

Abstract

The overactivation of transient receptor potential canonical 3 (TRPC3) is associated with neurodegenerative diseases and hypertension. Pyrazole 3 (Pyr3) is reported as the most selective TRPC3 inhibitor, but it has two inherent structural limitations: (1) the labile ester moiety leads to its rapid hydrolysis to the inactive Pyr8 in vivo, and (2) the alkylating trichloroacrylic amide moiety is known to be toxic. To circumvent these limitations, we designed a series of conformationally restricted Pyr3 analogues and reported that compound 20 maintains high potency and selectivity for human TRPC3 over its closely related TRP channels. It has significantly improved metabolic stability compared with Pyr3 and has a good safety profile. Preliminary evaluation of 20 demonstrated its ability to rescue Aβ-induced neuron damage with similar potency to that of Pyr3 in vitro. Collectively, these results suggest that 20 represents a promising scaffold to potentially ameliorate the symptoms associated with TRPC3-mediated neurological and cardiovascular disorders.
Keywords: transient receptor potential canonical 3, pyrazole 3, selective inhibitor, high metabolic stability, low toxicity
Transient receptor potential canonical (TRPC) channels are ubiquitously expressed in vertebrate cells.1 The TRPC channel subfamily consists of seven members designated as TRPC1–7. Except for pseudogene TRPC2, the other channels are further divided into two groups, TRPC1/4/5 and TRPC3/6/7, based on amino acid sequence homology and functional similarities.2 As nonselective cationic channels, they control the influx of Ca2+ and other cations like Na+ in response to activation of phospholipase C-coupled plasma membrane receptors and thus play critical roles in the regulation of intracellular Ca2+ concentration by hormones and growth factors.3,4 Among the TRPC family, TRPC3 plays a prominent functional role in basic cellular responses including proliferation, differentiation, and death in response to various environmental stimuli,5−7 implying a variety of diverse biological functions.3 TRPC3 is the most abundant TRPC channel in the brain.8 Recent studies have suggested that TRPC3 channels are critical for the signaling cascade of brain-derived neurotrophic factors,9,10 which has been postulated as a critical contributor to Alzheimer’s disease.11 TRPC3 is also expressed in cardiomyocytes, and its overexpression has been found to be involved in adverse stress responses, hypertrophy, and heart failure.12 In the immune system, it has been suggested to contribute to the restoration of Ca2+ influx and activation of T-cells, facilitating the response to antigen stimulation.13,14 Additionally, TRPC3 is involved in the proliferation and migration of a variety of tumor cells including melanoma, lung, and breast cancers.15−17
Current understanding of the role of TRPC3 in pathological studies suggests that selective TRPC3 channel inhibitors may be suitable for the treatment of diseases.15,18 Over the past decade, several TRPC3 inhibitors have been reported and are summarized in Figure 1. Among these inhibitors, Pyr3 is the most selective for TRPC3 inhibition, with a reported IC50 value of 0.7 μM, since most of the other inhibitors possess comparable or higher potency against both TRPC3 and TRPC6.19 Pyr3 has been reported in several pharmacological studies for the treatment of TRPC3-related diseases such as cardiac hypertrophy19 and smooth muscle proliferation.18
Figure 1.

