

Abstract
CRISPR gene therapy is one promising approach for treatment of Duchenne muscular dystrophy (DMD), which is caused by a large spectrum of mutations in the dystrophin gene. To broaden CRISPR gene editing strategies for DMD treatment, we report the efficient restoration of dystrophin expression in induced myotubes by SpCas9 and dual guide RNAs (gRNAs). We first sequenced 32 deletion junctions generated by this editing method and revealed that non-homologous blunt-end joining represents the major indel type. Based on this predictive repair outcome, efficient in-frame deletion of a part of DMD exon 51 was achieved in HEK293T cells with plasmids expressing SpCas9 and dual gRNAs. More importantly, we further corrected a frameshift mutation in human DMD (exon45del) fibroblasts with SpCas9-dual gRNA ribonucleoproteins. The edited DMD fibroblasts were transdifferentiated into myotubes by lentiviral-mediated overexpression of a human MYOD transcription factor. Restoration of DMD expression at both the mRNA and protein levels was confirmed in the induced myotubes. With further development, the combination of SpCas9-dual gRNA-corrected DMD patient fibroblasts and transdifferentiation may provide a valuable therapeutic strategy for DMD.
Keywords: CRISPR, Cas9, gene therapy, Duchenne muscular dystrophy, reprogramming, regenerative medicine, transdifferentiation, muscle regeneration
Graphical abstract
Duchenne muscular dystrophy (DMD) is caused by dystrophin deficiency and affects ∼1 in 5,000 boys. In this study, Yonglun Luo and colleagues report a CRISPR approach to efficiently correct DMD mutations and restore dystrophin expression in human muscle cells, providing a potential therapeutic approach for DMD.
Introduction
The simplicity and high efficiency of the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 gene editing system make the technology an attractive tool for gene therapy.1 The Cas9 endonuclease is guided to the genomic target site by a small guide RNA (gRNA) and introduces a DNA double strand break (DSB) at the target site. The Cas9 from Streptococcus pyogenes (SpCas9) predominately cleaves 3 bp upstream of the protospacer adjacent motif (PAM) NGG sequences.2 In mammalian cells, repair of a DSB is accomplished by the endogenous DNA repair machineries, which predominantly introduce small insertions or deletions, known as indels.3 It was generally thought that indels resulting from CRISPR editing are random. However, recent studies discovered that the indel profile for a given target site is predictable. Max W. Shen et al. analyzed the mutation profile of 2,000 gRNAs and developed a CRISPR-Cas9 indel predictor, inDelphi, by deep machine learning.4 Consistent with that, Allen et al.5 analyzed >40,000 gRNAs and created another, similar, algorithm. These predictors are valuable, but the indel profiles are still affected by CRISPR delivery methods and vary between different target sites.6
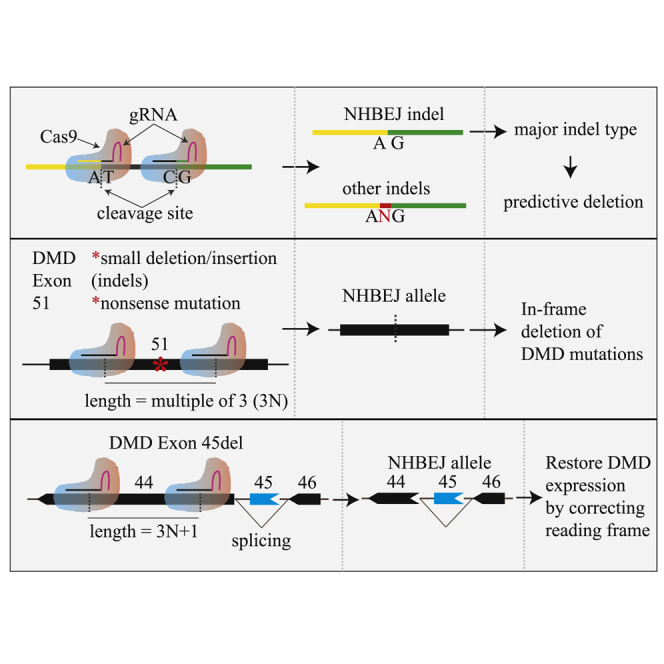
CRISPR gene editing with pairs of gRNAs (or dual gRNAs) has been used for gene knockout and deletion.7, 8, 9, 10 Compared with single gRNA, the dual gRNAs approach has several advantages. First, it is an efficient way to achieve small/large deletions11, 12, 13 and even mimic chromosomal aberrations.14 Second, the generation of CRISPR-edited cells/organisms is convenient since genotyping is based on PCR and gel electrophoresis. Third, the homology-directed repair (HDR) efficacy can be increased by dual-gRNA cleavages in either donor vectors15 or genome target loci.9 Most importantly, it has been found that the major indel type of the deletion junction is blunt end joining, by which the two DSBs are ligated together without introducing additional indels. We call this form of indel type “non-homologous blunt end joining” (NHBEJ). This predictive editing feature has been harnessed for precise and predictable deletions and insertions.16,17 Apart from deletions, we previously found that extrachromosomal circle DNA (eccDNA) ranging from several hundred base pairs to mega base scale can be generated in human cells by CRISPR editing with pairs of gRNAs.18 In analyses of the DSB repair at the junction of eccDNA and the deletion allele, we also observed that the majority of the repair events are NHBEJ.18,19. This suggests that NHBEJ is a common repair outcome for CRISPR editing with SpCas9 and dual gRNAs, representing an alternative, attractive, approach for gene therapy.
Duchenne muscular dystrophy (DMD) is a very severe degenerative disease of muscle cells caused by mutations in the dystrophin gene, which exhibits a large spectrum of mutations. The disease shows X-linked inheritance,20 affecting 1 in 3,600–6,000 live male births.21 The full-length DMD gene spans a genomic range of >2 Mb and consists of 79 exons. Mutations causing DMD comprise ∼70% frameshift mutations and 30% rare point mutations.22 It has been found that dystrophin proteins with small internal deletions retain most of their function compared with the normal full-length protein. Thus, controlled deletion is an attractive strategy for DMD gene therapy.23 Exon skipping, in-frame correction, and knockin correction mediated by CRISPR-Cas9 are thought to be potential strategies to restore the dystrophin protein.24 Several milestones in restoration of DMD expression based on in vivo CRISPR-Cas9 delivery have been achieved in preclinical models such as mouse,25 pig,26 and dog.27 In addition to the in vivo gene therapy, the ex vivo gene therapy, which involves CRISPR gene editing of patient cells outside the body and subsequent transplantation back to patients, is also considered clinically useful for treatment of DMD. For an extensive review of the current application of CRISPR-Cas9 treatment of DMD, we refer readers to recent reviews.28, 29, 30
According to the French UMD-DMD database http://www.umd.be/, the frameshift mutations in the DMD locus account for 2,594/2,898 of the DMD mutations, and 991 of them could be corrected by an exon skipping strategy. For instance, DMD exon 51 “skipping,” which is being tested in clinical trials, could in principle benefit ∼13% of DMD patients.31 However, alternative CRISPR gene editing strategies that could precisely introduce in-frame deletions of missense/nonsense mutations in exon 51, or correction of the frameshift mutation (e.g., exon 45 deletion), are needed to benefit the majority of DMD patients. Precision correction of the small DMD mutations by homology-directed repair and exon reframing is considered the most promising approach for such mutations.32 However, this approach is hampered by its low efficiency. The development of more efficient CRISPR gene editing and cell therapy methods is still needed to benefit more DMD patients with different mutations.
