

Abstract
Little is known about extracellular matrix (ECM) contributions to formation of the earliest cell lineages in the embryo. Here, we show that the proteoglycan versican and glycosaminoglycan hyaluronan are associated with emerging Flk1+ hematoendothelial progenitors at gastrulation. The mouse versican mutant Vcanhdf lacks yolk sac vasculature, with attenuated yolk sac hematopoiesis. CRISPR/Cas9-mediated Vcan inactivation in mouse embryonic stem cells reduced vascular endothelial and hematopoietic differentiation within embryoid bodies, which generated fewer blood colonies, and had an impaired angiogenic response to VEGF165. Hyaluronan was severely depleted in Vcanhdf embryos, with corresponding upregulation of the hyaluronan-depolymerase TMEM2. Conversely, hyaluronan-deficient mouse embryos also had vasculogenic suppression but with increased versican proteolysis. VEGF165 and Indian hedgehog, crucial vasculogenic factors, utilized the versican-hyaluronan matrix, specifically versican chondroitin sulfate chains, for binding. Versican-hyaluronan ECM is thus an obligate requirement for vasculogenesis and primitive hematopoiesis, providing a vasculogenic factor-enriching microniche for Flk1+ progenitors from their origin at gastrulation.
Keywords: Proteoglycan, Vasculogenesis, Angiogenesis, Endothelium, Hematopoiesis CRISPR-Cas9
Introduction
The first blood vessels in amniotes form de novo in the yolk sac prior to initiation of hemodynamic forces, indicating an in situ differentiation driven by the local environment, which includes extracellular matrix (ECM) [1]. This vasculogenic process is inextricably linked to subsequent primitive hematopoiesis, i.e., formation of the first erythroid and myeloid cells [2], implying a close lineage relationship between the first vascular endothelial and blood cells. The relevant Flk1+ hematoendothelial progenitor cells arise from mesodermal precursors at gastrulation [3], migrate from the primitive streak, and form blood islands in the proximal extra-embryonic yolk sac on the seventh day of gestation (E7) in mouse embryos [4,5]. Several key transcription factors and soluble effectors of vasculogenesis/angiogenesis and hematopoiesis are known [1]. Although ECM and ECM-derived proteolytic fragments are recognized as angiogenesis regulators [6] and sulfated glycosaminoglycans can promote endothelial differentiation of mesenchymal stem cells [7], much remains unknown about the influence of ECM on vasculogenesis. Another early cell lineage, primordial germ cells, are accompanied by a protective “traveling niche” of steel factor/stem cell factor-producing cells during migration to the gonads [8]. Whether a cell-associated ECM supports other early lineages such as hematoendothelial progenitors is unknown.
ECM surrounds all mammalian cells, forming a distinct pericellular matrix in some cell types, and the interstitial matrix of tissues. It influences cell behavior by modulating cell adhesion, migration and tissue mechanics, sequesters growth factors and cytokines, and its proteolysis can generate bioactive fragments. Proteoglycans, which are ECM and cell-surface molecules with one or more glycosaminoglycan chains covalently attached to a core protein, participate in all these mechanisms. For example, chondroitin sulfate proteoglycans (CSPGs) such as versican, which are bulky and inherently anti-adhesive, regulate focal adhesions and cell migration [9–11]. Heparan sulfate proteoglycans (HSPGs) sequester growth factors, including angiogenic growth factors such as VEGF165 and FGF2 through their HS chains and act as co-receptors in angiogenic signaling [12]. CSPGs can also bind VEGF165 and may overlap functionally with HSPGs in VEGF-induced sprouting angiogenesis [13], but physiological in vivo contexts for this regulatory activity remain unidentified.
Versican is a widely distributed CSPG that aggregates with the glycosaminoglycan hyaluronan (HA), through its N-terminal G1 domain [14–17]. Its C-terminal G3 domain binds to ECM components fibronectin, fibrillins and tenascins [18] and was shown to interact with VEGF165 in biochemical assays [19]. HA is anchored to cell-surface receptors such as CD44 and RHAMM as well as the membrane-localized hyaluronan synthases [20], providing a means by which versican can localize to the pericellular matrix. Pericellular versican was previously shown to modulate smooth muscle cell differentiation via regulation of cell adhesion [9,21,22]. Four versican isoforms (V0, V1–V3) arise from alternative splicing of large exons, numbered 7 and 8, encoding CS-bearing domains GAGα and GAGβ, respectively [16,23]. A versican insertional mutant mouse allele (Vcanhdf), which lacks all isoforms, demonstrated an essential role for versican in early cardiac development [24,25]. Has2 null embryos, lacking the major HA synthase, also die by 10 days of gestation with similar cardiac defects as Vcanhdf/hdf [26], consistent with the versican-HA complex being a major constituent of cardiac jelly. Here, upon identification of abnormal vasculature in Vcanhdf/hdf yolk sac, we investigated its role in vasculogenesis and early hematopoiesis, with investigation of the key findings in embryos with inactivation of hyaluronan synthase (Has) genes. An intimate association of versican and HA with Flk1+ hematoendothelial progenitor cells from their origin at gastrulation is demonstrated to have profound significance for vasculogenesis and primitive hematopoiesis.
Results
Vcanhdf/hdf yolk sacs lack a vascular plexus
Vcanhdf/hdf embryos did not survive past E10.5, and were recognizable by consistently smaller size and dilated pericardial sac at E9.5 (Fig. 1A, Supplemental Fig. 1). Although major developmental milestones, including axial rotation of the embryo and initiation of cardiac contraction were completed by E9.5, Vcanhdf/hdf yolk sacs and embryos were avascular (Fig. 1B, C). Whole mount CD31 immunostaining revealed the lack of a vascular network in E9.5 Vcanhdf/hdf yolk sacs, in place of which scattered CD31+ cells were present instead (Fig. 1C).
Fig. 1.
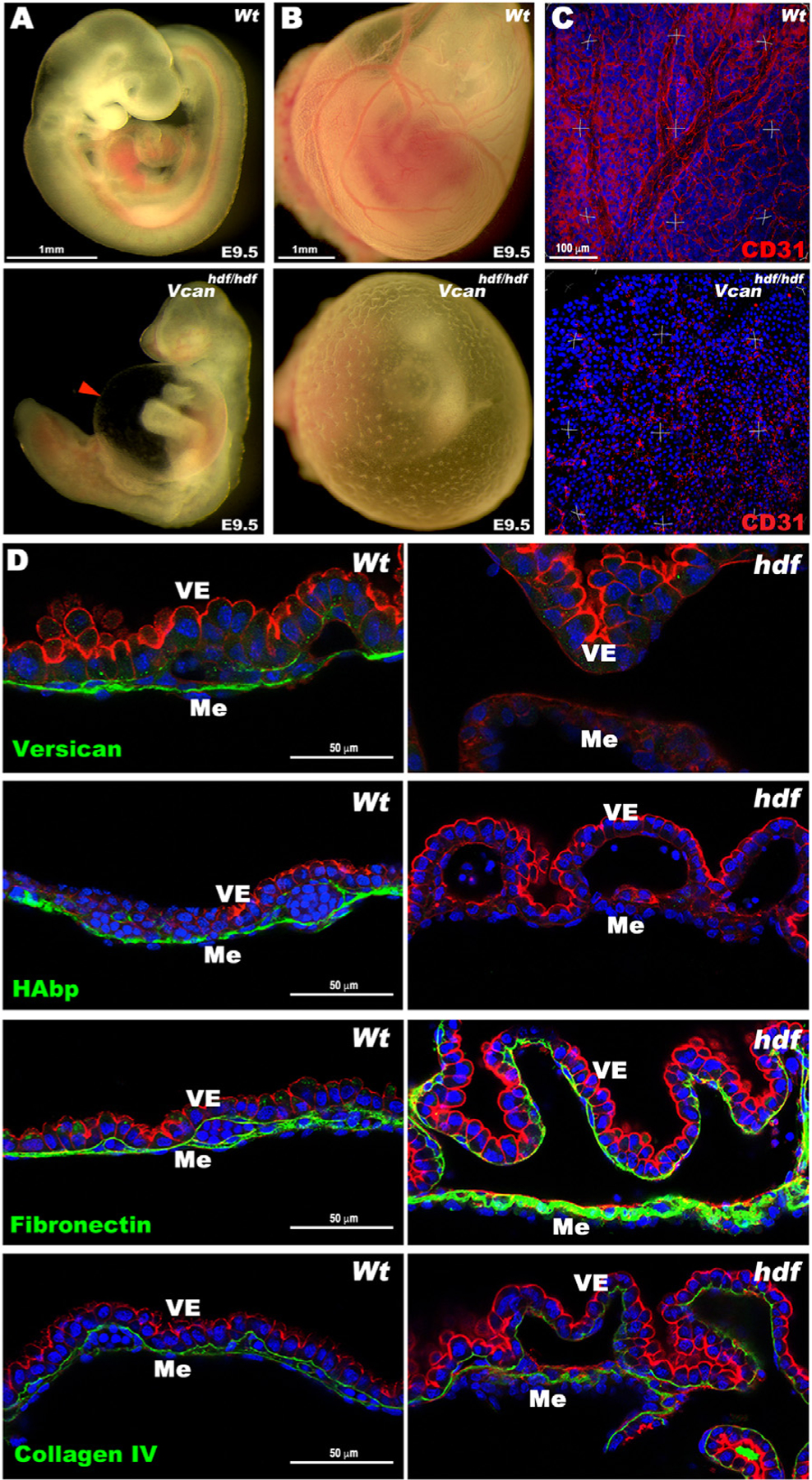
Vcanhdf/hdf yolk sacs are avascular and lack hyaluronan. (A) E9.5 wild type and Vcanhdf/hdf embryos. The red arrowhead shows the dilated pericardial sac in the mutant. (B) E9.5 yolk sacs imaged in situ demonstrate the absence of vasculature in the E9.5 Vcanhdf/hdf yolk sac. (C) Three-dimensional (3D) maximum-intensity projections of whole-mount yolk sacs stained with anti-CD31 (red) showing absence of the vascular network in E9.5 Vcanhdf/hdf yolk sacs (n=3 yolk sacs of each genotype). (D) Versican GAGβ staining (green) is present throughout the mesoderm (Me) of E9.5 wild type yolk sacs and absent in the Vcanhdf/hdf yolk sac. F-actin (red) staining highlights visceral endoderm (VE) detached from mesoderm in Vcanhdf/hdf yolk sacs. HA and fibronectin (green) were similarly distributed as versican in wild type yolk sac, but HA staining was absent in Vcanhdf/hdf yolk sac and fibronectin staining was more intense. Collagen IV staining intensity was similar in wild type and Vcanhdf/hdf yolk sacs (n=3 yolk sacs of each genotype). Scale bar in A, B=1mm, C= 100μm, D= 50μm.
In E9.5 wild-type yolk sac, versican localized to the mesoderm, but no staining was present in Vcanhdf/hdf yolk sacs (Fig. 1D). HA-binding protein (HAbp) staining overlapped with versican in wild-type yolk sac mesoderm but was absent in Vcanhdf/hdf yolk sacs (Fig. 1D). Fibronectin immunostaining in contrast, showed increased intensity in Vcanhdf/hdf yolk sacs, whereas collagen IV staining was unaffected (Fig. 1D), suggesting that loss of HA did not represent global ECM reduction in Vcanhdf/hdf yolk sacs. Similar to Has2, Itga5 and Fn1-null alleles, which have failed yolk sac vasculogenesis and hematopoiesis [26,27], Vcanhdf/hdf yolk sacs consistently showed mesoderm detachment from visceral endoderm with few evident blood islands (Fig. 1D, Supplemental Fig. 2A–C). Intense F-actin staining was consistently observed in Vcanhdf/hdf visceral endoderm (Fig. 1 D, Supplemental Fig. 2A), and rounded, rather than spindle-shaped nuclei were observed in the yolk sac mesothelial layer (Supplemental Fig. 2B). Transmission electron microscopy (TEM) undertaken with fixation conditions that preserved cell-matrix interactions [28] showed complete coverage of blood islands by vascular endothelium in wild-type yolk sac but discontinuous vascular endothelial cells with numerous membrane protrusions in Vcanhdf/hdf yolk sacs (Supplemental Fig. 2C). Thus, versican is essential for proper yolk sac morphogenesis.
