

Abstract
Background
This study sought to compare the therapeutic effects of angiotensin II (ANG II) and norepinephrine (NE) on cecal ligation and puncture (CLP)-induced septic acute kidney injury (AKI) in rats.
Methods
Sepsis shock was induced in anesthesia Sprague-Dawley male rats by CLP model for 24 hours. A total of 40 rats were divided into five groups, including control group, sham group, CLP group, CLP + ANG II group, and CLP + NE group. CLP + ANG II and CLP + NE group were administration of ANG II or NE after sepsis shock respectively, maintaining the MAP at 75–85 mmHg. CLP group was administration of saline for contrast. At 0, 18, 24 hours measured the renal blood grades and resistant index (RI) by ultrasound equipment. At 6, 12, 18 and 24 hours collected 0.5 mL blood sample for creatinine and lactic acid examination. Rats were observed for 24 hours after CLP procedure and then sacrificed for subsequent examination, rat serum were used to determine the levels of inflammatory response factors, kidney tissues were used to examine the oxidative stress factors and mitochondrial related proteins.” We added the sentence as following: “The AMPK, PGC-1α and NRF-2 expression in renal cortex was significantly increased in the CLP + ANG II group.
Results
Compared to the vehicle treatment, both ANG II and NE administration restored the decrease in the mean arterial pressure (MAP) and alleviated mitochondrial impairments in CLP rats. However, only ANG II alleviated CLP-induced abnormalities in serum creatinine and lactic acid concentrations, renal blood flow, the renal resistant index, renal histopathology, the production of proinflammatory cytokines, and oxidative stress markers in rats. ANG II was also found to be superior to NE in reversing the CLP-induced suppression of mitochondrial biogenesis-related protein expression in the kidneys of rats.
Conclusions
ANG II was better than NE in alleviating CLP-induced septic AKI in rats.
Keywords: Septic shock, acute kidney injury (AKI), angiotensin II, norepinephrine (NE), the AMPK/PGC-1α/NRF-2 pathway
Introduction
Sepsis is a fatal multiple organ dysfunction caused by an inflammatory response to infection. It has been reported that approximately 31.5 million sepsis cases and 19.4 million deaths due to sepsis occur worldwide each year (1). Nearly 50% of patients with sepsis develop acute kidney injury (AKI). Sepsis patients with AKI have a significantly higher mortality rate than patients without AKI. Sepsis leading to life-threatening low blood pressure is called septic shock (2). Effective fluid resuscitation is essential for maintaining organ perfusion during the treatment of AKI patients with septic shock (3,4). When fluid resuscitation fails to restore arterial blood pressure, vasopressor administration is required to treat hypotension. A drug that not only maintains the mean arterial pressure (MAP) but also alleviates the kidney injury would be an ideal treatment for patients with septic shock associated with AKI.
Norepinephrine (NE), a tyrosine-derived sympathomimetic amine, is a first-line agent for treating hypotension in septic shock; however, the long-term use of NE may lead to complications, such as myocardial infarction and arrhythmia (2). It has been reported that at doses that cause similar increases in arterial pressure during sepsis, the effects of NE on renal function are less potent than those of angiotensin II (ANG II) (5). ANG II is an endogenous vasopressin that belongs to the renin-angiotensin-aldosterone system. During sepsis shock, the decline in effective circulation volume and the MAP trigger the release of angiotensin, and the renin-angiotensin-mediated constriction of efferent arterioles maintains the MAP and glomerular filtration pressure without affecting glomerular perfusion pressure and renal oxygenation (6). In a pig sepsis model, researchers found that ANG II reversed hypotension caused by sepsis and that ANG II and NE were comparable in improving renal blood flow (7). Thus, we hypothesized that compared to NE, ANG II might provide better renal protection with similar vasoconstriction effects in the treatment of AKI resulting from septic shock.
During sepsis, pathogen-associated molecular patterns filter through the kidney glomerulus or peritubular capillaries, resulting in an increased production of proinflammatory cytokines, oxidative stress, and mitochondrial injury in tubular epithelial cells (8). Recent research has identified mitochondria dysfunction as a critical contributor to the pathogenesis of AKI; the restoration of mitochondrial mass and function is essential to the recovery of renal function (9). Peroxisome proliferator-activated receptor-γ coactivator-1-α (PGC-1α) is a master positive regulator of mitochondrial biogenesis. Adenosine monophosphate-activated protein kinase (AMPK) activates PGC-1α through phosphorylation, and the activated PGC-1α translocates into the nucleus where it activates nuclear respiratory factor 2 (NRF2), which promotes mitochondrial biogenesis (10). ANG II has been shown to contribute to mitochondrial dysfunction and cardiac injury in rats (11); however, a recent study demonstrated that ANG II has no adverse effects on mitochondrial respiration (12). In this study, the application of angiotensin II and norepinephrine in pig septic shock model found that both groups improved cardiac output, alleviate shock, improved renal blood flow and perfusion in septic pigs. In this study, animal ultrasound was used to evaluate the renal blood flow and renal resistance index of rats, so as to better simulate the examination method of clinical critical patients. At the same time, the renal mitochondrial function was studied from the aspect of molecular pathway to observe the changes of inflammatory response, oxidative stress and mitochondrial related protein expression after sepsis induced acute renal injury. Thus, the effects of ANG II on mitochondrial health and the underlying mechanisms warrant further exploration.
In the present study, we examined the effects of ANG II and NE on renal function, kidney structure, and renal hemodynamics in a rat model of septic AKI, and attempted to identify the potential mitochondrial mechanisms involved. We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-21-621).
