

Abstract

Herein we report the discovery of 2,4-1H-imidazole carboxamides as novel, biochemically potent, and kinome selective inhibitors of transforming growth factor β-activated kinase 1 (TAK1). The target was subjected to a DNA-encoded chemical library (DECL) screen. After hit analysis a cluster of compounds was identified, which was based on a central pyrrole-2,4-1H-dicarboxamide scaffold, showing remarkable kinome selectivity. A scaffold-hop to the corresponding imidazole resulted in increased biochemical potency. Next, X-ray crystallography revealed a distinct binding mode compared to other TAK1 inhibitors. A benzylamide was found in a perpendicular orientation with respect to the core hinge-binding imidazole. Additionally, an unusual amide flip was observed in the kinase hinge region. Using structure-based drug design (SBDD), key substitutions at the pyrrolidine amide and the glycine resulted in a significant increase in biochemical potency.
Keywords: Kinase, TAK1, MAP3K7, DNA-encoded chemical library (DECL), Structure-based drug design (SBDD)
The mitogen-activated protein kinase (MAPK) pathways consist of multiple conserved kinases, ultimately linking extracellular signals to crucial intracellular processes, and deregulation of its members has potential implications in both inflammatory1 and oncological disorders.2,3 After first being reported in 1995,4 the transforming growth factor β-activated kinase 1 (TAK1, MAP3K7) has in fact emerged as a potential therapeutic target for both inflammatory diseases and cancer.5,6 TAK1 is activated by transforming growth factor-β (TGF- β) and facilitates downstream signaling of multiple pro-inflammatory cytokines such as TNF-α, TLR ligands, LPS, and IL-1.7−10 Although the complex mechanisms through which TAK1 activation and signaling are controlled have not been fully elucidated, it has been recognized that TAK1 phospho-activates members of the MAP2K family, including MKK3/4/6/7, as well as IKK kinases, both of which are in turn activators of the downstream MAP kinases p38 and JNK and the transcription factor NFkB.11−13 Through activation of these pathways, TAK1 has a pro-survival role, by inducing expression of antiapoptotic proteins and cytokines, thus inhibiting apoptosis and promoting cell proliferation.14−16 Specifically, it has been demonstrated that inhibition of TAK1 activity sensitized tumor cells to TRAIL-induced apoptosis.17 Moreover, the therapeutic potential of TAK1 inhibitors has been investigated for the treatment of pancreatic,18 ovarian,19 and breast20 cancers as well as in inflammatory diseases.21
The first inhibitors described for TAK1 exploit the active site cysteine residue found in this kinase, and the most extensively studied example of these is the natural product 5Z-7-oxozeaenol 1(22,23) (Figure 1). This compound binds to the hinge with the phenol and reaction of the enone with a cysteine in the active site results in the formation of a covalent bond leading to irreversible inhibition of the enzyme. The compound was shown to be active in cells, but as many other kinases have a similarly located cysteine, the compound may not be very selective. More recently, both type I ATP hinge-binding inhibitors and type II DFG-out inhibitors have been described. Compound 2(24,25) is the result of a program pursued at OSI Pharmaceuticals, where the aminopyridine is binding to the hinge and the benzothiadiazole is forming an interaction with the catalytic lysine in the phosphate pocket. The compound showed good selectivity in a screen against 192 kinases and was found to be active in cells and in a mouse tumor xenograft model. Another type I inhibitor was developed by Chugai Pharmaceuticals. Optimization of a screening hit using SBDD resulted in compound 3.26 The inhibitor showed good activity and excellent selectivity. Unfortunately, poor cell activity was observed due to poor permeability. In a very nice display of SBDD using crystal structure overlays, the compound was converted into both DFG-out and c-helix-out type II inhibitors.27 The potential role of a TAK1 inhibitor AZ-TAK1 (4)28 in mantle cell lymphoma was described by Bugli et al. The compound was potent, but it was fairly unselective with an IC50 below 100 nM for 10 out of 30 enzymes in a small kinase panel. Recently, Takinib (5)29 was described to inhibit TAK1 selectively, resulting in TNF-α induced apoptosis. Finally, type II inhibitors have also been described for TAK1, such as NG-25 (6)30 as developed by Gray et al.
Figure 1.

Structures of TAK1 inhibitors.
