenerative therapies, enabling self-administration and thus expanding the potential impact of these exciting emerging therapeutics.
LNP formulations have been successfully applied to support clinical development of siRNA therapeutics due to their ability to both encapsulate RNA molecules and protect them from degradation by systemic RNase.35 These same considerations make them attractive formulations to be explored for mRNA delivery.13 Indeed, Pardi et al.36 demonstrated that mRNA, encoding the intracellular protein Luc encapsulated within LNPs, could be successfully translated to protein after administration using various routes of administration, including i.v. and s.c. Doses administered in this study were relatively low, up to 5 μg/injection (<150 μg/kg), and tolerability was not discussed.
LNPs, however, are recognized to have an immunostimulatory profile that is distinct from their cargo,14,37 and dose-limiting systemic inflammatory responses have been described for LNP formulations administered i.v.24 Indeed, recently de Groot et al.38 described a potential interaction of cationic lipids with Toll-like receptor 4 as a potential initiating factor in the immunogenic response to these types of nanoparticulate formulations. While the s.c. route of therapeutic administration has the benefits of convenience and allowing for self-administration when compared with i.v. administration, this administration route has the potential to display exaggerated inflammation due to the presence of multiple active defense mechanisms utilized by the skin to protect against microbial pathogens. These include components of both the innate and adaptive immune systems across a variety of cells capable of mounting a formidable inflammatory response.39 The response is likely amplified by the high local concentrations of mRNA LNPs at the s.c. injection site that appear to largely remain at the site of administration and not drained into the systemic circulation (Figure 2). Therefore, it is perhaps not unexpected that in the current study we observed a dose-limiting and pronounced local and systemic inflammation upon s.c. injection of mRNA LNP formulations, an effect that appeared exaggerated when compared with i.v. administration. Interestingly, the profile of cytokines measured systemically following s.c. administration was similar to that observed by others after i.v. administration of siRNA LNP formulations, with elevated levels primarily of IL-6, IP-10, KC, and MCP-1 suggesting similar underlying mechanisms.24,27,40
Pre-treatment with anti-inflammatory agents, both dexamethasone and Janus kinase inhibitor, have previously been shown to control adverse effects of siRNA LNPs after i.v. administration.24,41 Moreover, this strategy has been applied clinically to enable siRNA LNP delivery to patients where clinical trials for patisiran (Onpattro) included pretreatment with dexamethasone, paracetamol, and histamine H1 and H2 antagonists.42 However, this strategy is problematic, particularly when sub-chronic/chronic therapy is required. Steroids have been reported to induce hyperglycemia in non-diabetic patients, potentially leading to steroid-induced diabetes mellitus.43, 44, 45 Furthermore, steroids are also known to have unwanted side effects such as hypertension, cataracts, and an increased risk of fractures.46 We therefore hypothesized that formulation of a steroidal anti-inflammatory prodrug within an LNP would lead to precision delivery of the steroid compound to the precise time and location at which its action is required, i.e., the cells that are exposed to the mRNA LNP formulation.
We explored the relationship between inflammation, protein synthesis, and systemic steroid exposure when either rofleponide or budesonide was incorporated into the LNP formulation as either parent drug or ester prodrugs having different alkyl chain lengths to determine the value of the ester modification. We have shown that when the parent steroid is incorporated into the LNP formulation without any modifications, it is rapidly absorbed into the systemic circulation from the site of administration and then rapidly eliminated (Figure 8). As therefore might be expected, incorporation of the parent steroid within the mRNA LNP formulation gave limited protection, with an inflammatory response to this LNP formulation still being apparent (Figure 6). When the ester prodrugs of rofleponide and budesonide are incorporated within the LNP formulation, appearance of the parent steroid in the plasma is delayed and is associated with a lower Cmax and is sustained for a longer duration (Figure 8). Interestingly, there also appeared to be a relationship between exposure profile of the parent steroid in plasma and length of the alkyl chain of the prodrug, where a longer alkyl chain appeared to both delay and reduce Cmax and to extend the half-life (t1/2). It would thus seem that the rate of hydrolysis of the steroid prodrugs is inversely proportional to the alkyl chain length of the steroid prodrug, which in turn determines its retention at the site of administration. This was confirmed by measuring conversion rates of the steroid prodrug to the active parent in vitro using human adipocyte cultures (Figure S1; Table S2) and is consistent with what is known regarding hydrolysis rates of glycerides by lipases where cleavage of the ester and release of the fatty acid are reduced with increasing alkyl chain length of the acid.47