E8.5 Vcanhdf/hdf embryos had a normal shape, but appeared pale and their yolk sacs lacked a visible vascular network (Fig. 2A). Whole-mount CD31 immunostaining of E8.5 embryos identified severe, widespread attenuation of vasculature (Fig. 2B). En face imaging of whole-mount wild-type yolk sac stained with anti-CD31 demonstrated a well-formed vascular plexus which was lacking in Vcanhdf/hdf yolk sacs (Fig. 2C). Immunostaining of versican and CD31, together with HA-staining in E8.5 wild-type yolk sacs revealed versican and HA co-localization in patches corresponding to individual cells on the mesothelial aspect of blood islands (Fig. 2D). mRNA in situ hybridization with Vcan exon 7 (GAGα), or exon 8 (GAGβ)-specific probes revealed that only the exon 8 probe hybridized to E8.5 yolk sac mesoderm (Fig. 2E), suggesting exclusively versican V1 isoform expression at E8.5. Both Vcan probes hybridized strongly to E8.5 wild-type hearts (Fig. 2F) but not Vcanhdf/hdf hearts (e.g., exon 8 probe, Fig. 2G), indicative of specificity.
Fig. 2.
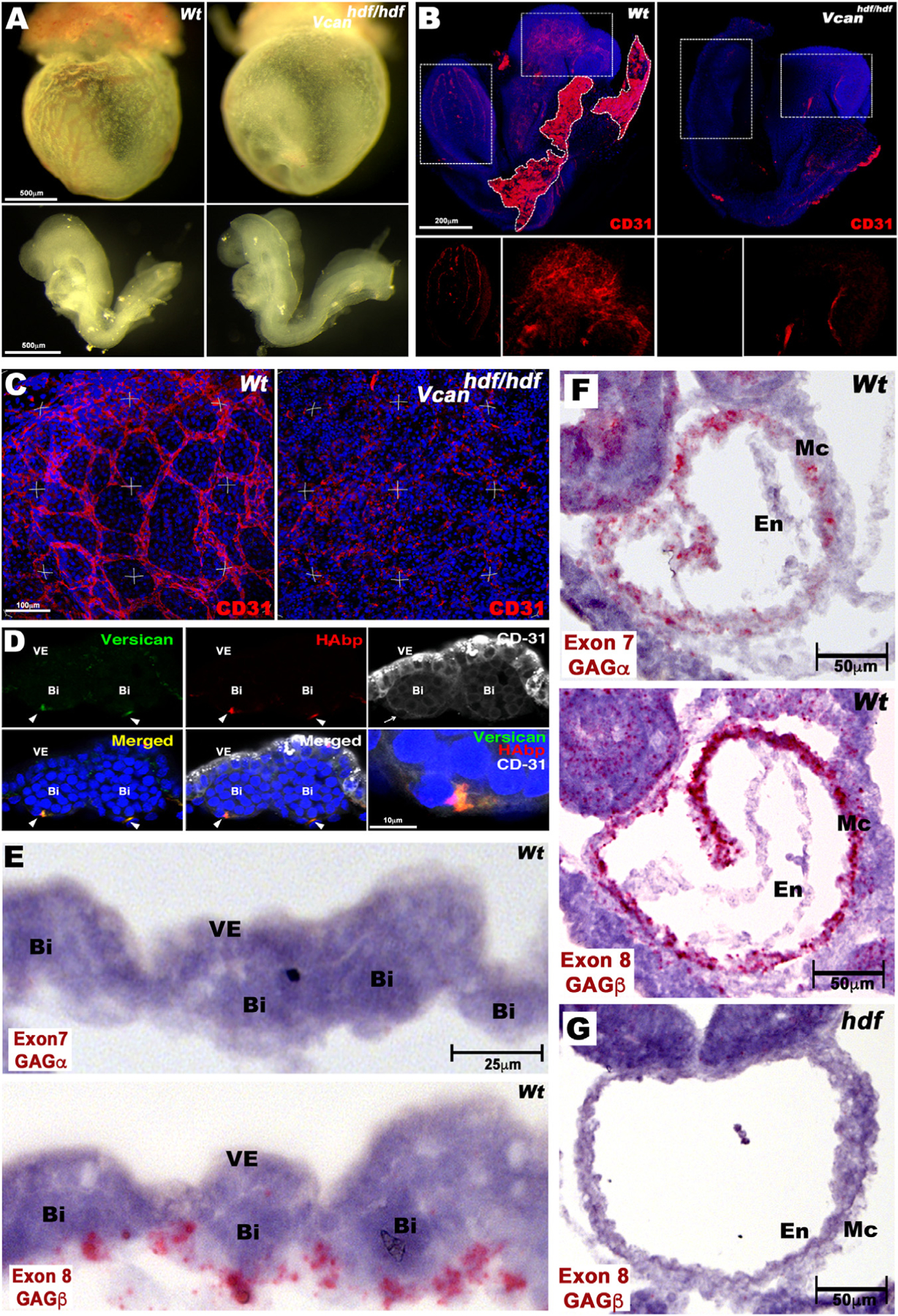
Impaired vasculogenesis in the Vcanhdf/hdf embryo and yolk sac. (A) E8.5 Vcanhdf/hdf yolk sacs are avascular yet embryos dissected out of the yolk sac appear morphologically similar to wild type. (B) Maximum intensity projections of whole mount E8.5 wild type and Vcanhdf/hdf embryos stained with anti-CD31 (red) and DAPI (blue). Boxed areas are shown at higher magnification with the red channel only in the lower panels. Residual wild-type yolk sac is marked by a white dotted line (n=3 embryos from each genotype). (C) Three-dimensional (3D) maximum intensity projections of yolk sacs stained en face with anti-CD31 (red) show a well-formed vascular plexus in wild type yolk sac and disorganized CD31+ cells in Vcanhdf/hdf yolk sac (n=3 yolk sacs from each genotype). (D) Cross section of E8.5 wild type yolk sac blood islands co-stained with versican (green), HAbp (red) and CD31 (white). Versican and HA co-localize with CD31+ cells on the mesothelial aspect of blood islands (arrowheads). The blood island imaged on the left is enlarged in the bottom right-hand panel (n=3 wild type yolk sacs). VE, visceral endoderm, Bi, blood island, (E) RNAscope in situ hybridization of E8.5 wild-type yolk sac shows expression of Vcan isoforms containing exon 8 (V0,V1), but not exon 7 (V0, V2) in mesoderm adjacent to blood islands (Bi), VE, visceral endoderm. (F) Vcan exon 7 and exon 8 probes both hybridize to myocardium (Mc) of E8.5 wild-type embryos. En, Endocardium. (G) Vcan probes (exon 8 shown) do not hybridize to Vcanhdf/hdf heart. Scale bar in D=10μm, E= 25μm, 50μm in F–G.
Versican and HA are associated with yolk sac Flk1+ cells
Flk1, a well characterized hematoendothelial progenitor marker indispensable for both vascular and blood lineage development [29], showed complete overlap with versican and HA on the mesothelial aspect of blood islands in wild-type E8.5 yolk sac (Fig. 3A). Vcanhdf/hdf yolk sacs, in contrast, showed small blood islands with dramatically attenuated HA and Flk1 staining (Fig. 3A). En face confocal microscopy of whole-mount wild-type E8.5 yolk sacs revealed discrete patches of versican throughout the mesoderm (Fig. 3B). CD41, which is expressed upon specific commitment to the blood lineage [30], was detected in blood islands in the mid-mesoderm plane in wild-type yolk sacs (Fig. 3B). Versican did not precisely overlap with CD41+ cells and was restricted to intense patches corresponding to individual endothelial cells in wild-type yolk sacs, evident both in the surface (mesothelial) plane and cross-sections, whereas Vcanhdf/hdf yolk sacs lacked CD41-stained blood islands entirely (Fig. 3B). High magnification images showed a few cells with weak CD41 staining in Vcanhdf/hdf yolk sac mesoderm (arrowheads in Fig. 3B), contrasting with well-demarcated CD41+ wild-type blood islands. Combined, these results show that versican and HA distribution at E8.5 is specifically associated with uncommitted Flk+ hemogenic endothelial cells, which are crucial for both blood and vascular development in the mouse embryo. However, versican and HA are not specifically associated with subsequently differentiated blood cells (Fig. 3 B,C).
Fig. 3.
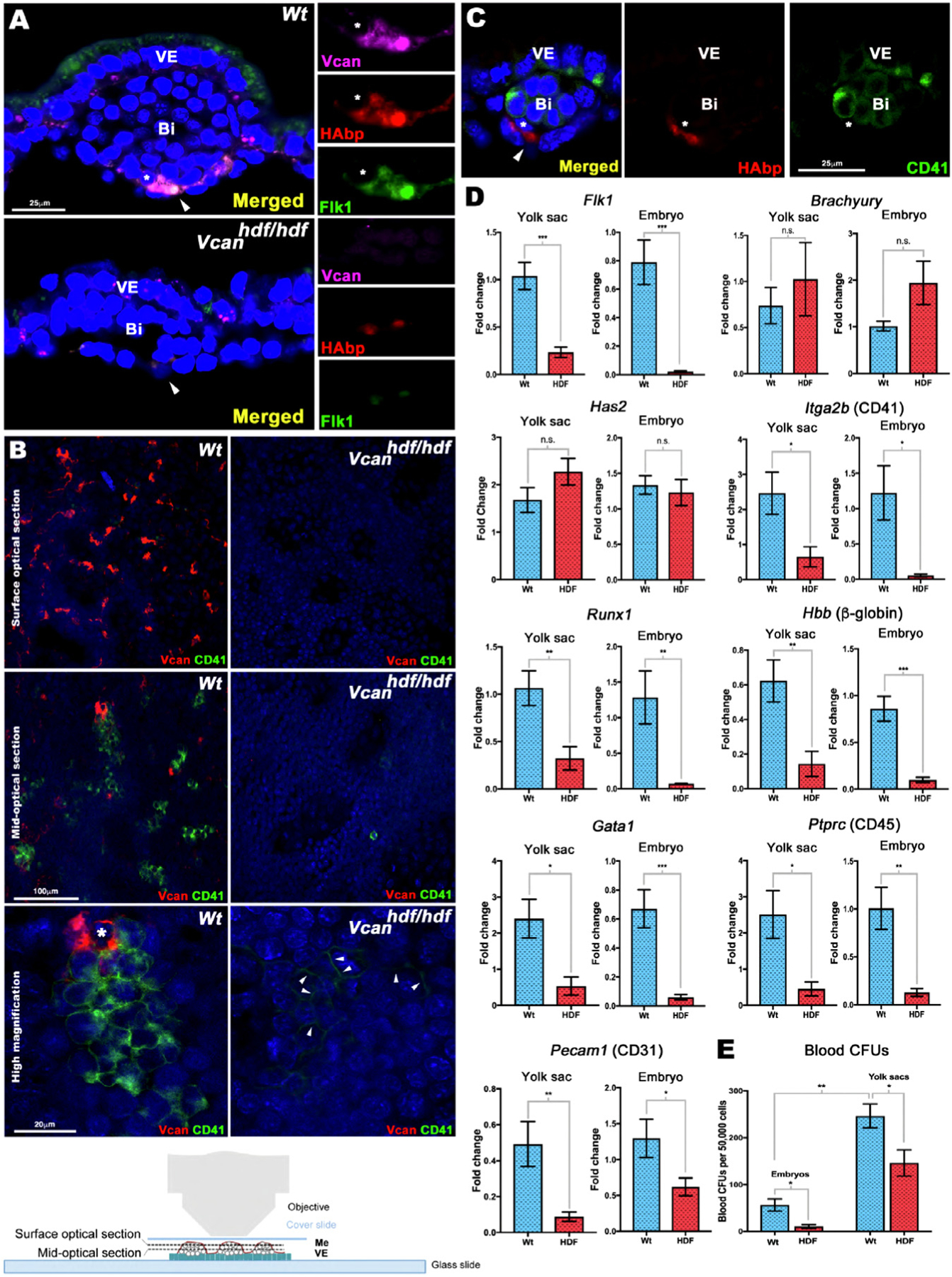
Versican and HA localize to Flk1+ cells and are essential for yolk sac blood island formation. (A) E8.5 wild type and Vcanhdf/hdf yolk sac cross-sections co-stained with versican (magenta), HAbp (red) and Flk1 (green). In wild type yolk sac, versican-HA staining colocalizes with Flk1. In Vcanhdf/hdf yolk sac, blood islands (Bi) are smaller and both HAbp staining and Flk1 staining are weak. Arrowhead: versican-HA-Flk1 co-stained patches, asterisks: cell shown at higher magnification in the right-hand panels (n=4 yolk sacs from each genotype). VE, visceral endoderm. (B) En face confocal imaging of E8.5 yolk sac versican (red) and CD41 (green) staining with the mesothelial aspect facing the objective (bottom). Surface optical sections show versican-rich foci throughout wild type yolk sac that are absent in Vcanhdf/hdf yolk sac. The mid-optical image shows versican and CD41 co-stained cells in blood islands. Versican is associated with wild-type blood islands but does not overlap with CD41. No CD41+ cells were observed in Vcanhdf/hdf yolk sac. The higher magnification image shows the distinct cell populations marked by versican and CD41. Arrowheads mark weak CD41 staining in Vcanhdf/hdf images (n=3 yolk sacs from each genotype). (C) Cross-section of an E8.5 wild type blood showing no overlap of HA (red) and CD41 (green) (n=3 yolk sacs). (D) qRT-PCR analysis of wild type and Vcanhdf/hdf yolk sacs and embryos shows significantly lower Flk1 expression but not Has2 or Brachyury expression in Vcanhdf/hdf mutants. CD41 (Itga2b) and Runx1 expression were significantly lower in Vcanhdf/hdf yolk sac and embryos. Blood markers β-globin, Gata1 and CD45 (Ptprc) and the vascular endothelial marker CD31 (Pecam1), were reduced in Vcanhdf/hdf yolk sacs and embryos (n=3 yolk sacs and embryos from each genotype, error bars= S.E.M., *, p<0.05; **, p<0.01; ***, p<0.001). (E) Methylcellulose assay shows significantly fewer blood colony-forming units (CFUs) in Vcanhdf/hdf yolk sacs and embryos (n=3 yolk sacs and three embryos from each genotype, error bars= S.D.,*, p<0.05; **, p<0.001). Scale bars=25μm in A, 100μm and 20μm in B.