Methods
Animals
The animal study was approved by the Ethics Committee for Laboratory Animals Care and Use of Hebei Medical University, Fourth Affiliated Hospital and Hebei Provincial Tumor Hospital (Approval Number: IACUC-4th Hos Hebmu). All the procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (1985, National Institutes of Health). Male Sprague-Dawley rats, aged 8–10 weeks, and weighing 230–250 g, were obtained from the Beijing Vital River Laboratory Animal Technology (China), and housed in standard conditions (12-h light/dark cycles at 22±2 °C, 60% humidity) with free access to food and water. After a week of acclimation and a day of starvation with access only to tap water, the rats were randomly divided into one of the following 5 groups (n=8 per group): the control, sham, CLP, CLP + ANG II, and CLP + NE groups.
All of the rats were anesthetized with sodium phenobarbital (50 mg/kg). An incision was made in the middle of the neck of the rat to expose the right internal carotid artery and the right external jugular vein. Catheters (Skillsmodel Biotechnology Company, Beijing, China) were inserted into the jugular vein for blood collection, and drug administration and into the carotid artery for blood pressure and heart rate monitoring. The CLP operation was performed as previously described (13) in the CLP, CLP + ANG II and CLP + NE groups. Briefly, an incision was made in the middle of the abdomen to expose the cecum. A ligation was made 0.5 cm below the ileocecal valve using a 4-0 silk ligature. The cecum was transversely punctured twice at opposite ends using an 18-gauge needle. A 1 cm3 volume of feces was extruded from each puncture point. After returning the cecum to the abdominal cavity, the incision was closed. The sham rats were subjected to the same procedures as the CLP rats, but without ligation or punctures. The control rats only received an incision to the right internal carotid artery and a catheterization of the right external jugular vein; no abdominal operation was performed.
At 18 h after surgery, when the MAP decreased to 20% of the baseline, the heart rate of increased by 20% over the control value, at this point, we evaluated the rat sepsis model was successful. The rats in the CLP, CLP + ANG II and CLP + NE groups received fluid resuscitation (20 mL/kg normal saline within 30 min) (14), and used the syringe pump (STOELTING company, USA) to treat with vehicle (normal saline), ANG II (initial dose: 66.7 ng/kg/min), or NE (initial dose: 0.05 µg/kg/min) via an intravenous catheter for 6 hours until euthanasia at 24 h, respectively. The mean blood pressure was maintained at 75–85 mmHg during the procedure. The rats were anesthetized with sodium phenobarbital (50 mg/kg), performed euthanasia by thoracotomy and take blood from the left ventricle.
Ultrasound examination of the kidneys
The left kidney of each rat was examined before the CLP and at 18 and 24 h after the CLP procedure using a Doppler ultrasonography (VEVO 2100; Visual Sonics, Canada) with a MS250 probe (24 MHz). Blood flow was assessed using a color doppler, and scored as follows: 0—no signals in the kidney; 1—some signals at the renal hilum; 2—obvious signals at the renal hilum and a few signals at the junction of the renal cortex and medulla; and 3—signals throughout the kidney, reaching the level of arcuate artery. The peak systolic velocity (PSV) and the end-diastolic velocity (EDV) of three continuous waves were measured using a pulse doppler, and the average values were calculated. Three different interlobar arteries were measured and averaged again. The resistance index (RI) was calculated using the following formula: (PSV – EDV)/PSV.
Measurement of hemodynamic and biochemical parameters
The MAP was continuously monitored using a physiological signal acquisition system (Powerlab, Australia), and data were recorded hourly. Venous blood samples (0.5 mL) were obtained at 0, 6, 12, 18, and 24 h after the experiment began, and were centrifuged at 4,000 rpm for 15 min at 4 °C for serum collection. A 20 µL volume of serum was used to measure the levels of lactic acid and creatinine using biochemical analysis kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) in accordance with the manufacturer’s instructions. The rats were euthanized using an overdose of pentobarbital (100 mg/kg, intravenously) at 24 h after the start of the experiment, and the kidneys were excised immediately. The capsule of one kidney was obtained for pathological analysis; the cortex and medulla of the other kidney were collected under a microscope and stored at −80 °C for further experiments.
Transmission microscopy examination
The rats were euthanized, and the kidneys were immediately excised and placed on ice. The renal cortex tissue samples (of approximately 1 mm3) were fixed with 2.5% glutaraldehyde in 0.01 mol/L phosphate buffer at 4 °C, followed by incubation with 2% osmium tetroxide. The sample was then dehydrated in graded ethanol solutions, embedded in paraffin, sliced, and stained with 3% uranyl acetate and lead citrate. The slices were observed using a transmission electron microscope (HT7700; HITACHI, Japan) at 80 kV.
Histological examination
The kidney tissue samples were fixed using embedding fixative for over 24 h, embedded in paraffin, cut into 4-µm-thick sections, and stained with hematoxylin and eosin. Tubular damage, including cellular degeneration and vacuolization, the reduction of the brush border epithelium, tubular obstruction, and cast formation, were evaluated in five randomly selected fields in the outer medulla by independent pathologists double-blinded to the experiment conditions. The tubular injury scores were defined as previously described (15): 0—normal; 1—<25% of the tubular area affected; 2—damage to 25–50% of the tubular area; 3—damage to 50–75% of the tubular area; and 4—damage to 75–100% of the tubular area.
Measurement of inflammation and oxidative stress indicators
Interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α) concentrations in kidney tissue samples were measured using enzyme-linked immunosorbent assay kits (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s instructions. Superoxide dismutase (SOD), malondialdehyde (MDA), glutathione peroxidase (GSH-PX) levels in the kidney tissue samples were measured using biochemical analysis kits (Nanjing Jiancheng Bioengineering Institute) in accordance with the manufactures’ instructions.
