

Abstract
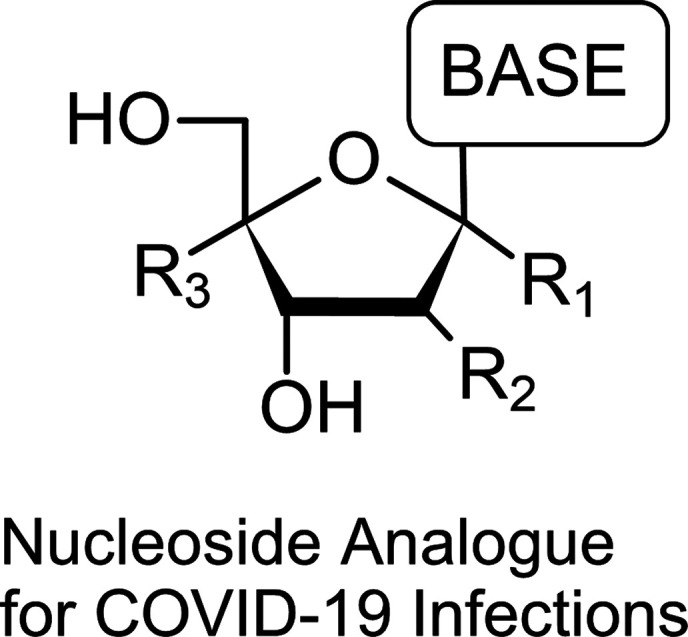
Great pioneers of nucleic acid chemistry had elucidated nucleic acid functions and structures and developed various antiviral modified nucleoside drugs. It is possible in theory that antiviral modified nucleosides prevent viral replication by inhibiting viral polymerases. However, biological phenomena far exceed our predictions at times. We describe the characteristics of the approved antiviral modified nucleosides from an organic chemistry perspective. Also, based on our experiences and findings through the development of the HIV-1 reverse-transcriptase inhibitor “Islatravir”, we provide the practical and approximate guidelines for the drug development of antiviral modified nucleosides against COVID-19.
Keywords: COVID-19, DNA virus, RNA virus, RNA polymerase inhibitor, antiviral modified nucleoside
The pandemic of the novel coronavirus infection (COVID-19) reminds us that “The merciless battle between humans and viruses never ends.” The discovery of viruses, especially RNA viruses, has had a significant impact on life sciences, forcing a substantial revision of the “Central Dogma,” a fundamental concept in molecular biology. The 2020 Nobel Prize in Physiology or Medicine was awarded to three virologists who discovered the hepatitis C virus (HCV). To confront the Global Virus Threat, many scientists struggle with research and the development of vaccines and antiviral drugs. Theoretically, antiviral modified nucleosides can prevent viral replication by inhibiting viral polymerases; in practice, though, things never work out as expected. In this Viewpoint, we describe the characteristics of the approved antiviral modified nucleosides on the basis of organic chemistry perspective. We refer to the possibility of the development of antiviral modified nucleosides against COVID-19. We hope that this Viewpoint will raise the researcher’s interest in the antiviral modified nucleosides.
Viruses that threaten humankind’s survival can be divided into DNA viruses and RNA viruses depending on their genomic nucleic acids.
DNA Virus
A DNA virus is a virus whose genome is stored in DNA, replicated, and proliferated by host (human) DNA polymerase. To give some examples, smallpox virus, varicella zoster virus (VZV), and herpes simplex virus (HSV) belong to this category. These viruses do not mutate because they use human DNA polymerase and benefit from its proof-reading capabilities. Since the smallpox virus is genetically stable and has few mutations, smallpox has been eradicated by a global vaccination program. Hepatitis B virus (HBV) is a unique DNA virus that utilizes human RNA polymerase to synthesize RNA from genomic DNA. It then uses the viral reverse transcriptase (RT) to replicate genomic DNA from the RNA. HBV is prone to mutation due to the nature of the viral nucleic acid polymerase (RT).
RNA Virus
Retroviruses use their own RTs to produce DNA from their RNA genomes. Thus, the viral DNA integrates into host DNA and forms a stable latent infection. Human Immunodeficiency Virus (HIV) belongs to them. On the other hand, many RNA viruses use viral RNA-dependent RNA polymerase to replicate and propagate genomic RNA. Coronavirus, influenza virus, Ebola virus, and hepatitis C virus (HCV) belong to this category. These viruses use viral RNA polymerase for replication and are therefore susceptible to mutations. (Replicative errors in DNA polymerases are suppressed to about once for every 108–1010 nucleotides by proof-reading, whereas RNA polymerases make mistakes at a rate of about 1 per every 10 000 nucleotides.)
Examples of Antiviral Modified Nucleoside Drugs for DNA Virus
Varicella-Zoster Virus (VZV) and Herpes Simplex Virus (HSV)
Acyclovir (ACV: 1)1 (Figure 1) and its analogues are considered silver bullets against herpes. As the name implies, acyclovir is acyclic and could be regarded as a 2′, 3′-dideoxyguanosine analogue.
Figure 1.

Chemical structure of acyclovir and guanosine.
1 is not phosphorylated by cellular thymidine kinase but is phosphorylated by viral thymidine kinase expressed in VZV-infected cells and further converted to the triphosphate by cellular phosphatases. The ACV-triphosphate is incorporated into the viral DNA instead of guanosine-5′-O-triphosphate. Therefore, human DNA polymerase is unable to elongate viral DNA fully. However, 1 does not undergo phosphorylation by human kinase and therefore is not toxic to uninfected cells.