To broaden the CRISPR gene therapy toolbox for DMD, we began the present study by validating that NHBEJ is the major indel type at deletion junctions introduced by SpCas9 and dual gRNAs based on 32 combinations of dual gRNAs in human cell lines. Using the NHBEJ strategy, we further demonstrated efficient in-frame deletion of a part of DMD exon 51 in human embryonic kidney 239T (HEK293T) cells and corrected a frameshift mutation in human DMD (exon45del) fibroblasts. Restoration of dystrophin expression at both mRNA and protein levels was achieved in the induced myotubes derived from CRISPR-edited DMD (exon45del) fibroblasts by transdifferentiation. Thus, editing with SpCas9 and dual gRNAs provides a potential therapeutic approach for DMD.
Results
NHBEJ is the major indel type at deletion junctions introduced by SpCas9 and dual gRNAs in human cells
To validate the indel profile at deletion junctions generated by SpCas9 and dual gRNAs, we generated eight pairs of gRNAs that targeted four human genes (TTR, CREB1, STAT2, IRF9; two pairs per gene) (see Table S1). We transfected HEK293T cells with plasmids expressing SpCas9 and each pair of gRNAs and amplified the allele with the intended deletion by PCR (Figures 1A and 1B). We sequenced the deletion allele from both directions in order to quickly evaluate the repairing events at junctions. Our results showed that the aberrant sequence signal began exactly from the expected cleavage sites (between the third and fourth base pair upstream of the PAM site) of the upstream and downstream gRNAs (Figure 1C; Figure S1). This suggests that the DSBs are prone to be repaired by blunt end ligation, by which the two DSBs are ligated together without introducing other indels. For simplification, we call this type of indel “NHBEJ” (non-homologous blunt end joining). To investigate whether this NHBEJ phenomenon is cell type dependent, we tested two additional cell lines (HepG2 and HeLa) and observed that NHBEJ is the major indel type in these two cell lines as well (Figures 1B and 1C; Figure S1).
Figure 1.

Repair outcomes of SpCas9-dual gRNA-induced deletions at 8 endogenous loci of 3 cell lines
(A) List of wild-type and NHBEJ alleles for 8 targeting loci. (B) PCR results of 8 loci in 3 cell lines edited with SpCas9 and the corresponding dual gRNAs (amplified from pool of triplicate transfections). (C) Sanger sequencing results of deletion alleles, indicated with white arrows in (B), of 2 representative loci (P1 and P8) from 3 human cell lines. Full sequencing results are shown in Figure S1.
Semiquantitative evaluation of NHBEJ frequency by Sanger sequencing and ICE analysis
We tried to quantify the NHBEJ frequency with conventional methods (such as tracking of indels by decomposition [TIDE], inference of CRISPR edits [ICE], and indel detection by amplicon analysis [IDAA]), which have been developed for indel profiling in gene editing with a single gRNA.33, 34, 35 We selected the ICE method for indel profiling because of its applicability and high throughput (batch analysis). To enable the use of ICE for indel profiling of the deletion junction by dual gRNAs, we first developed a simple method to generate a pseudo control sample. As shown in Figure 2A, the pseudo control sample is the deletion junction that simply joins the two DSBs without introducing other indels. The approach for quantifying indel profile at the deletion junction with ICE contains four steps. Step 1: the deletion allele was PCR amplified from cells transfected with SpCas9 and dual gRNAs. Step 2: the PCR product was cloned into a TA vector or topo vector, and competent E. coli cells were transformed. Step 3: a number of clones from step 2 were Sanger sequenced to identity at least one NHBEJ clone. Step 4: ICE analysis was performed with the pseudo control. The pseudo gRNA spacer used for ICE analysis was the 17 bp upstream and 3 bp downstream sequences flanking the NHBEJ junction. Using this approach, we quantified the NHBEJ efficiency for all eight pairs (P1–P8) of gRNAs in three cell lines. As shown in Figures 2B, 2C, and S2, our results showed that the NHBEJ was the major indel at deletion junctions introduced by SpCas9 and dual gRNAs (5 out of 8 pairs, with frequency above 50%). For the three pairs of gRNAs (P5, P6, P7) where the ICE-based NHBEJ frequency estimation was below 40%, the Sanger sequencing chromatogram clearly showed that the NHBEJ signal is above 50% (Figure S1), suggesting that the conventional ICE approach can only be used for semiquantitative estimation of NHBEJ frequency. For each pair of gRNAs, the NHBEJ frequency was consistent among all three cell lines, further corroborating the common phenomenon of NHBEJ.
Figure 2.

Quantification of NHBEJ with ICE
(A) Schematic illustration of NHBEJ. For ICE analysis, a NHBEJ control is generated by Topo cloning of the PCR product from the NHBEJ deletion allele. The pseudo gRNA spacer is the 20 nt sequence flanking the junction site. (B) Representative ICE analysis results of the P1 and P8 loci. Full ICE results are shown in Figure S2. (C) NHBEJ efficiency for the 8 endogenous loci in 3 cell lines.
Validation of NHBEJ frequency by deep sequencing
To more accurately quantify the NHBEJ frequency, we performed deep sequencing of the same PCR products available from P1–P8 HepG2 cells that had been used for the ICE analysis described above. Using the DNA nanoball-based and PCR-free next generation sequencing (NGS) strategy (DNB-seq; see Materials and methods), we generated DNA nanoballs for P1–P8 and sequenced each sample with a coverage of 5–40 million reads using the MGISEQ-2000 (Figure 3). Consistent with ICE-based indel and NHBEJ frequency estimations (Figure 2B; Figure S2), the top indel event is NHBEJ for all eight pairs of gRNAs as quantified by deep sequencing, with a frequency ranging from 45% to 70% (Figure 3). However, the accuracy of estimating NHBEJ frequency by ICE varies from 0.2- to 1.7-fold (Figure 3), suggesting a need for further improvement of the conventional ICE-based indel deconvolution program. However, the conventional ICE-based indel deconvolution and estimation method can still be used for semiquantitative estimation (ranking) of indel frequency, and thus it was used for all following analyses. Taken together, our deep sequencing results confirmed that NHBEJ is the major repair event at deletion junctions introduced by SpCas9 and dual gRNAs.