Another observation was that addition of rofleponide or budesonide prodrugs to the mRNA LNP formulations resulted in a sustained boost in the protein production levels and higher overall systemic protein exposures (Figure 6). It is recognized that transfected mRNA is able to activate cellular innate immune responses through pattern recognition receptors that detect nucleic acids as part of the cellular viral defense response.48 It is also known that LNPs can activate the innate immune system, thereby potentially initiating a cellular immune response to these nanoparticles.38 Coupled with observations that cellular innate immune activation is associated with downregulation of cellular translation,49,50 this could perhaps explain the unexpected increased and sustained protein levels resulting from inclusion of anti-inflammatory steroidal agents within LNPs that are able to suppress local inflammatory responses. Similar improvements in protein production with exogenous mRNA have recently been reported when dexamethasone was incorporated into LNPs and administered i.v.51
To summarize, incorporation of either rofleponide or budesonide ester prodrugs into two different LNP formulations was able to significantly improve the tolerability of mRNA LNPs, oftentimes entirely preventing the local edema response and the increase in systemic cytokine concentrations seen following s.c. administration. Due to the robust retention of steroid prodrug in the LNP, we think that the protection afforded by the steroid is likely due to a highly localized anti-inflammatory mechanism of action, probably at the cellular site of LNP uptake. Moreover, we demonstrate that inclusion of a steroid prodrug in a mRNA LNP formulation was able to dramatically increase the level and duration of protein production following s.c. administration. Taken together, these findings demonstrate that successful systemic protein exposure can be achieved through mRNA administered via the s.c. route, an observation that if repeated in humans could increase therapeutic application opportunities for this important emerging platform.
Materials and methods
mRNA synthesis
Modified mRNA encoding hFGF21 or Luc was synthesized as previously described.4 Briefly, the mRNA was codon optimized and synthesized in vitro by T7 RNA polymerase-mediated transcription. The uridine 5′-triphosphate (UTP) was substituted with 1-methylpseudo-UTP, using a linearized DNA template, which also incorporates 5′ and 3′ UTRs, including a poly(A) tail. A donor methyl group from S-adenosylmethionine was added to methylated capped RNA (cap-0), resulting in a cap-1 modification to increase mRNA translation efficiency. These chemical modifications to the mRNA are designed to both improve protein translation and reduce immunogenicity and are the same as used by Carlsson et al.9 and An et al.4
Synthesis of rofleponide and budesonide prodrugs
Rofleponide and budesonide were obtained from AstraZeneca. Rofleponide is a pure enantiomer, while budesonide is a diastereomeric mixture of approximately 55:45 at the acetal carbon. Rofleponide and budesonide prodrugs (in addition to the deuterated rofleponide-C14 prodrug used for SANS) were synthesized using a general procedure as exemplified in the following for the synthesis of partially deuterated rofleponide-C14 (d27). The chemical structures of rofleponide and budesonide and their respective prodrugs are illustrated in Figure 9.
Figure 9.

Chemical structures of rofleponide, budesonide (and their respective prodrugs), MC3, and L608 amino lipids
d27-Myristoic acid (75 mg, 0.29 mmol) was dissolved in dichloromethane (1.5 mL) and one drop of dimethylformamide. Oxalyl chloride (0.031 mL, 0.35 mmol) was added dropwise at room temperature and the mixture was left stirring overnight. Dichloromethane and excess oxalyl chloride were removed by evaporation to obtain the light yellow, oily d27-myristoyl chloride.
Triethylamine (0.045 mL, 0.32 mmol) was added to a solution of rofleponide (60 mg, 0.13 mmol) in dichloromethane (0.5 mL). d27-Myristoyl chloride (70.2 mg, 0.26 mmol) was dissolved in dichloromethane (0.5 mL) and then added to the rofleponide solution. The reaction mixture was allowed to stir at room temperature for 3 h. Liquid chromatography-mass spectrometry indicated >99% conversion to the desired product. All chemicals used for the manufacture and purification of the steroid prodrugs were purchased from Sigma-Aldrich (St. Louis, MO, USA).