The toxicity (side effect) of antiviral modified nucleoside drugs arises from the recognition of modified nucleoside triphosphate as a substrate by human nucleic acid polymerases.
Sorivudine (SRV: 3)2 (Figure 2), a synthetic analogue of thymidine, is approximately 2000–3000 times more potent than 1 against VZV and also shows activity against Epstein–Barr virus (EBV) for which there is no effective treatment. The 5′-OH of 3 is phosphorylated by the thymidine kinase of VZV. Therefore, 3 exhibits selective viral activity. 3 inhibits DNA polymerases as a 2′-deoxynucleoside derivative. Despite being a potent antiviral drug, 3 had significant drug interaction side effects when used with the common anticancer prodrug, 5FU (5). Phosphorolytic enzymes cleave the glycosidic bond of 3 to release 5-bromovinyluracil (4). At the same time, 3 loses its antiviral activity. 4 is an inhibitor of dihydrothymine dehydrogenase, the enzyme that catalyzes the hydrogenation of 5 (Figure 2), which is a highly toxic anticancer drug. Consequently, the plasma concentrations of 5 increase, causing severe side effects such as leukopenia and thrombocytopenia.
Figure 2.

Mechanism of lethal interactions between sorivudine and 5-fluorouracil.
In 1993, 15 cancer patients undergoing 5-FU chemotherapy died by the concomitant administration of SRV in Japan3,4 (Sorivudine Incident).
This case suggests that the glycosyl bond of nucleosides needs to be stable in vivo to prevent the loss of activity and the incident caused by the released base. It may also be necessary for the modified nucleosides that have no antiviral activity due to being not phosphorylated by human kinases to be re-examined the activity against VZV and HSV.
Hepatitis B Virus (HBV)
Infants are vaccinated to prevent HBV infection. RT inhibitors of HIV-1 are also used as anti-HBV drugs. The authors have developed a novel modified nucleoside analogue for the reverse-transcriptase inhibitor of HIV-1, EFdA (7)5 (Islatravir; details of this compound will be described later), which has an ethynyl group at the 4′-C-position (Figure 3). 7 exerts potent antiviral activity against HIV-1; however, it did not show the expected antiviral activity against HBV. Later, it was found that 2′-deoxynucleosides with a cyano group at the 4′-C-position showed good antiviral activity against HBV. It was also inferred that the ethynyl group at the 4′-C moiety is too large for the lipophilic pocket of the RT of HBV, but the cyano group at the 4′-C-position is just the right size to make the strong enzyme–substrate interactions.6
Figure 3.
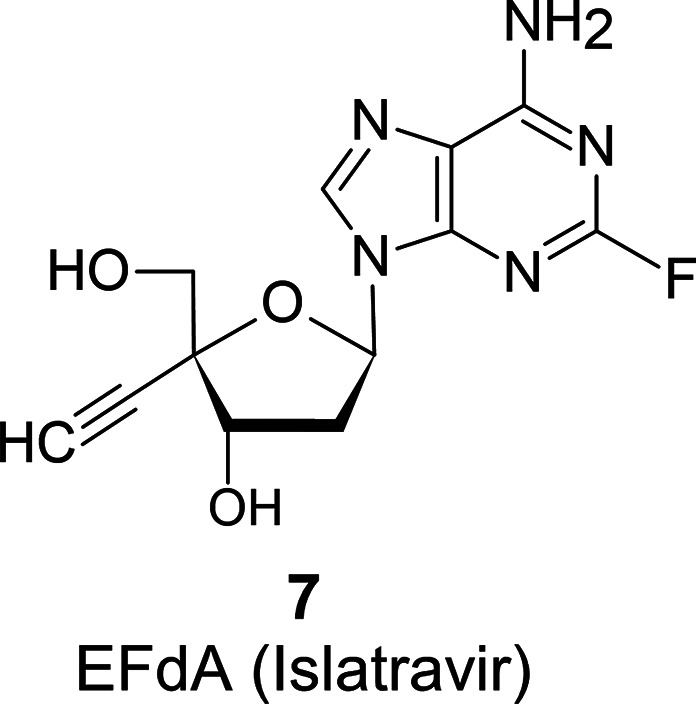
Chemical structure of EFdA (Islatravir).
It is reported that 4′-C-cyanoentecavir (8)7 and 4′-C-cyano-7-deaza-7-fluoro-2′-deoxyadenosine (9)8 have potent antiviral activity against HBV and prevent the emergence of resistant HBV strains (Figure 4).
Figure 4.

Anti-HBV-Nucleosides.
The above are examples of antiviral modified nucleoside drugs for DNA viruses.
Examples of Antiviral Modified Nucleoside Drugs for RNA Virus
Generally, the development of antiviral therapeutic agents for RNA viruses is considered difficult because RNA viruses have high mutation rates. However, the authors conceive that the mutation is the key to the creation of antiviral modified nucleosides. That is, the mutation is the process by which viruses alter their genes. Viral nucleic acid polymerases accept noncanonical nucleosides, which do not obey adenine-thymine and guanine-cytosine rules in canonical Watson–Crick base pairing, in place of normal nucleosides. This fact indicates that the substrate selectivity of viral nucleic acid polymerases is very lenient, and therefore, the viral nucleic acid polymerases will accept modified nucleosides.