Figure 3.

Next generation sequencing-based quantification of indel profiles
MGI-2000 sequencing of the amplicon products of deletion alleles from Figure 1B (HepG2 cells). Figure shows the top three indel types for each locus, with total reads for each sample and each indel type indicated. An asterisk (∗) indicates NHBEJ frequency predicted by ICE from Figure 2C.
The effect of PAM direction on NHBEJ efficiency
We wanted to investigate whether the PAM direction of the dual gRNA influences NHBEJ efficiency. To address this, we selected symmetric targeting regions that contain sequences with a 5′-CCNNNN|NNNNGG-3′ motif, which allows us to design two SpCas9 gRNAs that generate DSBs at the same site (Figure 4A). Using the DMD intron 44 locus as an example (Figure 4A), we selected four symmetric targeting regions and generated eight SpCas9 gRNAs. We transfected HEK293T cells with each pair of gRNAs (24 pairs in total) and genotyped the indel profile of the deletion junction by ICE analysis (Figure 4B; Figure S3; Table S3). Consistent with the observation, the major indel event of deletion junctions was NHBEJ (Figure S3). Based on the PAM direction of the pair gRNAs, the 24 pairs of dual-CRISPR editing were classified into three type groups (Figure 4C): (1) head to tail (H2T), in which both gRNAs target the same strand; (2) head to head (H2H), in which the PAMs are flanked by cleavage sites (also known as PAM-in); and (3) tail to tail (T2T), in which the cleavage sites are flanked by the PAMs (also known as PAM-out). To further analyze and compare the NHBEJ ratio among different combinations, we generated NHBEJ-TA vector clones and performed ICE analysis. Consistently, our results showed that the NHBEJ repair outcome comprised an average of ∼80% for all three groups (Figure 4D). The overall NHBEJ efficiency was similar between H2T, H2H, and T2T (Figures 4C and 4D), suggesting that PAM direction does not affect NHBEJ frequency.
Figure 4.

Effect of PAM directions on NHBEJ repair frequency
(A) Schematic outline of the experimental design. The gRNA 1 and 2 were pairs of gRNAs with opposite PAM directions but generating DSBs at the same position. Four regions in DMD intron 44 were selected. (B) PCR-based genotyping results in HEK293T cells targeted with SpCas9 and 24 pairs of gRNAs. PCR was done on a pool of transfected cells with triplicate transfections. An asterisk (∗) indicates DNA bands that were gel-purified and analyzed by Sanger sequencing and ICE. (C) Schematic presentation of the three PAM combinations. (D) Quantification and comparison of NHBEJ efficiency with different PAM orientations. There was no statistical difference between the three groups (ANOVA).
Efficient in-frame deletion of a part of exon 51 in the DMD gene in HEK293T cells
The NHBEJ represents an attractive approach for DMD therapy. Since the NHBEJ represents the major indel form for dual-gRNA editing, we tested whether the NHBEJ can be used for achieving precision in-frame deletion of a part of DMD exon 51. Many disease-causing mutations (both missense and nonsense mutations) have been reported in this exon (Figure 5A). We designed two pairs of gRNAs, which will generate in-frame deletion (180 bp and 168 bp, respectively) in exon 51 if repaired with NHBEJ (Figure 5B). We co-transfected HEK293T cells with plasmids encoding SpCas9 and a pair of gRNAs in triplicates and harvested cells for analysis 4 days after transfection (see Materials and methods) by PCRs. Flanking PCRs indicated >30% deletion efficiency by the designed gRNA pairs (Figures 5C and 5D). We gel-purified the deletion bands, Sanger sequenced, and analyzed the outcome with ICE (Figures 5E and 5F; Figure S4). Approximately 50% and 30% NHBEJ frequencies were estimated by ICE for pair gRNAs T1-T2 and T3-T4, respectively (Figure 5F), showing that efficient in-frame deletion of a part of DMD exon 51 can be achieved with SpCas9 and dual gRNAs.
Figure 5.

Exonic in-frame deletion of a part of DMD exon 51 by NHBEJ editing
(A) Presentation of human DMD exon 51. Nucleotides and amino acids in reading frame, with five previous reported disease-causing mutations highlighted. (B) Schematic presentation of gRNA (pair 1: T1 + T2, pair 2: T3 + T4) targeting positions, length of deletions, and the resulting amino acid sequences after NHBEJ-mediated in-frame deletion. (C) PCR-genotyping results of HEK293T cells transfected with SpCas9 and either pair 1 (T1 + T2) or pair 2 (T3 + T4) gRNAs. Controls were transfected with SpCas9 only. (D) Semi-quantification of deletion efficiency in (C) by ImageJ. (E) Representative semi-estimation of NHBEJ efficiency based on the deletion products by Sanger sequencing and ICE analysis. (F) Boxplot of NHBEJ efficiency as mean and 1 SD (n = 3).
Efficient correction of a frameshift mutation in human DMD fibroblasts
Encouraged by the high NHBEJ efficiency achieved by SpCas9 and dual gRNA, we investigated the approach for DMD therapy using a more clinically relevant cell model. Frameshift mutation caused by single exon 45 deletion is one of the common mutations causing DMD.36,37 As shown in Figure 6A, exon 45 deletion leads to a frameshift change from +2 to +1 in exon 46, introduction of a premature stop codon, and subsequent degradation of the mutated transcript by the nonsense-mediated decay pathway.38 In addition to exon 46 skipping, deletion of 3N+1 bp in exon 44 can also correct the frameshift mutation caused by exon 45 deletion. However, the 3N+1 bp deletion will introduce frameshift changes to exon 44. It is thus important to select dual gRNAs that will not introduce any premature stop codon in exon 44. Based on such criteria, we were able to design two pairs of gRNAs, which will delete 52 bp and 88 bp in exon 44 if repaired by NHBEJ. Using chemically modified gRNAs and ribonucleoprotein (RNP)-based CRISPR delivery into DMD exon45del fibroblasts (GM05114, EX45del), efficient targeted deletion efficiency (∼50% based on gel intensity) was achieved (Figure 6B). Semi-quantification of the deletion allele showed that NHBEJ represents >60% of all indels (Figure 6C; Figure S5). Our results showed that efficient correction of DMD exon 45 deletion can be achieved by SpCas9 and dual gRNAs in fibroblasts.
