

Abstract
Sulfite oxidase (SO) deficiency is a disorder caused either by isolated deficiency of SO or by defects in the synthesis of its molybdenum cofactor. It is characterized biochemically by tissue sulfite accumulation. Patients present with seizures, progressive neurological damage, and basal ganglia abnormalities, the pathogenesis of which is not fully established. Treatment is supportive and largely ineffective. To address the pathophysiology of sulfite toxicity, we examined the effects of intrastriatal administration of sulfite in rats on antioxidant defenses, energy transfer, and mitogen-activated protein kinases (MAPK) and apoptosis pathways in rat striatum. Sulfite administration decreased glutathione (GSH) concentration and glutathione peroxidase, glucose-6-phosphate dehydrogenase, glutathione S-transferase, and glutathione reductase activities in striatal tissue. Creatine kinase (CK) activity, a crucial enzyme for cell energy transfer, was also decreased by sulfite. Superoxide dismutase-1 (SOD1) and catalase (CAT) proteins were increased, while heme oxygenase-1 (HO-1) was decreased. Additionally, sulfite altered phosphorylation of MAPK by decreasing of p38 and increasing of ERK. Sulfite further augmented the content of GSK-3β, Bok, and cleaved caspase-3, indicating increased apoptosis. JP4-039 is a mitochondrial-targeted antioxidant that reaches higher intramitochondrial levels than other traditional antioxidants. Intraperitoneal injection of JP4-039 before sulfite administration preserved activity of antioxidant enzymes and CK. It also prevented or attenuated alterations in SOD1, CAT, and HO-1 protein content, as well as changes in p38, ERK, and apoptosis markers. In sum, oxidative stress and apoptosis induced by sulfite injection are prevented by JP4-039, identifying this molecule as a promising candidate for pharmacological treatment of SO-deficient patients.
Keywords: antioxidant defenses, apoptosis, JP4-039, striatum, sulfite, sulfite oxidase deficiency
1 |. INTRODUCTION
Sulfite oxidase (SO; EC 1.8.3.1), an enzyme located in the mitochondrial intermembrane space, catalyzes the oxidation of sulfite to sulfate, the final step in the degradation of sulfur-containing amino acids.1,2 It also degrades sulfite derived from exogenous sources, used extensively as preservatives and antimicrobial agents in foods and medications.3,4
Deficiency of SO is caused either by mutations in the SUOX gene, which encodes SO (isolated SO deficiency; iSOD; OMIM #272300), or by mutations in genes that encode enzymes of the molybdenum cofactor (MoCo) biosynthetic pathway (MoCo deficiency; MoCD; OMIM #242150, #242160, #615501).1,5 Mutations in MOCS1, MOCS2, and GPHN result in MoCD type A, B, and C, respectively. In addition to sulfite, both iSOD and MoCD patients present tissue accumulation and high urinary excretion of S-sulfocysteine, thiosulfate, and taurine. Since MoCo also is a cofactor for xanthine dehydrogenase and aldehyde oxidase, individuals with MoCD are deficient in the activity of these enzymes, and excrete high levels of xanthine and hypoxanthine in urine, and have low serum concentration of uric acid.1
Patients with iSOD and MoCD manifest severe and progressive neurological dysfunction, neonatal seizures, alterations in muscle tone, axial hypotonia, and failure to thrive. Both forms of SO deficiency often result in early childhood death, though MoCD is often more severe.1,6 Neuropathological findings include marked basal ganglia, cerebral cortex and cerebellum atrophy, dilated brain ventricles, and global white and gray matter abnormalities. Neuronal loss, gliosis, and demyelination are also observed.7,8
The mechanisms underlying the brain abnormalities in iSOD and MoCD are still poorly understood. However, it has been shown that sulfite, the main accumulating metabolite in both disorders, is neurotoxic. Sulfite undergoes auto-oxidation in vitro and in vivo in rat brain, leading to the formation of reactive species9 and impairing redox homeostasis and antioxidant defenses.9–14 Moreover, sulfite disrupts mitochondrial bioenergetics and biogenesis and induces mitochondrial permeability transition,11,15 as well as glial reactivity and neuronal loss in rat striatum.11
Treatment for iSOD and MoCD is based on the reduction of sulfite levels with a diet consisting of low content of sulfur-containing amino acids combined with the administration of antiepileptic drugs to control seizures, but with limited success.1,16 Cyclic pyranopterin monophosphate, an intermediate in the MoCo biosynthetic pathway, has been used to treat patients with MoCD type A, but is not effective for iSOD and other types of MoCD.17 Therefore, the evaluation of novel therapeutic strategies is critical to improve the prognosis of these disorders. JP4-039 is a mitochondria-targeted electron and reactive oxygen species (ROS) scavenger comprised of a nitroxide attached directly to an alkene-peptide isostere that exhibits 20-fold to 30-fold enrichment in the mitochondria over the cytosol.18 It has been shown to prevent radiation damage, lipid peroxidation, and apoptosis in irradiated rats.19,20 Treatment with JP4-039 has also been demonstrated to decrease ROS in cells from patients with MoCo, ACAD9, and very long chain acyl-CoA dehydrogenase deficiencies, defects of mitochondrial oxidative phosphorylation and fatty acid oxidation, respectively.21–23
In the present study, we evaluated the in vivo effect of intrastriatal administration of sulfite in rats on antioxidant defenses, creatine kinase (CK) activity, mitogen-activated protein kinases (MAPK) signaling pathways, and apoptosis markers. We also investigated the ability of JP4-039 to protect against the deleterious effects elicited by sulfite exposure.
2 |. MATERIAL AND METHODS
2.1 |. Reagents
All chemicals, including sodium sulfite (Na2SO3), were purchased from Sigma-Aldrich (St. Louis, Missouri), unless otherwise stated. JP4-039 was provided by Dr. Peter Wipf, Departments of Chemistry, Pharmaceutical Sciences and Bioengineering, University of Pittsburgh.24
2.2 |. Animals
Twenty-nine-day-old male Wistar rats were obtained from the Central Animal House of the Department of Biochemistry, Instituto de Ciências Básicas da Saúde (ICBS), Universidade Federal do Rio Grande do Sul, Brazil. The animals were maintained on a 12:12-hours light/dark cycle (lights on 07:00–19:00 hours) in air-conditioned constant temperature (22 ± 1°C) colony room, with free access to water and 20% (w/w) protein commercial chow (Nuvilab CR-1, Colombo, Brazil). The National Animal Rights Regulation (Law 11.794/2008) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication 85-23, revised 1966) were followed. All efforts were made to minimize the number of animals used, as well as their stress and suffering.