Hepatitis C Virus (HCV)
The Nobel Prize in Medicine 2020 was awarded for the discovery of HCV. The discovery has led to the development of superior therapeutic agents such as sofosbuvir (10), of which nucleoside part was invented by late Dr. Kyoichi A. Watanabe.9−11 The authors expected that he would be the Nobel laureate for the development of 10 (Figure 5).
Figure 5.

Anti-HCV-nucleosides.
Conventionally, the combination of interferon and ribavirin (11) (Figure 5) has been used to treat chronic HCV infection. Still, it has significant therapeutic challenges because of adverse events due to long-term administration. With the advent of 10, chronic HCV infection treatment has been revolutionized. The combination of 10 /11 and the combination of 10 with the NS5A inhibitor, ledipasvir (HARVONI tablets), have few side effects. They are therapeutically more effective than the combination of interferon and 11. In particular, 10 has the efficacy of achieving almost 100% sustained virological response rates (SVR) against HCV. Furthermore, it does not allow the emergence of resistant HCV strains.
10 is the HCV NS5B RNA polymerase inhibitor. 2′-C-Methyladenosine (12)12 (Figure 5) was known as an antiviral modified nucleoside that inhibits HCV RNA polymerase; however, it was not a clinically applicable drug due to its strong side effects resulting from inhibition of human RNA polymerase.
The nucleosides that are chemically modified at any single position of physiological nucleosides have high viral activity. At the same time, they are highly toxic for clinical use because they are indistinguishable from the original physiologic nucleosides for human nucleic acid polymerases. Tubercidin (7-deaza-adenosine: 13),13 with a single modification, is also highly active antibiotics but highly toxic against humans.
Olsen’s group synthesized a hybrid nucleoside 14 of 12 and 13. They also synthesized compound 15, which is a further modification of compound 14. Olsen’s group also found that compound 14 has lower side effects and higher anti-HCV activity than 12 and that 15 is superior to 14(14) (Figure 6).
Figure 6.

Olsen’s anti-HCV nucleotide to reduce cell toxicity.
Toxicity (side effects) of modified nucleosides is drastically reduced when they are modified more. In some cases, the activity of the further modified nucleosides may be even higher than the original one.
For example, 4′-C-ethynyl d4T (Ed4T: 17),15 which is a further modification of the anti-HIV clinical drug d4T (16), is less toxic and more active than 16 and 7 is much less harmful and much more anti-HIV active than 4′-C-ethynyl-2′-deoxyadenosine (EdA: 18) (Figure 7).
Figure 7.

Examples of reducing toxicity of modified nucleosides.
10 is a 2′-α-fluoro-2′-C-methyl-2′-deoxyuridine. Since nucleic acid polymerases of eukaryotes such as humans and bacteria do not recognize the 2′-α-F moiety of nucleosides as a 2′-OH group, the 2′-α-fluoro-2′-deoxynucleoside was recognized as a 2′-deoxynucleoside. However, RNA polymerase of HCV utilizes 10 as a substrate, and therefore, there is a possibility that the other viral RNA polymerases recognize the fluorine moiety of a nucleoside as 2′-OH. Also, 10 does not inhibit human RNA polymerase because human RNA polymerases do not recognize the 2′-α-F moiety of a nucleoside as the 2′-OH group. This is the difference between human RNA polymerase and HCV RNA polymerase. Since the base of 10 is uracil, it is not recognized as a substrate for human DNA polymerase. Therefore, 10 tends to have fewer side effects. It is very intriguing whether other RNA viruses recognize the 2′-α-F moiety of a nucleoside as the 2′-OH group. 10 is an antiviral drug that utilizes the difference in recognizing 2′-α-F moiety between human RNA polymerase and HCV RNA polymerase. Therefore, it is engaging to investigate how the unknown nucleosides such as 4′-C-cyano-2′-fluoro-2′-deoxyuridine (19), 4′-C-cyano-2′,7-difluoro-2′-deoxy-7-deazaadenosine (20), and 4′-C-cyano-2,2′-difluoro-2′-deoxyadenosine (21) (Figure 8) show activity against human and virus polymerases.
Figure 8.

4′-C-Cyano substituted nucleosides.
If human kinases do not phosphorylate the 5′-OH groups of these nucleosides, they need to be chemically modified to nucleotide prodrug like 10.
Human Immunodeficiency Virus (HIV)
HIV is a retrovirus that uses RT to synthesize DNA from genomic RNA, incorporates the DNA into host DNA, and proliferates genomic RNA using human RNA polymerase. Unlike other treatments for viral infections, the treatment of HIV infection requires a lifetime anti-HIV medication as the viral DNA incorporated into human DNA cannot be removed. Therefore, the side effects of drugs are a more severe problem. 2′, 3′-Dideoxynucleosides (ddN), which inhibit RT (Figure 9), have been used as anti-HIV drugs. This is because the ddN structure is thought to be essential to be a chain terminator for RT. However, the problems are the rapid emergence of drug-resistant HIV variants to ddN drugs and the side effects by them, which are the chain terminators of DNA polymerase, as shown by the Sanger method for DNA sequencing.
Figure 9.

Representative ddNs in clinical use.