In a program to identify novel and structurally distinct inhibitors of TAK1, we subjected the target to a DECL screen.31 Briefly, twenty-one different DNA-encoded chemical libraries were combined and this mixture was incubated with TAK1–TAB1 fusion protein. Separate incubations were set up with no target, TAK1–TAB1 at 7.8 μM, TAK1–TAB1 at 1.6 μM, and with TAK1–TAB1 at 7.8 μM and 5Z-7-oxozeaenol (1) at 40 μM. After a 1 h of incubation the mixtures were captured on the His-Select IMAC matrix, washed, and then heat-eluted at 85 °C. A second round of selection was performed using the round-one eluate as input and fresh proteins and 1 repeating the conditions of the first round. The encoding DNA in the output of the second round of selection was amplified, and the amplified sample was submitted for sequencing using an Illumina 2500 instrument in high-output mode. Ninety-seven million single-end reads were generated across all twenty-one libraries for these four selections. Sequence data were parsed, and each read was assigned to the library chemical scheme and the specific building blocks used to synthesize it and the experimental condition it was subjected to during selection. Statistical and demographic data were calculated for all building block combinations. One of the compounds prioritized for resynthesis was derived from a library which had been synthesized by installing 940 unique primary amines onto a DNA-linked aldehyde followed by reaction with 4,000 capping reagents including carboxylic acids and other secondary amine-reactive building blocks. This library contains 3.76 million different compounds, each of which is encoded by a unique DNA tag combination. This cluster of structurally related compounds was defined by the capping building block—(pyrrolidine-1-carbonyl)-1H-pyrrole-3-carboxylic acid—and was coenriched with a range of 2-substituted phenyl methanamines with 2-(difluoromethoxyphenyl)methanamine being the most enriched. These compounds showed a complete absence of enrichment in the selection with TAK1-TAB1 and a saturating concentration of 5Z-7-oxozeaenol, consistent with the hypothesis that their binding sites on the target protein overlap. It was this most enriched combination of building blocks that was chosen for resynthesis of the initial hit compound, compound 7 (Figure 2), along with the first few atoms of what during the screen was the linker to the encoding DNA.
Figure 2.

Primary hit compound 7.
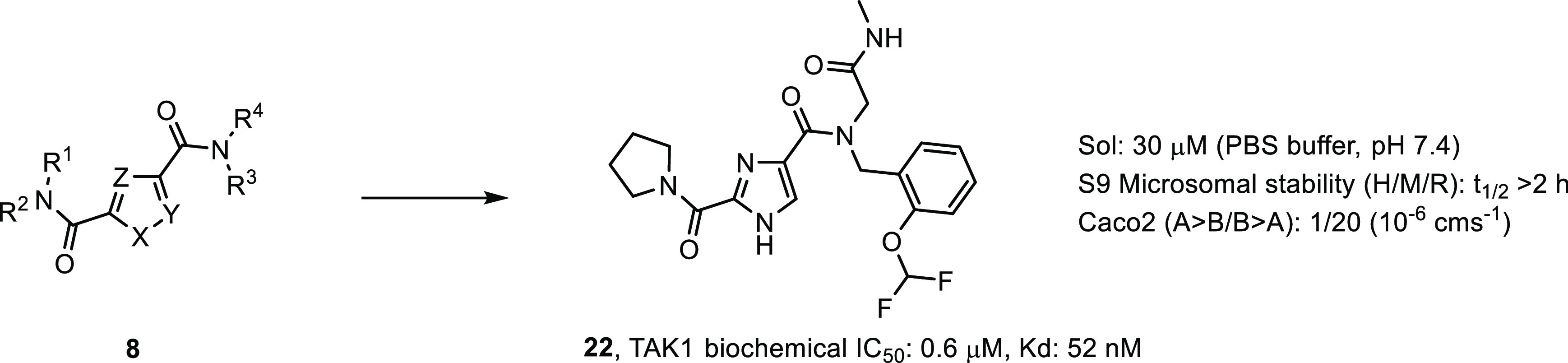
The primary hits were verified by synthesis of the label free analogs and subjected to a kinase inhibition assay. A LanthaScreen biochemical kinase inhibition assay using TAK1-TAB1 fusion protein in the presence of 10 μM ATP was used to assess the potency of the compounds. As a positive control Staurosporine was used and this had an IC50 value of 39 nM. To compare to known TAK1 inhibitors, the biochemical potency of compounds 2 and 3 was determined under these conditions. Compound 2 was found to have an IC50 value of 10 nM, which is lower than the 28 nM IC50 value reported,24 where an AlphaScreen biochemical assay was used in the presence of 100 μM ATP. Compound 3 had an IC50 value of 30 nM, where an IC50 value of 11 nM was reported26 using a radiometric biochemical kinase assay. In this assay, compound 7 (Figure 2) showed a biochemical IC50 value of 1.3 μM and it was used as a starting point for interrogating R1 to R4 and the pyrrole core in order to generate SAR (Table 1, compound 8). Although we had no structural information at this point, we assumed a type I binding mode, based on the binding modes described for compounds 2 and 3. We envisioned the pyrrolidine making hydrophobic interactions in the back of the pocket, the amide ketone and the pyrrole NH making hydrogen bond interactions with the hinge region of the kinase, and the N-methyl amide pointing to the solvent as this was the attachment point for the DNA tag. Indeed, structural changes at the DNA-linker region R4 (compounds 9–12) did not influence potency much, with IC50 values ranging from 0.9 to 3.5 μM. Some additional ortho-substituted benzyl amine derivatives (R3) were synthesized, but flat SAR was observed for compounds 13–16, indicating that there was some room for larger substituents at that position. Next, the pyrrolidine moiety was varied. Interestingly, the