The product was purified by automated silica flash column chromatography (10 g SNAP column, Biotage, Uppsala, Sweden) using an ethyl acetate/dichloromethane gradient (0% to 10%, respectively, over an 8× column volume) and a collection wavelength of 228 nm. The fractions containing product were pooled and evaporated, followed by co-evaporation with dichloromethane to remove any residual ethyl acetate. The identity of the deuterated (d27) rofleponide-C14 product (85 mg, 94%) was confirmed by 1H NMR (in deuterated chloroform, Bruker 500 MHz, Bruker, Billerica, MA, USA) and electrospray ionization mass spectrometry (positive ion mode, Waters Acquity UPC2, Waters, Milford, MA, USA): δ 0.85–0.98 (m, 6H), 1.44 (h, 2H), 1.55–1.86 (m, 7H), 1.98 (dd, 1H), 2.14–2.24 (m, 1H), 2.28 (m, 1H), 2.45 (m, 6H), 4.39 (d, 1H), 4.62–4.75 (m, 2H), 4.86 (d, 1H), 4.95 (d, 1H), 5.26 (m, 1H), 6.14 (s, 1H); m/z of 707.
LNP formulation
LNPs were prepared by microfluidic mixing as described previously.30 Briefly, an ethanolic solution of the lipid components and a solution of the mRNA in RNase-free citrate buffer (pH 3, 50 mM) were mixed at a ratio of 1:3, respectively, at a total flow rate of 12 mL/min using a NanoAssemblr (Precision NanoSystems, Vancouver, BC, Canada). Following microfluidic mixing, the LNPs were dialyzed overnight against 500× sample volume of PBS (pH 7.4) using Slide-A-Lyzer G2 dialysis cassettes with a molecular weight cutoff of 10,000 (Thermo Fisher Scientific, Waltham, MA, USA).
MC3 LNPs encapsulating hFGF21 or Luc mRNA were formulated using the amino lipid dilinoleylmethyl-4-dimethylaminobutyrate (DLin-MC3-DMA, synthesized as previously described),52 cholesterol (Chol), distearoylphosphatidylcholine (DSPC), and dimyristoylphosphatidylethanolamine polyethylene glycol 2000 (DMPE-PEG 2000) at a % molar composition of 50:38.5:10:1.5, respectively, and at a total lipid/mRNA weight ratio of 10:1 (nitrogen/phosphate (N:P) ratio of 3). L608 LNPs were formulated using the amino lipid (12Z,15Z)-N,N-dimethyl-2-nonylhenicosa-12,15-dien-l-amine (L608, synthesized as described for compound 32 in Brown et al.53), Chol, DSPC, and DMPE-PEG 2000 at the same % molar composition but at a total lipid/mRNA ratio of 17:1 (N:P ratio of 6). For incorporation of steroid prodrugs within the LNPs, the compounds were dissolved in the ethanolic solution of lipids prior to microfluidic mixing. To avoid losses during formulation, parent steroid was added to the formulation after LNP manufacture.
The size and polydispersity of LNPs were determined by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK), and the encapsulation and concentration of mRNA in the LNP formulations were determined using the RiboGreen assay.54 Typically, LNPs had a particle size (Z average) of 70–100 nm with a polydispersity index of <0.2 (which was not affected by the type of ionizable cationic lipid used or incorporation of AICs), and encapsulation of mRNA was >90% (see Table S3). Endotoxin levels within the mRNA LNP formulations were typically less than 1 endotoxin unit (EU)/mL (Endosafe-PTS, Charles River Laboratories, Wilmington, MA, USA) but were not routinely measured since values were consistently low. All formulations were prepared within 1 week of testing to ensure the chemical stability of the components.
Entrapment of steroid prodrug within LNPs
To estimate the entrapment of the steroid prodrugs within LNPs, any free prodrug (in solution or crystalline) and LNPs were separated following dialysis using a size exclusion gel column (PD-10; GE Healthcare, Chicago, Il, USA). The excluded volume containing the LNPs was diluted 2-fold with a solution containing 40 mM sodium dodecyl sulfate and 1% Triton X-100 to solubilize the LNPs. The steroid prodrug and lipid content of the resulting solution were then analyzed using ultra-performance liquid chromatography combined with charged aerosol detection (Corona CAD, ESA Biosciences, Chelmsford, MA, USA) fitted with a C18 column (Waters Acquity BEH, 1.7 μm, 2.1 × 50 mm; Waters, Milford, MA, USA) run at 80°C at a flow rate of 0.5 mL/min. A mobile phase of 100 mM aqueous ammonium acetate (A) and a 50:50 mix of acetonitrile/isopropyl alcohol (B) run at a gradient profile of 90% A for 1 min, 90% A to 90% B for 4 min, followed by 5 min of 90% B was used for compound elution. The entrapment efficiency of the steroid prodrug was calculated by comparing the ratio of the measured concentration of the steroid prodrug to the measured concentration of the amino lipid (DLin-MC3-DMA or L608) to the theoretical concentration of the two components. In all cases, entrapment of steroid prodrugs within either MC3 or L608 LNPs was greater than 75%.