Versican is required for blood island formation
RT-qPCR revealed significantly lower Flk1 expression in Vcanhdf/hdf yolk sac and embryos (Fig. 3D), in agreement with reduced Flk1 staining observed in the Vcanhdf/hdf mutants (Fig. 3A). Neither expression of Brachyury, a mesoderm marker, nor Has2, encoding the major HA synthase in the embryo, were altered in Vcanhdf/hdf yolk sac or embryos (Fig. 3D). Expression of genes encoding blood lineage commitment markers Itga2b (CD41) and Runx1 were similarly reduced in the Vcanhdf/hdf yolk sacs and embryos, as well as erythroid and myeloid transcripts Hbb (β-globin), Gata1 and Ptprc (CD45) (Fig. 3D). CD31 (Pecam1) mRNA expression was greatly reduced in the Vcanhdf/hdf yolk sacs and embryos (Fig. 3D), consistent with the observed lack of vasculature (Fig. 2B). Methylcellulose colony formation assays demonstrated fewer colony-forming units (CFUs) in E8.5 Vcanhdf/hdf embryos and yolk sacs (Fig. 3E), in agreement with fewer CD41-stained blood islands observed in the mutant yolk sacs.
Versican and HA are associated with the earliest Flk1+ cells at gastrulation
Since Flk1+ cells arise at gastrulation, we analyzed versican and HA distribution in wild-type E7.5 mouse embryos. Versican and HA co-localized to the extraembryonic mesoderm in the blood island ring (Fig. 4 A,B), and colocalized with Flk1+ cells (Fig. 4 C,D). Correspondingly, RNA in situ hybridization of serial sections from several embryos showed consistent overlap between Vcan-, Has2- and Flk1-expressing cells in the extraembryonic mesoderm (Fig. 4E, Supplemental Fig. 3). Vcan exon 7-containing probes gave a weaker signal than exon 8 probes. In contrast, Runx1, which identifies blood lineage-committed cells, was expressed by cells in the extraembryonic mesoderm distinct from those expressing Flk1, Vcan and Has2 (Fig. 4E, Supplemental Fig. 3). Thus, versican-HA ECM is intimately associated with the hematoendothelial lineage emerging at gastrulation.
Fig. 4.
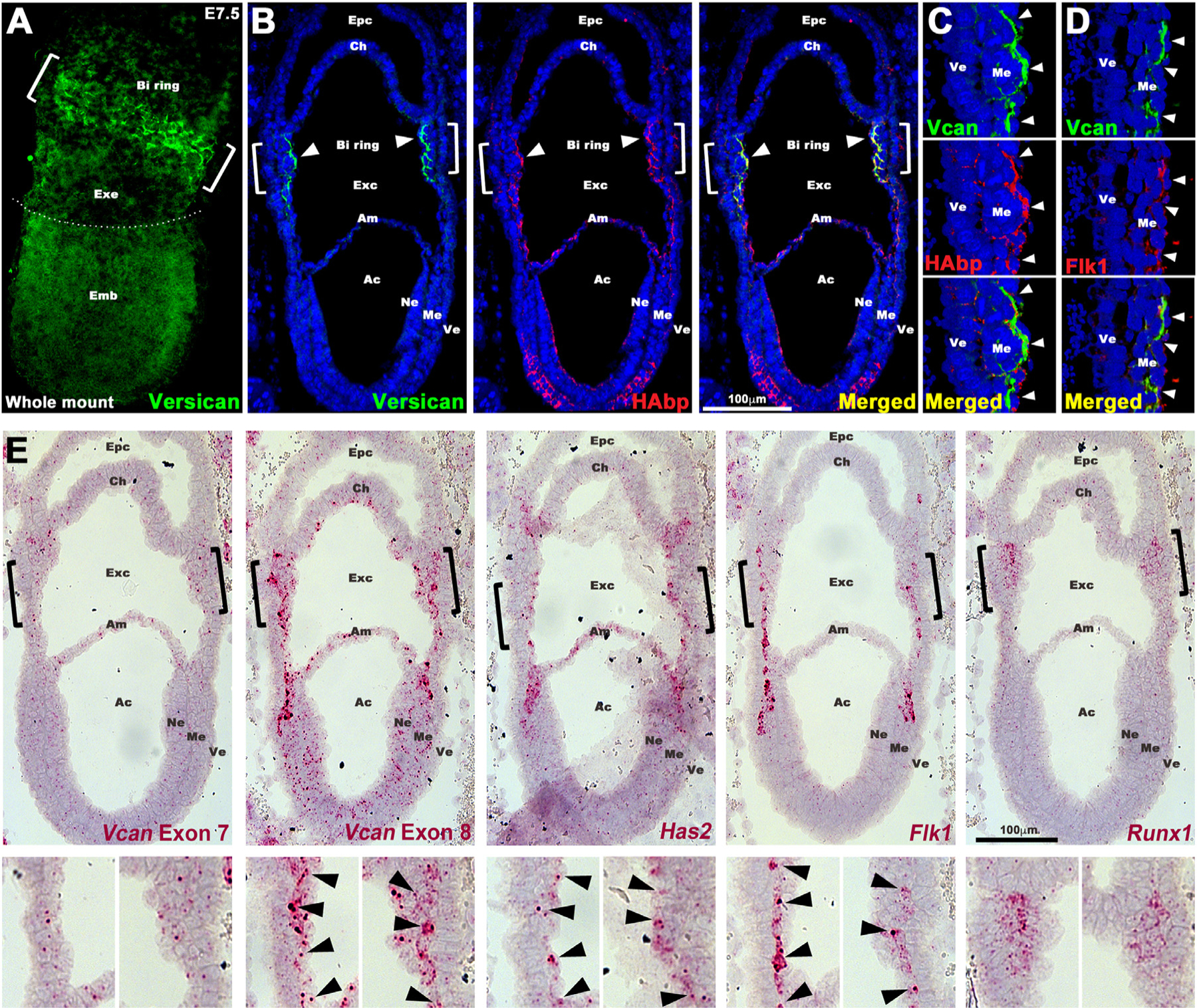
Versican and HA co-localize with Flk1+ cells from their origin at gastrulation. (A) Maximum intensity projection image of an E7.5 embryo shows strong versican staining (green) in the putative blood island (BI) ring (white brackets) in the proximal extraembryonic (Exe) region. Emb, embryo. (B-E) Serial sections of E7.5 wild type embryos analyzed by immunostaining (B–D), or by RNAscope in situ hybridization (E). (B–C) Versican (green) and HAbp (red) colocalize in extraembryonic mesoderm corresponding to the blood island ring (white brackets and arrowheads). The blood islands on the left are shown at high magnification in (C–D). Arrowheads show colocalization of versican, HAbp and Flk1 in extraembryonic mesoderm (N=4 embryos). Epc, ectoplacental cavity; Ch, chorion; Exc, exocelomic cavity; Am, amnion; Ac, amnion cavity; Ne, neural ectoderm; Me, mesoderm; Ve, visceral endoderm. (E) In situ hybridization for Vcan exon 7, Vcan exon 8, Has2, Flk1 and Runx1. Vcan exon 8, Has2 and Flk1 mRNAs have near-identical expression patterns (red) corresponding to primitive streak cells migrating toward extra-embryonic mesoderm. Vcan exon 7 (GAGα) shows weaker overlapping expression. Runx1 marks committed blood cells (n=4 embryos). Scale bars in B,E= 100μm.
Data mining of a single cell RNA sequencing (scRNA-seq) atlas of early (6.5 to 8.5 day-old) mouse embryos [31] supported specific Vcan expression by Flk1+ hematoendothelial progenitors. High Vcan expression was first detected in hematoendothelial progenitors emerging at E6.75, as well as in nascent and uncommitted mesoderm (Supplemental Fig. 4A–C). Vcan was expressed in Kdr (encoding Flk1)-expressing cells at E6.75, E7.0 and E7.5, spanning mouse gastrulation (Supplemental Fig. 4A–C). Kdr+ cells also expressed Has2 and the gene encoding the HA receptor, CD44 (Supplemental Fig. 4B). These observations suggest a potential cell-autonomous role for versican in Flk1+ cells, bound to HA and thus potentially localized to their pericellular matrix via CD44 and HAS2. After E7.5 (e.g., at E 8.5), Vcan was broadly expressed in mesoderm, the developing brain, allantois and cardiomyocytes (Supplemental Fig. 4C) and Kdr expression extended into endothelial cells, although hematoendothelial progenitors continued to express Vcan and Kdr subsequently (Supplemental Fig. 4C). scRNA-seq data also showed that HA link proteins (Hapln1–4) were not expressed in hematoendothelial progenitors (Supplemental Fig. 5A), and the major versican-binding link protein, HAPLN1, was not detected on immunostaining of E7.5 embryo sections (Supplemental Fig. 5B). Staining for ADAMTS-cleaved versican (using anti-DPEAAE) revealed no staining in wild-type blood islands, suggesting that versican does not normally undergo proteolysis by ADAMTS proteases at this developmental stage (Supplemental Fig. 5C).
Loss of versican dramatically reduces yolk sac and embryo HA levels
In addition to reduced HA staining in Vcanhdf/hdf yolk sac blood islands, we observed dramatic reduction of HAbp staining throughout Vcanhdf/hdf embryos (Fig. 5A). Since Has2 mRNA expression was unaltered in Vcanhdf/hdf embryos and yolk sacs (Fig. 3D), reduced HAbp staining may have resulted from increased breakdown in situ, or alternatively, extraction of HA (which is water soluble) from tissue sections during staining. Therefore, fluorophore-assisted carbohydrate electrophoresis (FACE) [32–34] was undertaken on snap-frozen whole embryos to detect and quantify HA content (Fig. 5B). HA-FACE showed reduction of HA compared to wild-type littermates, supporting HA loss intrinsically rather than during sample preparation (Fig. 5B). In addition, FACE analysis demonstrated that versican is a major CSPG in E8.5 embryos, contributing nearly all unsulfated (0S) CS, since both the overall level of CS and its 0S form were dramatically reduced in Vcanhdf/hdf embryos (Fig. 5B). Consistent with the role of versican and HA as an expansile complex, craniofacial mesenchyme and myocardium were severely compacted in Vcanhdf/hdf embryos (Fig. 5C). We also asked whether the absence of versican affected fibronectin, a major embryonic ECM component that is essential for angiogenesis and observed a dramatic increase both in fibronectin staining and mRNA in Vcanhdf/hdf embryos (Fig. 5D, Supplemental Fig. 6A,B), which is presently unexplained. In combination with unaltered Has2 expression, these findings suggested increased HA turnover in Vcanhdf/hdf embryos. TMEM2 was recently identified as a major extracellular hyaluronan-depolymerizing enzyme [35–37]. RT-qPCR revealed significantly raised Tmem2 expression in Vcanhdf/hdf yolk sac and embryos (Fig. 5E), indicating that hyaluronan catabolism was activated in the absence of versican.