The authors predicted that the reason for the emergence of HIV drug-resistant mutant strains to ddN drugs was the ability of RT to discriminate ddN drugs from the physiological 2′-deoxynucleoside (dN: 25) and not to incorporate the ddN drugs into the active center of RT. Since the difference between ddN and 25 is whether they have 3′-OH, HIV can discriminate them by the 3′-OH. In other words, for a modified nucleoside drug to prevent the emergence of HIV drug-resistant mutant strains, it must have the 3′-OH group in the molecule to be misidentified as 25 by RT.
Furthermore, we figured that for the ddN nucleoside drugs with 3′-OH to be the chain-terminator of RT, a substituent should be introduced at the 4′-position of 25. The reason is that when a substituent is introduced at the 4′-position of 25, the 3′-OH becomes a neopentyl-type secondary hydroxyl group, which results in an extremely low reactive OH and would stop DNA chain synthesis. However, when a substituent at the 4′-position is introduced into 25, the 5′-OH becomes an unreactive neopentyl primary hydroxyl group, raising whether the kinase phosphorylates the 5′-OH. The side effects of modified nucleoside drugs are thought to occur because the triphosphates of them are recognized and incorporated as the substrates by human DNA polymerases. Therefore, we considered it necessary to modify the physiological nucleosides at two or more positions to prevent the modified nucleosides from being recognized as the substrates for human DNA polymerases. Furthermore, we expected that the introduction of a substituent at the 4′-position makes the glycosyl bond of the nucleosides less susceptible to the decomposition by acids and enzymes, thus improving the stability of the 4′-substituted nucleosides and the persistence of antiviral activity of the nucleosides in vivo.
Based on these working hypotheses, we designed a 4′-C-substituted-2′-deoxynucleoside (4′SdN: 26) as a RT inhibitor that might prevent the emergence of drug-resistant HIV strains and evaluated its biological activities (Figure 10).
Figure 10.
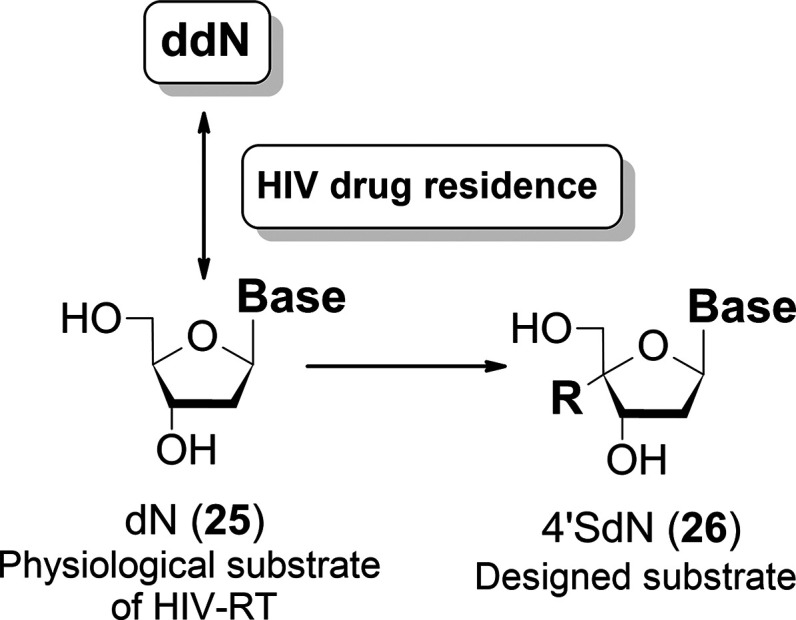
HIV drug resistance refers to the phenomenon of discrimination between ddN and dN and prevents ddN from being incorporated into the active center of RT. 4′SdN is a designed RT inhibitor to be recognized by human DNA polymerase.
The ribonucleosides with a substituent at the 4′-C-position showed no biological activity because 5′-OH is not phosphorylated by kinase. The 2′,3′-dideoxy (dd: 27), and 2′,3′-didehydrodideoxy (d4: 28) nucleosides with a substituent at the 4′-C-position generally showed much lower anti-HIV activity than the original 27 and 28, nucleosides respectively (Figure 11).
Figure 11.

Anti-HIV activities of 4′-C-substituted nucleosides.
We speculated that the reason for these results is that the neopentyl alcohol moiety at the 5′-position is difficult to be phosphorylated by the kinase, but, fortunately, the 5′-OH group of 26, which has 3′-OH group in the molecule, was phosphorylated and showed high anti-HIV activity. However, 26 with a natural base which is modified at one position of the physiologic nucleoside showed high toxicity.
In vivo mice studies showed the 2′-deoxyadenosine derivatives of 26 were deaminated by adenosine deaminase (ADA), but the precise toxicity could not be evaluated (data not shown). These findings indicated that ADA’s deamination of the 6-position of the purine base poses an essential issue in developing antiviral modified nucleosides. Montgomery and Hewson reported that the introduction of halogen at the 2-position of adenine makes it less susceptible to hydrolysis by adenosine deaminase due to the electronegativity of the halogens.16 Therefore, fluorine was introduced into the adenine at the 2-position of 18, and we finally developed 4′-C-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA: 7), which was modified at two positions of 2′-deoxyadenosine (Figure 3).
EFdA (7) has unique characteristics as shown below:
-
(1)
It does not allow the emergence of resistant HIV strains. This is because the 3′-OH group in the molecule prevents HIV from distinguishing it from physiological 2′-deoxyadenosine.
-
(2)
It is more than 400 times potent than AZT and several orders of magnitude more potent than other anti-HIV drugs.5 This is because it is firmly combined to RT by both the 4′-ethynyl group and 3′-OH to make it translocation defective RT inhibitor.