2.3 |. JP4-039 treatment
Rats were divided in four treatment groups: vehicle + NaCl (control), vehicle + sulfite (Na2SO3), JP4-039 + NaCl, and JP4-039 + sulfite. JP4-039 was prepared in 0.1 M phosphate-buffered saline, pH 7.4, containing 5% DMSO (vehicle), and administered intraperitoneally twice, at 24 hours (3 or 5 μg/g) and 2 hours (5.0 μg/g) prior to sulfite intrastriatal injection.25
2.4 |. Sulfite administration
The experimental protocol was based on previous work from our group.11 At the age of 30 days, rats were deeply anesthetized with an intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg), and placed in a stereotaxic apparatus. Two small holes were drilled in the skull and 2 μL of 1 M sulfite (2 μmol) or 1 M NaCl (2 μmol) were injected bilaterally into striatum over 4 minutes. After each injection, the needle was left in place for 1 minute and then carefully removed. The aqueous solutions for injection were freshly prepared and had their pH adjusted to pH 7.4 immediately prior the procedure. The coordinates used for the injection were: 0.6 mm posterior to the bregma, 2.6 mm lateral to the midline, and 4.5 mm ventral from dura.26 Animals were euthanized 30 minutes after the procedure and the striatum was dissected for subsequent evaluation.
2.5 |. Measurement of reduced glutathione
Striatal tissue was homogenized in 1:10 w/v 20 mM sodium phosphate buffer, pH 7.4, 140 mM KCl. The homogenate was centrifuged at 750g for 10 minutes at 4°C, and the supernatant was assayed for glutathione (GSH) concentration.27 One hundred microliters of tissue supernatant were deproteinized with an equal volume of 2% metaphosphoric acid and centrifuged at 7000g for 10 minutes at 4°C. Thirty microliters of supernatant were added to the well of a 96-well plate and brought to 100 μL with 100 mM sodium phosphate buffer, pH 8.0, 5 mM EDTA. Subsequently, 185 μL of the same buffer and 15 μL of o-phthaldialdehyde (1 mg/mL in methanol) were added, and the plates were incubated in absence of light for 15 minutes at room temperature. The fluorescence was then measured at 350 (excitation) and 420 (emission) nm on a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, California). A calibration curve was prepared using a GSH standard solution (0.001–1 mM) and the results were expressed as nmol GSH/mg protein.
2.6 |. CK activity
The striatum was homogenized (1:10 w/v) in isosmotic saline solution and centrifuged at 750g for 10 minutes at 4°C to discard nuclei and cell debris. The supernatant was collected, diluted in a proportion of 1:30 in isosmotic saline solution, and used for enzymatic activity determination. The assay was carried out according to Hughes28 using ADP and phosphocreatine as substrates.
2.7 |. Antioxidant enzyme activities
For the determination of the antioxidant enzyme activities, the striatum was homogenized (1:10 w/v) in 20 mM sodium phosphate buffer, pH 7.4, 140 mM KCl. The homogenate was centrifuged at 750g for 10 minutes at 4°C and the supernatant was used for the assays. The absorbance in all assays was read on a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, California). The specific activities were calculated as U/mg protein.
Glutathione peroxidase (GPx) activity was measured according to Wendel29 by monitoring the oxidation of NADPH at 25°C at 340 nm in a medium containing striatum supernatant (approximately 75 μg of protein), 100 mM potassium phosphate buffer, 1 mM EDTA, pH 7.7, 2 mM GSH, 0.1 U/mL glutathione reductase, 0.4 mM azide, 0.5 mM tert-butyl-hydroperoxide, and 0.1 mM NADPH.
Glutathione reductase (GR) activity was measured according to Carlberg and Mannervik30 by monitoring NADPH oxidation at 25°C at 340 nm. The reaction medium contained tissue supernatants (approximately 90 μg of protein), 200 mM sodium phosphate buffer, pH 7.5, 6.3 mM EDTA, 1 mM oxidized glutathione (GSSH), and 0.1 mM NADPH.
Glutathione S-transferase (GST) activity was measured according to Mannervik and Guthenberg31 using 1-chloro-2,4-dinitrobenzene as substrate and monitoring the formation of dinitrophenyl-S-glutathione at 25°C at 340 nm. The reaction medium contained tissue supernatants (approximately 60 μg of protein), 50 mM potassium phosphate, pH 6.5, 1 mM GSH, and 1 mM 1-chloro-2,4-dinitrobenzene.
Glucose-6-phosphate dehydrogenase (G6PDH) activity was determined according to Leong and Clark32 following NADPH formation at 25°C at 340 nm. The reaction medium contained striatum supernatants (approximately 45 μg of protein), 100 mM Tris-HCl buffer, pH 7.5, 1 mM MgCl2, 0.05 NADP+, and 0.1 mM glucose-6-phosphate.
2.8 |. Preparation of samples and western blot analysis
Striatum was homogenized in RIPA buffer containing protease and phosphatase inhibitors (1 mM sodium orthovanadate, 1 mM aprotinin, and 1% protease inhibitor cocktail) and centrifuged at 10000g for 10 minutes at 4°C. Supernatants were used for western blotting, as described in da Rosa-Junior.33 The following primary antibodies were used: anti-phospho-JNK and anti-JNK were purchased from R&D Systems, Minnesota; anti-Bok, anti-Bcl-xL, anti-caspase-9, anti-caspase-3, anti-cleaved caspase-3, anti-phospho-p38, anti-p38, anti-phospho-ERK1/2, and anti-ERK1/2 from Cell Signaling, Massachusetts; anti-superoxide dismutase 1 (SOD1), anti-heme oxygenase-1 (HO-1), and anti-α-synuclein from Abcam, Cambridge, UK; anti-catalase (CAT) was from Merk Millipore, Massachusetts; anti-GSK-3β from Santa Cruz Biotechnology, Texas; anti-β-actin from Sigma-Aldrich, Missouri.
2.9 |. Statistical analysis
Results are presented as mean ± SD. Assays were performed in replicates and the mean was used for statistical analysis. Data were analyzed by one-way ANOVA followed by the post hoc Duncan multiple range test when F was significant. Only significant F values are shown in the text. Differences between groups were rated significant at P < .05. All analyses were carried out using the IBM SPSS software (Armonk, New York).
3 |. RESULTS
3.1 |. JP4-039 mitigates alterations in antioxidant defenses in striatum injected with sulfite
Striatal injection in rat brains with sulfite has previously been shown to alter antioxidant defenses.11 To examine the effect of JP4-039 on this process, it was administered at 3 μg/g 24 hours and 5.0 μg/g 2 hours prior to sulfite injection. JP4-039 attenuated the reduction of GPx [F(3,22) = 3.254; P < .05] (Figure 1A) and G6PDH [F(3,18) = 2.737; P < .05] (Figure 1G) induced by sulfite, but did not affect the activities of GR (Figure 1C) and GST (Figure 1E). In contrast, administrations of 5.0 μg/g of JP4-039 at 24 hours and again at 2 hours prior to sulfite injection protected against the reduction of GST [F(3,15) = 3.968; P < .05] (Figure 1F) and G6PDH [F(3,14) = 6.018; P < .01] (Figure 1H), but failed to protect against reduction of GPx (Figure 1B) and GR (Figure 1D). JP4-039 did not protect against a sulfite-induced decrease of GSH levels at either dosing regimen (Figure 2). Sulfite administration decreased HO-1 protein [F(3,8) = 17.808; P < .001] and increased CAT [F(3,10) = 6.755; P < .05] and SOD1 [F(3,10) = 88.474; P < .001] protein, all of which was prevented by pretreatment with JP4-039 at the higher dose (Figure 3).
FIGURE 1.