corresponding dimethyl analog 17 showed a 20-fold drop in potency, while the piperidine amide 18 retained significant potency, which suggested steric repulsion. The more polar primary amide 19 was completely inactive, possibly caused by hydrophobic repulsion. Apparently, this amide is involved in positioning key interactions with the target kinase. Finally, some changes to the core pyrrole were made. The pyrazole derivative 20 showed a large drop in potency. Methylation of the nitrogen gave derivative 21, which was inactive. Interestingly, imidazole 22 resulted in a 2-fold increase in potency. This steep SAR suggested that the core is of prime importance for inhibitory activity, rather than just being a scaffold with orientation function for the peripheral R-groups and would be consistent with the assumption it was making hydrogen bond interactions with the hinge of the kinase. As the imidazole showed promising potency, additional assay data were collected. The Kd was determined to be 55 nM (KdELECT, DiscoverX). The compound had a 30 μM solubility in PBS buffer and showed good metabolic stability in microsome assays. A Caco-2 assay showed low A to B permeability and an efflux ratio of 20. Most striking was the selectivity profile of imidazole 22. Upon screening at 10 μM in a 468 kinase panel containing 453 human and 403 wildtype kinases (KINOMEscan, DiscoverX), only 4 additional kinases were inhibited >65%: ABL1(H369P), EIF2AK1, TNK2, and YANK1. No other kinases in the TKL subfamily were inhibited. As we were expecting the selectivity to change during the optimization of the compound, we did not further investigate these off-targets. Assuming a type I binding mode, it is hard to explain this selectivity as the molecule is relatively small. At that point, 22 became our front-runner compound.
Table 1. In Vitro Biochemical Potency Data for Initial Derivatives of Compound 7.
| Compd | R1, R2N | R3 | R4 | X, Y, Z | TAK1 IC50a |
|---|---|---|---|---|---|
| 9 | 1-pyrrolidinyl | 2-(OCHF2)benzyl | 2-amino-2-oxoethyl | N, CH, CH | 0.9 |
| 10 | 1-pyrrolidinyl | 2-(OCHF2)benzyl | 2-(isopropylamino)-2-oxoethyl | N, CH, CH | 1.7 |
| 11 | 1-pyrrolidinyl | 2-(OCHF2)benzyl | oxazol-2-ylmethyl | N, CH, CH | 3.2 |
| 12 | 1-pyrrolidinyl | 2-(OCHF2)benzyl | methyl | N, CH, CH | 3.5 |
| 13 | 1-pyrrolidinyl | 2-(OMe)benzyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | 2.8 |
| 14 | 1-pyrrolidinyl | 2-(OEt)benzyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | 2.6 |
| 15 | 1-pyrrolidinyl | (3,4-dihydro-2H-benzo[b][1,4]dioxepin-6-yl)methyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | 3.4 |
| 16 | 1-pyrrolidinyl | (1H-indol-4-yl)methyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | 1.8 |
| 17 | N,N-dimethyl | 2-(OCHF2)benzyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | 26 |
| 18 | 1-piperidinyl | 2-(OCHF2)benzyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | 1.8 |
| 19 | N-ethyl | 2-(OCHF2)benzyl | 2-(methylamino)-2-oxoethyl | N, CH, CH | >100 |
| 20 | 1-pyrrolidinyl | 2-(OCHF2)benzyl | 2-(methylamino)-2-oxoethyl | N, N, CH | 59 |
| 21 | 1-pyrrolidinyl | 2-(OCHF2)benzyl | 2-(methylamino)-2-oxoethyl | NMe, CH, CH | >100 |
Biochemical Lanthascreen assay with TAK1–TAB1 fusion protein in the presence of 10 μM ATP, mean IC50 values in μM, n = 2.
As the imidazole core was readily accessible and easily amenable to synthetic derivatization, a targeted library approach was initiated to further explore the potential of compound 22 (Scheme 1). For this purpose, the diester 23 was synthesized on multigram scale as an advanced intermediate.32 The orthogonally protected esters conveniently allowed for stepwise derivatization and synthesis of a plethora of analogs. Two routes were devised toward the desired products. Basic hydrolysis of the ethyl ester, followed by pyrrolidine coupling and acid induced deprotection, afforded key building block 24. The carboxylic acid was then converted to amides 25 by coupling with an amine or via an Ugi multicomponent reaction (Route A). Alternatively, the tBu ester was deprotected and converted into the amide, followed by hydrolysis of the ethyl ester to afford intermediate 26. This was then converted into the amide to afford products 27 (Route B).
Scheme 1. Synthetic Routes (A, B) for the Synthesis of 2,4-1H-Imidazole Carboxamides.