Fig. 5.
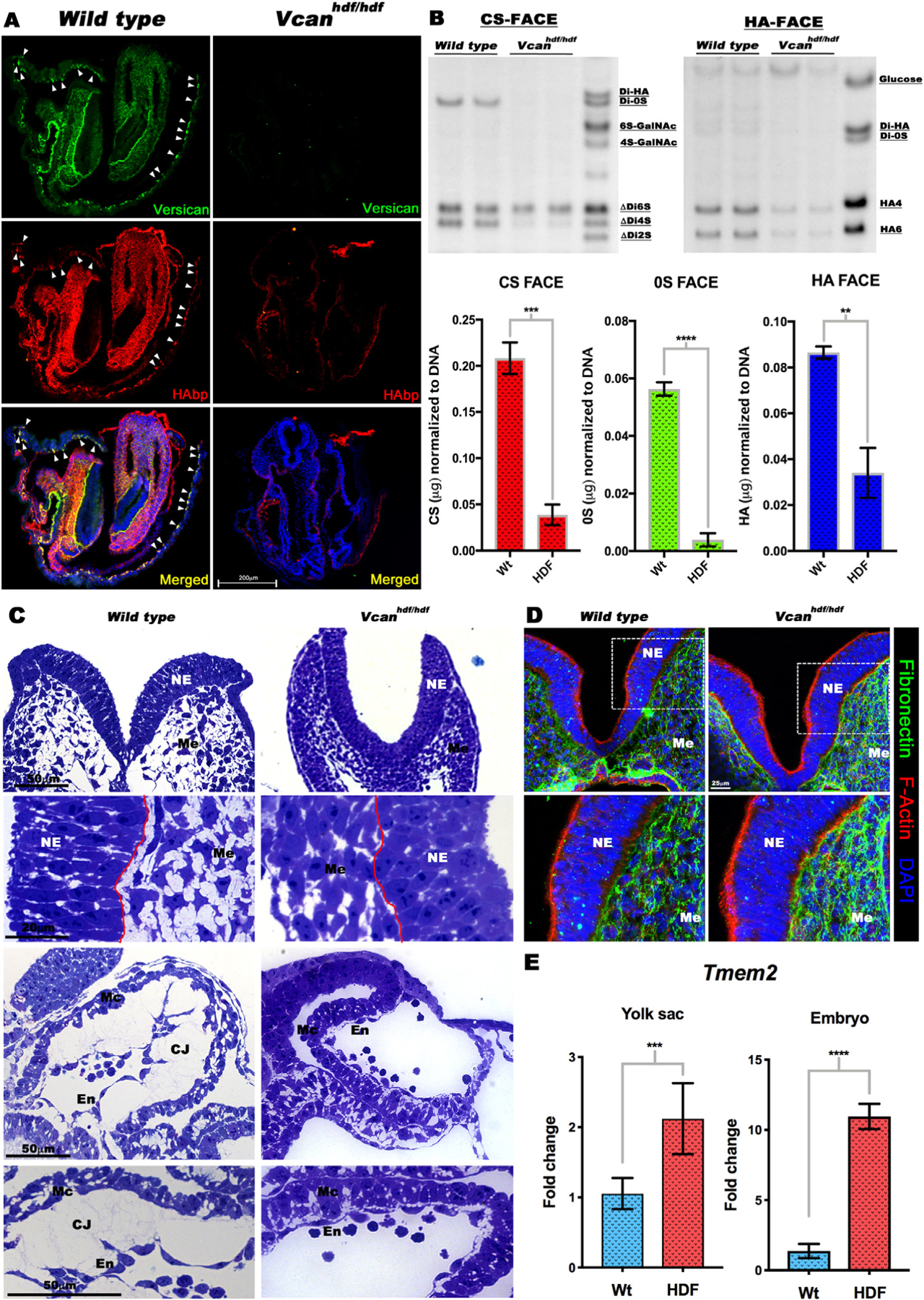
Vcan inactivation results in loss of hyaluronan. (A) Versican (green) and HAbp (red) staining of E8.5 wild type and Vcanhdf/hdf embryos showing severely reduced HAbp staining in Vcanhdf/hdf embryos. Arrowheads indicate versican and HAbp-stained yolk sac blood islands (n=3 embryos each genotype). (B) (Top) Fluorophore-assisted carbohydrate electrophoresis (FACE) analysis for chondroitin sulfate (CS-FACE) and hyaluronan (HA-FACE) in E8.5 littermate embryos. Versican is the major CS-proteoglycan in E8.5 embryos. HA content is significantly reduced in Vcanhdf/hdf embryos. (Bottom) Quantification of CS and HA FACE, normalized to DNA (n=4 embryos from each genotype, error bars= S.E.M.,**, p<0.01; ***, p<0.001; ****, p<0.0001). (C) 1 μm thick, toluidine blue-stained Eponate 12 sections from E8.5 wild type and Vcanhdf/hdf embryos showing compaction of craniofacial mesenchymal cells (Me) and loss of cardiac jelly (CJ) between the myocardium (Mc) and endocardium (En). The red line indicates the boundary of neural epithelium (NE) with mesenchyme (n=3 embryos each genotype). (D) E8.5 wild Vcanhdf/hdf embryo sections stained with F-actin (red) and fibronectin (green) showing stronger fibronectin staining (n=3 embryos each genotype). (E) RT-qPCR shows increased Tmem2 mRNA in Vcanhdf/hdf embryos and yolk sacs (n=3 embryos and yolk sacs from each genotype, error bars= S.E.M., ***, p<0.001; ****, p<0.0001). Scale bar in A= 200μm, 50μm, 20μm in C D=25 μm.
HA deficiency impairs vasculogenesis
Since HA was lost in the absence of versican, we inquired if the converse were true, and whether HA-deficiency indeed led to defective vasculogenesis, as previously suggested by morphology of Has2 germline mutant yolk sac [26]. Has2−/− embryos die by E10 due to heart defects [26] whereas Has1−/− and Has3−/− mice are both viable and do not exhibit noticeable embryonic phenotypes [38,39]. To completely deplete HA from developing embryos, we generated triple knockout embryos (TKO) lacking all three Has genes (Has1−/−; Has2−/−; Has3−/−; referred to as Has1–3TKO). We generated HA-deficient mouse embryos by intercrossing Has1+3−/−; Has2+/− mice, as well as by epiblast-specific conditional deletion of Has2 using Sox2-Cre mice. At E9.5, Has1–3TKO embryos, like Vcanhdf/hdf and Has2-null embryos [26] showed loss of yolk sac vasculature (Fig. 6A). HA staining in the Has1–3TKO yolk sac and embryos was absent and the sections also showed a near-complete lack of versican staining (Fig. 6B,C). In contrast, immunostaining for cleaved versican (anti-DPEAAE) showed robust and widespread versican processing in the absence of HA (Fig. 6 B,D). Epiblast-specific Has2 conditional deletion (Sox2-Cre; Has2Fl/Fl) showed a similar loss of yolk sac vasculature and cardiac defects (Supplemental Fig. 7A). RNA in situ hybridization showed a low level of Has2 expression in these embryos and slightly reduced Vcan transcript levels (Supplemental Fig. 7B). Phenocopying Has1–3TKO embryos, the Sox-2Cre; Has2Fl/Fl embryos and yolk sacs showed loss of both HA and versican staining and increased ADAMTS-mediated versican cleavage (Supplemental Fig. 7C, E). Taken together, these data suggest that HA may protect versican from ADAMTS-mediated degradation.
Fig. 6.
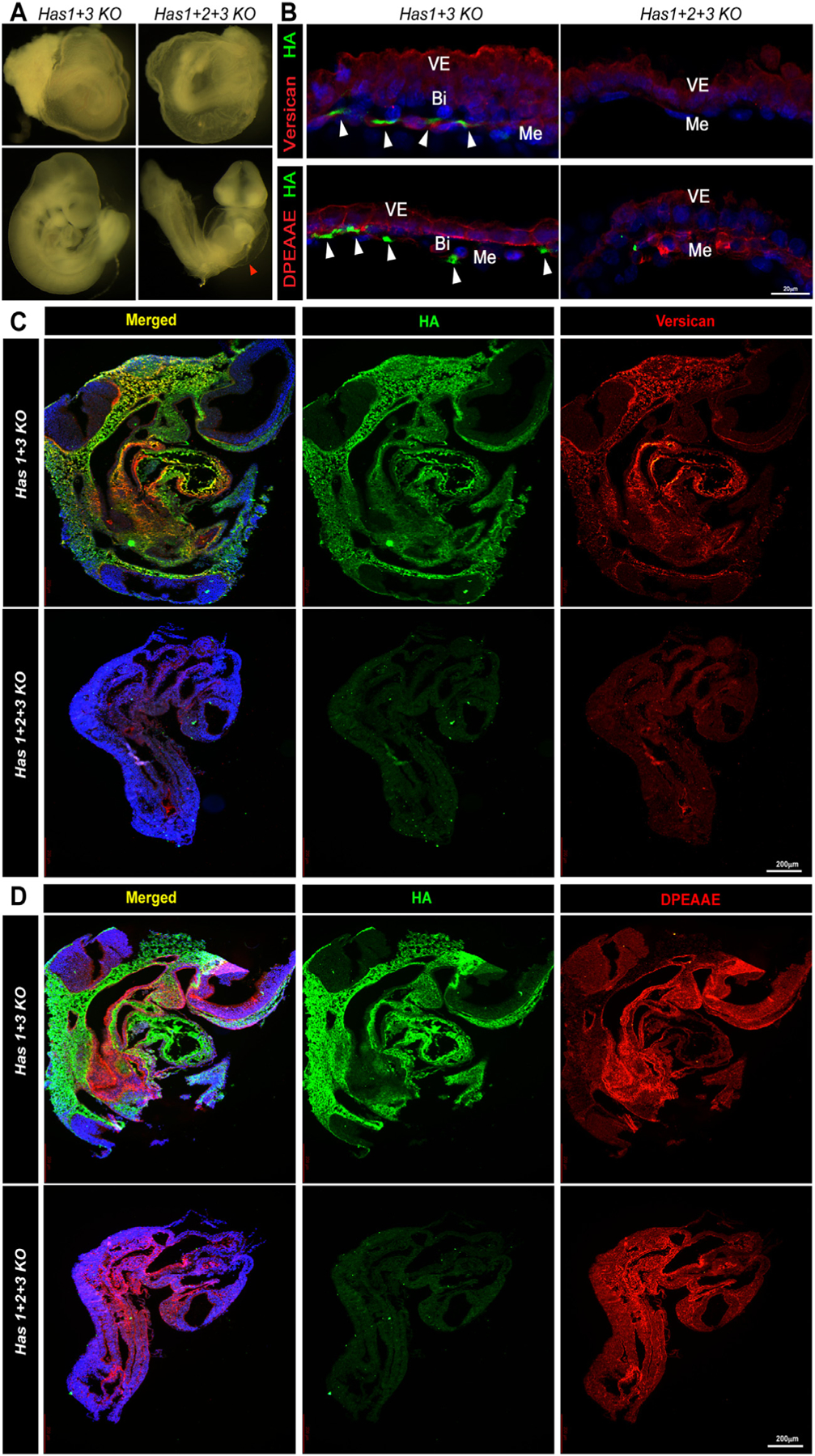
Versican content is severely reduced in Has1+2+3 null embryos. (A) E9.5 Has1+3 null embryos (control) and Has1+2+3 null embryos showing the dilated pericardial sac in the latter (arrowhead) similar to Vcanhdf/hdf (n=3 embryos each genotype). (B) Has1+3 null yolk sac (control) and Has1+2+3 null yolk sac co-stained with HAbp (green) and versican or cleaved versican (DPEAAE) (red) show loss of both HA and versican, although cleaved versican (DPEAAE, red, bottom) is present in Has1+2+3 null yolk sac (n=3 yolk sacs each genotype). (C-D) Has1 3 null (control) and Has1+2+3 null embryos co-stained with HAbp and versican (C) or cleaved versican (DPEAAE) (D), show loss of HA and versican staining and weaker DPEAAE staining in the Has1+2+3 null embryos (n=3 embryos each genotype). Scale bars = 2200μm in C,D.