Figure 6.

Correction of DMD exon 45 deletion mutation by NHBEJ editing
(A) Schematic illustration of the DMD exon 45 deletion locus, transcription, and CRISPR NHBEJ targeting. NMD, nonsense-mediated decay. (B) PCR-genotyping results from a pool of DMD ex45del fibroblasts edited with SpCas9 protein and either the pair 1 (T1 + T2) or pair 2 (T3 + T4) gRNAs. Controls were transfected with SpCas9 only (n = 3). (C) Semi-quantification of NHBEJ frequency based on Sanger sequencing and ICE analysis.
Restoration of dystrophin expression in induced myotubes
To investigate whether we have successfully restored dystrophin expression, we generated a lentiviral vector expressing the human MYOD transcription factor and transdifferentiated fibroblasts into myotubes based on methods described previously39,40 and as shown in Figure 7A. We modified the myotube reprograming vector to enhance MYOD transgene expression (see Materials and methods and Figure S6A). Significant changes in cell morphology appear ∼4–5 days after transdifferentiation (Figure S6B). Most importantly, expression of DMD at the mRNA level was only detected for CRISPR-edited DMD(EX45del) and healthy control cells (Figure 7B) but not the DMD(EX45del) cells. Fluorescence immunostaining of the dystrophin and myosin heavy chain (MHC, part of the thick filaments expressed in differentiated skeletal muscle cells) further confirmed that multi-nucleated myotubes co-expressing human MYOD, MHC, and Dystrophin were only detected in the CRISPR-edited DMD(EX45del) and healthy control cells (Figure 7C; Figure S6C). Finally, to investigate whether off-target cleavage had been introduced by these gRNAs, we performed off-target analysis using CRISPRspec41 and Sanger sequencing analysis of the top four predicted off-target sites of each gRNA. Our results show that no off-target cleavage was introduced in any of the sites analyzed (Figure S7). Taken together, our results show that the NHBEJ approach can successfully restore dystrophin expression in myotubes, thus providing a potential cell therapy strategy for DMD based on SpCas9 and dual gRNA editing.
Figure 7.

Restoration of DMD expression in transdifferentiated DMD EX45del myotubes
(A) Schematic illustration of CRISPR editing and transdifferentiation of edited cells into myotubes by overexpressing human MYOD transcription factor. Since the cells are primary fibroblasts that can only be passaged a few times without growth retardation, myotube reprogramming was performed for a pool of CRISPR-edited cells without pre-selection or enrichment of any indel types. (B) Examination of DMD mRNA expression by reverse transcription PCR in the transdifferentiated DMD Ex45del myotubes (day 12) edited by SpCas9 and dual gRNAs. Control is DMD Ex45del cells treated with SpCas9 only. Positive control: transdifferentiated normal human dermal fibroblasts (NHDFs). NTC, negative control for PCR reaction. (C) Representative immunofluorescence staining results for MHC (myosin heavy chain) and DMD (dystrophin protein) expression. MYOD expression was detected with dsRED signal. Nuclei are stained with DAPI. Magnification, 40×. Extended figures are shown in Figure S6.
Discussion
The CRISPR Cas9 technology has been widely used for gene knockout applications.42 Increasing knowledge tells us that the indel outcomes introduced by a single gRNA are to some extent predictable. However, prediction is hampered by differences between different gRNAs and targets.4,5,43 In this study, we have shown that the major DNA repair outcome of deletion by SpCas9 and dual gRNAs is NHBEJ. This is consistent with previous findings,16,17 thus providing a more predictive precision editing approach for gene therapy. Compared with gene editing with single gRNA, gene editing by dual gRNAs is easier for genotyping and selection of edited clones. In addition, the method is convenient for distinguishing biallelic and monoallelic edited clones based on PCR analysis.44,45 We acknowledge that one disadvantage of using dual gRNAs is that they might increase the risk of introducing off-target effects,46 as the off-target issue is still a concern for all CRISPR Cas9 applications.47 However, better methods to optimize the design of gRNAs and modifications of the Cas9 protein have significantly minimized the off-target potential.48,49
Correction of DMD causative mutations by CRISPR-Cas9 has been regarded as one of the most promising applications of the CRISPR technology. This is indeed evidenced by a growing number of studies both in vitro (mostly based on induced pluripotent stem cells [iPSCs])24,50,51 and in vivo (mouse, dog, and pig).26,27,52,53 For comprehensive reviews of the current applications of CRISPR correction of DMD, we refer readers to several recent articles.23,29,30,32 In this study, we have demonstrated that efficient correction of DMD frameshift mutation can be achieved by using the NHBEJ approach, which offers a precise deletion with predicted length. Several research groups (including us) have observed that the indel profile introduced by SpCas9 and a single gRNA is not random.4,16,54 More specifically, 1 bp deletion and 1 bp insertion are the two most frequently observed indels for CRISPR editing with single gRNA.6 Thus, correction of DMD exon45del frameshift mutation can also be achieved by using SpCas9 and one single gRNA targeting DMD exon 44. Indeed, in the evaluation of DMD expression from MYOD reprogramming cells from a mixture of CRISPR-edited DMD(EX45del) fibroblasts (Figure 6B), we observed the expression of a second restored DMD transcript not caused by NHBEJ. Taken together, our NHBEJ-based method combined with direct reprogramming of the SpCas9-dual gRNA-edited fibroblasts into differentiated muscle cells (myotubes) gave results that clearly demonstrate an alternative approach for DMD therapy.
We foresee that many inherited disorders could benefit from our NHBEJ-based therapy. One example is Leber congenital amaurosis type 10 (LCA10) caused by disruption of the CEP290 gene, which encodes nephrocystin-6 (NPHP6).55 The most frequent mutation found in patients with LCA10 is an A-to-G transition 5 bp downstream of a cryptic exon, which could be precisely deleted by Cas9 and dual gRNAs.56 In other forms of CEP290 loss-of-function mutations, such as the nonsense mutation (2249T-G) and the 5-bp deletion (1260delTAAAG),57 the protein function could be partially restored by in-frame deletion of the nonsense mutations based on NHBEJ with SpCas9 and dual gRNAs. Exon skipping is a way to restore partial function of the dystrophin protein,31,58 and this strategy has been approved by the FDA.59 Most exon skipping strategies are based on altering splicing sites (which gives very limited space for designing the gRNA) or deletion of a large piece of DNA (which will lead to lower efficiency). The NHBEJ by SpCas9 and dual gRNAs provides an alternative strategy. Our combination approach with NHBEJ editing of fibroblasts and transdifferentiation of the edited cells into myotubes should provide an attractive approach for DMD therapy and broaden the categories of DMD patients who can benefit.