Chemical structures of reported TRPC3 inhibitors.
However, Pyr3 has two structural liabilities. First, while structure–activity relationship (SAR) studies showed that the trichloroacrylic amide group in Pyr3 (tail moiety in Figure 2) is critical for its TRPC3 inhibition specificity,20 it is a reactive group with high toxic liabilities. Second, the ester head moiety in Pyr3 can be quickly hydrolyzed in vivo, leading to the inactive acid derivative Pyr8.21 To circumvent these two structural limitations, we hypothesized that incorporating the tail group in a conformationally restricted ring will significantly reduce the alkylating toxicity, and isosterically replacing the ester with a stable linker will improve the metabolic stability. Toward this direction, we designed and synthesized a series of modified pyrazoles as illustrated in Figure 2. Among these molecules, compound 20 shows a significantly improved metabolic stability and safety profile, while maintaining high potency and selectivity for TRPC3 inhibition similar to that of Pyr3. Preliminary evaluation of compound 20 demonstrated its ability to rescue Aβ-induced neuron damages with similar potency to Pyr3.
Figure 2.
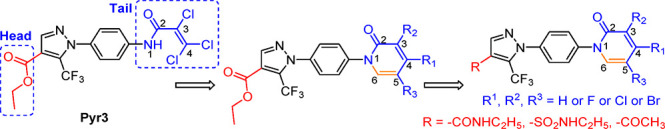
Design of metabolically stable and low toxic selective TRPC3 inhibitors by addressing structural liabilities in Pyr3.
As the first step of modification, we only replaced the tail moiety with a 6-membered pyridone ring while keeping the rest of the Pyr3 structure intact, to determine whether restricting the conformation in the tail region still maintains TRPC3 inhibition activity at all. To synthesize compounds 2–8, we started from the condensation of ethyl-2-(ethoxymethylene)-4,4,4-trifluoro-3-oxobutanoate and 4-bromophenylhydrazine hydrochloride to produce the pyrazole intermediate 1. Coupling of 1 with 4-chloropyridone or pyridone afforded 2 or 3, respectively (Scheme 1a). Treatment of 2 with N-chlorosuccinimide (NCS) gave the multichlorinated compounds 4–6 (Scheme 1b). Finally, 3,5-dichlorinated 7 and 5-chlorinated 8 were obtained using copper catalyst mediated coupling reactions as shown in Scheme 1c.
Scheme 1. Synthesis of Ester Compounds 2–8 with Different Chlorine Substitution Patterns.

Reagents and conditions: (i) EtOH, 60 °C, 6 h; (ii) CuI, DMCDA, K2CO3, toluene, reflux, 12 h; (iii) NCS, DMF, 100 °C, 8 h.
To make compounds 9 and 10, fluorine or dibromine was incorporated to compound 2 by the treatment with selectfluor or N-bromosuccinimide, respectively (Scheme 2a and b). The corresponding carboxylic acids 11–16 were obtained through hydrolysis of the parent ester compounds before they were converted to their corresponding amides 17–22 (Scheme 2c). Other compounds 27, 32, 37, and 38 were synthesized by following the procedures shown in Supporting Information (Schemes S1–S3). All compounds have purities ≥ 95% based on analytical chemistry characterization (Supporting Information).
Scheme 2. Synthesis of Amide Compounds 17–22.

Reagents and conditions: (i) selectfluor, MeCN, 50 °C, 3 h; (ii) NBS, DMF, r.t., 8 h. (iii) 1 M KOH (aq.), EtOH, H2O, rt, 8 h; (iv) SOCl2, CH2Cl2, reflux for 4 h; (v) ethylamine, TEA, r.t., 12 h.
We first evaluated the ability of the Pyr3 analogues to inhibit TRPC3 using the patch-clamp technique in the whole-cell configuration (Figure 3a). We activated human TRPC3 (hTRPC3) with a specific agonist, GSK1702934A (GSK170), in HEK293 cells and determined the inhibitory effect of each compound at 10 μM concentration in the presence of the agonist (Figures 3b and S1a). We recorded TRPC3 currents in the absence of Ca2+ to evaluate compound inhibition with a minimal contribution of Ca2+-desensitized channels. As expected, Pyr3 inhibited 91 ± 4% (mean ± SD) of hTRPC3 currents. Notably, compound 20 was as efficient as Pyr3 inhibiting hTRPC3 (89 ± 3%), followed by compounds 17 (84 ± 2%) and 2 (71 ± 5%). Further modification of compound 20 by replacing 3-chlorine with 3-fluorine to produce 21 or replacing the amide by a sulfonamide moiety to provide 32 failed to further improve the inhibitory potency. Since compound 20 best mimics the structure of Pyr3 (the C5 carbon in compound 20 occupies the position for one of the chlorines at C4 in Pyr3), the overall SAR for this new scaffold is largely consistent with that of the Pyr3 scaffold.20 It is also interesting to note that the chlorine at C4 in the compound 20 scaffold plays a more important role than the chlorine at C3 or C5, since analogues lacking this C4 chlorine (e.g., 3, 7, and 8) are significantly less potent. As an added benefit, the amide analogues which are predicted to be more metabolically stable also showed higher potency than the corresponding ester analogues (Figure 3b).
Figure 3.