Distribution of steroid prodrug within LNPs
The location/distribution of one selected AIC, namely rofleponide-C14, within the hFGF21 mRNA MC3 LNP was further evaluated using SANS with isotropic contrast variation. The technique of contrast variation SANS is based on the distinct interaction that neutrons have with hydrogen (H) and deuterium (D) atoms, such that by substituting D for H (selective deuteration), it is possible to highlight different regions of the LNP structure.30 MC3 LNPs incorporating rofleponide-C14 at a prodrug/mRNA ratio of 1:1 (w/w) were therefore formulated with partially deuterated rofleponide-C14 (d27, synthesized as described above). SANS measurements were performed using the KWS-2 instrument operated by the Jülich Centre for Neutron Science (JCNS) at Forschungs-Neutronenquelle, Heinz Maier-Leibnitz.55 The measurements were performed at three different sample-to-detector distances, that is, 2, 8, and 20 m, and two neutron wavelengths, λ = 5 Å and 10 Å, with a wavelength resolution of 10%. These configurations cover the scattering vector (q) range of 0.00133 < q (Å−1) < 0.411. The measurements were done in quartz disc-shaped (“banjo”) cuvettes (Hellma, Jena, Germany) of 1- and 2-mm path length, maintained at 25°C. LNPs were diluted in the appropriate solvent ratio of H2O/D2O (15%–100% [v/v]) to a final concentration of 0.3 mg/mL mRNA (3 mg/mL lipid). The generated data were corrected for detector sensitivity/noise and solvent/empty cell contribution, taking into account the measured sample transmission.
Scattering profiles obtained in the buffers containing various proportions of D2O were fitted simultaneously (SasView software, NIST Center for Neutron Research, Gaithersburg, MD, USA) using the “core-shell sphere” model. The model describes a spherical particle comprised of a hydrated core (composed of DLin-MC3-DMA, Chol, mRNA, and 24 vol % water) and a lipid monolayer shell (composed of DSPC, DLin-MC3-DMA, Chol, and the DMPE part of DMPE-PEG 2000) as previously described for the composition/structure of MC3 LNPs.30 The parameters fitted were the radius of the core, the thickness of the outer shell, and the scattering length densities of the core and the shell. As the presence of deuterated rofleponide-C14 in either the core or in the shell of the LNP alters the scattering length density of that region, it is possible to identify the location and distribution of AIC within the LNP. The experimental SANS profiles were compared to three models generated using different values for the scattering length densities of the core and shell, calculated based on the deuterated rofleponide-C14 being only in the shell (“AIC in the shell”), only in the core (“AIC in the core”), or to have no preferential location in the particle (“AIC distributed throughout”). The best fit was obtained for the “AIC in the shell” model, where the core has a radius of approximately 27 nm and is surrounded by a shell layer of 2.4 nm, which contains most of the AIC. No layer representing the diffuse PEG surrounding the particle was included, as it did not improve the model. Schultz polydispersity (0.16) was included in the model to describe the size distribution of the LNPs.
In vivo studies
Twelve-week-old female CD1 mice were obtained from Charles River Laboratories (Sulzfeld, Germany). Mice were kept in communal cages with aspen wood chip bedding in a holding facility with a controlled environment (12-h light/12-h dark cycle, room temperature 21°C–22°C, and relative humidity 40%–60%, with free access to water and standard rodent chow [R70, Lantmännen]). Mice were acclimatized to these conditions for at least 5 days before use. All in vivo studies were approved by the Local Ethics Review Committee on Animal Experiments (Gothenburg Region).