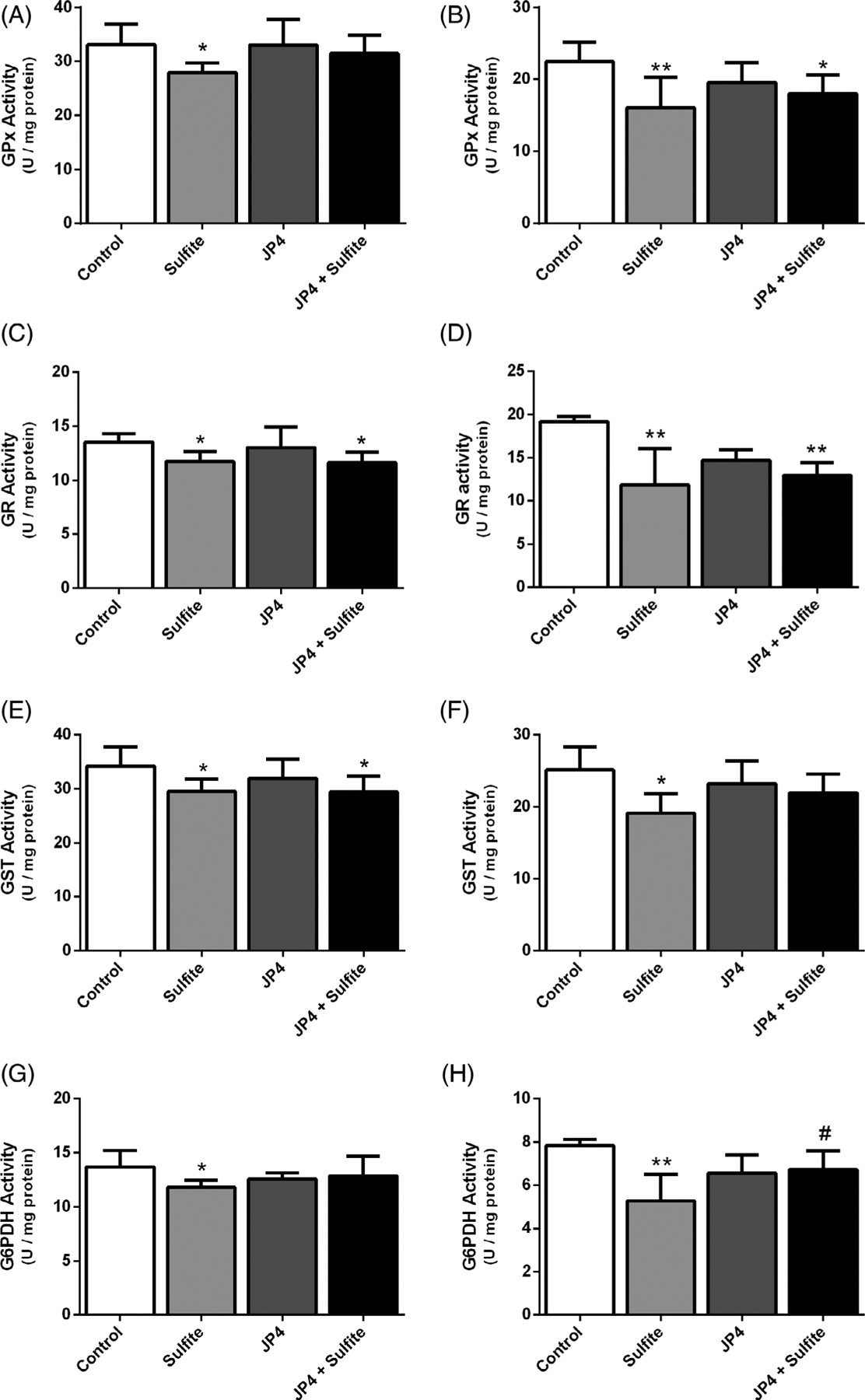
Effect of JP4-039 on sulfite-induced decrease in glutathione peroxidase (GPx), A, B, glutathione reductase (GR), C, D, glutathione transferase (GST), E, F, and glucose-6-phosphate dehydrogenase (G6PDH), G, H, activities in rat striatum 30 minutes after sulfite injection (2 μmol). Pretreatment with JP4-039 at the doses of 3.5 and 5.0 μg/g, A, C, E, G, or 5.0 and 5.0 μg/g, B, D, F, H, was performed via intraperitoneal injection 24 and 2 hours prior to sulfite administration. Values are means ± SD for four to seven independent experiments (animals) per group. *P < .05, **P < .01, compared to rats receiving NaCl (2 μmol) (control group); #P < .05, compared to rats receiving sulfite (sulfite group) (Duncan multiple range test)
FIGURE 2.
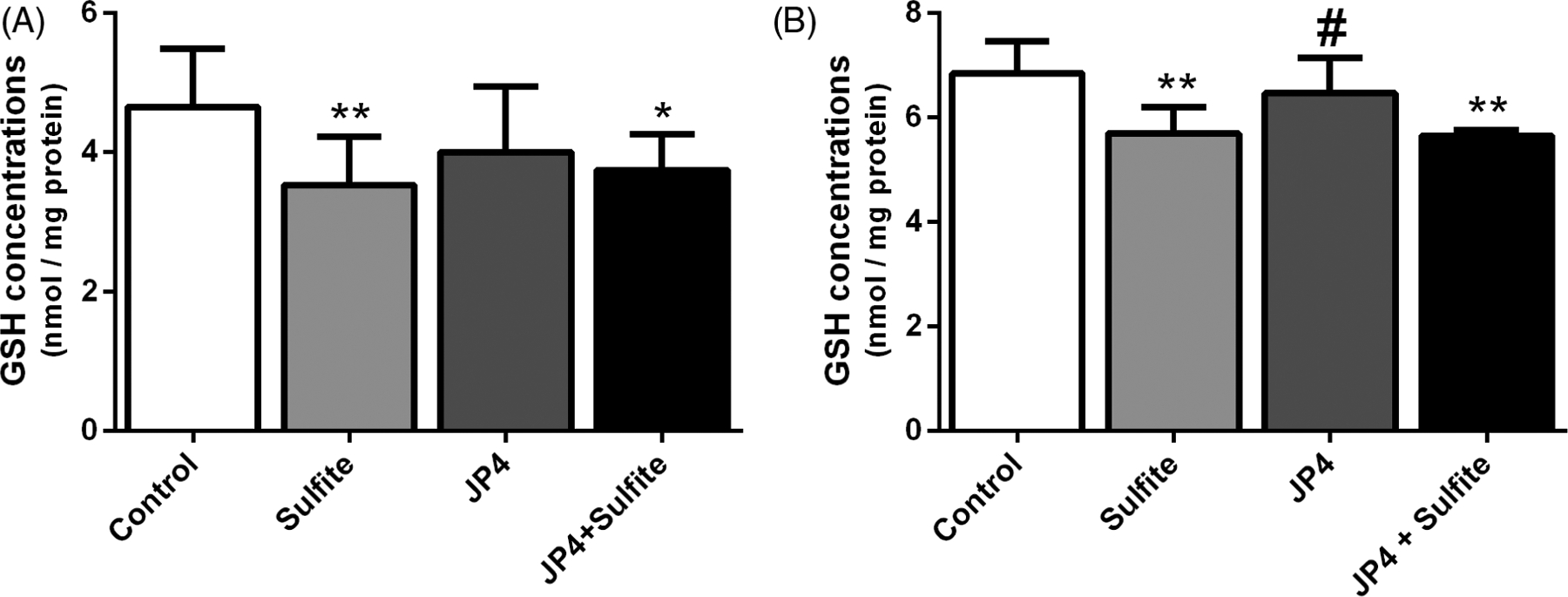
Effect of JP4-039 on sulfite-induced decrease in reduced glutathione concentrations (GSH) in rat striatum 30 minutes after sulfite injection (2 μmol). Pretreatment with JP4-039 at the doses of 3.5 and 5.0 μg/g, A, or 5.0 and 5.0 μg/g, B, was performed via intraperitoneal injection 24 and 2 hours prior to sulfite administration. Values are means ± SD for four to seven independent experiments (animals) per group. *P < .05, **P < .01, compared to rats receiving NaCl (2 μmol) (control group); #P < .05 compared to rats receiving sulfite (sulfite group) (Duncan multiple range test)
FIGURE 3.