Materials and methods
Small guide RNA design and vector construction
All oligonucleotides used in this study were either synthesized by the synthetic biology platform of BGI-Qingdao (China) or ordered from Merck (Darmstadt, Germany). Sequences for all oligonucleotides can be found in Tables S1 and S2.
The CRISPR-SpCas9 gRNA spacers were designed with the online CRISPR design tool “CRISPOR” (http://crispor.tefor.net/crispor.py). To generate the gRNA expression vectors, we used the Golden Gate Assembly (GGA) protocol previously optimized by us.3 Briefly, 1 μL each of the sense (100 μM) and antisense (100 μM) strand of each gRNA spacer were mixed with 2 μL of 10× NEB Buffer 2.1 and 16 μL of double-distilled water (ddH2O) to a total volume of 20 μL. Using a thermal cycler, we first denature the oligos at 95°C for 5 min and decrease to 25°C at a ramping rate of −5°C/min. Then we prepare the GGA reaction system as follows: For one reaction, add 1 μL of annealed oligo, 1 μL of T4 ligase (NEB), 2 μL of 10× T4 ligase buffer, 1 μL of Esp3I (Thermo Fisher Scientific), and ddH2O in a total volume of 20 μL. Perform GGA with the thermocycle program: 10 cycles of 37°C for 5 min and 22°C for 10 min; 1 cycle of 37°C for 30 min; 1 cycle of 75°C for 15 min. Save the ligation product at −20°C or transform competent cells directly. For transformation, 50 μL of DH5a competent cells were transformed with 1 μL of the ligation product. We normally analyzed 3 bacterial colonies by PCR using rTaq (5 μL rTaq, 2 μL lentiCRISPR-U6-F primer [primer specific for the U6 primer], 2 μL gRNA antisense strand oligonucleotide [spacer], 0.5 μL bacterial liquid, 4.1 μL ddH2O) with the PCR program (32 cycles of 98°C for 10 s, 55°C for 10 s, 72°C for 30 s; 1 cycle of 72°C for 5 min; 1 cycle of 20°C for 2 min). The PCR positive colonies were further validated by Sanger sequencing.
Cell culture
HEK293T cells, a liver hepatocellular carcinoma cell line (HepG2), HeLa cells, and human melanoma cells (HME) were used in this study. All the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza) supplemented with 10% fetal bovine serum (FBS) (Gibco), 1% GlutaMAX (Gibco), and penicillin-streptomycin (100 units penicillin and 0.1 mg streptomycin/mL). The normal human dermal fibroblasts (NHDFs) are primary fibroblasts established from skin biopsies. The cells were a gift from Dr. Kristine Freude of Copenhagen University. The DMD patient-derived primary fibroblasts with the DMD exon 45 deletion (GM05114) were purchased from Coriell Institute. Both NHDFs and GM05114 cells were cultured in DMEM/Nutrient Mixture F-12 Ham (DMEM/F-12) supplemented with 10% FBS (Gibco) and penicillin-streptomycin (100 units penicillin and 0.1 mg streptomycin/mL). All the cells were growing in a 37°C incubator with 5% CO2 atmosphere and maximum humidity. Cells were passaged every 2–3 days when the confluence reached 80%–90%.
PEI transfection
PEI40,000 (Polysciences) was used for cell transfection. One day before transfection, a total number of 0.5 × 106 cells per well were seeded in a 6-well plate, with 2 mL of DMEM-10% FBS medium. Before transfection, the cells were given fresh culture medium. For each transfection, 3 μg of plasmid DNA (pair-CRISPR gRNA plasmids were co-transfected within a ratio of 1:1) was mixed with 9 μg of PEI solution in Opti-MEM (Gibco) to a final volume of 200 μL. The PEI-DNA mixtures were stirred gently and incubated at room temperature for 15 min before being added to the cells in a dropwise manner. Twenty-four hours after transfection, transfected cells were given puromycin (1 μg/mL)-containing medium and cultured for 3 days (or until the wild-type [WT] control cells were dead). All surviving cells were harvested for further assays.
PCR, Sanger sequencing, and ICE analysis
Genomic DNA of cells was extracted with a TIANamp Genomic DNA Kit (TIANGEN) according to the manufacturer’s instructions. All PCR reactions were conducted with rTaq polymerase (TaKaRa). All primers for the PCRs can be found in Table S2. We gel purified the PCR bands with the proper deletions (smaller band with the correct size), using a gel extraction kit (Omega). TA cloning of the PCR product of interest was performed with a T-vector pMD19 kit (TaKaRa). For each amplicon, 8 bacterial colonies were picked and screened by PCR. Note that the purpose of TA cloning is to generate a pure deletion product (pseudo control) with the junction of the deletion repaired by NHBEJ so that it can be used as the control sample for ICE-TIDE analyses. The ICE analysis was then used to evaluate the total indel profiles and NHBEJ efficiencies (https://ice.synthego.com/).
CRISPR-Cas9 RNP nucleofection
gRNAs for DMD(EX45del) correction were synthesized by Synthego (California, USA). For nucleofection, 0.6 μL of gRNA (3.2 μg/μL in nuclease-free water) with 0.6 μL of spCas9 protein (10 μg/μL, Integrated DNA Technologies, Iowa, USA) were mixed gently in a PCR tube and kept at room temperature for 10 min to ∼1 h to form gRNA-spCas9 complexes. 200,000 fibroblast cells were trypsinized and spun down, washed with PBS and resuspended in 20 μL of Opti-MEM (Gibco). Then the cell suspension was mixed with RNP gently. Nucleofection was performed with a Lonza 4D-nucleofector (Lonza, Switzerland) with program CM138. After nucleofection, the cells were transferred to a 12-well plate with prewarmed culture medium. We harvested the cells ∼48–72 h after nucleofection, performed PCR for genotyping, or passaged the cells for transdifferentiation into myotubes. The forward primer for genotyping was DMD-exon44-F: 5′-TGCAGGAAACTATCAGAGTGAT-3′, and the reverse primer was DMD-exon44-R: 5′-ATCACCCTTCAGAACCTGATCT-3′.