Embryoid bodies formed by Vcan-null ES cells have impaired angiogenesis and hematopoiesis
Since the dramatic loss of vasculature could result from perturbed hemodynamics due to cardiac defects in Vcanhdf/hdf embryos, we undertook an orthogonal in vitro approach for evaluation of versican in vasculogenesis and hematopoiesis. We used gene editing by CRISPR/Cas9 [40,41] to introduce Vcan-null mutations in R1 mouse embryonic stem cells (mESC) [42]. Vcan exon 2 (containing the start codon) and exon 3 (start of the G1 domain) were targeted independently to obtain mESC clones D8 and F9 respectively, each with defined frameshift mutations that generated null alleles (Fig. 7A). Since exons 2 and 3 are included in all Vcan splice isoforms, no versican was produced, as shown by western blot with anti-GAGβ antibody (Fig. 7B). The mutated clones retained normal expression of Oct4, Sox2, Nanog and C-myc (Fig. 7C) indicating unaltered pluripotency, validating their suitability for in vitro differentiation. Vcan-null and wild-type ES cells were allowed to form embryoid bodies (EBs), in which random differentiation into various lineages occurs. RT-qPCR analysis of Has2 showed no change in Vcan-null EBs (Supplemental Fig. 8A) while the mesoderm differentiation marker Brachyury/T showed a significant decrease in the F9 but not the D8 Vcan-null EBs (Supplemental Fig. 8B). Similar to Vcanhdf/hdf embryos, Vcan-null embryoid bodies showed reduced Kdr expression (Supplemental Fig. 8C), and the blood lineage commitment markers Itga2b (CD41) and Runx1 (Supplemental Fig. 8D), as well as of differentiated blood and vascular endothelial markers (Supplemental Fig. 8 E,F). Since EB differentiation is random and independent of a closed pulsatile circulation, the data from embryoid bodies and the mutant mice together suggests that versican acts directly on Flk1+ hematoendothelial progenitors rather than indirectly via abnormal cardiac development.
Fig. 7.
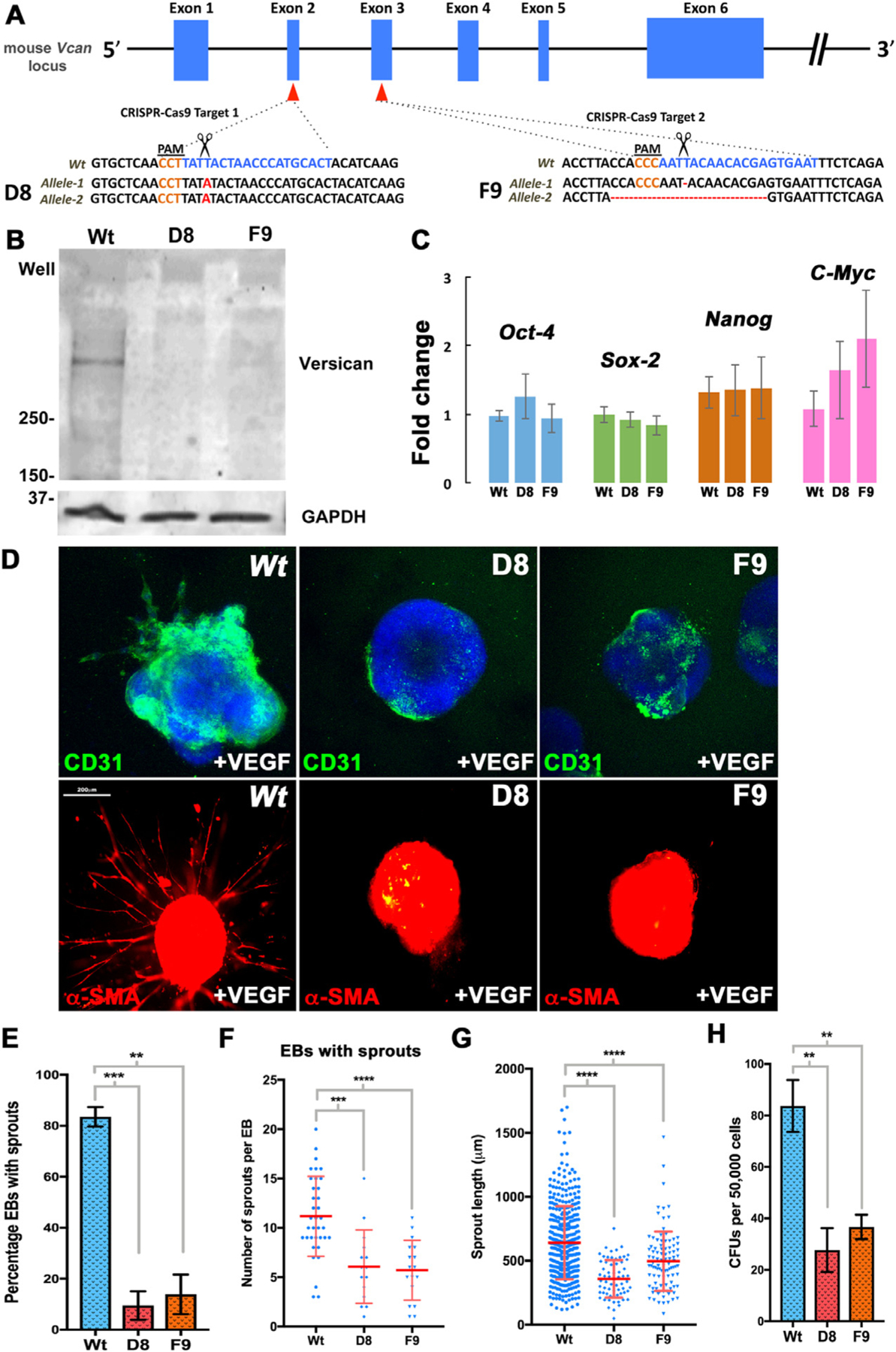
Embryoid bodies (EBs) from Vcan-null mouse embryonic stem cells (mESCs) are poorly responsive to VEGF and form few blood colonies. (A) Vcan locus showing targeting by independent guide RNAs (gRNAs) targeting exons 2 and 3. Scissors indicate the Cas9 cleavage site 3 bp from the protospacer adjacent motif (PAM, orange lettering). An exon 2 mutant mESC clone (D8) had a homozygous 1 bp insertion (bold red letters) and an exon 3 mutant clone (F9) had heterozygous targeting: deletion of T/A in one allele and a 19-bp deletion in the other, each resulting in frame-shifts. (B) Western blot of 10-day differentiated EBs shows no versican in EBs derived from clones D8 and F9 (n=10 pooled embryoid bodies each group). (C) RT-qPCR analysis showing unaffected expression of pluripotency markers Oct4, Sox2, Nanog and C-myc in D8 and F9 (n=3 independent batches of EBs from each genotype, error bars= S.E.). (D) 4-day-old Vcan-null embryoid bodies embedded in collagen I and treated with VEGF165 for 12 days have reduced vascular sprouting identified by CD31 (green) or a-SMA (red) immunostaining (n=3 independent batches of EBs from each genotype). (E) D8 and F9 EBs show significantly fewer vascular sprouts (n=3 independent batches of EBs from each genotype, error bars= S.E. **, p<0.01; ***, p<0.001). (F) Fewer sprouts/EB were seen in D8 and F9 EBs. (n=3 independent batches of EBs from each genotype, error bars= S.D., ***, p<0.001; ****, p<0.0001). (G) Significantly shorter sprouts in Vcan null lines (n=3 independent batches of EBs from each genotype, error bars= S.D., ****, p<0.0001). (H) Methylcellulose colony assay using dissociated 10-day old EBs shows significantly fewer colonies in Vcan null EBs. (n=3 independent batches of EBs from each genotype, error bars= S.D., **, p<0.01). Scale bar in D is 200 μm.
We evaluated the angiogenic and hematopoietic potential of Vcan-null ESC using additional approaches. Sprouting angiogenesis was induced by culturing 4-day differentiated EBs in 3-dimensional collagen I gels and treatment with VEGF-A165 for 12 days [43]. Robust sprouts positive for CD31 and smooth muscle α-actin staining were seen in wild-type EBs, but not in the majority of Vcan-null EBs (Fig. 7D,E). The few Vcan-null EBs with sprouting had fewer sprouts per EB, and these were significantly shorter than wild-type sprouts (Fig. 7F,G). When 10-day old EBs were disaggregated and the cells were plated in a 3-dimensional methylcellulose matrix containing a complete set of blood differentiation cytokines, significantly fewer blood colonies were formed by Vcan null EBs (Fig. 7H).
The versican-HA complex sequesters growth factors essential for vasculogenesis and primary hematopoiesis
These observations raised questions about the underlying mechanisms by which the versican-HA matrix supported Flk1+ cells. To investigate this, we turned to an ADAMTS9 null RPE1 cell line (D12) which lacks two key versican-degrading proteases, ADAMTS9 and ADAMTS20 [22]. Although RPE1 cells are not relevant to hematoendothelial progenitors, they were chosen for the relevance of their matrix, which demonstrates constitutively strong versican staining as a result of reduced ADAMTS activity, in order to determine how versican may modulate vasculogenesis (Fig. 8A). HAbp staining of these cells demonstrated stronger HA staining than wild type RPE1 cultures, with formation of long HA-stained cables decorated with versican (Fig. 8A, Supplemental Fig. 9A,B). Such cables were previously noted to occur in the presence of 25 mM glucose-containing medium, which was used here [44,45]. RT-qPCR revealed reduced HAS2 & HAS3 expression in RPE-1 D12 cells compared to wild-type (Fig. 8B) with no change in HAS1 expression and drastic reduction of TMEM2 expression was observed (Fig. 8B). Upon siRNA-mediated versican depletion [46,47], RPE1-D12 cells showed dramatically reduced versican and HA staining and absence of HA/versican cables (Fig. 8C, Supplemental Fig. 10A). Conversely, RT-qPCR showed increased TMEM2 expression after VCAN knockdown (Supplemental Fig. 10B), suggesting upregulation of the HA-depolymerase TMEM2 by reduction of versican as a possible mechanism of HA loss.
Fig. 8.