Western blotting analysis
Excised samples of renal cortical tissue were homogenized using an ice-cold Radio Immunoprecipitation Assay (RIPA) buffer containing phosphatase inhibitors. Proteins were isolated using a total protein extraction kit (Solarbio, Beijing, China) and quantified using a bicinchoninic acid (BCA) kit (Solarbio). After 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels, proteins were transferred onto polyvinylidene fluoride membranes (Millipore-Upstate, Beijing, China, 0.45 µm). The membranes were blocked with 5% albumin (BioShop Canada Inc., Burlington, ON, USA) in tris-buffered solution containing 0.1% Tween-20 (TBST) for 1 h at 37 °C, and incubated with primary antibody against AMPK (1:500 catalog number 10929-2-AP AMPK alpha 1 Rabbit Polyclonal antibody, Proteintech), P-AMPK [1:500; phospho-AMPK (Thr172) (D4D6D), Cell Signaling Technology, USA], PGC-1α, (1:1,000; catalog number A-12348, PGC-1α Polyclonal Antibody, Affinity Company, China), nuclear factor erythroid 2-related factor 2 (NRF-2) (1:1,000; catalog number YT3189, NRF2 Polyclonal Antibody, Immunoway Company, China), or beta (β)-actin (1:2,000; catalog number 20536-1-AP Beta-Actin Rabbit Polyclonal antibody, Proteintech, China) overnight at 4 °C. After washing with TBST (15 min ×3), the membranes were incubated with the appropriate secondary antibody (Proteintech) at room temperature for 2 h and washed with TBST (15 min ×3). The protein bands were developed using an enhanced chemiluminescence (ECL) detection kit and visualized using an Amersham Imager 600. The band intensities were quantified using Image J software (Scion Corporation).
Statistical analysis
Data are presented as mean ± standard error (SE) of the mean. A survival analysis was conducted using Kaplan-Meier and Log rank tests. Physiological parameters, renal blood flow, and the RI among different groups were compared using an independent sample t-test. An intra-group comparison was performed using a paired t-test. Other parameters, including inflammation and oxidative stress indicators, renal tubular injury scores, and protein levels detected by Western blotting, were analyzed by a one-way analysis of variance, followed by the Newman-Keuls post-hoc test. A P value <0.05 was considered statistically significant.
Results
ANG II or NE administration restored the decreased the MAP in CLP rats
In the CLP group, 3 rats died at 19, 20, and 23 h following CLP, respectively. In the CLP + ANG II group, 2 rats died at 19 and 23 h after CLP, respectively. In the CLP + NE group, 2 rats died at 19 and 23 h after CLP, respectively. All rats in the control and sham groups survived. No significant differences were observed in overall survival among the groups (P>0.05; see Figure 1).
Figure 1.
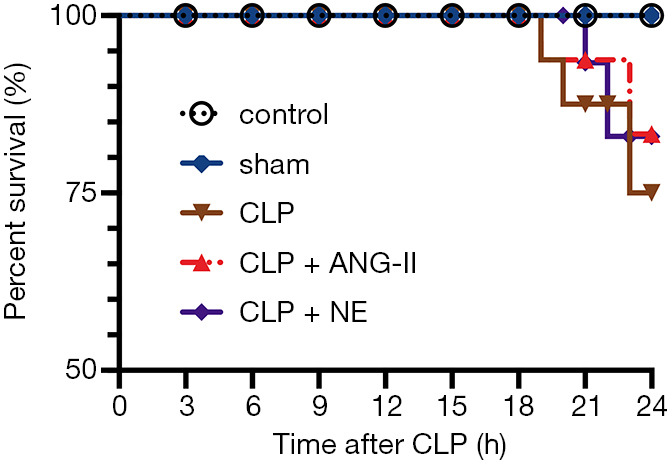
Comparison of the survival rates. Rats were randomly divided into 5 groups (n=8 per group), including the control, sham, cecal ligation and puncture (CLP) + vehicle, CLP + angiotensin II (ANG II), and CLP + norepinephrine (NE) groups. Rats were treated with vehicle, ANG II, or NE 18 h after CLP and euthanized 24 h after CLP. The survival rates were analyzed using Kaplan-Meier curves. Data are expressed as the mean percentage of rats that survived at the end of the experiment.
Compared with the control or sham rats, the MAP was significantly decreased in all CLP rats at 18 h after CLP (P<0.001). The rats were treated with vehicle, ANG II (66.7–1,000 ng/kg/min), or NE (0.05–1.0 µg/kg/min) 18 h after CLP to maintain the MAP at 75–85 mmHg. Compared with vehicle treatment, ANG II or NE administration rapidly and significantly restored the MAP (P<0.01) within 2 h after treatment. No significant differences were observed in the MAP between the ANG II- and NE-treated groups (see Figure 2), suggesting that ANG II showed comparable vasoconstrictive effects in response to NE in rats with septic shock.
Figure 2.

The effects of ANG II and NE on the MAPs and biochemical parameters in rats. (A) The MAPs were recorded every 2 hours after the experiment start; (B,C) doses of ANG II and NE were recorded every hour after treatment; (D,E) serum levels of creatinine and lactic acid were measured every 6 h after the experiment start. The Lac concentration of CLP group increased continuously at 24 h after the CLP procedure (P=0.004 vs. 18 h after procedure). The After administration of ANG II, Lac concentration decreased at 24 h after CLP procedure (P=0.005 vs. CLP group), but the effect was not the same in the CLP + NE group (P=1.000 vs. CLP group). Data are expressed as mean ± SEM. CLP; n=8 in the control or sham, n=5 in CLP, n=6 in CLP + ANG II, n=6 in CLP + NE. **, at the 24 h after procedure, compare with the CLP group, P<0.01; ##, at the 24 h after procedure, compare with the CLP group, P<0.05.