On the day of dosing, mice were lightly anesthetized with 5% isoflurane and were administered the test treatment either i.v. or s.c. (in the intra-scapular region) at an administration volume of 5 mL/kg. Following dose administration, blood samples were collected from the saphenous vein from alternating legs at various time points up to 24 h. At termination, mice were anesthetized using isoflurane, examined for any clinical abnormalities, including edema at the injection site, and terminal blood samples were collected. Plasma was prepared by centrifugation (3,000 × g for 10 min at 4°C) and the samples were quantified for hFGF21 protein using a multiplex Luminex assay (Merck Millipore, Burlington, MA, USA, catalog no. HLPPMAG-57K). The terminal sample was also used for quantification of haptoglobin using the Milliplex map mouse acute phase panel 2 kit (Merck Millipore, Burlington, MA, USA, catalog no. MAP2MAG-76K) and quantification of cytokines/chemokines (IL-6, KC, IP-10, MCP-1) using a Milliplex Luminex assay (Merck Millipore, Burlington, MA, USA, catalog no. MCYTOMAG-70K). At termination, the injection site was collected, fixed in formalin and sectioned, stained with hematoxylin and eosin, and examined microscopically. Statistical analyses for all of the experiments were performed using a one-way ANOVA with unequal variance and adjusted p values to compare all conditions. The p values were adjusted to control the false discovery rate using the Benjamini-Hochberg procedure.56
Biodistribution study
MC3 LNPs encapsulating Luc mRNA were administered to CD1 mice either i.v. (tail vein) or s.c. (intrascapular) at a dose of 0.3 mg/kg and a dosing volume of 5 mL/kg while under light anesthesia (isoflurane). At 8, 12, 24, and 48 h (8 h only for i.v. dosing) post-administration, whole-body scans of the mice were collected using an IVIS Spectrum (PerkinElmer, Waltham, MA, USA). Twenty minutes prior to imaging each mouse received a 150 mg/kg dose of luciferin (RediJect d-luciferin, PerkinElmer, Waltham, MA, USA) administered s.c. at a dosing volume of 5 mL/kg. At the 8, 24, and 48 h time points, n = 5–8 mice were euthanized and organs were extracted (liver, spleen, kidneys, lung, axillary/brachial lymph nodes, and tissue surrounding injection site) and imaged ex vivo using the IVIS Spectrum. After imaging, the organs were separately stored in formalin for immunohistological evaluation to evaluate cellular distribution of transfection.
Studies to explore systemic exposure and pharmacokinetics of steroids
The pharmacokinetic properties of rofleponide and budesonide after i.v. tail vein (parent steroid only) or s.c. intrascapular injection (parent steroid and selected ester prodrugs) were evaluated in CD1 mice. Serial blood samples were collected from the saphenous vein at varying time points after dosing and a terminal sample was collected at 24 h after dosing. Blood samples were quantified for rofleponide and budesonide using an Acquity UPLC I-class system and a Xevo TQ-XS triple quadrupole mass spectrometer (Waters, Milford, MA, USA). The lower limit of quantification was 0.1 nmol/L for both rofleponide and budesonide. Pharmacokinetic parameters were calculated using non-compartment analysis in Phoenix 6.4 (Certara, St. Louis, MO, USA).
Acknowledgments
We thank Moderna, Inc., for the provision of mRNA.
Author contributions
N.D., D.H., N.E., A. Dahlén, A.H., F.S., E.H., S.B., E.A., N.B., and S.A. were members of the cross-functional team within AstraZeneca who were involved in conceptualizing and designing all studies, in data analysis/interpretation, and in overall execution of the project. N.E., A.H., E.H., A. Dahlén, and S.A. were additionally responsible for resourcing and coordinating the work. A. Dahlén and P.N. synthesized the ionizable lipids and the rofleponide/budesonide prodrugs used in the investigations. T.K., Y.J., L.L., A.H., and N.D. were responsible for coordination, manufacturing, characterization, and supply of LNP formulations to the work described. A.R., M.Y.A., A. Dabkowska, and L.L. contributed to the design, preparation of samples, and analytics as well as the execution and interpretation of the data for the SANS investigations. S.B. was responsible for the design and conduct of all in vitro studies used to support the investigations and summarizing/interpreting the results. D.H., N.E., C.J., E.H., J.L., L.B., M.J., and A.-S.S. coordinated and conducted in vivo studies and were responsible for bioanalysis and result generation. L.H. and M.J. were responsible for and conducted the biodistribution study. F.S. and N.E. analyzed and summarized the data from in vivo studies with regard to safety, immunology, and histology. N.D. was largely responsible for authoring the manuscript, with contributions from all co-authors according to areas of expertise. S.A. was the project leader and coordinated the project together with E.A.
Declaration of interests
L.L. and T.K. are authors on the patent WO2017/194454 (A1) relating to lipid nanoparticles comprising lipophilic anti-inflammatory agents and methods of use thereof. All authors were at the time of study employees of AstraZeneca.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.03.008.