Reagents and conditions: [i] LiOH, THF/H2O, RT, 16 h; [ii] pyrrolidine, HATU, DMF, RT, 16 h, 36% over two steps; [iii] formic acid, CHCl3, 2 h, reflux, 95%; [iv] amine, HATU, Et3N, DMF, RT, 16 h, 20–67%; [v] R3NH2, R5CHO, methylisocyanide, MeOH, RT, 16 h, 9–64%; [vi] formic acid, CHCl3, 2 h, reflux, 88%; [vii] 2-((2-(difluoromethoxy)benzyl)amino)-N-methylacetamide, HATU, Et3N, DMF, RT, 16 h, 77%; [viii] LiOH, THF/H2O, 40 °C, 16 h, 98%; [ix] R1R2NH, HATU, Et3N, DMF, RT, 16 h, 18–80%.
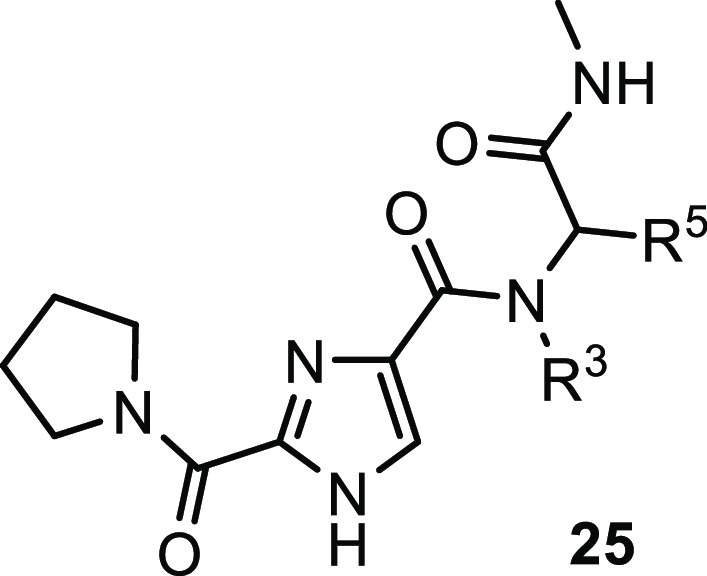
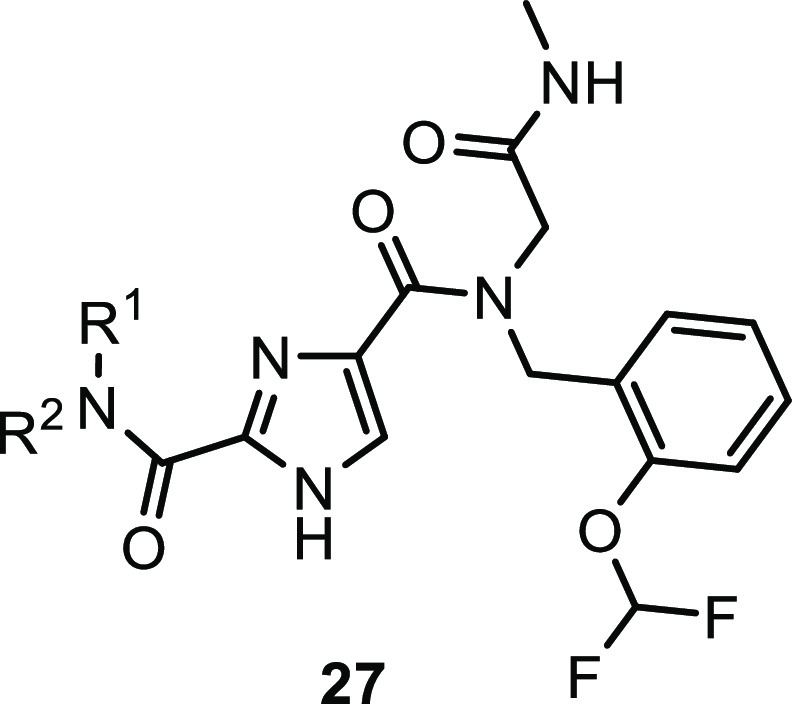
With the chemistry set up, a closer look was taken at the N-benzyl group (R3) and the glycine (R5) moiety (Table 2) and a small diversity set was made. Introduction of an α-methyl on the benzyl (28) abolished the activity, possibly caused by changing the orientation of the benzyl group. Removal of or shifting the OCHF2 group (29, 30) also rendered the compound to be inactive, indicating that this group makes an essential interaction. While keeping the 2-OCHF2 on the benzyl in place, additional substitution on the benzyl ring was investigated. It turned out the phenyl ring was sensitive toward further substitution. Introduction of a methoxy group on the 3- (31) or 4-postion (32) as well as fluorination or chlorination on the 5- (33, 34) or 6-position (35, 36) resulted in inactive compounds, suggesting steric occlusion. Next, substitution on the glycine carbon was investigated. Introduction of a methyl was allowed and the compound retained potency (37), while larger groups like an ethyl (38) or 2-propyl (39) resulted in loss of potency. The enantiomers of 37 were synthesized separately, and it was found that the R-enantiomer 40 was considerably more active than the S-enantiomer 41, effectively increasing the potency of compound 22 by a factor of 2. This difference was hard to explain without structural information, but suggested limited room at that position with the S-methyl making an unfavorable interaction. As the pyrrolidine amide was also found to be sensitive to changes in the first hit expansion campaign, this moiety was further probed. First, the ring size was investigated (Table 3). Change of the pyrrolidine for a smaller azetidine resulted in a 4-fold drop in potency (42). Ring expansion to a 6- (43) or 7-membered ring (44) resulted in a slight drop in potency, indicating limited room as was also observed for the pyrrole analogs 17 and 18. As the pyrrolidine 22 showed the best biochemical potency, substitution on the five-membered ring was probed. Introduction of a 2-methyl resulted in a large drop in potency (45) indicating steric congestion, but substitution on the 3-position of the pyrrolidine was tolerated. Lipophilic groups like fluoro, methyl, or phenyl groups retained potency (46–48), but more polar moieties like a methoxy (49) or an N-acetyl (50) resulted in loss of potency, which is consistent with the hydrophobic nature of the residues in the back part of the pocket. Interestingly, an isoindoline (51) yielded a 2-fold increase in potency, but the corresponding fully reduced form lost 10-fold in activity (52), again indicating steric congestion.