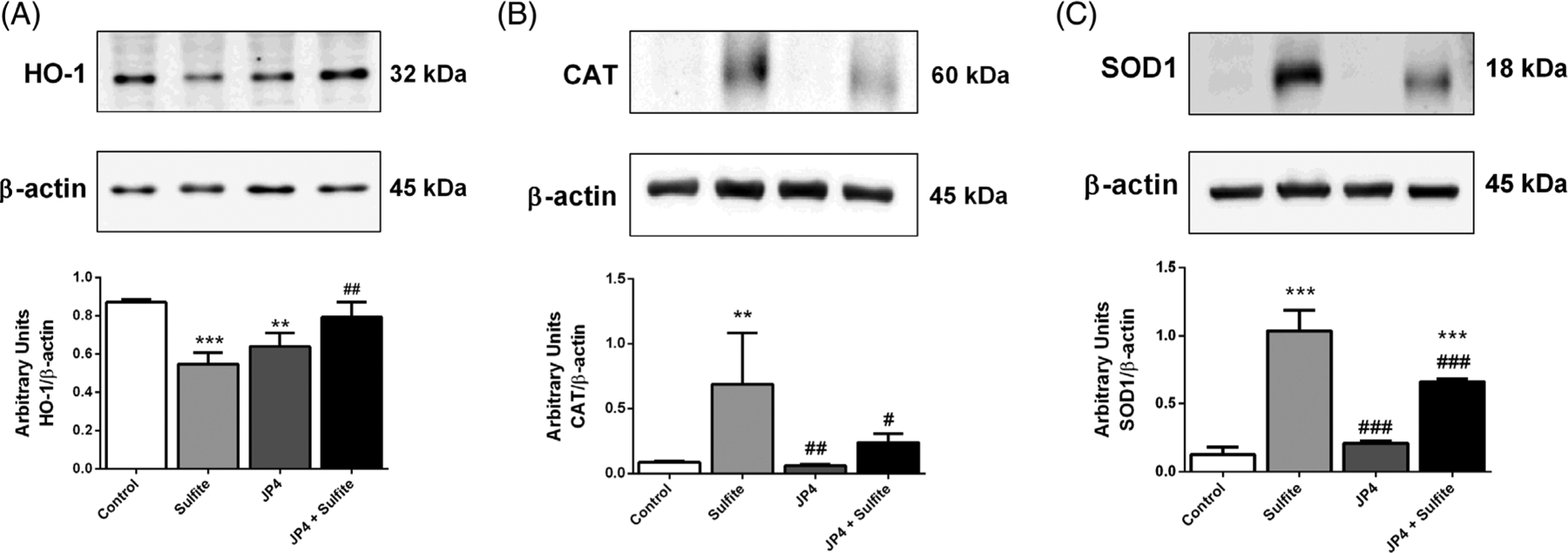
Effect of JP4-039 on sulfite-induced alterations on heme oxygenase-1 (HO-1), A, catalase (CAT), B, and superoxide dismutase 1 (SOD1), C, content in rat striatum 30 minutes after sulfite injection (2 μmol). Pretreatment with JP4-039 was performed via intraperitoneal injection 24 (5.0 μg/g) and 2 hours (5.0 μg/g) prior to sulfite administration. Values are means ± SD for three to four independent experiments (animals) per group. **P < .01, ***P < .001, compared to rats receiving NaCl (2 μmol) (control group); #P < .05, ##P < .01, ###P < .001, compared to rats receiving sulfite (sulfite group) (Duncan multiple range test)
3.2 |. JP4-039 prevents sulfite-induced decrease of CK activity
Previous data from our group showed that sulfite injection decreased the activity of CK in striatum, an enzyme that controls the homeostasis of energy transfer between cytosolic and mitochondrial compartments.11 Pretreatment with high dose JP4-039 led to preservation of CK activity [F(3,20) = 7.350; P < .01], while the low dose regimen did not (Figure 4).
FIGURE 4.
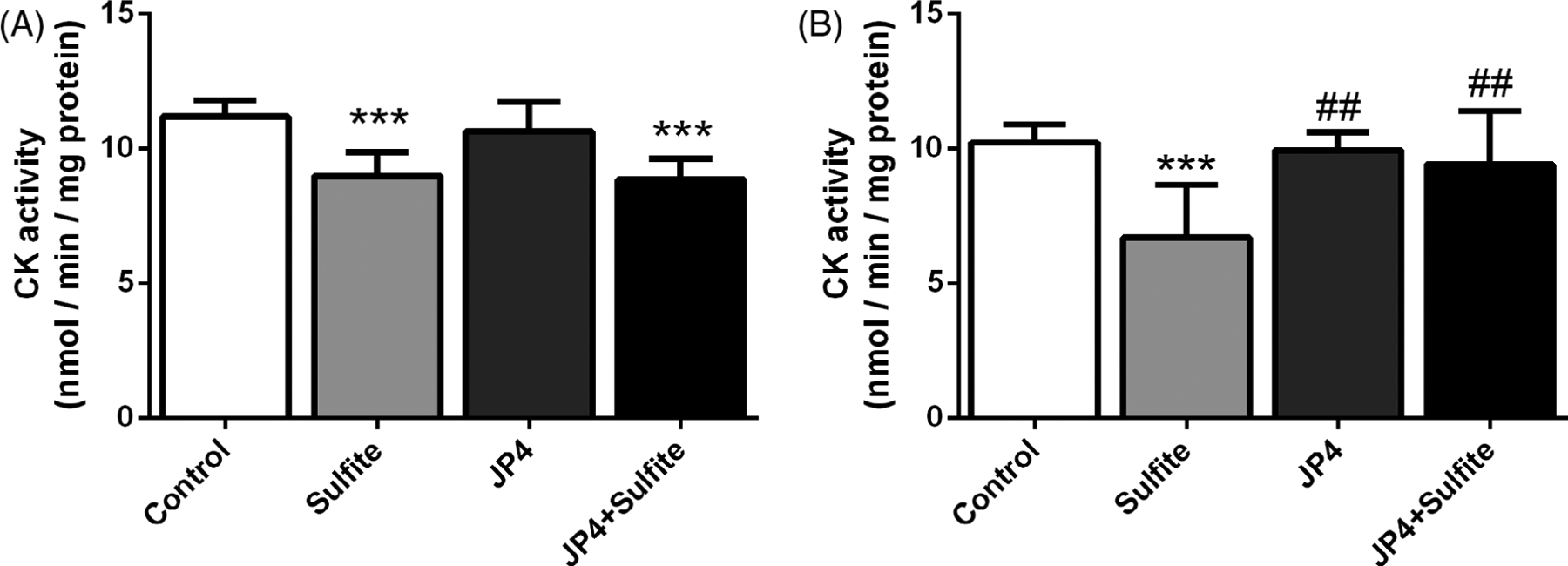
Effect of JP4-039 on sulfite-induced decrease in creatine kinase (CK) activity in rat striatum 30 minutes after sulfite injection (2 μmol). Pretreatment with JP4-039 at the doses of 3.5 and 5.0 μg/g, A, or 5.0 and 5.0 μg/g, B, was performed via intraperitoneal injection 24 and 2 hours prior to sulfite administration. Values are means ± SD for four to seven independent experiments (animals) per group. ***P < .001, compared to rats receiving NaCl (2 μmol) (control group); ##P < .01, compared to rats receiving sulfite (sulfite group) (Duncan multiple range test)
3.3 |. JP4-039 mitigates sulfite-induced alterations on MAPK phosphorylation and protein content
MAPK signaling pathways are essential for cell homeostasis, being involved in proliferation, growth, and survival processes,34 so we examined the effects of sulfite and JP4-039 on phosphorylation and protein levels of MAPK pathway members p38, ERK1/2, and JNK. Sulfite exposure decreased p38 phosphorylation, a change that was prevented by JP4-039 [F(3,10) = 19.144; P < .001] (Figure 5A). Conversely, sulfite injection increased ERK1/2 phosphorylation (Figure 5B), which was attenuated by JP4-039 [F(3,10) = 3.761; P < .05] (Figure 5B). JNK phosphorylation, total p38, ERK1/2, and JNK protein levels were not altered by sulfite exposure (Figures 5C–F). Of note, JP4-039 treatment alone increased the level of total p38 (Figure 5D).