-
(3)
Due to a two-position modified nucleoside, it has very low toxicity.
-
(4)
It exhibits a long-acting anti-HIV activity because of the stability against both ADA and phosphorylase.
-
(5)
It is effective not only for the treatment of infection but also for prophylaxis.
The supremely high anti-HIV activity is ascribed to the fact that the ethynyl group at the 4′-position of 7 forms a strong bond with RT by fitting precisely into the previously unknown lipophilic pocket of HIV RT.17,18 The recent structural analysis of protein–ligand interactions unveiled that the 4′-ethynyl moiety of the EFdA-triphosphate has formed strong van der Waals interactions with both wild-type HIV and drug-resistant HIV strains in the active site cavity of RT.19
An efficient synthesis of 7 has been difficult due to the control of stereoselectivity.20 Schürmann et al. dramatically improved the stereoselective synthesis of 7 by developing a multistep enzymatic cascade reaction combining five engineered enzymes and four auxiliary enzymes, generating a single isomer.21
7 was named “Islatravir” by Merck, and the clinical trials began in 2013. A clinical study reported that single doses of 7 as low as 0.5 mg significantly suppressed HIV-1 plasma RNA for at least 7 days with tolerability.22 With regard to infection prevention, Merck announced a collaboration with the Bill & Melinda Gates Foundation to jointly conduct a Phase III Trial to evaluate 7 as once-monthly oral PrEP (pre-exposure prophylaxis) for women and adolescent girls in Africa (IMPOWER 22). This trial is aimed to end the HIV pandemic and eradicate it further.
COVID-19 (SARS-CoV-2)
Attention has been drawn to favipiravir (Avigan), remdesivir (Veklury), and morunupiavir, which are used for the treatment of COVID-19. We will discuss these therapeutic drugs.
Favipiravir (27)23 was developed by Toyama Chemical as a new type of anti-influenza drug, but it has a severe side effect of teratogenicity. According to Toyama Chemical, 27 is converted directly into ribonucleotide (28a) in the body, is further converted to trisphosphate (29), and inhibits viral RNA polymerase. Administration of the ribonucleoside of Favipiravir analogue (28b) has no activity because it is not phosphorylated by a kinase24 (Figure 12). The uptake of triphosphates of modified nucleosides by human nucleic acid polymerases mediates the side effects, and the teratogenicity of modified nucleosides is unknown so far. Hence, the teratogenicity will come from the Favipiravir itself before it is converted to nucleotides. Nucleosides (or nucleotides) of 27 are presumed to be quite unstable because they are formed by losing the aromaticity of 27. Therefore, the enzymatic reactions highly skew the chemical equilibrium between the 27 and 28a. This is probably why a high dose of 27 may be required for treatment.
Figure 12.

Chemical equilibrium between favipiravir and the favipiravir nucleotides.
The Favipiravir nucleotide prodrug (30) (Figure 12) will be a potential drug candidate with no teratogenic side effects and high antiviral activity.
Remdesivir (31)25 (Figure 13), a 1′-C-CN modified adenosine C-nucleoside, was initially developed for the treatment of the Ebola virus. This is the sole FDA-approved drug for the treatment of COVID-19. The CN group at the 1′-position seems to be the best substituent. This is because the CN may fit into a lipophilic pocket of COVID-19 coronavirus RNA polymerase.26
Figure 13.
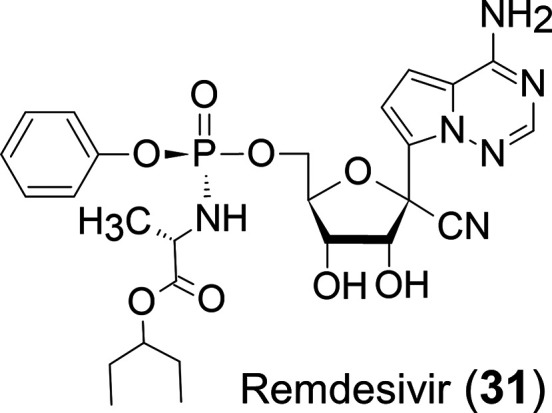
Chemical structure of remdesivir.
Reported side effects of 31 include liver dysfunction, diarrhea, skin rash, and renal dysfunction. 31 could cause more severe side effects, including multiple organ dysfunction syndromes (MODS), septic shock, acute kidney injury (AKI), and hypotension.27 These side effects would be acceptable for the treatment of lethal Ebola virus infection. However, the chemical structure will need to be improved to be used as a therapeutic agent for other viral infections.
In our experience, further modifications of the modified nucleoside have reduced toxicity and, in several cases, enhanced the antiviral activity of the compounds (Figure 7). Hence, 4′-C-cyanoremdesibir (32), a further modification of 31, may be a compound that attracts a great deal of attention. In addition, 4′-C-cyano-2′-deoxyremdesivir (33) and 4′-C-cyano-2′-fluoro-2′-deoxyremdesivir (34) (Figure 14) are also attractive modified nucleosides. Compound 33 is expected to reduce the side effects and enhance the antiviral activity of 31. Compound 33 is expected to have antiviral activity against HIV and HBV, and compound 34 is expected to be active against RNA viruses, including HCV. It is speculated that compounds 33 and 34 do not need to be prodrug nucleotides because human kinases would phosphorylate these nucleosides.
Figure 14.

Further modification of remdesivir.