Supplemental information
References
- 1.Sahin U., Karikó K., Türeci Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 2.Kaczmarek J.C., Kowalski P.S., Anderson D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017;9:60. doi: 10.1186/s13073-017-0450-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zangi L., Lui K.O., von Gise A., Ma Q., Ebina W., Ptaszek L.M., Später D., Xu H., Tabebordbar M., Gorbatov R. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013;31:898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.An D., Schneller J.L., Frassetto A., Liang S., Zhu X., Park J.S., Theisen M., Hong S.J., Zhou J., Rajendran R. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21:3548–3558. doi: 10.1016/j.celrep.2017.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schrom E., Huber M., Aneja M., Dohmen C., Emrich D., Geiger J., Hasenpusch G., Herrmann-Janson A., Kretzschmann V., Mykhailyk O. Translation of angiotensin-converting enzyme 2 upon liver- and lung-targeted delivery of optimized chemically modified mRNA. Mol. Ther. Nucleic Acids. 2017;7:350–365. doi: 10.1016/j.omtn.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J.B., Karmali P.P., Chivukula P., Verma I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA. 2017;114:E1941–E1950. doi: 10.1073/pnas.1619653114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richner J.M., Himansu S., Dowd K.A., Butler S.L., Salazar V., Fox J.M., Julander J.G., Tang W.W., Shresta S., Pierson T.C. Modified mRNA vaccines protect against Zika virus infection. Cell. 2017;168:1114–1125.e10. doi: 10.1016/j.cell.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson S., Antonsson M., Elebring M., Jansson-Löfmark R., Weidolf L. Drug metabolism and pharmacokinetic strategies for oligonucleotide- and mRNA-based drug development. Drug Discov. Today. 2018;23:1733–1745. doi: 10.1016/j.drudis.2018.05.030. [DOI] [PubMed] [Google Scholar]
- 9.Carlsson L., Clarke J.C., Yen C., Gregoire F., Albery T., Billger M., Egnell A.C., Gan L.M., Jennbacken K., Johansson E. Biocompatible, purified VEGF-A mRNA improves cardiac function after intracardiac injection 1 week post-myocardial infarction in swine. Mol. Ther. Methods Clin. Dev. 2018;9:330–346. doi: 10.1016/j.omtm.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weng Y., Li C., Yang T., Hu B., Zhang M., Guo S., Xiao H., Liang X.J., Huang Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020;40:107534. doi: 10.1016/j.biotechadv.2020.107534. [DOI] [PubMed] [Google Scholar]
- 11.Kanasty R., Dorkin J.R., Vegas A., Anderson D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 12.Kotterman M.A., Schaffer D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajj K.A., Whitehead K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017;2:17056. [Google Scholar]
- 14.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kauffman K.J., Dorkin J.R., Yang J.H., Heartlein M.W., DeRosa F., Mir F.F., Fenton O.S., Anderson D.G. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 2015;15:7300–7306. doi: 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- 16.Pardi N., Parkhouse K., Kirkpatrick E., McMahon M., Zost S.J., Mui B.L., Tam Y.K., Karikó K., Barbosa C.J., Madden T.D. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018;9:3361. doi: 10.1038/s41467-018-05482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroeder A., Levins C.G., Cortez C., Langer R., Anderson D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010;267:9–21. doi: 10.1111/j.1365-2796.2009.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue H.Y., Guo P., Wen W.-C., Wong H.L. Lipid-based nanocarriers for RNA delivery. Curr. Pharm. Des. 2015;21:3140–3147. doi: 10.2174/1381612821666150531164540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 20.Alberer M., Gnad-Vogt U., Hong H.S., Mehr K.T., Backert L., Finak G., Gottardo R., Bica M.A., Garofano A., Koch S.D. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet. 2017;390:1511–1520. doi: 10.1016/S0140-6736(17)31665-3. [DOI] [PubMed] [Google Scholar]