Human MyoD (hMyoD) lentivirus package and transdifferentiation of fibroblasts to myotubes
The pLV hMyoD-IRES-dsRed was a gift from Charles Gersbach (Addgene plasmid #66631). To enhance the expression of hMyoD in fibroblasts, we replaced the UbC promoter in the pLV hMyoD-IRES-dsRed plasmid with a cytomegalovirus promoter (Addgene plasmid # 132775). For lentivirus packaging, 4 × 106 cells were seeded into a 10 cm culture dish. Transfection for viral packaging was performed with PEI40000 when the cells reached 60% confluency. Briefly, 13 μg of pLV-cytomegalovirus-hMyoD-IRES-dsRed, 3 μg of pRSV-REV, 3.75 μg of pMD.2G, and 13 μg of pMDGP-Lg/p-RRE were mixed with 98 μL of PEI40000 (1 μg/μL) in Opti-MEM to a final volume of 1 mL. The mixture was kept at room temperature for 15 min and was added to the cells in a dropwise manner. Transfected cells were given fresh medium 24 h later. Then virus was harvested from the medium 48 h after transfection and filtered with a 0.45 μm filter. We added polybrene (Sigma-Aldrich, Germany) into the lentivirus crude to a final concentration of 8 μg/mL and stored it at −80°C.
The hMyoD-lentivirus transduction and transdifferentiation of fibroblast to myotubes were conducted as described previously.39,40 Briefly, fibroblasts were seeded in a 6-well plate. The growth medium was changed to 8 μg/mL polybrene-containing medium (2 mL) when cells reached 80% confluence. 1 mL of hMyoD-lentivirus crude was added to each well, and the cells were kept in virus-containing medium in 5% CO2 at 37°C for 48 h. The medium was changed to fresh growth medium and incubated at 37°C for another 48 h. The transduced fibroblasts were then seeded into chamber slide flasks (Thermo Fisher Scientific, USA). After attaching in the flask, the cells were given differentiation medium that contained DMEM/F-12, 2% horse medium (Sigma-Aldrich, Germany), 1% insulin-transferrin selenium supplement (ITS, Thermo Fisher Scientific, USA) and 1% penicillin-streptomycin (100 units penicillin and 0.1 mg streptomycin/mL). The medium was changed every 3 days (4 mL per flask). We cultured the hMyoD-transduced fibroblast for 12 days for myotube transdifferentiation.
RNA isolation and reverse transcription PCR (RT-PCR)
Total RNA was isolated from transdifferentiated cells at day 12 after reprogramming according to the manufacturer’s protocol for TRI Reagent (Sigma-Aldrich, Germany). First-strand cDNA was synthesized from the total RNA in a 20 μL reaction system using the iScript cDNA Synthesis Kit (Bio-Rad, USA). Detection of DMD expression by PCR was performed with AccuPrime Pfx DNA polymerase (Invitrogen, USA) according to the manufacturer’s instructions. Briefly, the 20 μL PCR reaction system consisted of 3 μL of cDNA, 2 μL of 10× AccuPrime Pfx reaction buffer, 1.2 μL of forward primer (5 μM), 1.2 μL of reverse primer (5 μM), and 0.16 μL of AccuPrime Pfx polymerase, supplemented with ddH2O. The thermocycle program was 95°C for 2 min, (95°C for 15 s, 58°C for 30 s, 68°C for 30 s) with 35 cycles, 68°C for 5 min, and 4°C hold. For dystrophin RT-PCR, the forward primer was DMD-EX45del-RT-F: 5′-GCAAGAAGACAGCAGCATTGCA-3′ and the reverse primer was DMD-EX45del-RT-R: 5′-CAGGTTCAAGTGGGATACTAGC-3′. For glyceraldehyde 3-phosphate dehydrogenase (GAPDH) RT-PCR, the forward primer was hGAPDH-RT-F: 5′-TGGTATCGTGGAAGGACTCATGAC-3′ and the reverse primer was hGAPDH-RT-R: 5′-ATGCCAGTGAGCTTCCCGTTCAGC-3′.
Immunofluorescence staining
The cells in the slide flasks were washed twice with PBS, fixed with 4% paraformaldehyde (Sigma-Aldrich, Germany) at room temperature for 4 min, and then washed with PBS for 5 min three times. The cells were incubated with blocking buffer at room temperature for 1.5 h. The blocking buffer consisted of PBS, 2% bovine serum albumin (BSA, Sigma-Aldrich, Germany), 5% FBS, 2% donkey serum (Sigma-Aldrich, Germany), 0.2% Triton X-100 (Sigma-Aldrich, Germany), and 0.1% sodium azide (Sigma-Aldrich, Germany). After blocking, cells were washed with PBS twice and incubated with primary antibodies overnight at 4°C. We diluted 1:500 anti-dystrophin (ab15277, Abcam, USA) and 1:10 anti-myosin heavy chain (anti-MHC, clone MF-20, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) in 1% BSA for use as primary antibodies. After overnight incubation the cells were washed with PBS for 3 × 5 min and incubated with secondary antibodies at room temperature for 1.5 h in the dark. Alexa Fluor 488 donkey-anti-mouse IgG (1:1,000, Thermo Fisher Scientific, USA) and Alexa Fluor 647 donkey-anti-rabbit IgG (1:300, Thermo Fisher Scientific, USA) in 1% BSA were used as secondary antibodies. Finally, the cells were washed again with PBS for 3 × 5 min and mounted in ProLong Diamond Antifade Mountant with DAPI (P36962, Thermo Fisher Scientific, USA). All the slides were stored at −20°C. Images of immunocytochemistry were taken by the Inverted Zeiss LSM710 confocal microscope system, and we conducted image processing utilizing ZEN 2 (blue edition) software.
Deep sequencing
MGISEQ-2000 (MGI Tech, China) was used to perform amplicon deep sequencing following the standard operation protocol. Amplicon PCR-free library was prepared with the MGIeasy FS PCR-Free DNA Library Prep Kit (MGI Tech, China), following the manufacturer’s instructions. Briefly, each sample was diluted to 10 ng/μL. Ten microliters of diluted PCR product was mixed with an A-tailing reaction that contained A-tailing enzyme and buffer, incubated at 37°C for 30 min and then 65°C for 15 min to inactivate the enzyme. The A-tailed sample was mixed with PCR-Free index adapters, T4 DNA ligase, and T4 ligase buffer to add index adapters at both 3′ and 5′ ends of the PCR products. The reaction was run at 23°C for 30 min, and then the products were purified with XP beads. The PCR products were denatured to become single-stranded DNA (ssDNA) by incubating at 95°C for 3 min and were then immediately kept at 4°C for the subsequent steps. The denatured ssDNA was transformed into circles with cyclase at 37°C for 30 min and digested with Exo enzyme at 37°C for 30 min to remove linear DNA. The products were purified with XP beads. The concentration of the library was assayed with a Qubit 4 fluorometer. To avoid sequencing bias induced by base unbalance of PCR products, 12 ng of whole-genome DNA library (balance library) was mixed with the 8 PCR samples to a final concentration of 1.5 ng/μL and sequenced in one lane. All the samples were subjected to pair-ended 150 bp deep sequencing on the MGISEQ-2000 platform.