Table 2. In Vitro Potency Data for Compounds 28–41.
| Compd | R3 | R5 | TAK1 IC50a |
|---|---|---|---|
| 28 | α-methyl-2-(OCHF2)benzyl | H | >10 |
| 29 | benzyl | H | >10 |
| 30 | 3-(OCHF2)benzyl | H | >10 |
| 31 | 2-(OCHF2)-3-(OMe)benzyl | H | >10 |
| 32 | 2-(OHCF2)-4-(OMe)benzyl | H | >10 |
| 33 | 2-(OCHF2)-5-fluorobenzyl | H | >10 |
| 34 | 2-(OCHF2)-5-chlorobenzyl | H | >10 |
| 35 | 2-(OCHF2)-6-fluorobenzyl | H | >10 |
| 36 | 2-(OCHF2)-6-chlorobenzyl | H | >10 |
| 37 | 2-(OCHF2)benzyl | Me | 0.5 |
| 38 | 2-(OCHF2)benzyl | Et | 1.0 |
| 39 | 2-(OCHF2)benzyl | iPr | >10 |
| 40 | 2-(OCHF2)benzyl | R-Me | 0.3 |
| 41 | 2-(OCHF2)benzyl | S-Me | 4.5 |
Biochemical Lanthascreen assay with TAK1–TAB1 fusion protein in the presence of 10 μM ATP, mean IC50 values in μM, n = 2.
Table 3. In Vitro Potency Data for Compounds 42–52.
| Compd | R1R2N | TAK1 IC50a |
|---|---|---|
| 42 | 1-azetidinyl | 2.6 |
| 43 | 1-piperidinyl | 1.2 |
| 44 | 1-azepinyl | 2.9 |
| 45 | 1-(2-methylpyrrolidinyl) | 5.7 |
| 46 | 1-(3-methylpyrrolidinyl) | 0.9 |
| 47 | 1-(3-fluoropyrrolidinyl) | 1.2 |
| 48 | 1-(3-phenylpyrrolidinyl) | 2.3 |
| 49 | 1-(3-(OMe)-pyrrolidinyl) | >10 |
| 50 | 1-(3-(N-Ac)-pyrrolidinyl) | >10 |
| 51 | 1-isoindolinyl | 0.3 |
| 52 | 1-octahydro-1H-isoindolinyl | 3.4 |
Biochemical Lanthascreen assay with TAK1–TAB1 fusion protein in the presence of 10 μM ATP, mean IC50 values in μM, n = 2.
Having made only moderate gains in potency, further guidance for compound optimization needed to be obtained from structural information. At this point a crystal structure of compound 22 bound to TAK1 was acquired (Figure 3, top). The structure confirmed binding to the hinge in a type-I fashion and showed the imidazole and the carbonyl to bind as a donor–acceptor pair with the backbone functionalities of Ala107. As envisioned, the pyrrolidine is pointing to the back, and now the SAR observed for this moiety can be rationalized. There is no room for substitution of the 2-position, and it is clear why substitution at the 3-position is allowed, with more lipophilic residues (46–48, 51) retaining potency as this part of the pocket contains mainly aliphatic residues. The glycine carbonyl makes an interaction at the edge of the pocket with the amine hydrogen of Ser111. Finally, the orientation of the benzyl group is observed to be perpendicular to the imidazole core with the OCHF2 moiety nicely filling up a small lipophilic subpocket and the phenyl group making a lipophilic interaction. This explains the steep SAR observed for compounds 28–36 as removal of the OCHF2 or substitution of the phenyl would thwart these interactions. Additionally, the difference observed between the R- and S-enantiomers 40 and 41 can be explained. The R-methyl would fit, whereas the S-methyl would result in a steric clash with the protein. As the most striking observation, an overlay of the structure with some examples of crystal structures of other type-I TAK1 inhibitors showed this to be a unique binding mode for TAK1 (Figure 3, bottom). In the hinge region a peptide flip occurs between Glu108 and Gly109, thereby moving the backbone trace of the hinge when compared to the structures of other type-I inhibitors bound to TAK1. These conformational differences of the TAK1 protein induced by compound 22 potentially contribute to the observed selectivity in the kinase panel.33,34 With the structure in hand, a rational design campaign was enabled. For compounds 2 and 3, structures were reported and both papers stated a jump in potency was reached by targeting the Lys63-Asp175 pair. An overlay of these structures with the compound 22 structure (Figures 4 and S-1) suggested we should be able to target this interaction from the pyrrolidine. Docking indicated the isoindoline 51 to be a suitable platform for this and compound 53 was synthesized (Figure 5).