FIGURE 5.
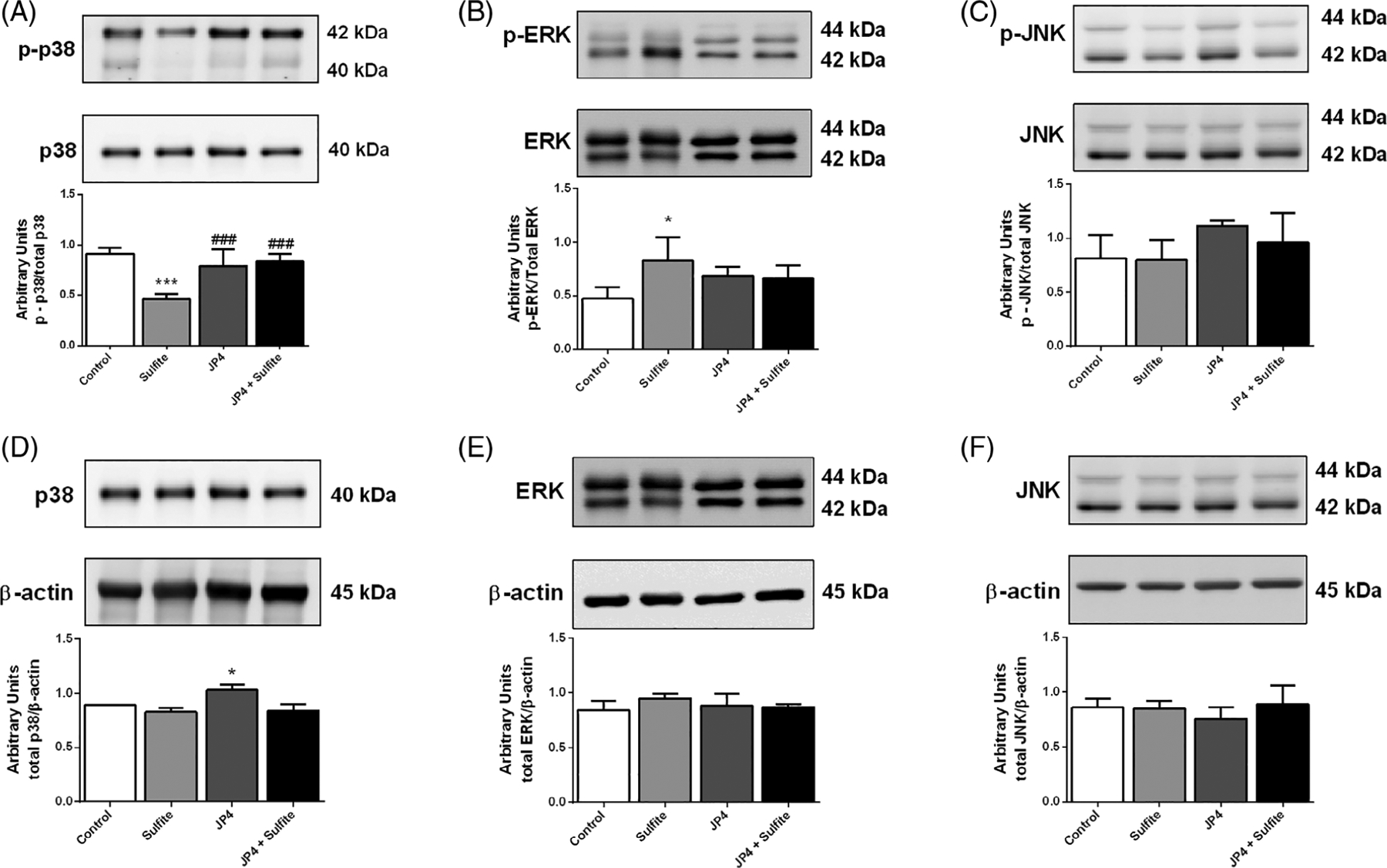
Effect of JP4-039 on sulfite-induced alterations on p38, A, ERK 1/2, B, and JNK, C, phosphorylation in rat striatum 30 minutes after sulfite injection (2 μmol). Pretreatment with JP4-039 was performed via intraperitoneal injection 24 (5.0 μg/g) and 2 hours (5.0 μg/g) prior to sulfite administration. Total content of p38, D, ERK 1/2, E, and JNK, F, is also shown. Values are means ± SD for three to four independent experiments (animals) per group. *P < .05, ***P < .001, compared to rats receiving NaCl (2 μmol) (control group); ###P < .001, compared to rats receiving sulfite (sulfite group) (Duncan multiple range test)
3.4 |. JP4-039 mitigates sulfite-induced apoptosis
In order to determine the effect of sulfite exposure on cell death by apoptosis, we assessed the levels of some proteins involved in the apoptosis cascade. Sulfite injection significantly increased the striatal GSK-3β [F(3,10) = 9.106; P < .01] (Figure 6A), Bok [F(3,10) = 11.658; P < .001] (Figure 6C) and cleaved caspase-3 [F(3,10) = 112.794; P < .001] (Figure 6F). These changes were prevented or mitigated by pretreatment with JP4-039. In contrast, sulfite did not change content of caspase-9 (Figure 6D), total caspase-3 (Figure 6E), and Bcl-xL (Figure 6B). Moreover, sulfite did not alter the content of α-synuclein (Figure 6G), a marker of cell injury.