Molnupiravir (MK-4482/EIDD-2801: 35),28 an oral anti-COVID-19 drug, is currently in a clinical trial with Merck (Figure 15).
Figure 15.
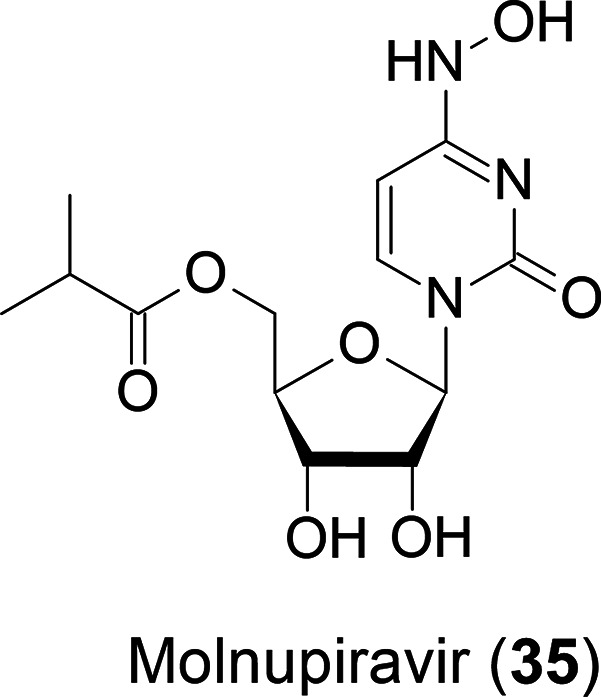
Chemical structure of molnupiravir.
This is a prodrug of N4-hydroxycytidine with an isobutyryl ester, and the active species is its 5′-O-trisphosphate. According to our experience, the nucleosides modified at any single position of physiological nucleosides may have high antiviral activity but severe side effects. Therefore, monomodified nucleosides may not be suitable for clinical agents. Hence, we are very interested in the efficacy and side effects of 35.
If 35 is found to have severe side effects and does not become a clinical drug, further modifications could be made to reduce the side effects. Therefore, it will be intriguing to investigate the efficacy of compounds 36–39 against COVID-19 (Figure 16).
Figure 16.

Further modification of molnupiravir.
Ideas are conceived in the research process. We hope that this Viewpoint inspires researchers on COVID-19 and better drugs can be developed by them as soon as possible.
Acknowledgments
The authors are grateful to Dr. Yoshio Hamada for preparing the manuscript.
Views expressed in this viewpoint are those of the author and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Elion G. B.; Furman P. A.; Fyfe J. A.; de Miranda P.; Beauchamp L.; Schaeffer H. J. Selectivity of action of an antiherpetic agent, 9- (2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. U. S. A. 1977, 74 (12), 5716–5720. 10.1073/pnas.74.12.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida H.; Nishitani M.; Ashida N. Effect of BV-araU and Acyclovir on Varicella-Zoster Virus Replication with Various Length and Timing of Drug Exposure. Microbiol. Immunol. 1994, 38 (2), 109–115. 10.1111/j.1348-0421.1994.tb01751.x. [DOI] [PubMed] [Google Scholar]
- Watabe T.; Ogura K.; Nishiyama T. Molecular Toxicological Mechanism of the Lethal Interactions of the New Antiviral Drug, Sorivudine, with 5 Fluorouracil Prodrugs and Genetic Deficiency of Dihydropyrimidine Dehydrogenase. Yakugaku Zasshi 2002, 122 (8), 527–535. 10.1248/yakushi.122.527. [DOI] [PubMed] [Google Scholar]
- Diasio R. B. Sorivudine and 5-fluorouracil; a clinically significant drug-drug interaction due to inhibition of dihydropyrimidine dehydrogenase. Br. J. Clin. Pharmacol. 1998, 46, 1–4. 10.1046/j.1365-2125.1998.00050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ohrui H. 2’-deoxy-4’-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem. Rec. 2006, 6, 133–143. 10.1002/tcr.20078. [DOI] [PubMed] [Google Scholar]; b Ohrui H. Development of modified nucleosides that have supremely high anti-HIV activity and low toexicity and prevent the emergence of resistant HIV mutants. Proc. Jpn. Acad., Ser. B 2011, B87, 53–65. 10.2183/pjab.87.53. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ohrui H. A New Paradigm for Developing Antiviral Drugs Exemplified by the development of Supremely High Anti-HIV active EFdA. Antivir Antiretrovir. 2014, 6, 32–39. 10.4172/jaa.1000092. [DOI] [Google Scholar]; d Ohrui H. Kagaku to Seibutsu 2017, 12, 833–840. 10.1271/kagakutoseibutsu.55.833. [DOI] [Google Scholar]
- Takamatsu Y.; Tanaka Y.; Kohgo S.; Murakami S.; Singh K.; Das D.; Venzon D. J.; Amano M.; Higashi-Kuwata N.; Aoki M.; Delino N. S.; Hayashi S.; Takahashi S.; Sukenaga Y.; Haraguchi K.; Sarafianos S. G.; Maeda K.; Mitsuya H. 4’-modified nucleoside analogs: Potent inhibitors active against entecavir-resistant hepatitis B virus. Hepatology 2015, 62 (4), 1024–1036. 10.1002/hep.27962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi-Kuwata N.; Hayashi S.; Das D.; Kohgo S.; Murakami S.; Hattori S.-i.; Imoto S.; Venzon D. J.; Singh K.; Sarafian’s S.; Tanaka Y.; Mitsuya H. CMCdG, a Novel Nucleoside Analog with Favorable Safety Features, Exerts Potent Activity against Wild-Type and Entecavir-Resistant Hepatitis B Virus. Antimicrob. Agents Chemother. 2019, 63 (4), e02143. 10.1128/AAC.02143-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S.; Higashi-Kuwata N.; Das D.; Tomaya K.; Yamada K.; Murakami S.; Venzon D. J.; Hattori S.