Figure 3.

(Top) Detailed view of compound 22 bound to the ATP binding site of TAK1. (Bottom) Overlay of several X-rays of TAK1 with type-I binding compounds (gray) (PDB ID codes: 2EVA, 4L53, 5JGD, and 5V5N) and compound 22 structure (green).
Figure 4.

Overlay of the structures of compound 22 (orange and green) with 2 (gray and cyan, PDB ID code: 4L53). Key lysine residue depicted.
Figure 5.

Compounds 53 and 54. a Biochemical Lanthascreen assay with TAK1–TAB1 fusion protein in the presence of 10 μM ATP, mean IC50 values in μM, n = 2.
Gratifyingly, a >100-fold jump in biochemical potency compared to 22 was obtained. Additionally, selectivity was maintained; in a screen at 1 μM in a 468 kinase panel, apart from TAK1, only 6 additional kinases were inhibited >65%: CSNK2A1, SCNK2A2, FLT3(D835 V), PFCDPK1, PIK3CD, and PIP5K1C. Interestingly, none of these kinases were inhibited by compound 22 and selectivity in the TKL family was retained. Finally, addition of the privileged R-methyl group onto the glycine resulted in compound 54, with a 2 nM TAK1 biochemical IC50 value, which marks the detection limit of the biochemical assay. A crystal structure was obtained for compound 54 that confirmed the binding mode (Figure 6). The isoindoline carboxamide showed a dual interaction with Lys63 as well as the Asp175. Again, the hinge was moved as was observed in the first structure. To assess the potency in a cellular context, TNF-α stimulated HCT-15 cells were treated with compound 54 to determine its effect on cell viability, but no effect was observed using concentrations up to 10 μM, whereas control compound 1 showed an EC50 value of 2 nM. Some additional ADME parameters were collected for 54. Compared to 22, the solubility of 54 had dropped 6-fold and introduction of additional substituents resulted in a decrease in metabolic stability as observed in the S9 microsome stability assay. Finally, the permeability was checked using a Caco-2 assay and A to B permeability had dropped to 0.1 × 10–6 cms–1, which could explain the results observed in the cell based assay.
Figure 6.

Detailed view of compound 54 bound to the ATP binding site of TAK1.
In conclusion, a DECL library hit against TAK1 was found and partially optimized by a library approach. Next, the binding mode was determined by X-ray and the potency was improved using SBDD. The resulting compound 54 has good biochemical potency and kinome selectivity, but the physchem properties need further optimization, which will be reported in due course.
Glossary
Abbreviations
- TAK1
transforming growth factor β-activated kinase 1
- DECL
DNA-encoded chemical library
- SBDD
structure based drug design
- TGF-β
transforming growth factor-β
- TNF-α
tumor necrosis factor alpha
- TLR
Toll-like receptor
- LPS
lipopolysaccharide
- IL-1
interleukin 1
- TAB
TAK1-binding protein
- MAP
mitogen-activated protein
- NFkB
nuclear factor-κB
- TRAIL
TNF-related apoptosis-inducing ligand
- ATP
adenosine triphosphate
- SAR
structure–activity relationship
- RT
room temperature
- HATU
(1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- Et3N
triethylamine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00547.
Experimental procedures for the synthesis of compounds 7–24, 26, and 28–54. A description of the biochemical, cell, solubility, S9 and permeability assays, DECL-screen, crystallization conditions, kinase panel data and Supplementary Figure S-1 (PDF)
The authors declare no competing financial interest.
Notes
The structures of TAK1:22 and TAK1:54 will be released upon publication.