FIGURE 6.
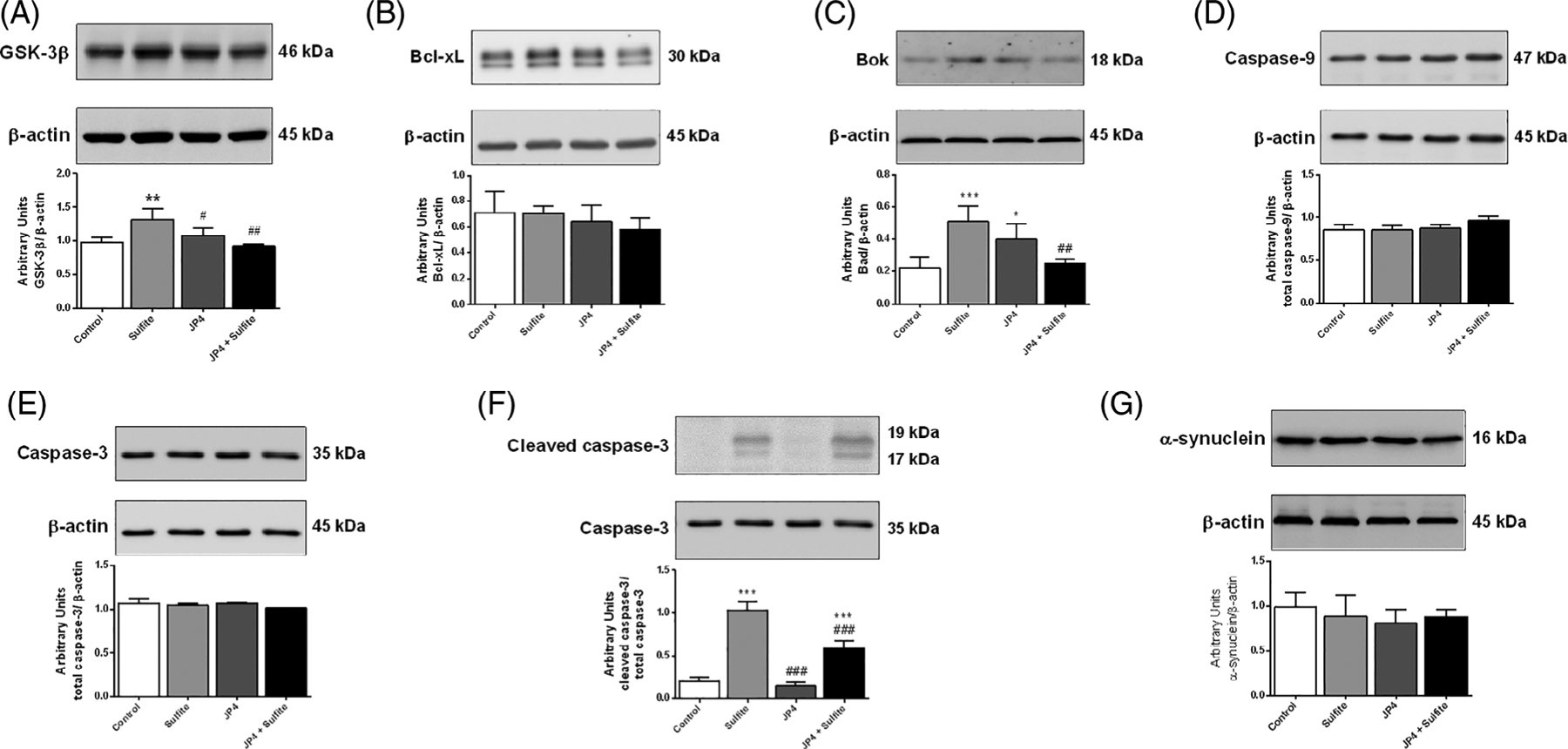
Effect of JP4-039 on sulfite-induced alterations on GSK-3β, A, Bcl-xL, B, Bok, C, caspase-9, D, caspase-3, E, cleaved caspase-3, F, α-synuclein, G, content in rat striatum 30 minutes after sulfite injection (2 μmol). Pretreatment with JP4-039 was performed via intraperitoneal injection 24 (5.0 μg/g) and 2 hours (5.0 μg/g) prior to sulfite administration. Values are means ± SD for three to four independent experiments (animals) per group. *P < .05, **P < .01, ***P < .001, compared to rats receiving NaCl (2 μmol) (control group); #P < .05, ##P < .01, ###P < .001, compared to rats receiving sulfite (sulfite group) (Duncan multiple range test)
4 |. DISCUSSION
iSOD and MoCD are clinically characterized by neurodeterioration and untreatable seizures.1 While the pathophysiology has been attributed to the accumulation of sulfite in the CNS, the mechanisms of damage have not been elucidated.11–13,15 Moreover, therapy is largely ineffective with a high mortality rate. The goal of the present study was to evaluate the mechanisms of toxicity induced by administration of sulfite into the striatum of rats and investigate the possible neuroprotective effect of the mitochondria-targeted ROS scavenger JP4-039. Sulfite exposure was found to perturbate the striatal antioxidant system, reducing the activities of GPx, GR, GST, and G6PDH and decreasing GSH level, likely related to the overproduction of reactive species induced by this metabolite.9,11,12 In this regard, it has been reported that hydrogen peroxide, peroxyl, hydroxyl, and superoxide radicals can cause oxidative attack to these protein structures leading to impaired activity.35–38 Also of note, glutathione S-sulfonate, a compound originated from the lysis of GSSG by sulfite, has been shown to act as a competitive inhibitor of GST.39
Increased CAT and SOD1 protein in striatum induced by sulfite exposure likely represents compensatory mechanisms countering increased levels of hydrogen peroxide and superoxide radical, respectively,9,12,40 but this hypothesis should be further investigated. An increase in CAT protein correlates with a similar increase in CAT activity previously reported by our group.11 On the other hand, in the same study, we have shown that sulfite does not alter SOD activity11 suggesting that, even though sulfite may cause oxidative damage to SOD, the increased level of enzyme protein compensates for and is capable of maintaining its activity at normal levels. A reduction in HO-1, which catabolizes heme and prevents the production of hydroxyl radicals, suggests that the ubiquitin-proteasome system is activated, since this system degrades damaged proteins that are commonly generated by ROS.41
Most sulfite-induced alterations of antioxidant defense components were prevented by pretreatment of animals with JP4-039. These findings are in line with other studies reporting beneficial effects of JP4-039 on reducing superoxide levels in fibroblasts from patients with MOCS1 deficiency and other hereditary disorders of energy metabolism.21–23
It was previously shown that sulfite reduces CK activity in vitro and in vivo in rat brain.11,12 This enzyme is involved in the transport of high-energy phosphate from sites of ATP production in the mitochondrial matrix to sites of consumption in the cytosol, acting as an energy buffer.42 As expected, administration of sulfite into rat striatum reduced CK activity, which was prevented by the lower dose JP4-039. This is consistent with the fact that CK contains critical cysteinyl residues that are vulnerable to oxidative damage.43
MAPKs regulate many cell responses to external stimuli, such as proliferation, differentiation, and apoptosis, thereby regulating the activity of many enzymes, transcription factors, and other bioactive proteins.44,45 Sulfite exposure led to altered phosphorylation of p38 and ERK 1/2, indicating impairment of cellular homeostasis. It similarly increased the level of GSK-3β, Bok and cleaved caspase-3, an executioner caspase, indicating that apoptosis is indeed increased in the striatum of exposed rat brains.46 These findings may explain the neuronal loss seen in these disorders8,47 and are in agreement with previous works demonstrating that sulfite induces mitochondrial permeability transition pore opening and cytochrome c release15 that is a trigger to the mitochondrial pathway of apoptosis.48
Changes in MAPK and apoptosis markers were prevented or attenuated by JP4-039, which is consistent with prior publications showing that ROS may modulate phosphorylation of MAPKs49,50 and trigger mitochondrial permeability transition.51 These findings are of particular interest given that JP4-039 and closely related analogs, such as XJB-5-131, have been shown to cross the blood-brain barrier in mice, and that both iSOD and MoCD are characterized by severe neurological dysfunction and basal ganglia abnormalities.47,52
In summary, we have demonstrated that sulfite exposure impairs the striatal antioxidant system and energy transfer in rat brain, altering critical cellular signaling pathways and leading to apoptosis. These changes were mitigated by JP4-039, implicating the importance of elevated ROS in the cellular pathophysiology of iSOD and MoCD. Our findings identify JP4-039 as a promising candidate for treatment of SO deficiency.