Administration of ANG II, but not NE, alleviated CLP-induced changes in serum biochemical parameters in rats
Septic AKI exhibited biochemical abnormalities indicative of impaired renal functions (16). Compared to control or sham-operated rats, all CLP-treated rats exhibited significantly higher serum creatinine and lactic acid concentrations 18 h after CLP (all P<0.05). Compared with vehicle treatment, the administration of ANG II (P<0.01), but not NE (P>0.05), significantly reduced the elevated serum creatinine and lactic acid concentrations in CLP rats (see Figure 2D,E). These data suggested that ANG II, but not NE, ameliorated renal function in CLP rats.
Administration of ANG II, but not NE, improved renal blood flow and the renal RI in CLP rats
We then examined the effects of ANG II and NE on renal blood flow and the renal RI in CLP rats. At 18 h after CLP, all CLP rats showed remarkably reduced renal blood flow and a higher RI than the control or sham-operated rats (all P<0.01). Compared to the vehicle treatment, the administration of ANG II, but not NE, significantly restored the renal blood flow while reducing the RI in CLP rats (for ANG II, all P<0.01; for NE, all P>0.05; see Figure 3A,B,C,D). These data suggested that ANG II increased renal perfusion more than NE in CLP rats.
Figure 3.

Effects of ANG II and NE on renal blood flow and the renal RI. The renal blood flow (A and C) and RI (B and D) of each rat were measured 18 and 24 h after the start of the experiment. Representative ultrasound images are shown. Data are expressed as the mean ± SEM. n=8 in the control or sham, n=5 in CLP, n=6 in CLP + ANG II, n=6 in CLP + NE.
Administration of ANG II, but not NE, mitigated renal histopathological damage in CLP rats
Next, we examined the effects of ANG II and NE on CLP-induced histopathological damage in rat kidneys. As Figure 4A and B show, the CLP rats exhibited vacuolar degeneration, renal tubular dilatation, and inflammatory infiltration, and had dramatically higher tubular injury scores than the control or sham rats (all P<0.001). These results indicated that severe renal damage was induced by CLP. Compared to the vehicle treatment, ANG II administration significantly alleviated the histopathological damage and reversed increases in tubular injury scores (P<0.01) in CLP rats. No significant changes were observed in the NE-treated rats compared to the vehicle-treated rats (P>0.05). These results indicated that the administration of ANG II, but not NE, mitigated the renal histopathological damage induced in CLP rats.
Figure 4.

Effects of ANG II and NE on histopathological injury in rat kidneys. Rats were euthanized 24 hours after the experiment commenced. (A) Hematoxylin and eosin staining was performed to evaluate renal injury. Representative images are shown. Magnification ×400. (B) The histopathological scores were assessed in 5 randomly selected fields. Data are expressed as the mean ± SEM. n=8 in the control or sham, n=5 in CLP, n=6 in CLP + ANG II, n=6 in CLP + NE.
Administration of ANG II, but not NE, abrogated the CLP-induced production of proinflammatory cytokine and oxidative stress markers
Sepsis triggers an excessive production of proinflammatory cytokines in the circulating blood, inducing oxidative stress and cell apoptosis in the kidney. As Figure 5A and B show, CLP-treated rats had significantly higher serum IL-6 and TNF-α levels than control or sham rats (IL-6, P<0.05; TNF-α, P<0.001). Treatment with ANG II, but not NE, partially, but significantly abrogated the CLP-induced elevation of IL-6 and TNF-α production, compared to vehicle treatment alone (for ANG II, all P<0.05; for NE, all P>0.05). A similar trend was observed for MDA activity (see Figure 5C). Contrasting results were observed in both SOD and GSH-PX activities (see Figure 5D,E). These data suggested that ANG II treatment, but not NE treatment, abrogated the CLP-induced inflammatory response and oxidative stress in rats.
Figure 5.

Effects of ANG II and NE on the production of proinflammatory cytokines and oxidative stress markers. The rats were euthanized 24 h after the start of the experiment. (A,B) Serum levels of IL-6 and TNF-α were measured; (C,D,E) the MDA, SOD, and GSH-PX activities were measured. Data are expressed as the mean ± SEM. n=8 in the control or sham, n=5 in the CLP, n=6 in the CLP + ANG II, n=6 in the CLP + NE. IL-6 groups, interleukin 6; TNF-α, tumor necrosis factor alpha; MDA, malondialdehyde; SOD, superoxide dismutase; GSH-PX, glutathione peroxidase.
ANG II and NE alleviated mitochondrial impairment in CLP rats
To examine the effects of ANG II and NE treatment on CLP-induced mitochondrial dysregulation, we examined the ultrastructure of the mitochondrial reticulum in the renal cortex. Transmission electron microscopy revealed that CLP-induced mitochondrial impairment, such as mitochondria swelling, outer membrane protrusion, and crista structure disruption, were significantly more alleviated in ANG II- or NE-treated rats than vehicle-treated rats (see Figure 6).
Figure 6.