- 21.Hassett K.J., Benenato K.E., Jacquinet E., Lee A., Woods A., Yuzhakov O., Himansu S., Deterling J., Geilich B.M., Ketova T. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids. 2019;15:1–11. doi: 10.1016/j.omtn.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sedic M., Senn J.J., Lynn A., Laska M., Smith M., Platz S.J., Bolen J., Hoge S., Bulychev A., Jacquinet E. Safety evaluation of lipid nanoparticle-formulated modified mRNA in the Sprague-Dawley rat and cynomolgus monkey. Vet. Pathol. 2018;55:341–354. doi: 10.1177/0300985817738095. [DOI] [PubMed] [Google Scholar]
- 23.Zhong Z., Mc Cafferty S., Combes F., Huysmans H., De Temmerman J., Gitsels A., Vanrompay D., Portela Catani J., Sanders N.N. mRNA therapeutics deliver a hopeful message. Nano Today. 2018;23:16–39. [Google Scholar]
- 24.Abrams M.T., Koser M.L., Seitzer J., Williams S.C., DiPietro M.A., Wang W., Shaw A.W., Mao X., Jadhav V., Davide J.P. Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: Effect of dexamethasone co-treatment. Mol. Ther. 2010;18:171–180. doi: 10.1038/mt.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen S., Zaifman J., Kulkarni J.A., Zhigaltsev I.V., Tam Y.K., Ciufolini M.A., Tam Y.Y.C., Cullis P.R. Dexamethasone prodrugs as potent suppressors of the immunostimulatory effects of lipid nanoparticle formulations of nucleic acids. J. Control. Release. 2018;286:46–54. doi: 10.1016/j.jconrel.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Lindfors L., Kjellman T. 2017. Lipid nanoparticles comprising lipophilic anti-inflammatory agents and methods of use thereof. WIPO patent application publication WO2017194454A1, filed May 8, 2017, published November 16, 2017. [Google Scholar]
- 27.Kumar V., Qin J., Jiang Y., Duncan R.G., Brigham B., Fishman S., Nair J.K., Akinc A., Barros S.A., Kasperkovitz P.V. Shielding of lipid nanoparticles for siRNA delivery: impact on physicochemical properties, cytokine induction, and efficacy. Mol. Ther. Nucleic Acids. 2014;3:e210. doi: 10.1038/mtna.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnes P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. (Lond.) 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 29.Edsbäcker S., Johansson C.J. Airway selectivity: An update of pharmacokinetic factors affecting local and systemic disposition of inhaled steroids. Basic Clin. Pharmacol. Toxicol. 2006;98:523–536. doi: 10.1111/j.1742-7843.2006.pto_355.x. [DOI] [PubMed] [Google Scholar]
- 30.Yanez Arteta M., Kjellman T., Bartesaghi S., Wallin S., Wu X., Kvist A.J., Dabkowska A., Székely N., Radulescu A., Bergenholtz J., Lindfors L. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA. 2018;115:E3351–E3360. doi: 10.1073/pnas.1720542115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cray C., Besselsen D.G., Hart J.L., Yoon D., Rodriguez M., Zaias J., Altman N.H. Quantitation of acute phase proteins and protein electrophoresis in monitoring the acute inflammatory process in experimentally and naturally infected mice. Comp. Med. 2010;60:263–271. [PMC free article] [PubMed] [Google Scholar]
- 32.Akinc A., Querbes W., De S., Qin J., Frank-Kamenetsky M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Cantley W.L., Dorkin J.R. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraemer F.B., Sather S.A., Park B., Sztalryd C., Natu V., May K., Nishimura H., Simpson I., Cooper A.D., Cushman S.W. Low density lipoprotein receptors in rat adipose cells: subcellular localization and regulation by insulin. J. Lipid Res. 1994;35:1760–1772. [PubMed] [Google Scholar]
- 34.Blakney A.K., Deletic P., McKay P.F., Bouton C.R., Ashford M., Shattock R.J., Sabirsh A. Effect of complexing lipids on cellular uptake and expression of messenger RNA in human skin explants. J. Control. Release. 2021;330:1250–1261. doi: 10.1016/j.jconrel.2020.11.033. [DOI] [PubMed] [Google Scholar]
- 35.Zatsepin T.S., Kotelevtsev Y.V., Koteliansky V. Lipid nanoparticles for targeted siRNA delivery—Going from bench to bedside. Int. J. Nanomedicine. 2016;11:3077–3086. doi: 10.2147/IJN.S106625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang F., Lindgren G., Lin A., Thompson E.A., Ols S., Röhss J., John S., Hassett K., Yuzhakov O., Bahl K. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol. Ther. 2017;25:2635–2647. doi: 10.1016/j.ymthe.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Groot A.M., Thanki K., Gangloff M., Falkenberg E., Zeng X., van Bijnen D.C.J., van Eden W., Franzyk H., Nielsen H.M., Broere F. Immunogenicity testing of lipidoids in vitro and in silico: Modulating lipidoid-mediated TLR4 activation by nanoparticle design. Mol. Ther. Nucleic Acids. 