ACKNOWLEDGMENTS
This study is funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Grant 409009/2016-4), Programa de Apoio a Núcleos de Excelência (PRONEX II) (Grant 16/2551-0000465-0), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS) (Grant 16/2551-0000192-8), and Instituto Nacional de Ciência e Tecnologia em Excitotoxicidade e Neuroproteção (INCT-EN) (Grant 465671/2014-4).
The author(s) confirm(s) independence from the sponsors; the content of the article has not been influenced by the sponsors.
Funding information
Conselho Nacional de Desenvolvimento Científico e Tecnológico (BR), Grant/Award Number: 409009/2016-4; Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (BR), Grant/Award Number: 16/2551-0000192-8; Instituto Nacional de Ciência e Tecnologia para Excitotoxicidade e Neuroproteção (BR), Grant/Award Number: 465671/2014-4; Programa de Apoio a Núcleos de Excelência (PRONEX II), Grant/Award Number: 16/2551-0000465-0
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest. Peter Wipf has the patents of US 10526368 B2 and WO 2010/009389 A1 issued.
DATA AVAILABILITY STATEMENT
Raw data will be available when requested.
ETHICS STATEMENT
The study was approved by the local Animal Ethics Commission (Universidade Federal do Rio Grande do Sul) and the National Animal Rights Regulations (Law 11.794/2008). The guidelines of National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication n°. 80-23, revised 1996) and Directive 2010/63/EU were also followed. All institutional and national guidelines for care and use of laboratory animals were followed.
REFERENCES
- 1.Johnson JL, Duran M. Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. Scriver CR, Beaudet AL, Valle D, Sly WS. (eds.), The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001;3181–3217. [Google Scholar]
- 2.Feng C, Tollin G, Enemark JH. Sulfite oxidizing enzymes. Biochim Biophys Acta Proteins Proteomics. 2007;1774:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman K Sulfite-containing pharmaceuticals. CMAJ. 1993;148:714. [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor SL, Higley NA, Bush RK. Sulfites in foods: uses, analytical methods, residues, fate, exposure assessment, metabolism, toxicity, and hypersensitivity. Adv Food Res. 1986;30:1–76. [DOI] [PubMed] [Google Scholar]
- 5.Chen L-W, Tsai Y-S, Huang C-C. Prenatal multicystic encephalopathy in isolated Sulfite oxidase deficiency with a novel Mutaion. Pediatr Neurol. 2014;51:181–182. [DOI] [PubMed] [Google Scholar]
- 6.Bayram E, Topcu Y, Karakaya P, et al. Molybdenum cofactor deficiency: review of 12 cases (MoCD and review). Eur J Paediatr Neurol. 2013;17:1–6. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi R, Sugimoto T, Tamura A, et al. Early features in neuroimaging of two siblings with molybdenum cofactor deficiency. Pediatrics. 2014;133:267–271. [DOI] [PubMed] [Google Scholar]
- 8.Bosley TM, Alorainy IA, Oystreck DT, et al. Neurologic injury in isolated sulfite oxidase deficiency. Can J Neurol Sci. 2014;41:42–48. [DOI] [PubMed] [Google Scholar]
- 9.Mottley C, Mason RP. Sulfate anion free radical formation by the peroxidation of (bi) sulfite and its reaction with hydroxyl radical scavengers. Arch Biochem Biophys. 1988;267:681–689. [DOI] [PubMed] [Google Scholar]
- 10.Grings M, Moura AP, Parmeggiani B, et al. Higher susceptibility of cerebral cortex and striatum to sulfite neurotoxicity in sulfite oxidase-deficient rats. Biochim Biophys Acta Mol Basis Dis. 2016;1862:2063–2074. [DOI] [PubMed] [Google Scholar]
- 11.Grings M, Moura AP, Parmeggiani B, et al. Bezafibrate prevents mitochondrial dysfunction, antioxidant system disturbance, glial reactivity and neuronal damage induced by sulfite administration in striatum of rats: implications for a possible therapeutic strategy for sulfite oxidase deficiency. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2135–2148. [DOI] [PubMed] [Google Scholar]
- 12.Grings M, Moura AP, Parmeggiani B, et al. Disturbance of brain energy and redox homeostasis provoked by sulfite and thiosulfate: potential pathomechanisms involved in the neuropathology of sulfite oxidase deficiency. Gene. 2013;531:191–198. [DOI] [PubMed] [Google Scholar]
- 13.Parmeggiani B, Moura AP, Grings M, et al. In vitro evidence that sulfite impairs glutamatergic neurotransmission and inhibits glutathione metabolism-related enzymes in rat cerebral cortex. Int J Dev Neurosci. 2015;42:68–75. [DOI] [PubMed] [Google Scholar]
- 14.de Moura Alvorcem L, da Rosa MS, Glänzel NM, et al. Disruption of energy transfer and redox status by Sulfite in hippocampus, striatum, and cerebellum of developing rats. Neurotox Res. 2017;32:264–275. [DOI] [PubMed] [Google Scholar]
- 15.Grings M, Moura AP, Amaral AU, et al. Sulfite disrupts brain mitochondrial energy homeostasis and induces mitochondrial permeability transition pore opening via thiol group modification. Biochim Biophys Acta Mol Basis Dis. 2014;1842:1413–1422. [DOI] [PubMed] [Google Scholar]
- 16.Touati G, Rusthoven E, Depondt E, et al. Dietary therapy in two patients with a mild form of sulphite oxidase deficiency. Evidence for clinical and biological improvement. J Inherit Metab Dis. 2000;23:45–53. [DOI] [PubMed] [Google Scholar]
- 17.Schwahn BC, Van Spronsen FJ, Belaidi AA, et al. Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type a: a prospective cohort study. Lancet. 2015;386:1955–1963. [DOI] [PubMed] [Google Scholar]