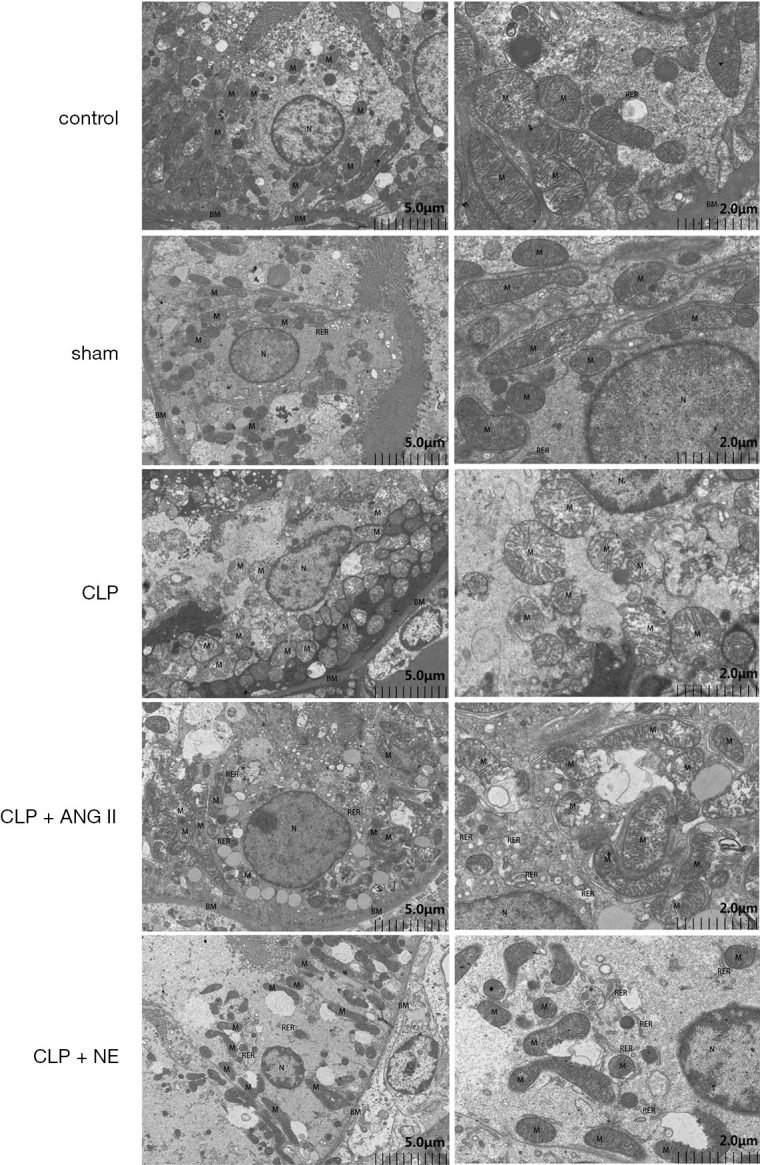
Effects of ANG II and NE on mitochondrial impairment. Rats were euthanized 24 h after the experiment commenced. A transmission electron microscopy analysis was performed to assess mitochondrial impairment in rat renal cortex tissue samples. Representative images are shown. control and sham group: proximal tubular epithelial cells, with abundant cytoplasmic matrix, continuous and complete basement membrane (BM), uniform thickness and relatively normal structure. The mitochondria (M) were round or rod-shaped, with normal size, large number, clear structure, clear cristae and well-arranged matrix. CLP: epithelial cells of proximal tubules, with severe cytoplasmic edema, obvious weakening of intracellular matrix, local hyperplasia and multilayered basement membrane (BM). Mitochondria (M) swelled and became larger and rounder, more mitochondria cristae arranged irregularly, some mitochondria membrane damaged, inner cristae ruptured, matrix became shallower, and a few mitochondria appeared vacuolar degeneration. CLP + ANG II group: proximal tubular epithelial cells showed moderate cytoplasmic edema, slightly lighter intracellular matrix, more vacuoles, and continuous and complete basement membrane (BM). Mitochondrion (M) swelled moderately, became larger and rounder, most mitochondria swelled, internal cristae became shorter and less, matrix became shallower, a few severe membranes damaged, matrix overflow. CLP + NE group: proximal tubular epithelial cells showed moderate cytoplasmic edema, a large area of low electron density edema area, few organelles, and continuous and complete basement membrane (BM). Mitochondrion (M) was moderately swollen, most of the mitochondria swelled locally, the internal cristae became shorter and less, and the matrix became shallower. In a few area, the membrane damaged and the matrix overflowed. BM: basement membrane. M: mitochondrion. N: nucleus. RER: rough endoplasmic reticulum. Scale bar = 2 µm, 5 µm.
We further measured the levels of mitochondrial biogenesis-related proteins in the rat kidneys. A Western blotting analysis (see Figure 7) showed that compared to the vehicle treatment, ANG II administration significantly reversed CLP-induced suppression of P-AMPK (P<0.05), PGC-1α (P<0.01), and NRF-2 (P<0.01) protein expression in the rat kidneys. Similar trends were observed in NE-treated rats; however, a significant reversal was only observed in NRF-2 protein expression (P<0.05). These findings suggested that both ANG II and NE alleviated mitochondrial dysregulation in mice with septic AKI, and that ANG II was superior to NE in promoting the expressions of mitochondrial biogenesis-related proteins.
Figure 7.

Effects of ANG II and NE on the AMPK/PGC-1α/NRF-2 pathway. Rats were euthanized 24 h after the start of the experiment. (A,B,C) A Western blotting analysis was performed to determine protein levels of PGC-1α, NRF-2, p-AMPK, and AMPK in the rat kidneys. β-actin was used as an internal control. Data are expressed as the mean ± SEM. n=8 in the control or sham, n=5 in the CLP, n=6 in the CLP + ANG II, n=6 in the CLP + NE groups. PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1-α; NRF2, nuclear respiratory factor; AMPK, AMP-activated protein kinase; p-AMPK, phosphorylated AMPK.