NGS data analysis
Raw data quality was evaluated by FastQC 0.11.3, and low-quality sequencing reads were filtered out by fastp 0.19.6. Clean amplicon reads were aligned with the theoretical NHBEJ reference using the Burrows-Wheeler Aligner-Maximal Exact Match algorithm. Reads that were identical to the NHBEJ reference were counted, and the percentage was determined.
Data statistics
The webtool “CRISPOR” (http://crispor.tefor.net/crispor.py) was used for design of CRISPR-spCas9 gRNAs. Prism 7 was used for the analysis with boxplot and point chart. SnapGene Viewer was used for the analysis of Sanger sequencing results, and the webtool ICE Analysis (https://ice.synthego.com/#/) was used for genotype percentage analysis with Sanger data. All experiments were performed in at least triplicates. ANOVA was used for statistic tests, with a p value <0.05 considered significant.
Acknowledgments
This project is partially supported by the Sanming Project of Medicine in Shenzhen (SZSM201612074) to L.B. and Y.L., the National Natural Science Foundation of China (31911530148) to H.T., and the DFF Sapere Aude Starting grant (8048-00072A) to L.L. We thank other colleagues from LBI for providing technical assistance for carrying out the study. Professor Niels Jessen from SDCA is thanked for his insightful comments on the study.
Author contributions
Y.L. and X.X. conceived the idea. Y.L., H.T., L.L., L.B., and J.F. oversaw the study. X.X and X.Z. performed most of the work and contributed equally to the study. All other co-authors contributed to the performance of experiments, discussion, presentation, and interpretation of the results. X.X. and Y.L. drafted the manuscript, and all authors read and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.03.005.
Contributor Information
Huajing Teng, Email: tenghj@biols.ac.cn.
Lin Lin, Email: linlin@biomed.au.dk.
Yonglun Luo, Email: alun@biomed.au.dk.
Supplemental information
References
- 1.Wang H., La Russa M., Qi L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016;85:227–264. doi: 10.1146/annurev-biochem-060815-014607. [DOI] [PubMed] [Google Scholar]
- 2.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiang X., Luo L., Nodzyński M., Li C., Han P., Dou H., Petersen T.S., Liang X., Pan X., Qu K. LION: a simple and rapid method to achieve CRISPR gene editing. Cell. Mol. Life Sci. 2019;76:2633–2645. doi: 10.1007/s00018-019-03064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen M.W., Arbab M., Hsu J.Y., Worstell D., Culbertson S.J., Krabbe O., Cassa C.A., Liu D.R., Gifford D.K., Sherwood R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 2018;563:646–651. doi: 10.1038/s41586-018-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018;37:64–72. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosicki M., Rajan S.S., Lorenzetti F.C., Wandall H.H., Narimatsu Y., Metzakopian E., Bennett E.P. Dynamics of Indel Profiles Induced by Various CRISPR/Cas9 Delivery Methods. Prog. Mol. Biol. Transl. Sci. 2017;152:49–67. doi: 10.1016/bs.pmbts.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Chen X., Xu F., Zhu C., Ji J., Zhou X., Feng X., Guang S. Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci. Rep. 2014;4:7581. doi: 10.1038/srep07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng Q., Cai X., Tan M.H., Schaffert S., Arnold C.P., Gong X., Chen C.Z., Huang S. Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques. 2014;57:115–124. doi: 10.2144/000114196. [DOI] [PubMed] [Google Scholar]
- 9.Do P.T., Nguyen C.X., Bui H.T., Tran L.T.N., Stacey G., Gillman J.D., Zhang Z.J., Stacey M.G. Demonstration of highly efficient dual gRNA CRISPR/Cas9 editing of the homeologous GmFAD2-1A and GmFAD2-1B genes to yield a high oleic, low linoleic and α-linolenic acid phenotype in soybean. BMC Plant Biol. 2019;19:311. doi: 10.1186/s12870-019-1906-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho T.T., Zhou N., Huang J., Koirala P., Xu M., Fung R., Wu F., Mo Y.Y. Targeting non-coding RNAs with the CRISPR/Cas9 system in human cell lines. Nucleic Acids Res. 2015;43:e17. doi: 10.1093/nar/gku1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J., Zhang J., Chen L., Shen B., Zhou J., Hu B., Du Y., Tate P.H., Huang X., Zhang W. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol. 2014;11:829–835. doi: 10.4161/rna.29624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandal P.K., Ferreira L.M., Collins R., Meissner T.B., Boutwell C.L., Friesen M., Vrbanac V., Garrison B.S., Stortchevoi A., Bryder D. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J., Wang J., Shen B., Chen L., Su Y., Yang J., Zhang W., Tian X., Huang X. Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J. 2014;281:1717–1725. doi: 10.1111/febs.12735. [DOI] [PubMed] [Google Scholar]
- 14.Maddalo D., Manchado E., Concepcion C.P., Bonetti C., Vidigal J.A., Han Y.C., Ogrodowski P., Crippa A., Rekhtman N., de Stanchina E. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516:423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J.P., Li X.L., Li G.H., Chen W., Arakaki C., Botimer G.D., Baylink D., Zhang L., Wen W., Fu Y.W. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017;18:35. doi: 10.1186/s13059-017-1164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shou J., Li J., Liu Y., Wu Q. Precise and Predictable CRISPR Chromosomal Rearrangements Reveal Principles of Cas9-Mediated Nucleotide Insertion. Mol. Cell. 2018;71:498–509.e4. doi: 10.1016/j.molcel.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 17.Guo T., Feng Y.L., Xiao J.J., Liu Q., Sun X.N., Xiang J.F., Kong N., Liu S.C., Chen G.Q., Wang Y. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018;19:170. doi: 10.1186/s13059-018-1518-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Møller H.D., Lin L., Xiang X., Petersen T.S., Huang J., Yang L., Kjeldsen E., Jensen U.B., Zhang X., Liu X. CRISPR-C: circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res. 2018;46:e131. doi: 10.1093/nar/gky767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J., Xiang X., Huang J., Liang X., Pan X., Dong Z., Petersen T.S., Qu K., Yang L., Zhao X. Haplotyping by CRISPR-mediated DNA circularization (CRISPR-hapC) broadens allele-specific gene editing. Nucleic Acids Res. 2020;48:e25. doi: 10.1093/nar/gkz1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ousterout D.G., Kabadi A.M., Thakore P.I., Majoros W.H., Reddy T.E., Gersbach C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015;6:6244. doi: 10.1038/ncomms7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L., Kaul A., Kinnett K., McDonald C., Pandya S., DMD Care Considerations Working Group Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 22.Salmaninejad A., Valilou S.F., Bayat H., Ebadi N., Daraei A., Yousefi M., Nesaei A., Mojarrad M. Duchenne muscular dystrophy: an updated review of common available therapies. Int. J. Neurosci. 2018;128:854–864. doi: 10.1080/00207454.2018.1430694. [DOI] [PubMed] [Google Scholar]