Supplementary Material
References
- Arthur J. S. C.; Ley S. C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- Burotto M.; Chiou V. L.; Lee J.-M.; Kohn E. C. The MAPK pathway across different malignancies: a new perspective. Cancer 2014, 120, 3446–3456. 10.1002/cncr.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon A. S.; Hagan S.; Rath O.; Kolch W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K.; Shirakabe K.; Shibuya H.; Irie K.; Oishi I.; Ueno N.; Taniguchi T.; Nishida E.; Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 1995, 270, 2008–2011. 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Kilty I.; Jones L. H. TAK1 selective inhibition: state of the art and future opportunities. Future Med. Chem. 2015, 7, 23–33. 10.4155/fmc.14.138. [DOI] [PubMed] [Google Scholar]
- Sakurai H.; Shigemori N.; Hasegawa T.; Sugita T. TGF-beta-activated kinase 1 stimulates NF-kappa B activation by an NF-kappa B-inducing kinase-independent mechanism. Biochem. Biophys. Res. Commun. 1998, 243, 545–549. 10.1006/bbrc.1998.8124. [DOI] [PubMed] [Google Scholar]
- Shirakabe K.; Yamaguchi K.; Shibuya H.; Irie K.; Matsuda S.; Moriguchi T.; Gotoh Y.; Matsumoto K.; Nishida E. TAK1Mediates the Ceramide Signaling to Stress-activated Protein Kinase/c-Jun N-terminal Kinase. J. Biol. Chem. 1997, 272, 8141–8144. 10.1074/jbc.272.13.8141. [DOI] [PubMed] [Google Scholar]
- Sakurai H.; Miyoshi H.; Toriumi W.; Sugita T. Functional interactions of transforming growth factor beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB activation. J. Biol. Chem. 1999, 274, 10641–10648. 10.1074/jbc.274.15.10641. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J.; Kishimoto K.; Hiyama A.; Inoue J.; Cao Z.; Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999, 398, 252–256. 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- Adhikari A.; Xu M.; Chen Z. J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007, 26, 3214–3226. 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- Shibuya H.; Yamaguchi K.; Shirakabe K.; Tonegawa A.; Gotoh Y.; Ueno N.; Irie K.; Nishida E.; Matsumoto K. TAB1: An Activator of the TAK1MAPKKK in TGF-β Signal Transduction. Science 1996, 272, 1179–1182. 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- Takaesu G.; Kishida S.; Hiyama A.; Yamaguchi K.; Shibuya H.; Irie K.; Ninomiya-Tsuji J.; Matsumoto K. TAB2, a novel adaptor protein, mediates activation of TAK1MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 2000, 5, 649–658. 10.1016/S1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- Mihaly S. R.; Ninomiya-Tsuji J.; Morioka S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676. 10.1038/cdd.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M. S.; Ghosh S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Zhang W.; Shi M.; Yu Y.; Wang H.; Cao W.-M.; Zhao Y.; Zhang H. TAK1 inhibitor NG25 enhances doxorubicin-mediated apoptosis in breast cancer cells. Sci. Rep. 2016, 6, 32737. 10.1038/srep32737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo M.-K.; Kawasaki N.; Singhirunnusorn P.; Koizumi K.; Sato S.; Akira S.; Saiki I.; Sakurai H. Blockade of transforming growth factor-beta-activated kinase 1 activity enhances TRAIL-induced apoptosis through activation of a caspase cascade. Mol. Cancer Ther. 2006, 5, 2970–2976. 10.1158/1535-7163.MCT-06-0379. [DOI] [PubMed] [Google Scholar]
- Melisi D.; Xia Q.; Paradiso G.; Ling J.; Moccia T.; Carbone C.; Budillon A.; Abbruzzese J. L.; Chiao P. J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. 10.1093/jnci/djr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying L.; Chunxia Y.; Wei L. Inhibition of ovarian cancer cell growth by a novel TAK1 inhibitor LYTAK1. Cancer Chemother. Pharmacol. 2015, 76, 641–650. 10.1007/s00280-015-2822-8. [DOI] [PubMed] [Google Scholar]
- Zonneville J.; Wong V.; Limoge M.; Nikiforov M.; Bakin A. V. TAK1 signaling regulates p53 through a mechanism involving ribosomal stress. Sci. Rep. 2020, 10, 2517. 10.1038/s41598-020-59340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarneo S. A.; Mansourati A.; Eibschutz L. S.; Totzke J.; Roques J. R.; Loiselle D.; Carlson D.; Hughes P.; Haystead T. A. J. Genetic and pharmacological validation of TAK1 inhibition in macrophages as a therapeutic strategy to effectively inhibit TNF secretion. Sci. Rep. 2018, 8, 17058. 10.1038/s41598-018-35189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J.; Kajino T.; Ono K.; Ohtomo T.; Matsumoto M.; Shiina M.; Mihara M.; Tsuchiya M.; Matsumoto K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1MAPK kinase kinase. J. Biol. Chem. 2003, 278, 18485–18490. 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- Wu J.; Powell F.; Larsen N. A.; Lai Z.; Byth K. F.; Read J.; Gu R. F.; Roth M.; Toader D.; Saeh J. C.; Chen H. Mechanism and In Vitro Pharmacology of TAK1 Inhibition by (5Z)-7-Oxozeaenol. ACS Chem. Biol. 2013, 8, 643–650. 10.1021/cb3005897. [DOI] [PubMed] [Google Scholar]