Discussion
Recent studies have reported that ANG II can restore and maintain the MAP observed in patients experiencing septic shock with little adverse effects (7,14,17). In this study, we compared the therapeutic effects of ANG II and NE on CLP-induced shock and subsequent AKI. We found that ANG II was superior to NE in alleviating the dysregulated biochemical parameters, in improving renal perfusion, in mitigating renal tubular injury, in inhibiting inflammation and oxidative stress, and in promoting the expression of mitochondrial biogenesis-related proteins in septic kidneys. Our results suggested that ANG II outperformed NE in alleviating the symptoms of AKI caused by septic shock.
NE remains the first-line vasopressor for patients with septic shock (15). However, accumulating evidence has shown that there are numerous adverse effects associated with the NE treatment of septic shock. NE preserves renal blood flow, renal cortical perfusion, and partial oxygen pressure; however, it exacerbates renal medulla ischemia and hypoxia in animal models. Research has shown that NE fails to improve sepsis-induced altered microcirculatory blood flow in the kidney (16,18). In septic AKI patients, NE does not improve microcirculatory dysfunction in the sublingual circulation, and the long-term effects of NE-induced hyper-ischemia and hypoxic injury in septic kidneys remain unclear (19). Further, NE may affect immune function, induce thrombosis, and decrease metabolic efficiency, and myocardium function.
Conversely, treatment with ANG II has not been found to have any negative effects on the renal cortex and medulla perfusion (20). Thus, ANG II may serve as a promising alternative to NE in the treatment of hypotension in septic shock. ANG II binds to its receptor, leading to systemic arteriole constriction and elevation in peripheral resistance and blood pressure. ANG II also enhances the release of NE from sympathetic nerve endings (6). The exogenous application of ANG II improves the sensitivity of blood vessels to catecholamines, constricts the glomerular efferent arteries, increases the glomerular perfusion pressure, and elevates the glomerular filtration rate (20-22).
Consistent with the findings described above, our results showed that compared with the vehicle treatment, the administration of ANG II rapidly and significantly restored the MAP within 2 h after treatment, and maintained the MAP at 75–85 mmHg for at least 4 h. No significant differences were observed in the MAP between the ANG II and NE-treated groups. Thus, ANG II and NE appear to have comparable vasoconstrictive effects in the treatment of septic shock. A clinical study has shown that ANG II markedly reduced NE dosing and was well-tolerated in patients with septic shock, while the cessation of the ANG II infusion resulted in a concomitant rebound of NE dosing (23). Notably, we found that the administration of ANG II, but not NE, significantly restored renal blood flow, while reducing the RI, reducing the serum creatinine and lactic acid levels, mitigating the renal histopathological damage, and suppressing the production of proinflammatory cytokines and oxidative stress markers in CLP rats. Thus, ANG II outperformed NE in terms of increasing renal perfusion and alleviating renal injury in septic shock. Collectively, these findings suggest that ANG II is an ideal substitute of NE, as it targets both hypotension and AKI in septic shock.
Mitochondria play an important role in maintaining normal renal function. Renal mitochondria are most concentrated in the renal cortex (24). Hypoxia and ischemia are prone to occur at the junction of the cortex and medulla where high energy-demanding renal tubular cells reside (25). The important function of mitochondria is to provide energy (ATP), which is used by the kidney to remove wastes from the blood and regulate the balance of body fluids and electrolytes. In the process of mitochondrial biogenesis, PGC-1α is a positive regulator of mitochondrial biogenesis. During sepsis, transient ischemia and elevated cytokine levels, especially TNF-α, can reduce the expression of PGC-1 α in renal tubular cells and inhibit the recovery of kidney (10,26). Trump et al. (27) found significant mitochondrial swelling in renal tubular cells in patients who died of septic shock. In both AKI rodent models and in patients, researchers have observed a reduction in the number of mitochondria, mitochondrial swelling, vacuole formation, as well as mitochondrial cristae disorder, dissolution, and even rupture in the proximal tubular epithelial cells using an electron microscope. Importantly, structural changes of mitochondria can be visually observed in the ischemic kidney before the clinical manifestations of AKI occur, which suggests that mitochondrial dysfunction may lead to renal injury (27-30).
Consistent with previous findings, our results showed that rats subjected to CLP exhibited significant mitochondrial ultrastructural impairment in the renal cortex. To investigate the mechanisms underlying the protective effects of ANG II in septic AKI, we used Western blotting to assess the expression of components of the AMPK/PGC-1α/NRF-2 pathway in the renal cortex. The results showed that the expression of the AMPK/PGC-1α/NRF-2 pathway was significantly more reduced in the CLP rats than the control or sham-treated rats. This effect on protein expression was consistent with the results of a previous study that showed that messenger ribonucleic acid and protein expression of PGC-1α were decreased in the proximal renal tubular cells of a cisplatin-induced rat model of AKI, which was also accompanied by concomitant mitochondrial dysfunction, structural, and regeneration disorders. Similar results have been observed in both ischemia-reperfusion– and sepsis-induced AKI models (30,31). Our results showed that both ANG II and NE alleviated the mitochondrial impairments observed in CLP rats; however, ANG II performed better than NE in reversing the CLP-induced suppression of AMPK/PGC-1α/NRF-2 signaling. Thus, in septic AKI, it may be that ANG II alleviates mitochondrial impairments by activating the AMPK/PGC-1α/NRF-2 pathway, while NE improves mitochondrial function via other signaling pathways.