-i.; Isogawa M.; Sarafianos S. G.; Mitsuya H.; Tanaka Y. 7-Deaza-7-fluoro modification confers on 4’-cyano-nucleosides potent activity against entecavir/adefovir-resistant HBV variants and favorable safety. Antiviral Res. 2020, 176, 104744. 10.1016/j.antiviral.2020.104744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. L.; Hollecker L.; Mason J. C.; Stuyver L. J.; Tharnish P. M.; Lostia S.; McBrayer T. R.; Schinazi R. F.; Watanabe K. A.; Otto M. J.; Furman P. A.; Stec W. J.; Patterson S. E.; Pankiewicz K. W. Design, synthesis, and antiviral activity of 2’-deoxy-2’-fluoro-2’-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 2005, 48, 5504–5508. 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- Shi J.; Du J.; Ma T.; Pankiewicz K. W.; Patterson S. E.; Tharnish P. M.; McBrayer T. R.; Stuyver L. J.; Otto M. J.; Chu C. K. Synthesis and anti-viral activity of a series of d- and l-2’-deoxy-2’-fluororibonucleosides in the subgenomic HCV replicon system. Bioorg. Med. Chem. 2005, 13, 1641–1652. 10.1016/j.bmc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Patterson S. E. Dedication to Kyoichi A. Watanabe. Heterocycl. Commun. 2015, 21 (5), 245–248. 10.1515/hc-2015-0206. [DOI] [Google Scholar]
- Carroll S. S.; Tomassini J. E.; Bosserman M.; Getty K.; Stahlhut M. W.; Eldrup A. B.; Bhat B.; Hall D.; Simcoe A. L.; LaFemina R.; Rutkowski C. A.; Wolanski B.; Yang Z.; Migliaccio G.; De Francesco R.; Kuo L. C.; MacCoss M.; Olsen D. B. Olsen, Inhibition of Hepatitis C Virus RNA Replication by 2′-Modified Nucleoside Analogs. J. Biol. Chem. 2003, 278 (14), 11979–11984. 10.1074/jbc.M210914200. [DOI] [PubMed] [Google Scholar]
- Olsen D. B.; Eldrup A. B.; Bartholomew L.; Bhat B.; Bosserman M. R.; Ceccacci A.; Colwell L. F.; Fay J. F.; Flores O. A.; Getty K. L.; Grobler J. A.; LaFemina R. L.; Markel E. J.; Migliaccio G.; Prhavc M.; Stahlhut M. W.; Tomassini J. E.; MacCoss M.; Hazuda D. J.; Carroll S. S. A 7-Deaza-Adenosine Analog Is a Potent and Selective Inhibitor of Hepatitis C Virus Replication with Excellent Pharmacokinetic Properties. Antimicrob. Agents Chemother. 2004, 48 (10), 3944–3953. 10.1128/AAC.48.10.3944-3953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldrup A. B.; Prhavc M.; Brooks J.; Bhat B.; Prakash T. P.; Song Q.; Bera S.; Bhat N.; Dande P.; Cook P. D.; Bennett C. F.; Carroll S. S.; Ball R. G.; Bosserman M.; Burlein C.; Colwell L. F.; Fay J. F.; Flores O. A.; Getty K.; LaFemina R. L.; Leone J.; MacCoss M.; McMasters D. R.; Tomassini J. E.; von Langen D.; Wolanski B.; Olsen D. B. Structure–Activity Relationship of Heterobase-Modified 2‘-C-Methyl Ribonucleosides as Inhibitors of Hepatitis C Virus RNA Replication. J. Med. Chem. 2004, 47 (21), 5284–5297. 10.1021/jm040068f. [DOI] [PubMed] [Google Scholar]
- Haraguchi K.; Takeda S.; Tanaka H.; Nitanda T.; Baba M.; Dutschman G.E.; Cheng Y.-C. Synthesis of a highly active new anti-HIV agent 2′,3′-Didehydro-3′-deoxy-4′-ethynylthymidine. Bioorg. Med. Chem. Lett. 2003, 13 (21), 3775–3777. 10.1016/j.bmcl.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Montgomery J.; Hewson K. Nucleosides of 2-Fluoroadenine. J. Med. Chem. 1969, 12 (3), 498–504. 10.1021/jm00303a605. [DOI] [PubMed] [Google Scholar]
- Yang G.; Wang J.; Cheng Y.; Dutschman G. E.; Tanaka H.; Baba M.; Cheng Y.-C. Mechanism of Inhibition of Human Immunodeficiency Virus Type 1 Reverse Transcriptase by a Stavudine Analogue, 4’-Ethynyl Stavudine Triphosphate. Antimicrob. Agents Chemother. 2008, 52 (6), 2035–2042. 10.1128/AAC.00083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidis E.; Marchand B.; Kodama E. N.; Singh K.; Matsuoka M.; Kirby K. A.; Ryan E. M.; Sawani A. M.; Nagy E.; Ashida N.; Mitsuya H.; Parniak M. A.; Sarafianos S. G. Sarafianos, Mechanism of Inhibition of HIV-1 Reverse Transcriptase by 4’-Ethynyl-2-fluoro-2’-deoxyadenosine Triphosphate, a Translocation-defective Reverse Transcriptase Inhibitor. J. Biol. Chem. 2009, 284 (51), 35681–35691. 10.1074/jbc.M109.