- Hornberger K. R.; Chen X.; Crew A. P.; Kleinberg A.; Ma L.; Mulvihill M. J.; Wang J.; Wilde V. L.; Albertella M.; Bittner M.; Cooke A.; Kadhim S.; Kahler J.; Maresca P.; May E.; Meyn P.; Romashko D.; Tokar B.; Turton R. Discovery of 7-aminofuro[2,3-c]pyridine inhibitors of TAK1: optimization of kinase selectivity and pharmacokinetics. Bioorg. Med. Chem. Lett. 2013, 23, 4511–4516. 10.1016/j.bmcl.2013.06.054. [DOI] [PubMed] [Google Scholar]
- Hornberger K. R.; Berger D. M.; Crew A. P.; Dong H.; Kleinberg A.; Li A.-H.; Medeiros M. R.; Mulvihill M. J.; Siu K.; Tarrant J.; Wang J.; Weng F.; Wilde V. L.; Albertella M.; Bittner M.; Cooke A.; Gray M. J.; Maresca P.; May E.; Meyn P.; Peick W. Jr.; Romashko D.; Tanowitz M.; Tokar B. Discovery and optimization of 7-aminofuro[2,3-c]pyridine inhibitors of TAK1. Bioorg. Med. Chem. Lett. 2013, 23, 4517–4522. 10.1016/j.bmcl.2013.06.053. [DOI] [PubMed] [Google Scholar]
- Muraoka T.; Ide M.; Morikami K.; Irie M.; Nakamura M.; Miura T.; Kamikawa T.; Nishihara M.; Kashiwagi H. Discovery of a potent and highly selective transforming growth factor β receptor-associated kinase 1 (TAK1) inhibitor by structure based drug design (SBDD). Bioorg. Med. Chem. 2016, 24, 4206–4217. 10.1016/j.bmc.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Muraoka T.; Ide M.; Irie M.; Morikami K.; Miura T.; Nishihara M.; Kashiwagi H. Development of a Method for Converting a TAK1 Type I Inhibitor into a Type II or c-Helix-Out Inhibitor by Structure-Based Drug Design (SBDD). Chem. Pharm. Bull. 2016, 64, 1622–1629. 10.1248/cpb.c16-00606. [DOI] [PubMed] [Google Scholar]
- Buglio D.; Palakurthi S.; Byth K.; Vega F.; Toader D.; Saeh J.; Neelapu S. S.; Younes A. Essential role of TAK1 in regulating mantle cell lymphoma survival. Blood 2012, 120, 347–355. 10.1182/blood-2011-07-369397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totzke J.; Gurbani D.; Raphemot R.; Hughes P. F.; Bodoor K.; Carlson D. A.; Loiselle D. R.; Bera A. K.; Eibschutz L. S.; Perkins M. M.; Eubanks A. L.; Campbell P. M.; Fox D. A.; Westover K. D.; Haystead T. A. J.; Derbyshire E. R. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chemical Biology 2017, 24, 1029–1039. 10.1016/j.chembiol.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L.; Nomanbhoy T.; Gurbani D.; Patricelli M.; Hunter J.; Geng J.; Herhaus I.; Zhang J.; Pauls E.; Ham Y.; Choi H. G.; Xie T.; Deng X.; Buhrlage S. J.; Sim T.; Cohen P.; Sapkota G.; Westover K. D.; Gray N. S. Discovery of type II inhibitors of TGFβ-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase kinase kinase 2 (MAP4K2). J. Med. Chem. 2015, 58, 183–196. 10.1021/jm500480k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow R. A. Jr.; Dumelin C. A.; Keefe A. D. Dna-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discovery 2017, 16, 131–147. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Imidazo[1,2-A]indeno[1,2-E]pyrazine-2-carboxylic acid derivatives, preparation thereof and drugs containing same. US5726175 A1, 1998.
- Wrobleski S. T.; Lin S.; Murali Dhar T. G.; Dyckman A. J.; Li T.; Pitt S.; Zhang R.; Fan Y.; Doweyko A. M.; Tokarski J. S.; Kish K. F.; Kiefer S. E.; Sack J. S.; Newitt J. A.; Mark R.; Witmer M. R.; McKinnon M.; Barrish J. C.; Dodd J. H.; Schieven G. L.; Leftheris K. The identification of novel p38α isoform selective kinase inhibitors having an unprecedented p38α binding mode. Bioorg. Med. Chem. Lett. 2013, 23, 4120–4126. 10.1016/j.bmcl.2013.05.047. [DOI] [PubMed] [Google Scholar]
- Röhm S.; Berger B.-T.; Schröder M.; Chaikuad A.; Winkel R.; Hekking K. F. W.; Benningshof J. J. C.; Müller G.; Tesch R.; Kudolo M.; Forster M.; Laufer S.; Knapp S. Fast Iterative Synthetic Approach toward Identification of Novel Highly Selective p38 MAP Kinase Inhibitors. J. Med. Chem. 2019, 62, 10757–10782. 10.1021/acs.jmedchem.9b01227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.