The suppression of PGC-1α signaling is closely associated with inflammation during sepsis. Tran et al. demonstrated (31) increased endotoxin and inflammatory cytokine inhibition of PGC-1α transcription in septic AKI. Similarly, Tran et al. showed that interferon gamma inhibited PGC-1α transcription in a septic cell model (32). Signals from innate inflammatory pathways show the downregulation of PGC-1α during infection (33-35). In the present study, we found that ANG II abrogated the CLP-induced excessive production of proinflammatory cytokines and oxidative stress markers. Thus, ANG II appears to alleviate renal inflammation and oxidative stress, which in turn activates the PGC-1α signaling to promote mitochondrial biogenesis in septic AKI. Further studies examining the proximal tubular epithelial cells should be conducted to investigate the involvement of the AMPK/PGC-1α/NRF-2 pathway in the mechanism through which ANG II improves renal function in the setting of septic AKI.
The present study had a number of limitations. First, given the premature deaths dictated by the experimental protocol requiring tissue sampling at 24 hours, there is a risk of survivor bias in our study. However, as no significant differences were observed in overall survival, especially among the CLP, CLP + ANG II, and CLP + NE treatment groups, any such survivor bias may not have significantly affected our results. Second, sham + NE and sham + ANG II control groups were not included in this study. To simulate the treatment process of patients with septic shock and better guide clinical treatment decisions, these groups should be examined in future experiments. In our study, the blood pressure of the sham rats did not decrease, as ANG II and NE exerted strong vasoactive effects on blood vessels. Treatment with vasoactive agents leads to a series of abnormal changes in blood pressure and renal perfusion. It is a limitation of this study that the application of vasoactive drugs in the sham operation group was not observed. In future experiments, treatment with vasoactive drugs in combination with a sham operation should be performed to observe the effects of vasoactive drugs on the mitochondria and hemodynamics of rats in the sham operation group. Third, renal perfusion color Doppler grading is a semi-quantitative index with certain subjectivity. In addition, due to the limitations of ultrasonic equipment, including the color Doppler, the imaging of renal blood flow may not have been sufficiently clear or accurate. Finally, survival was only monitored for 24 h, which is a short time interval for survival study.
In conclusion, in addition to having comparable vasoconstriction effects in response to NE, ANG II outperforms NE in increasing renal perfusion, ameliorating renal function, mitigating renal histopathological damages, alleviating inflammation and oxidative stress, and promoting the expression of mitochondrial biogenesis-related proteins in septic kidneys. Thus, ANG II appears to represent a better option than NE in the treatment of AKI in septic shock.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The animal study was approved by the Ethics Committee for Laboratory Animals Care and Use of Hebei Medical University, Fourth Affiliated Hospital and Hebei Provincial Tumor Hospital (Approval Number: IACUC-4th Hos Hebmu). All the procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (1985, National Institutes of Health).
Reporting Checklist: The authors have completed the ARRIVE reporting checklist. Available at http://dx.doi.org/10.21037/atm-21-621
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-21-621
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-21-621). The authors have no conflicts of interest to declare.
(English Language Editor: L. Huleatt)
References
- 1.Fleischmann C, Scherag A, Adhikari NKJ, et al. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am J Respir Crit Care Med 2016;193:259-72. 10.1164/rccm.201504-0781OC [DOI] [PubMed] [Google Scholar]
- 2.Kellum JA, Chawla LS, Keener C, et al. The Effects of Alternative Resuscitation Strategies on Acute Kidney Injury in Patients with Septic Shock. Am J Respir Crit Care Med 2016;193:281-7. 10.1164/rccm.201505-0995OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Xing G, Wang L, et al. Acute kidney injury in China: a cross-sectional survey. Lancet 2015;386:1465-71. 10.1016/S0140-6736(15)00344-X [DOI] [PubMed] [Google Scholar]
- 4.ProCESS Investigators , Yealy DM, Kellum JA, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014;370:1683-93. 10.1056/NEJMoa1401602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antonucci E, Fiaccadori E, Donadello K, et al. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care 2014;29:500-11. 10.1016/j.jcrc.2014.03.028 [DOI] [PubMed] [Google Scholar]
- 6.Di Giantomasso D, May CN, Bellomo R. Norepinephrine and vital organ blood flow during experimental hyperdynamic sepsis. Intensive Care Med 2003;29:1774-81. 10.1007/s00134-003-1736-9 [DOI] [PubMed] [Google Scholar]
- 7.Jadhav AP, Sadaka FG. Angiotensin II in septic shock. Am J Emerg Med 2019;37:1169-74. 10.1016/j.ajem.2019.03.026 [DOI] [PubMed] [Google Scholar]
