Redefining the Histone Deacetylase Inhibitor Pharmacophore: High Potency with No Zinc Cofactor Interaction
Histone deacetylases
(HDACs) enzymatically regulate gene transcription
of proteins through the cleavage of their N-acetylated lysine residues
and remodeling of chromatin. While several FDA-approved HDAC inhibitors
(HDACis) target proliferating tumor cells of certain cancers, the
putatively required HDACi pharmacophoric elements of these drugs that
involve enzymatic zinc atom-engagement also induce mutagenicity, toxicity
or present pharmacokinetic hurdles that are challenging to overcome,
thereby limiting their development for broader application. In this
Featured letter (DOI: 10.1021/acsmedchemlett.1c00074), Beshore and co-workers report on the development of natural-product-inspired,
indole-acetamide-based HDAC inhibitors that, by X-ray crystallographic
evidence, do not coordinate to the catalytic zinc atom. Nonetheless,
members of the structural class showed potent biochemical enzymatic
inhibition and cell-based efficacy that is similar to that of marketed
HDAC inhibitor drugs containing traditional zinc-binding moieties.
The authors explored structure–activity relationships that
probed binding capability, HDAC isozyme selectivity, and efficacy
in cell models evaluating HIV latency reversal. The resulting lead
compound 19 was profiled for off-target liability, was
shown to be nonmutagenic, and exhibited promising pharmacokinetic
properties. Based on the collective results, this report marks an
important leap in the understanding of HDAC inhibitor development,
thus reconceptualizing a pharmacophoric HDAC inhibition model that
traditionally requires a metal binding structural moiety.
Discovery of the First Orally Available, Selective KNa1.1 Inhibitor: In Vitro and In Vivo Activity of an Oxadiazole Series
The sodium-activated potassium channel KNa1.1 is expressed
throughout the central nervous system and is encoded by the KCNT1 gene. Mutations in KCNT1 that confer
enhanced protein function are associated with multiple forms of drug-resistant
infant and childhood seizure disorder. Therapeutics are urgently sought
to address inadequate efficacy and target selectivity associated with
investigational drugs. To discover novel KNa1.1 channel
modulators, Griffin and co-workers (DOI: 10.1021/acsmedchemlett.0c00675) screened a custom commercial compound library to reveal oxadiazole
appended pyrazole 5-carboxamides as a hit scaffold. Structure–activity
relationships based on human KNa1.1 channel inhibition
were explored across four scaffold regions in a dose response format
with attention paid to potency, lipophilicity, solubility, stereochemical
influence, clearance, and validation of response on the equivalent
mouse KNa1.1 channel. The best candidate resulting from
this effort was further profiled against human KCNT1 variants that
result in a gain of function, along with an 80-member off-target panel
that included other ion channels for which cross-activity may be of
concern. Pharmacokinetic evaluation in mice showed reasonable brain
exposure and complementary parameters that permitted oral dosing in
a mouse model of epileptic encephalopathy. The authors show that mice
dosed orally with the candidate compound showed reduced seizure activity
compared to control mice, suggesting that this class of compounds
may provide insights into the development of therapeutic options for
severe forms of epileptic syndromes in children.
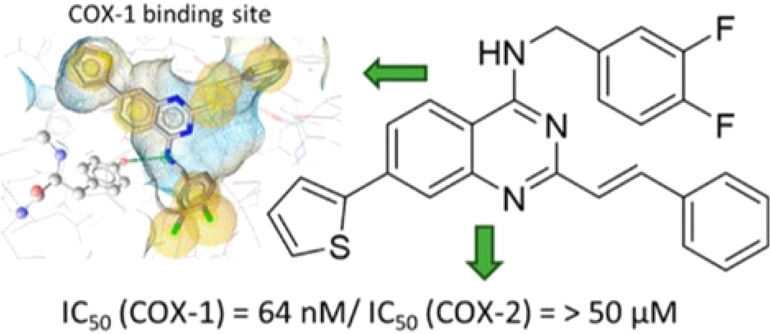
Synthesis, Inhibitory Activity, and In Silico Modeling of Selective COX-1 Inhibitors with a Quinazoline Core
Cyclooxygenase (COX)
isozymes, COX-1 and COX-2, are integral to
inflammatory mechanisms, and their inhibition by nonsteroidal anti-inflammatory
drugs (NSAIDs) treats pain, fever, and inflammation. While classical
NSAIDs are unselective between the two isozymes, newer agents have
focused on targeting COX-2 selectively for these therapeutic properties.
While some selective COX-1 inhibitors have been developed, interest
in selective COX-1 inhibitors with appropriate pharmacokinetic characteristics
has been renewed as the roles of this enzyme in multiple cancers,
cardiovascular and neurological inflammatory processes have been better
elucidated. With the intent of designing selective COX-1 inhibitors
with improved properties, Dvorakova and co-workers (DOI: 10.1021/acsmedchemlett.1c00004) generated structurally
inspired analogues of quinazoline-based third generation NSAIDs such
as fluproquazone and proquazone but which feature augmented structural
elements intended to exploit key differences between the binding pockets
of COX-1 and COX-2. After multiple rounds of structure–activity
relationship development and evaluation of COX inhibition and selectivity,
a styrene-containing aminoquinazoline 9b resulted that
potently and selectively inhibited COX-1 through a substrate competitive
mechanism. In silico docking of the analogues was
performed to provide a rationale for the observed selectivity and
suggests that selective COX-1 inhibition may be achieved and probed
for therapeutic advantage.