- 23.Sun C., Shen L., Zhang Z., Xie X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes (Basel) 2020;11:837. doi: 10.3390/genes11080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H.L., Fujimoto N., Sasakawa N., Shirai S., Ohkame T., Sakuma T., Tanaka M., Amano N., Watanabe A., Sakurai H. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim K.R.Q., Yoon C., Yokota T. Applications of CRISPR/Cas9 for the Treatment of Duchenne Muscular Dystrophy. J. Pers. Med. 2018;8:38. doi: 10.3390/jpm8040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu-Motohashi Y., Komaki H., Motohashi N., Takeda S., Yokota T., Aoki Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J. Pers. Med. 2019;9:1. doi: 10.3390/jpm9010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai A., Kong X. Development of CRISPR-Mediated Systems in the Study of Duchenne Muscular Dystrophy. Hum. Gene Ther. Methods. 2019;30:71–80. doi: 10.1089/hgtb.2018.187. [DOI] [PubMed] [Google Scholar]
- 31.Aartsma-Rus A., Fokkema I., Verschuuren J., Ginjaar I., van Deutekom J., van Ommen G.J., den Dunnen J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009;30:293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 32.Min Y.L., Bassel-Duby R., Olson E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019;70:239–255. doi: 10.1146/annurev-med-081117-010451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsiau T., Conant D., Rossi N., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., Enzmann B.L., Stoner R. Inference of CRISPR Edits from Sanger Trace Data. bioRxiv. 2019 doi: 10.1101/251082. [DOI] [PubMed] [Google Scholar]
- 35.Lonowski L.A., Narimatsu Y., Riaz A., Delay C.E., Yang Z., Niola F., Duda K., Ober E.A., Clausen H., Wandall H.H. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat. Protoc. 2017;12:581–603. doi: 10.1038/nprot.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thakur N., Abeysekera G., Wanigasinghe J., Dissanayake V.H.W. The spectrum of deletions and duplications in the dystrophin (DMD) gene in a cohort of patients with Duchenne muscular dystrophy in Sri Lanka. Neurol. India. 2019;67:714–715. doi: 10.4103/0028-3886.263235. [DOI] [PubMed] [Google Scholar]
- 37.Wang R.T., Barthelemy F., Martin A.S., Douine E.D., Eskin A., Lucas A., Lavigne J., Peay H., Khanlou N., Sweeney L. DMD genotype correlations from the Duchenne Registry: Endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum. Mutat. 2018;39:1193–1202. doi: 10.1002/humu.23561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khajavi M., Inoue K., Lupski J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 39.Kameyama T., Ohuchi K., Funato M., Ando S., Inagaki S., Sato A., Seki J., Kawase C., Tsuruma K., Nishino I. Efficacy of Prednisolone in Generated Myotubes Derived From Fibroblasts of Duchenne Muscular Dystrophy Patients. Front. Pharmacol. 2018;9:1402. doi: 10.3389/fphar.2018.01402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J.J.A., Saito T., Duddy W., Takeda S., Yokota T. Direct Reprogramming of Human DMD Fibroblasts into Myotubes for In Vitro Evaluation of Antisense-Mediated Exon Skipping and Exons 45-55 Skipping Accompanied by Rescue of Dystrophin Expression. Methods Mol. Biol. 2018;1828:141–150. doi: 10.1007/978-1-4939-8651-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alkan F., Wenzel A., Anthon C., Havgaard J.H., Gorodkin J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018;19:177. doi: 10.1186/s13059-018-1534-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 43.Wang D., Zhang C., Wang B., Li B., Wang Q., Liu D., Wang H., Zhou Y., Shi L., Lan F., Wang Y. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019;10:4284. doi: 10.1038/s41467-019-12281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giuliano C.J., Lin A., Girish V., Sheltzer J.M. Generating Single Cell-Derived Knockout Clones in Mammalian Cells with CRISPR/Cas9. Curr. Protoc. Mol. Biol. 2019;128:e100. doi: 10.1002/cpmb.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li K., Wang G., Andersen T., Zhou P., Pu W.T. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS ONE. 2014;9:e105779. doi: 10.1371/journal.pone.0105779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Editorial Keep off-target effects in focus. Nat. Med. 2018;24:1081. doi: 10.1038/s41591-018-0150-3. [DOI] [PubMed] [Google Scholar]
- 47.Benston S. Everything in moderation, even hype: learning from vaccine controversies to strike a balance with CRISPR. J. Med. Ethics. 2017;43:819–823. doi: 10.1136/medethics-2016-103666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tycko J., Myer V.E., Hsu P.D. Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity. Mol. Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hajiahmadi Z., Movahedi A., Wei H., Li D., Orooji Y., Ruan H., Zhuge Q. Strategies to Increase On-Target and Reduce Off-Target Effects of the CRISPR/Cas9 System in Plants. Int. J. Mol. Sci. 2019;20:3719. doi: 10.3390/ijms20153719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long C., Li H., Tiburcy M., Rodriguez-Caycedo C., Kyrychenko V., Zhou H., Zhang Y., Min Y.L., Shelton J.M., Mammen P.P.A. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018;4:eaap9004. doi: 10.1126/sciadv.aap9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kyrychenko V., Kyrychenko S., Tiburcy M., Shelton J.M., Long C., Schneider J.W., Zimmermann W.H., Bassel-Duby R., Olson E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight. 2017;2:e95918. doi: 10.1172/jci.insight.95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y., Long C., Li H., McAnally J.R., Baskin K.K., Shelton J.M., Bassel-Duby R., Olson E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017;3:e1602814. doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin L., Luo Y. Tracking CRISPR’s Footprints. Methods Mol. Biol. 2019;1961:13–28. doi: 10.1007/978-1-4939-9170-9_2. [DOI] [PubMed] [Google Scholar]
- 55.den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruan G.X., Barry E., Yu D., Lukason M., Cheng S.H., Scaria A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cideciyan A.V., Aleman T.S., Jacobson S.G., Khanna H., Sumaroka A., Aguirre G.K., Schwartz S.B., Windsor E.A., He S., Chang B. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum. Mutat. 2007;28:1074–1083. doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- 58.Aartsma-Rus A., van Ommen G.J. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aartsma-Rus A., Straub V., Hemmings R., Haas M., Schlosser-Weber G., Stoyanova-Beninska V., Mercuri E., Muntoni F., Sepodes B., Vroom E., Balabanov P. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017;27:251–259. doi: 10.1089/nat.2017.0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.