2018;11:159–169. doi: 10.1016/j.omtn.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kupper T.S., Fuhlbrigge R.C. Immune surveillance in the skin: Mechanisms and clinical consequences. Nat. Rev. Immunol. 2004;4:211–222. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitehead K.A., Dorkin J.R., Vegas A.J., Chang P.H., Veiseh O., Matthews J., Fenton O.S., Zhang Y., Olejnik K.T., Yesilyurt V. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun. 2014;5:4277. doi: 10.1038/ncomms5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao W., Mao X., Davide J.P., Ng B., Cai M., Burke P.A., Sachs A.B., Sepp-Lorenzino L. Mechanistically probing lipid-siRNA nanoparticle-associated toxicities identifies Jak inhibitors effective in mitigating multifaceted toxic responses. Mol. Ther. 2011;19:567–575. doi: 10.1038/mt.2010.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suhr O.B., Coelho T., Buades J., Pouget J., Conceicao I., Berk J., Schmidt H., Waddington-Cruz M., Campistol J.M., Bettencourt B.R. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015;10:109. doi: 10.1186/s13023-015-0326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonaventura A., Montecucco F. Steroid-induced hyperglycemia: An underdiagnosed problem or clinical inertia? A narrative review. Diabetes Res. Clin. Pract. 2018;139:203–220. doi: 10.1016/j.diabres.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 44.van Raalte D.H., Ouwens D.M., Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? Eur. J. Clin. Invest. 2009;39:81–93. doi: 10.1111/j.1365-2362.2008.02067.x. [DOI] [PubMed] [Google Scholar]
- 45.Elena C., Chiara M., Angelica B., Chiara M.A., Laura N., Chiara C., Claudio C., Antonella F., Nicola G. Hyperglycemia and diabetes induced by glucocorticoids in nondiabetic and diabetic patients: Revision of literature and personal considerations. Curr. Pharm. Biotechnol. 2018;19:1210–1220. doi: 10.2174/1389201020666190102145305. [DOI] [PubMed] [Google Scholar]
- 46.Veenstra D.L., Best J.H., Hornberger J., Sullivan S.D., Hricik D.E. Incidence and long-term cost of steroid-related side effects after renal transplantation. Am. J. Kidney Dis. 1999;33:829–839. doi: 10.1016/s0272-6386(99)70414-2. [DOI] [PubMed] [Google Scholar]
- 47.Tsuzuki W., Ue A., Nagao A., Akasaka K. Fluorimetric analysis of lipase hydrolysis of intermediate- and long-chain glycerides. Analyst (Lond.) 2002;127:669–673. doi: 10.1039/b110038f. [DOI] [PubMed] [Google Scholar]
- 48.Roers A., Hiller B., Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44:739–754. doi: 10.1016/j.immuni.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Anderson B.R., Muramatsu H., Nallagatla S.R., Bevilacqua P.C., Sansing L.H., Weissman D., Karikó K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010;38:5884–5892. doi: 10.1093/nar/gkq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohto T., Konishi M., Tanaka H., Onomoto K., Yoneyama M., Nakai Y., Tange K., Yoshioka H., Akita H. Inhibition of the inflammatory pathway enhances both the in vitro and in vivo transfection activity of exogenous in vitro-transcribed mRNAs delivered by lipid nanoparticles. Biol. Pharm. Bull. 2019;42:299–302. doi: 10.1248/bpb.b18-00783. [DOI] [PubMed] [Google Scholar]
- 52.Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown D., Cunningham J.J., Gindy M., Pickering V., Stanton M.G., Stirdivant S.M., Strapps W. 2012. RNA interference mediated inhibition of Catenin (cadherin-associated protein), beta 1 (CTNNB1) gene expression using short interfering nucleic acid (siNA). WIPO patent application publication WO2012018754, filed February 8, 2011, published September 2, 2012. [Google Scholar]
- 54.Jones L.J., Yue S.T., Cheung C.-Y., Singer V.L. RNA quantitation by fluorescence-based solution assay: RiboGreen reagent characterization. Anal. Biochem. 1998;265:368–374. doi: 10.1006/abio.1998.2914. [DOI] [PubMed] [Google Scholar]
- 55.Radulescu A., Pipich V., Frielinghaus H., Appavou M.S. KWS-2, the high intensity / wide Q-range small-angle neutron diffractometer for soft-matter and biology at FRM II. J. Phys. Conf. Ser. 2012;351:012026. [Google Scholar]
- 56.Benjamini Y., Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.