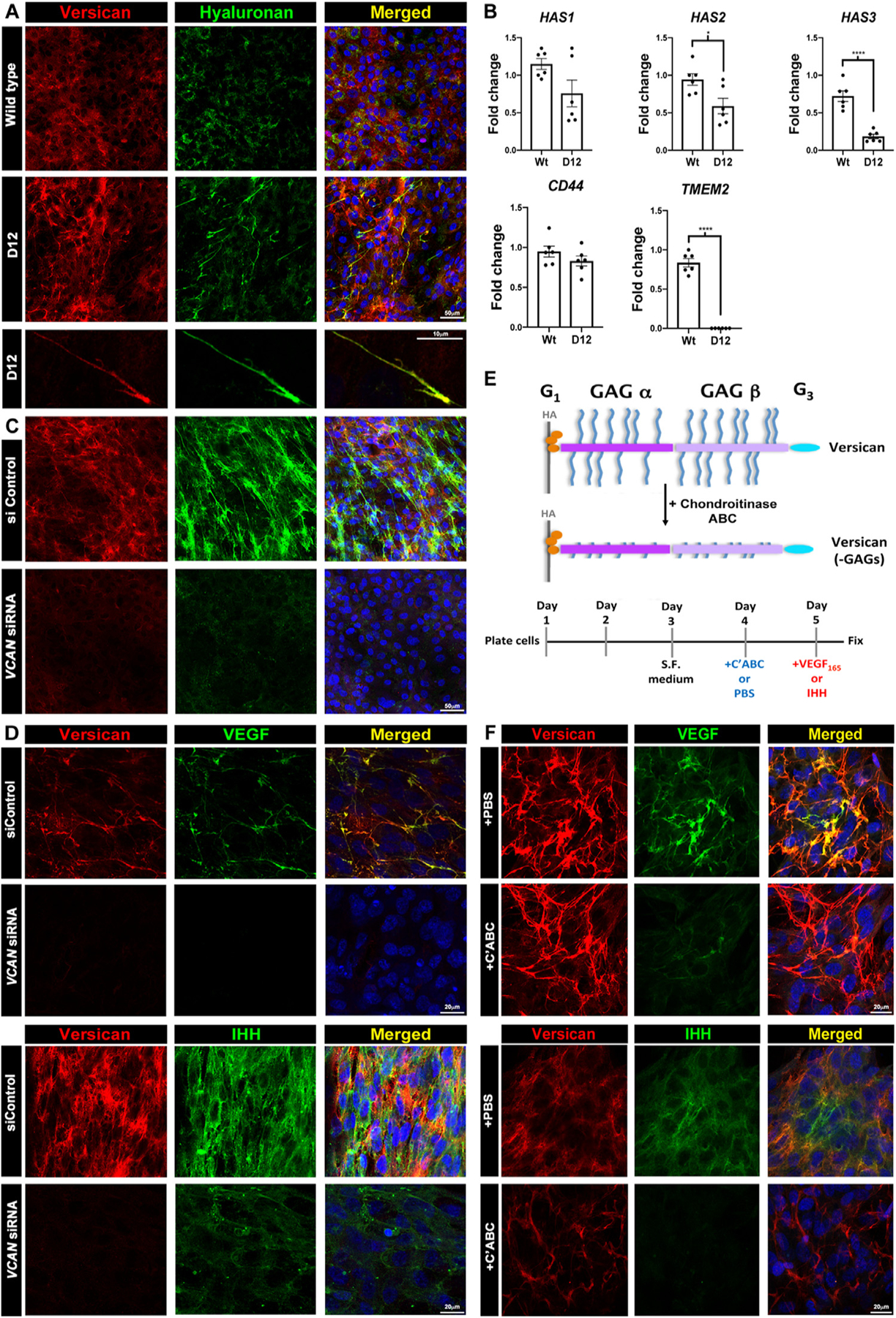
Versican regulates HA abundance and cable formation and sequesters VEGF and Ihh via chondroitin sulfate (CS) chains in a cell culture model. (A) Increased versican (red) and HAbp staining (green) in ADAMTS9-deficient (D12) RPE-1 cells. Versican and HA co-stained cables are formed in D12 cells. (B) RT-qPCR showing reduced HAS2 and HAS3 mRNA but not HAS1 or CD44 and dramatically reduced TMEM2 expression in D12 cells. (n=3 independent RNA extractions, error bars= S.E.M., *, p<0.05; ****, p<0.0001). (C) VCAN knockdown reduces both versican (red) and HA staining in D12 cells. (D) Recombinant VEGF165 or Ihh (green) co-staining with versican (red) in control siRNA-transfected D12 cells is lost in VCAN siRNA-transfected D12 cells. (E) The schematic illustrates chondroitinase ABC removal of versican GAG chains and the experimental timeline used. (F) Fluorescence microscope of VEGF and Ihh with versican, with or without chondroitinase ABC treatment prior to addition of recombinant VEGF165 or Ihh (green) shows reduced binding of both growth factors after CS-chain removal, which does not affect versican core protein staining (red). Scale bars in A are 50μm and 10μm, 20μm in D and F and 50μm in C.
Next, to test if versican could sequester growth factors known to regulate vasculogenesis, we treated wild-type and RPE1-D12 cells with increasing concentrations of recombinant VEGF165 and Ihh. Each showed dose-dependent punctate staining in RPE1-D12 monolayers which co-localized with the versican-HA cables, imaged by confocal fluorescent and super-resolution microscopy (Fig. 8D, Supplemental Fig. 11A–B, Supplemental Fig. 12A–B). Upon VCAN mRNA depletion in RPE1-D12 cells, both VEGF165 and Ihh staining were significantly reduced (Fig. 8D). Enzymatic removal of proteoglycan chondroitin sulfate chains prior to addition of recombinant VEGF165 and Ihh to D12 cultures dramatically reduced VEGF165 and Ihh staining intensity without affecting versican core protein staining intensity or distribution (Fig. 8E,F), suggesting that versican CS chains mediated VEGF and Ihh binding.
Discussion
Although versican is widely expressed during organogenesis and in adult tissues [14], the chronology of its expression in early vascular development undertaken here and independently derived from recently published single cell RNA-seq data, shows a specific association with Flk1+ cells from their earliest origin in the gastrulating embryo until establishment of the yolk sac vasculature and primitive hematopoiesis. Analysis of Vcan mutant mice in vivo and Vcan-null embryoid bodies in vitro suggests that versican has an indispensable role in vasculogenesis and primitive hematopoiesis. Taken together, RNA in situ hybridization data, scRNA-seq analysis of Flk1+ cells and immunostaining of embryos suggests that versican is a product of Flk1+ cells that associates with them via HA and CD44 to form a crucial, possibly cell-autonomously acting pericellular ECM. Furthermore, in vitro analysis undertaken in RPE-1 D12 cells for their high versican levels, showed dose-dependent binding of VEGF and Ihh to the CS-chains of versican.
Two novel, unexpected findings of this study were the substantial loss of embryonic and extraembryonic HA in the absence of versican and reciprocally, of versican in the absence of HA. Also unexpected, and presently unexplained, is the transcriptional effect of versican levels on TMEM2. As in Vcanhdf/hdf yolk sac, avascularity was previously noted in Has2 null yolk sac [26], although it was not studied further. Has2 and Vcan null cardiac defects are similar, which suggested that an obligate versican-HA complex in cardiac jelly and endocardial cushions was required for cardiac morphogenesis [24–26]. The present work similarly strongly suggests that a versican-HA complex, rather than versican alone, is necessary for vasculogenesis and primitive hematopoiesis (Fig. 9). HA-versican aggregates have a net negative charge, and exert swelling pressure via absorption of water (the Gibbs-Donnan effect) [48]. As shown here and previously, embryonic tissues compact in their absence [24,49]. Although versican-deficient yolk sac showed extensive structural disorganization at E9.5, it is clear that the association and effect of versican vis-à-vis Flk1+ cells occurs much earlier, i.e., shortly after gastrulation. Previous work identified both versican and HA in association with mesoderm formed in vitro in embryoid bodies arising from mouse ES cells [50], as well as expression in human ES cells [51]. Reduced vasculogenesis, angiogenesis and hematopoiesis in Vcan-null embryoid bodies supports a direct, local role of versican on hematoendothelial progenitors rather than a secondary effect of cardiac anomalies on vasculogenesis, or of yolk sac disorganization on blood island formation. We therefore conclude that versican-HA aggregates act directly and as early as E6.75 within the microenvironment of Flk1+ cells.
Fig. 9.
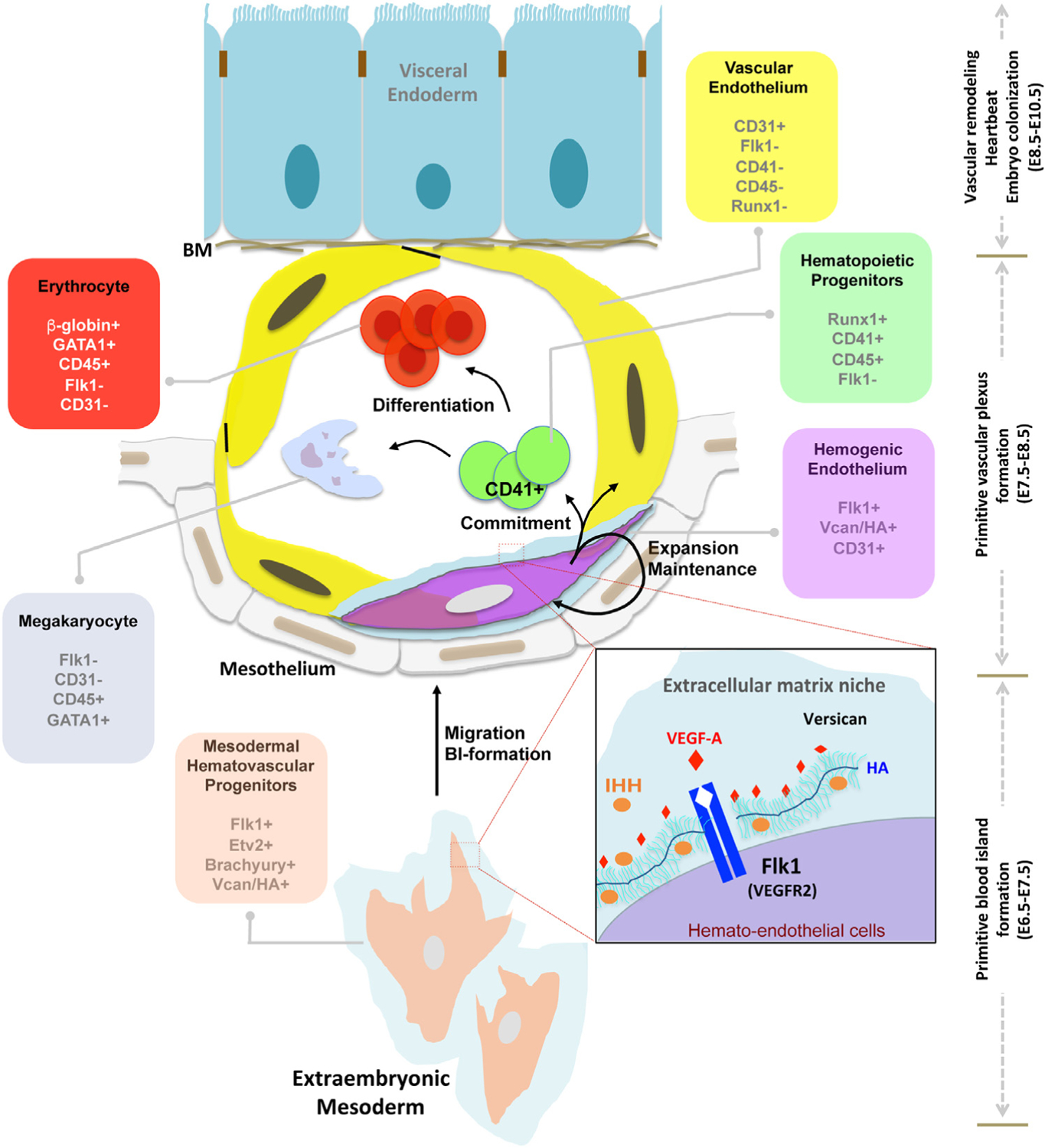
HA-versican pericellular matrix functions as an essential niche for Flk1+ cells. Model depicting the chronology of versican and HA expression and proposed functions in murine extraembryonic mesoderm during vasculogenesis and hematopoiesis. The present work localizes HA-versican to the pericellular matrix of Flk1+ cells, but not the downstream CD41+ committed progenitors or to other CD31 vascular endothelial cells. The inset box proposes that the negatively charged HA-versican pericellular matrix sequesters pro-vasculogenic factors such as VEGF and Ihh.
HA, which is extruded directly from HA synthases on the plasma membrane, is decorated with versican, and retained at the cell surface via HA synthases or CD44 and other HA receptors [52]. Cd44 is coordinately expressed with Vcan and Has2 by Flk1+ cells during early embryogenesis. Nevertheless, Cd44 null mice survive and are not known to have vasculogenesis or primitive erythropoiesis defects [53], suggesting that HAS2 acting as a de facto receptor, may participate in forming a cell-associated versican-HA complex. The versican-HA pericellular matrix has a well-established impact on different cells. We previously found that the amount of versican in the pericellular matrix of fibroblasts and vascular and myometrial smooth muscle cells [9,21,54] was a determinant of phenotype modulation. Versican is anti-adhesive [9,54], which may allow Flk1+ cells to migrate and proliferate efficiently after their emergence at gastrulation. Another possible role of versican-HA is sequestration of vasculogenic factors such as VEGF-A and Ihh. Versican binding to VEGF-A165 and Ihh via its CS-chains (this study) or G3 domain [19], may generate high concentrations around Flk1+ cells. Versican is associated with Flk1+CD41− cells, but not with the Flk1−CD41+ yolk sac cells subsequently, suggesting that the versican-HA-rich pericellular ECM specifically regulates the fate of Flk1+ CD41− cells. We conclude that as the dominant CSPG in the embryo, specifically localized to the vicinity of Flk1+ cells, versican may sequester essential factors such as VEGF-A and Ihh to provide a high local concentration that sustains the Flk1+ population.