- 8.Wan L, Langenberg C, Bellomo R, et al. Angiotensin II in experimental hyperdynamic sepsis. Crit Care 2009;13:R190. 10.1186/cc8185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dellepiane S, Marengo M, Cantaluppi V. Detrimental cross-talk between sepsis and acute kidney injury: new pathogenic mechanisms, early biomarkers and targeted therapies. Crit Care 2016;20:61. 10.1186/s13054-016-1219-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun J, Zhang J, Tian J, et al. Mitochondria in Sepsis-Induced AKI. J Am Soc Nephrol 2019;30:1151-61. 10.1681/ASN.2018111126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fontecha-Barriuso M, Martin-Sanchez D, Martinez-Moreno JM, et al. The Role of PGC-1alpha and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020;10:347. 10.3390/biom10020347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mariappan N, Elks CM, Haque M, et al. Interaction of TNF with angiotensin II contributes to mitochondrial oxidative stress and cardiac damage in rats. PLoS One 2012;7:e46568. 10.1371/journal.pone.0046568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corrêa TD, Jeger V, Pereira AJ, et al. Angiotensin II in septic shock: effects on tissue perfusion, organ function, and mitochondrial respiration in a porcine model of fecal peritonitis. Crit Care Med 2014;42:e550-9. 10.1097/CCM.0000000000000397 [DOI] [PubMed] [Google Scholar]
- 14.Lankadeva YR, Booth LC, Kosaka J, et al. Clonidine Restores Pressor Responsiveness to Phenylephrine and Angiotensin II in Ovine Sepsis. Crit Care Med 2015;43:e221-9. 10.1097/CCM.0000000000000963 [DOI] [PubMed] [Google Scholar]
- 15.Lankadeva YR, Kosaka J, Evans RG, et al. Urinary Oxygenation as a Surrogate Measure of Medullary Oxygenation During Angiotensin II Therapy in Septic Acute Kidney Injury. Crit Care Med 2018;46:e41-e48. 10.1097/CCM.0000000000002797 [DOI] [PubMed] [Google Scholar]
- 16.Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med 2017;43:304-77. 10.1007/s00134-017-4683-6 [DOI] [PubMed] [Google Scholar]
- 17.Treacy O, Brown NN, Dimeski G. Biochemical evaluation of kidney disease. Transl Androl Urol 2019;8:S214-S223. 10.21037/tau.2018.10.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrara G, Kanoore Edul VS, Caminos Eguillor JF, et al. Effects of fluid and norepinephrine resuscitation in a sheep model of endotoxin shock and acute kidney injury. J Appl Physiol (1985) 2019;127:788-97. 10.1152/japplphysiol.00172.2019 [DOI] [PubMed] [Google Scholar]
- 19.Ma S, Evans RG, Iguchi N, et al. Sepsis-induced acute kidney injury: A disease of the microcirculation. Microcirculation 2019;26:e12483. 10.1111/micc.12483 [DOI] [PubMed] [Google Scholar]
- 20.Tumlin JA, Murugan R, Deane AM, et al. Outcomes in Patients with Vasodilatory Shock and Renal Replacement Therapy Treated with Intravenous Angiotensin II. Crit Care Med 2018;46:949-57. 10.1097/CCM.0000000000003092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khalique SC, Ferguson N, Angiotensin II. (Giapreza): A Distinct Mechanism for the Treatment of Vasodilatory Shock. Cardiol Rev 2019;27:167-9. 10.1097/CRD.0000000000000247 [DOI] [PubMed] [Google Scholar]
- 22.Sacha GL, Bauer SR, Lat I. Vasoactive Agent Use in Septic Shock: Beyond First-Line Recommendations. Pharmacotherapy 2019;39:369-81. 10.1002/phar.2220 [DOI] [PubMed] [Google Scholar]
- 23.Wakefield BJ, Busse LW, Khanna AK. Angiotensin II in Vasodilatory Shock. Crit Care Clin 2019;35:229-45. 10.1016/j.ccc.2018.11.003 [DOI] [PubMed] [Google Scholar]
- 24.Chawla LS, Busse L, Brasha-Mitchell E, et al. Intravenous angiotensin II for the treatment of high-output shock (ATHOS trial): a pilot study. Crit Care 2014;18:534. 10.1186/s13054-014-0534-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens 2010;19:393-402. 10.1097/MNH.0b013e32833aa4ac [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plotnikov EY, Pevzner IB, Zorova LD, et al. Mitochondrial Damage and Mitochondria-Targeted Antioxidant Protection in LPS-Induced Acute Kidney Injury. Antioxidants (Basel) 2019;8:176. 10.3390/antiox8060176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trump BF, Valigorsky JM, Jones RT, et al. The application of electron microscopy and cellular biochemistry to the autopsy. Observations on cellular changes in human shock. Hum Pathol 1975;6:499-516. 10.1016/S0046-8177(75)80068-2 [DOI] [PubMed] [Google Scholar]
- 28.Aparicio-Trejo OE, Reyes-Fermin LM, Briones-Herrera A, et al. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic Biol Med 2019;130:379-96. 10.1016/j.freeradbiomed.2018.11.005 [DOI] [PubMed] [Google Scholar]
- 29.Choi YM, Kim HK, Shim W, et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS One 2015;10:e0135083. 10.1371/journal.pone.0135083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takasu O, Gaut JP, Watanabe E, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med 2013;187:509-17. 10.1164/rccm.201211-1983OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alkhamis MA, Arruda AG, Morrison RB, et al. Novel approaches for Spatial and Molecular Surveillance of Porcine Reproductive and Respiratory Syndrome Virus (PRRSv) in the United States. Sci Rep 2017;7:4343. 10.1038/s41598-017-04628-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran M, Tam D, Bardia A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 2011;121:4003-14. 10.1172/JCI58662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sweeney TE, Suliman HB, Hollingsworth JW, et al. Differential regulation of the PGC family of genes in a mouse model of Staphylococcus aureus sepsis. PLoS One 2010;5:e11606. 10.1371/journal.pone.0011606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sweeney TE, Suliman HB, Hollingsworth JW, et al. A toll-like receptor 2 pathway regulates the Ppargc1a/b metabolic co-activators in mice with Staphylococcal aureus sepsis. PLoS One 2011;6:e25249. 10.1371/journal.pone.0025249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casemayou A, Fournel A, Bagattin A, et al. Hepatocyte Nuclear Factor-1beta Controls Mitochondrial Respiration in Renal Tubular Cells. J Am Soc Nephrol 2017;28:3205-17. 10.1681/ASN.2016050508 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as