- 18.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krainz T, Gaschler MM, Lim C, Sacher JR, Stockwell BR, Wipf P. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent Sci. 2016;2:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernard ME, Kim H, Berhane H, et al. GS-Nitroxide (JP4-039)-mediated radioprotection of human Fanconi Anemia cell lines. Radiat Res. 2011;176:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leipnitz G, Mohsen A-W, Karunanidhi A, et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci Rep. 2018;8:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grings M, Seminotti B, Karunanidhi A, et al. ETHE1 and MOCS1 deficiencies: disruption of mitochondrial bioenergetics, dynamics, redox homeostasis and endoplasmic reticulum-mitochondria crosstalk in patient fibroblasts. Sci Rep. 2019;9:12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seminotti B, Leipnitz G, Karunanidhi A, et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum Mol Genet. 2019;28:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frantz M-C, Pierce JG, Pierce JM, et al. Large-scale asymmetric synthesis of the bioprotective agent JP4-039 and Analogs. Org Lett. 2011;13:2318–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goff JP, Epperly MW, Dixon T, et al. Radiobiologic effects of GS-nitroxide (JP4-039) on the hematopoietic syndrome. In Vivo. 2015;25:315–323. [PMC free article] [PubMed] [Google Scholar]
- 26.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th ed. London: Elsevier Inc; 2007. [Google Scholar]
- 27.Browne RW, Armstrong D. Reduced glutathione and glutathione disulfide. Methods Mol Biol. 1998;108:347–352. [DOI] [PubMed] [Google Scholar]
- 28.Hughes BP. A method for the estimation of serum creatine kinase and its use in comparing creatine kinase and aldolase activity in normal and pathological sera. Clin Chim Acta. 1962;7:597–603. [DOI] [PubMed] [Google Scholar]
- 29.Wendel A Glutathione peroxidase. Methods Enzymol. 1981;77:325–333. [DOI] [PubMed] [Google Scholar]
- 30.Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113:484–490. [DOI] [PubMed] [Google Scholar]
- 31.Mannervik B, Guthenberg C. Glutathione transferase (human placenta). Methods Enzymol. 1981;77:231–235. [DOI] [PubMed] [Google Scholar]
- 32.Leong SF, Clark JB. Regional development of glutamate dehydrogenase in the at brain. J Neurochem. 1984;43:106–111. [DOI] [PubMed] [Google Scholar]
- 33.da Rosa-Junior NT, Parmeggiani B, da Rosa MS, et al. Bezafibrate in vivo administration prevents 3-Methylglutaric acid-induced impairment of redox status, mitochondrial biogenesis, and neural injury in brain of developing rats. Neurotox Res. 2019;35(4):809–822. [DOI] [PubMed] [Google Scholar]
- 34.Braicu C, Buse M, Busuioc C, et al. A comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers. 2019;11: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ninfali P, Ditroilo M, Capellacci S, Biagiotti E. Rabbit brain glucose-6-phosphate dehydrogenase: biochemical properties and inactivation by free radicals and 4-hydroxy-2-nonenal. Neuroreport. 2001;12:4149–4153. [DOI] [PubMed] [Google Scholar]
- 36.Murakami K, Tsubouchi R, Fukayama M, Yoshino M. Copper-dependent inhibition and oxidative inactivation with affinity cleavage of yeast glutathione reductase. Biometals. 2014;27:551–558. [DOI] [PubMed] [Google Scholar]
- 37.Shen H, Tsuchida S, Tamai K, Sato K. Identification of cysteine residues involved in disulfide formation in the inactivation of glutathione transferase P-form by hydrogen peroxide. Arch Biochem Biophys. 1993;300:137–141. [DOI] [PubMed] [Google Scholar]
- 38.Pigeolet E, Corbisier P, Houbion A, et al. Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech Ageing Dev. 1990; 51:283–297. [DOI] [PubMed] [Google Scholar]
- 39.Leung KH, Post GB, Menzel DB. Glutathione S-sulfonate, a sulfur dioxide metabolite, as a competitive inhibitor of glutathione S-transferase, and its reduction by glutathione reductase. Toxicol Appl Pharmacol. 1985;77:388–394. [DOI] [PubMed] [Google Scholar]
- 40.Inglés M, Serra-Añó P, Gambini J, et al. Active paraplegics are protected against exercise-induced oxidative damage through the induction of antioxidant enzymes. Spinal Cord. 2016;54:830–837. [DOI] [PubMed] [Google Scholar]
- 41.Cervellati C, Sticozzi C, Romani A, et al. Impaired enzymatic defensive activity, mitochondrial dysfunction and proteasome activation are involved in RTT cell oxidative damage. Biochim Biophys Acta Mol Basis Dis. 2015;1852:2066–2074. [DOI] [PubMed] [Google Scholar]
- 42.Du F, Cooper A, Lukas SE, Cohen BM, Öngür D. Creatine kinase and ATP synthase reaction rates in human frontal lobe measured by 31P magnetization transfer spectroscopy at 4T. Magn Reson Imaging. 2013;31:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wendt S, Schlattner U, Wallimann T. Differential effects of per-oxynitrite on human mitochondrial creatine kinase isoenzymes: inactivation, octamer destabilization, and identification of involved residues. J Biol Chem. 2003;278:1125–1130. [DOI] [PubMed] [Google Scholar]
- 44.Bryk D, Olejarz W, Zapolska-Downar D. Mitogen-activated protein kinases in atherosclerosis. Postepy Hig Med Dosw. 2014;68:10–22. [DOI] [PubMed] [Google Scholar]
- 45.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–2849. [DOI] [PubMed] [Google Scholar]
- 46.Maurer U, Preiss F, Brauns-Schubert P, Schlicher L, Charvet C. GSK-3 -at the crossroads of cell death and survival. J Cell Sci. 2014;127:1369–1378. [DOI] [PubMed] [Google Scholar]
- 47.Hobson EE, Thomas S, Crofton PM, Murray AD, Dean JCS, Lloyd D. Isolated sulphite oxidase deficiency mimics the features of hypoxic ischaemic encephalopathy. Eur J Pediatr. 2005;164:655–659. [DOI] [PubMed] [Google Scholar]
- 48.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100. [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Li P, Xuan J, et al. Cholesterol enhances colorectal cancer progression via ROS elevation and MAPK Signaling pathway activation. Cell Physiol Biochem. 2017;42:729–742. [DOI] [PubMed] [Google Scholar]
- 50.Santabárbara-Ruiz P, López-Santillán M, Martínez-Rodríguez I, et al. ROS-induced JNK and p38 Signaling is required for unpaired cytokine activation during drosophila regeneration. PLoS Genet. 2015;11:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rottenberg H, Hoek JB. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell. 2017;16:943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christner S, Guo J, Parise RA, et al. Liquid chromatography–tandem mass spectrometric assay for the quantitation of the novel radiation protective agent and radiation mitigator JP4-039 in murine plasma. J Pharm Biomed Anal. 2018;150:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]