Although lack of versican led to higher Fn1 expression, Vcanhdf/hdf defects are unlikely to result from excess fibronectin, because previous work has shown that deficiency, not excess of fibronectin or the fibronectin receptor subunit a5 integrin results in reduced embryo and yolk sac vascularity [27,55]. Since versican binds fibronectin through the G3 domain [19], we conclude that the three major components of the provisional embryonic ECM (versican, HA and fibronectin) are each crucial for establishment of the first blood vessels in the embryo. In the absence of versican, the HA catabolism rate via upregulated TMEM2 likely exceeds the HA synthesis rate. It was previously noted that HA deposition was reduced to 85% of normal in fibroblasts taken from a mouse hypomorphic Vcan mutant (VcanΔ3/Δ3) having 75% reduction of versican [56]. In association with reduced HA deposition, VcanΔ3/Δ3 fibroblasts had accelerated senescence, suggesting a possible mechanism for the lack of Flk1+ cells in Vcanhdf/hdf yolk sac, although this study did not specifically address the fate of Flk1+ cells.
The data suggested stronger association of exon 8-containing Vcan transcripts than exon 7-containing transcripts with blood islands at gastrulation, and exon 7 transcripts were absent at E8.5. Autosomal dominant splice site mutations affecting exon 8 (leading to its exclusion) and exon 8 deletions in humans cause Wagner syndrome [57–59], which is characterized by impaired vision and defects of the ocular vitreous and retina, but lacks consistent extra-ocular manifestations. Exon 7 inactivation in mice led to specific neural anomalies and subtle cardiac anomalies [60,61]. Thus, neither individual exon mutation in mice or humans is associated with embryonic lethality, defective vasculogenesis or impaired hematopoiesis. We conclude that both exon 7 and exon 8-containing transcripts, possibly included in V0, the transcript containing both exons, are required for vasculogenesis and hematopoiesis.
The GAGβ domain encoded by exon 8 has a unique N-terminal sequence, which is cleaved by ADAMTS proteases at the E441-A442 peptide bond in several contexts, notably cardiac valve development, interdigital web regression, umbilical cord development, neural tube and palate closure and myometrial activation [9,22,62,63]. The resulting N-terminal versican V1 fragment, G1-DPEAAE441, named versikine, has bioactivity in interdigital web regression and myeloma growth [64,65]. However, Vcan knock-in mouse mutants in which E441-A442 was mutated to render it uncleavable can complete gestation and have normal yolk sac avascularity, suggesting that E441-A442 cleavage is not involved in hematoendothelial development ([66] and Nandadasa et al., manuscript in preparation).
Both phylogenetically and ontogenetically, basement membranes are accepted to be the earliest organized matrices formed, and indeed, basement membrane components such as laminins have a well-established significance for maintenance of stemness and for epiblast organization [67–69]. The versican-HA matrix appears to be a later evolutionary innovation, and our data suggests that it may be representative of a second wave of matrix expansion and utilization for cellular regulation during mammalian evolution. With the roles in vasculogenesis and hematopoiesis elucidated here, and given their established significance for cardiac development, it is not an exaggeration to state that the versican-HA matrix is crucial for development of the entire circulatory system. Indeed, versican-HA matrix may also have a broad role in formation of vasculature by angiogenesis in other physiological and disease settings. Recent work found that syngeneic B16F10 tumors in adult Vcanhdf/+ mice had significantly impaired angiogenesis and reduced growth [70]. Relevant to the overlap of versican-HA with Flk1 expression, a recent study utilizing syngeneic B16F10 tumors in Flk1+/− mice also found reduced angiogenesis during tumor growth [71]. Furthermore, VEGF binding of the versican-HA ECM which is quantitatively increased in the presence of high glucose, may be relevant to diabetic retinopathy, a common complication of diabetes where the increased activity of VEGF is well-established and indeed, a current target of treatment [72].
Methods
Mice
VcanTg(Hoxa1)1Chm (Vcanhdf) mice [25] (Supplemental Fig. 1) were obtained under a material transfer agreement from Roche. Generation of Has1−/−; Has3−/− double knockout mice was described previously [73]. A Has2-null allele (Has2−) was created from a Has2flox allele [74] by crossing Has2flox/flox mice with the germline deleter Meox2-Cre mice [75]. All of these mouse lines were backcrossed to C57BL/6J for more than 10 generations. Has1−/−; Has2+/−; Has3−/− mice were bred from these mutant mice. Triple Has knockout (TKO) embryos were generated by crossing Has1−/−; Has2+/−; Has3−/− female and male mice. In a second approach to generate HA-deficient mouse embryos, Has2Fl/Fl; Sox2Cretg mice were generated by crossing Has2Fl/Fl males with Has2+/Fl; Sox2Cretg females. Mouse experiments were conducted with IACUC approval (Cleveland clinic protocols 2015:1530 and 2018:2045). Mice were maintained in a fixed light-dark cycle with food and water ad libitum. For genotyping E8.5 embryos with intact yolk sacs, the allantois was dissected out and lysed in 10 μl DirectPCR (Tail) digest reagent (Qiagen, catalog no. 102-T) supplemented with 1 ml of proteinase K overnight at 55°C. Tails were used to genotype E9.5 embryos. Vcanhdf and wild-types were identified with a specific genotyping strategy based on the genetic interruption (Supplemental Fig. 1). Details of the Has1–3 mutant mouse genotyping is provided in the Supplemental Methods. Vcanhdf/hdf embryos were compared to wild-type littermates in all experiments.
Mouse embryonic stem cell (mESC) culture
R1 mESC [42] (Case Transgenic and Targeting Facility) were cultured on 0.3% type B gelatin (Sigma-Aldrich, catalog no. G9382) coated 60 mm cell culture plates in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 4mM L-glutamine and 1mM sodium pyruvate, supplemented with 20% fetal bovine serum (Hyclone, catalog no. SH30071), 0.1 mM cell culture grade 2-mercaptoethanol (Gibco, Life Technologies, catalog no. 21985), 0.1mM nonessential amino acids (Gibco, Life Technologies, catalog no. 11140–050), 50 μg/mL penicillin/streptomycin and 1 × 106 units/mL leukemia inhibitory factor (LIF; ESGRO, EMD Millipore, catalog no. ESG1106) in a humidified 5% CO2, 37°C environment. mESC were maintained at 60–80% confluence, with daily medium change and passaged every other day in a 1:5 split.
CRISPR/Cas9 targeting of mESC Vcan
2.5 μg of CRISPR/Cas9 plasmids in the U6gRNA-Cas9–2A-GFP vector, targeting Vcan exon 2 or exon 3 (Sigma-Aldrich, target IDs MM0000080027 and MM0000080028) were transfected into R1 mESCs at 60–80% confluence in 6-well plates coated with 0.3% gelatin, using FuGene 6 (Promega, catalog no. E2691). 24 hrs post-transfection, individual GFP+ mESCs were sorted into 96-well plates coated with 0.3% gelatin using a FACSAria-II cell sorter (BD Biosciences). Fast-growing wells (containing >1000 cells/well) were trypsinized and expanded to 24-well cell culture plates after 7–10 days. Culture medium was replaced daily. Genomic DNA from clones was isolated using DirectPCR (Tail) reagent (Viagen, catalog no. 102-T) and Vcan exon 2 and exon 3 were amplified using Phusion Taq (NEB, catalog no. F530L) (see SI). Amplicons were excised from 2% agarose gels, purified using the QIAquick Gel Extraction kit (QIAGEN, catalog no. 28704) and cloned into pCR-Blunt II-TOPO vector using the Zero blunt PCR cloning kit (Life Technologies, Invitrogen, catalog no. K2800–40). Plasmid DNA was harvested from bacterial colonies for Sanger-sequencing to determine the precise mutations. Two independent Vcan-null mESC clones, D8 and F9, were established. Western blotting and immunostaining of 10-day old embryoid bodies from these lines (see below) confirmed Vcan inactivation. Embryoid body formation and induction of vascular sprouts is described in the Supplemental Methods.
Immunostaining and fluorescence microscopy
Immunostaining of E 9.5 and E 8.5 yolk sac was carried out on 30 μm thick vibratome sections [22] or paraffin-embedded 7 μm sections. Immunostaining of collagen-embedded embryoid bodies were carried out in 4-chamber cell culture slides (Fisher Scientific, catalog no. 354114). Confocal microscopy images of whole-mount mouse embryos and sections were acquired using a Leica TCS SP5 II multiphoton confocal microscope equipped with a 25X water immersion objective (Leica Microsystems, Wetzlar, Germany). For 3D-projection of whole-mount Z-stacks, the Volocity 3D imaging software was used (version 6.3, PerkinElmer, Inc., Waltham, MA) in maximum intensity projection method.
Methylcellulose colony formation assay
Single cell suspensions of E8.5 embryos, yolk sacs or day-10 embryoid bodies were generated by incubation with trypsin for 10 minutes followed by disaggregation by pipetting with a 200 μL pipette tip until complete. 50,000 cells from each experimental group were transferred to a single 35 mm culture dish containing 1 mL of MethoCult GF M3434 culture medium (Methylcellulose medium with recombinant cytokines for mouse cells, Stem Cell Technologies, Vancouver, CA, catalog no. 03434) using a 3 mL syringe and 16-gauge needle, following the manufacturer’s protocol. Triplicate cultures from each genotype were incubated for 14 days in a humidified, 5% CO2, 37°C cell culture incubator. Blood colonies were counted using an inverted microscope. Aggregates with >50 cells were considered a colony-forming unit (CFU).
Additional details of reagents and procedures including primary antibodies used, RNAscope in situ hybridization, western blotting, transmission electron microscopy, and fluorophore-assisted carbohydrate electrophoresis (FACE) are described in the Supplemental Methods
Supplementary Material
Acknowledgments
This work was supported by the NIH-NHLBI Program of Excellence in Glycosciences award HL107147 (to S.S.A., R.J.M.,), by the Allen Distinguished Investigator Program, through support made by The Paul G. Allen Frontiers Group and the American Heart Association (award 17DIA33820024 to S.S.A), NIH RF1AG057579 (to Y.Y.) and the David and Lindsay Morgenthaler Postdoctoral Fellowship and the Mark Lauer Pediatric Research grant (to S.N.). Purchase of the Leica SP8 confocal microscope was supported by NIH SIG grant 1S10RR026820-01. We thank Dr. David LePage and Dr. Ron Conlon at the Case Transgenic and Targeting Facility for R1 mES cells, Eric Schultz and Joseph Gerow of the LRI Flow Cytometry Core for mES cell sorting, Valbona Cali for FACE assays, Dr. Judy Drazba and Mei Yin of the LRI Imaging Core for guidance with confocal and electron microscopy and the Apte laboratory members for valuable discussions.
Footnotes
Declaration of Competing Interests
The authors declare no competing interests.
Supplementary materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.matbio.2021.01.002.