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu Y.; Das D.; Kohgo S.; Hayashi H.; Delino N. S.; Sarafianos S. G.; Mitsuya H.; Maeda K. The High Genetic Barrier of EFdA/MK-8591 Stems from Strong Interactions with the Active Site of Drug-Resistant HIV-1 Reverse Transcriptase. Cell Chemical Biology 2018, 25, 1268–1278.e3. 10.1016/j.chembiol.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemical synthesis of EFdA.; a Fukuyama K.; Ohrui H.; Kuwahara S. Synthesis of EFdA via a diastereoselective aldol reaction of a protected 3-keto furanose. Org. Lett. 2015, 17 (4), 828–831. 10.1021/ol5036535. [DOI] [PubMed] [Google Scholar]; b Nawrat C. C.; Whittaker A. M.; Huffman M. A.; McLaughlin M.; Cohen R. D.; Andreani T.; Ding B.; Li H.; Weisel M.; Tschaen D. M. Tschaen, Nine-Step Stereoselective Synthesis of Islatravir from Deoxyribose. Org. Lett. 2020, 22 (6), 2167–2172. 10.1021/acs.orglett.0c00239. [DOI] [PubMed] [Google Scholar]
- Huffman M. A.; Fryszkowska A.; Alvizo O.; Borra-Garske M.; Campos K. R.; Canada K. A.; Devine P. N.; Duan D.; Forstater J. H.; Grosser S. T.; Halsey H. M.; Hughes G. J.; Jo J.; Joyce L. A.; Kolev J. N.; Liang J.; Maloney K. M.; Mann B. F.; Marshall N. M.; McLaughlin M.; Moore J. C.; Murphy G. S.; Nawrat C. C.; Nazor J.; Novick S.; Patel N. R.; Rodriguez-Granillo A.; Robaire S. A.; Sherer E. C.; Truppo M. D.; Whittaker A. M.; Verma D.; Xiao L.; Xu Y.; Yang H. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366 (6470), 1255–1259. 10.1126/science.aay8484. [DOI] [PubMed] [Google Scholar]
- Schürmann D.; Rudd D. J.; Zhang S.; De Lepeleire I.; Robberechts M.; Friedman E.; Keicher C.; Huser A.; Hofmann J.; Grobler J. A; Stoch S A.; Iwamoto M.; Matthews R. P Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK-8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single-dose administration to treatment-naive adults infected with HIV-1: an open-label, phase 1b, consecutive-panel trial. Lancet HIV 2020, 7 (3), e164–e172. 10.1016/S2352-3018(19)30372-8. [DOI] [PubMed] [Google Scholar]
- a Kiso M.; Takahashi K.; Sakai-Tagawa Y.; Shinya K.; Sakabe S.; Le Q. M.; Ozawa M.; Furuta Y.; Kawaoka Y. T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (2), 882–887. 10.1073/pnas.0909603107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Furuta Y.; Gowen B. B.; Takahashi K.; Shiraki K.; Smee D. F.; Barnard D. L. Barnard, T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Antiviral Res. 2013, 100 (2), 446–54. 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y.; Takahashi K.; Kuno-Maekawa M.; Sangawa H.; Uehara S.; Kozaki K.; Nomura N.; Egawa H.; Shiraki K. Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 2005, 49 (3), 981–986. 10.1128/AAC.49.3.981-986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel D.; Hui H. C.; Doerffler E.; Clarke M. O.; Chun K.; Zhang L.; Neville S.; Carra E.; Lew W.; Ross B.; Wang Q.; Wolfe L.; Jordan R.; Soloveva V.; Knox J.; Perry J.; Perron M.; Stray K. M.; Barauskas O.; Feng J. Y.; Xu Y.; Lee G.; Rheingold A. L.; Ray A. S.; Bannister R.; Strickley R.; Swaminathan S.; Lee W. A.; Bavari S.; Cihlar T.; Lo M. K.; Warren T. K.; Mackman R. L. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60 (5), 1648–1661. 10.1021/acs.jmedchem.6b01594. [DOI] [PubMed] [Google Scholar]
- A private letter from Dr. Tomas Cihlar, Gilead Sciences.
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; Lopez de Castilla D.; Finberg R. W.; Dierberg K.; Tapson V.; Hsieh L.; Patterson T. F.; Paredes R.; Sweeney D. A.; Short W. R.; Touloumi G.; Lye D. C.; Ohmagari N.; Oh M.-d.; Ruiz-Palacios G. M.; Benfield T.; Fatkenheuer G.; Kortepeter M. G.; Atmar R. L.; Creech C. B.; Lundgren J.; Babiker A. G.; Pett S.; Neaton J. D.; Burgess T. H.; Bonnett T.; Green M.; Makowski M.; Osinusi A.; Nayak S.; Lane H. C. Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 2020, 383 (19), 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R. M.; Wolf J. D.; Plemper R. K. Plumper, Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nature Microbiology 2021, 6 (1), 11–18. 10.1038/s41564-020-00835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]