References
- [1].Qiu J, Hirschi KK, Endothelial cell development and its application to regenerative medicine, Circ. Res 125 (4) (2019) 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Risau W, Flamme I, Vasculogenesis, Annu. Rev. Cell Dev. Biol 11 (1995) 73–91. [DOI] [PubMed] [Google Scholar]
- [3].Lugus JJ, et al. , Both primitive and definitive blood cells are derived from Flk-1+ mesoderm, Blood 113 (3) (2009) 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ferkowicz MJ, Yoder MC, Blood island formation: long-standing observations and modern interpretations, Exp. Hematol 33 (9) (2005) 1041–1047. [DOI] [PubMed] [Google Scholar]
- [5].Dzierzak E, Speck NA, Of lineage and legacy: the development of mammalian hematopoietic stem cells, Nat. Immunol 9 (2) (2008) 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Red-Horse K, et al. , Endothelium-microenvironment interactions in the developing embryo and in the adult, Dev. Cell 12 (2) (2007) 181–194. [DOI] [PubMed] [Google Scholar]
- [7].Lozito TP, et al. , Human mesenchymal stem cells express vascular cell phenotypes upon interaction with endothelial cell matrix, J. Cell. Biochem 107 (4) (2009) 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gu Y, et al. , Steel factor controls primordial germ cell survival and motility from the time of their specification in the allantois, and provides a continuous niche throughout their migration, Development 136 (8) (2009) 1295–1303. [DOI] [PubMed] [Google Scholar]
- [9].Mead TJ, et al. , ADAMTS9-regulated pericellular matrix dynamics governs focal adhesion-dependent smooth muscle differentiation, Cell Rep 23 (2) (2018) 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Perris R, et al. , Inhibitory effects of PG-H/aggrecan and PG-M/versican on avian neural crest cell migration, FASEB J 10 (2) (1996) 293–301. [DOI] [PubMed] [Google Scholar]
- [11].Yamagata M, et al. , Regulation of cell-substrate adhesion by proteoglycans immobilized on extracellular substrates, J. Biol. Chem 264 (14) (1989) 8012–8018. [PubMed] [Google Scholar]
- [12].van Wijk XM, van Kuppevelt TH, Heparan sulfate in angiogenesis: a target for therapy, Angiogenesis 17 (3) (2014) 443–462. [DOI] [PubMed] [Google Scholar]
- [13].Le Jan S, et al. , Functional overlap between chondroitin and heparan sulfate proteoglycans during VEGF-induced sprouting angiogenesis, Arterioscler. Thromb. Vasc. Biol 32 (5) (2012) 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bode-Lesniewska B, et al. , Distribution of the large aggregating proteoglycan versican in adult human tissues, J. Histochem. Cytochem 44 (4) (1996) 303–312. [DOI] [PubMed] [Google Scholar]
- [15].Kimata K, et al. , A large chondroitin sulfate proteoglycan (PG-M) synthesized before chondrogenesis in the limb bud of chick embryo, J. Biol. Chem 261 (29) (1986) 13517–13525. [PubMed] [Google Scholar]
- [16].Shinomura T, et al. , cDNA cloning of PG-M, a large chondroitin sulfate proteoglycan expressed during chondrogenesis in chick limb buds. Alternative spliced multiforms of PG-M and their relationships to versican, J. Biol. Chem 268 (19) (1993) 14461–14469. [PubMed] [Google Scholar]
- [17].Zimmermann DR, Ruoslahti E, Multiple domains of the large fibroblast proteoglycan, versican, EMBO J 8 (10) (1989) 2975–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu YJ, et al. , The interaction of versican with its binding partners, Cell Res 15 (7) (2005) 483–494. [DOI] [PubMed] [Google Scholar]
- [19].Zheng PS, et al. , Versican/PG-M G3 domain promotes tumor growth and angiogenesis, FASEB J 18 (6) (2004) 754–756. [DOI] [PubMed] [Google Scholar]
- [20].Slevin M, et al. , Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways, Matrix Biol 26 (1) (2007) 58–68. [DOI] [PubMed] [Google Scholar]
- [21].Hattori N, et al. , Pericellular versican regulates the fibroblast-myofibroblast transition: a role for ADAMTS5 protease-mediated proteolysis, J. Biol. Chem 286 (39) (2011) 34298–34310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nandadasa S, Nelson CM, Apte SS, ADAMTS9-mediated extracellular matrix dynamics regulates umbilical cord vascular smooth muscle differentiation and rotation, Cell Rep 11 (10) (2015) 1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dours-Zimmermann MT, Zimmermann DR, A novel glycosaminoglycan attachment domain identified in two alternative splice variants of human versican, J. Biol. Chem 269 (52) (1994) 32992–32998. [PubMed] [Google Scholar]
- [24].Mjaatvedt CH, et al. , The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation, Dev. Biol 202 (1) (1998) 56–66. [DOI] [PubMed] [Google Scholar]
- [25].Yamamura H, et al. , A heart segmental defect in the anterior-posterior axis of a transgenic mutant mouse, Dev. Biol 186 (1) (1997) 58–72. [DOI] [PubMed] [Google Scholar]
- [26].Camenisch TD, et al. , Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme, J. Clin. Invest 106 (3) (2000) 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Francis SE, et al. , Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies, Arterioscler. Thromb. Vasc. Biol 22 (6) (2002) 927–933. [DOI] [PubMed] [Google Scholar]
- [28].Hunziker EB, Herrmann W, Schenk RK, Ruthenium hexammine trichloride (RHT)-mediated interaction between plasmalemmal components and pericellular matrix proteoglycans is responsible for the preservation of chondrocytic plasma membranes in situ during cartilage fixation, J. Histochem. Cytochem 31 (6) (1983) 717–727. [DOI] [PubMed] [Google Scholar]
- [29].Shalaby F, et al. , A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis, Cell 89 (6) (1997) 981–990. [DOI] [PubMed] [Google Scholar]
- [30].Ferkowicz MJ, et al. , CD41 expression defines the onset of primitive and definitive hematopoiesis in the murine embryo, Development 130 (18) (2003) 4393–4403. [DOI] [PubMed] [Google Scholar]
- [31].Pijuan-Sala B, et al. , A single-cell molecular map of mouse gastrulation and early organogenesis, Nature 566 (7745) (2019) 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Calabro A, et al. , Microanalysis of enzyme digests of hyaluronan and chondroitin/dermatan sulfate by fluorophore-assisted carbohydrate electrophoresis (FACE), Glycobiology 10 (3) (2000) 273–281. [DOI] [PubMed] [Google Scholar]
- [33].Calabro A, Hascall VC, Midura RJ, Adaptation of FACE methodology for microanalysis of total hyaluronan and chondroitin sulfate composition from cartilage, Glycobiology 10 (3) (2000) 283–293. [DOI] [PubMed] [Google Scholar]
- [34].Midura RJ, et al. , Quantification of hyaluronan (HA) using a simplified fluorophore-assisted carbohydrate electrophoresis (FACE) procedure, Methods Cell Biol 143 (2018) 297–316. [DOI] [PubMed] [Google Scholar]
- [35].De Angelis JE, et al. , Tmem2 regulates embryonic vegf signaling by controlling hyaluronic acid turnover, Dev. Cell 40 (2) (2017) 123–136. [DOI] [PubMed] [Google Scholar]
- [36].Schinzel RT, et al. , The hyaluronidase, TMEM2, promotes ER homeostasis and longevity independent of the UPR(ER), Cell 179 (6) (2019) 1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yamamoto H, et al. , A mammalian homolog of the zebrafish transmembrane protein 2 (TMEM2) is the long-sought-after cell-surface hyaluronidase, J. Biol. Chem 292 (18) (2017) 7304–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bai KJ, et al. , The role of hyaluronan synthase 3 in ventilator-induced lung injury, Am. J. Respir. Crit. Care Med 172 (1) (2005) 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mack JA, et al. , Enhanced inflammation and accelerated wound closure following tetraphorbol ester application or full-thickness wounding in mice lacking hyaluronan synthases Has1 and Has3, J. Invest. Dermatol 132 (1) (2012) 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Doudna JA, Charpentier E, Genome editing. The new frontier of genome engineering with CRISPR-Cas9, Science 346 (6213) (2014) 1258096. [DOI] [PubMed] [Google Scholar]
- [41].Jinek M, et al. , A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity, Science 337 (6096) (2012) 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nagy A, et al. , Derivation of completely cell culture-derived mice from early-passage embryonic stem cells, Proc Natl Acad Sci U S A, 90 (18) (1993) 8424–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Baker M, et al. , Use of the mouse aortic ring assay to study angiogenesis, Nat. Protoc 7 (1) (2012) 89–104. [DOI] [PubMed] [Google Scholar]
- [44].Wang A, Hascall VC, Hyaluronan structures synthesized by rat mesangial cells in response to hyperglycemia induce monocyte adhesion, J. Biol. Chem 279 (11) (2004) 10279–10285. [DOI] [PubMed] [Google Scholar]
- [45].Selbi W, et al. , Characterization of hyaluronan cable structure and function in renal proximal tubular epithelial cells, Kidney Int 70 (7) (2006) 1287–1295. [DOI] [PubMed] [Google Scholar]
- [46].Gueye NA, et al. , Versican proteolysis by ADAMTS proteases and its influence on sex steroid receptor expression in uterine leiomyoma, J. Clin. Endocrinol. Metab 102 (5) (2017) 1631–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Keire PA, et al. , Inhibition of versican expression by siRNA facilitates tropoelastin synthesis and elastic fiber formation by human SK-LMS-1 leiomyosarcoma smooth muscle cells in vitro and in vivo, Matrix Biol 50 (2016) 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Buschmann MD, Grodzinsky AJ, A molecular model of proteoglycan-associated electrostatic forces in cartilage mechanics, J. Biomech. Eng 117 (2) (1995) 179–192. [DOI] [PubMed] [Google Scholar]
- [49].Szabo A, et al. , In vivo confinement promotes collective migration of neural crest cells, J. Cell Biol 213 (5) (2016) 543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shukla S, et al. , Synthesis and organization of hyaluronan and versican by embryonic stem cells undergoing embryoid body differentiation, J. Histochem. Cytochem 58 (4) (2010) 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chan CK, et al. , Differentiation of cardiomyocytes from human embryonic stem cells is accompanied by changes in the extracellular matrix production of versican and hyaluronan, J. Cell. Biochem 111 (3) (2010) 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Evanko SP, et al. , Hyaluronan-dependent pericellular matrix, Adv. Drug. Deliv. Rev 59 (13) (2007) 1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Protin U, et al. , CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets, J. Immunol 163 (9) (1999) 4917–4923. [PubMed] [Google Scholar]
- [54].Evanko SP, Angello JC, Wight TN, Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells, Arterioscler. Thromb. Vasc. Biol 19 (4) (1999) 1004–1013. [DOI] [PubMed] [Google Scholar]
- [55].George EL, et al. , Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin, Development 119 (4) (1993) 1079–1091. [DOI] [PubMed] [Google Scholar]
- [56].Suwan K, et al. , Versican/PG-M assembles hyaluronan into extracellular matrix and inhibits CD44-mediated signaling toward premature senescence in embryonic fibroblasts, J. Biol. Chem 284 (13) (2009) 8596–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kloeckener-Gruissem B, et al. , Identification of the genetic defect in the original Wagner syndrome family, Mol. Vis 12 (2006) 350–355. [PubMed] [Google Scholar]
- [58].Kloeckener-Gruissem B, et al. , Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome, Eur. J. Hum. Genet 21 (3) (2013) 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rothschild PR, et al. , De novo splice mutation in the versican gene in a family with Wagner syndrome, JAMA Ophthalmol 131 (6) (2013) 805–807. [DOI] [PubMed] [Google Scholar]
- [60].Burns TA, et al. , Imbalanced expression of Vcan mRNA splice form proteins alters heart morphology and cellular protein profiles, PLoS One 9 (2) (2014) e89133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dours-Zimmermann MT, et al. , Versican V2 assembles the extracellular matrix surrounding the nodes of ranvier in the CNS, J. Neurosci 29 (24) (2009) 7731–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nandadasa S, Foulcer S, Apte SS, The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis, Matrix Biol 35 (2014) 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nandadasa S, et al. , Vascular dimorphism ensured by regulated proteoglycan dynamics favors rapid umbilical artery closure at birth, bioRxiv (2020) 2020.07.02.184978. [DOI] [PMC free article] [PubMed]
- [64].Hope C, et al. , Immunoregulatory roles of versican proteolysis in the myeloma microenvironment, Blood 128 (5) (2016) 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].McCulloch DR, et al. , ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression, Dev. Cell 17 (5) (2009) 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Islam S, et al. , Accumulation of versican facilitates wound healing: implication of its initial ADAMTS-cleavage site, Matrix Biol (2019). [DOI] [PubMed]
- [67].Fidler AL, et al. , Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues, Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Miner JH, et al. , Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation, Development 131 (10) (2004) 2247–2256. [DOI] [PubMed] [Google Scholar]
- [69].Rodin S, et al. , Long-term self-renewal of human pluripotent stem cells on human recombinant laminin-511, Nat. Biotechnol 28 (6) (2010) 611–615. [DOI] [PubMed] [Google Scholar]
- [70].Asano K, et al. , Stromal versican regulates tumor growth by promoting angiogenesis, Sci. Rep 7 (1) (2017) 17225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Oladipupo SS, et al. , Impaired tumor growth and angiogenesis in mice heterozygous for Vegfr2 (Flk1), Sci. Rep 8 (1) (2018) 14724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhao Y, Singh RP, The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy, Drugs Context 7 (2018) 212532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Arranz AM, et al. , Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space, J. Neurosci 34 (18) (2014) 6164–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Matsumoto K, et al. , Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb, Development 136 (16) (2009) 2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tallquist MD, Soriano P, Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function, Genesis 26 (2) (2000) 113–115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.