Inhibition of TRPC3-mediated currents by synthesized compounds. (a) Cartoon depicting the experimental conditions used to test the effect of the synthesized compounds on HEK293 cells overexpressing hTRPC3. TRPC3 currents were measured in the whole-cell configuration of the patch-clamp technique, and cells were challenged with agonists/antagonists through perfusion. (b) Percentage of hTRPC3 currents elicited by GSK170 and inhibited by the synthesized compounds in transfected HEK293 cells. Bars represent mean ± SD; n is denoted above the x-axis. (c) Representative traces of hTRPC3 currents activated by 1 μM GSK170 and inhibited at different concentrations of compound 20 (blue traces). Each concentration was tested on independent cells and normalized against its internal control (maximum amplitude) to avoid tachyphylaxis. (d) Dose–response profile of hTRPC3 currents elicited by 1 μM GSK170 and inhibited at different concentrations of compound 20. Circles represent mean ± SD. (e) Representative traces of hTRPC3 currents activated by 1 μM GSK170 and inhibited at different concentrations of Pyr3 (gray traces). Each concentration was tested on independent cells and normalized against its internal control (maximum amplitude) to avoid tachyphylaxis. (f) Dose–response profile of hTRPC3 currents elicited by 1 μM GSK170 and inhibited at different concentrations of Pyr3. Circles represent mean ± SD. (g) Representative traces of hTRPC3 currents activated by 1 μM GSK170 and inhibited at different concentrations of compound 20 (blue traces) in the presence of extracellular Ca2+ (2 mM). Each concentration was tested on independent cells and normalized against its internal control (maximum amplitude) to avoid tachyphylaxis. (h) Dose–response profile of hTRPC3 currents elicited by 1 μM GSK170 and inhibited at different concentrations of compound 20 in the presence of extracellular Ca2+ (2 mM). Circles represent mean ± SD. (i) Representative traces of hTRPC3 currents activated by 1 μM GSK170 and inhibited at different concentrations of Pyr3 (gray traces) in the presence of extracellular Ca2+ (2 mM). Each concentration was tested on independent cells and normalized against its internal control (maximum amplitude) to avoid tachyphylaxis. (j) Dose–response profile of hTRPC3 currents elicited by 1 μM GSK170 and inhibited at different concentrations of Pyr3 in the presence of extracellular Ca2+ (2 mM). Circles represent mean ± SD. GSK170: GSK1702934A.
Since compound 17 had a slower time course of inhibition than those of Pyr3 and compound 20 (Figure S1b and c), we focused our subsequent studies using compound 20. We determined the IC50 of compound 20 for hTRPC3 inhibition in the absence of extracellular Ca2+ as 0.37 ± 0.03 μM (mean ± SD), which is slightly lower than that of Pyr3 (0.53 ± 0.05 μM, Figure 3c–f). We also compared their inhibition potency in the presence of extracellular Ca2+ (Figures 3g–j and S2e,f). As expected, 20 showed a comparable potency with Pyr3 at a 2 mM Ca2+ concentration. To further confirm the inhibitory activity of compound 20, we performed a separate experiment with a commonly used TRPC3 agonist, 1-oleoyl-2-acetyl-sn-glycerol (OAG, a membrane-permeable analogue of the endogenous agonist diacylglycerol). Compound 20 and Pyr3 showed similar inhibitory effects and a similar trend in the dose–response profile compared to that of GSK170 (Figures S1d–f and S2a−d). We next determined the ability of compound 20 to inhibit members of the TRP channel superfamily hTRPA1, rat TRPV1 (rTRPV1), rTRPV4, hTRPC3, mouse TRPC6 (mm TRPC6), hTRPC7, or hTRPM8 overexpressed in HEK293 cells (Figure 4a). The experiments were performed using solutions without Ca2+ to avoid TRP channel desensitization.22 After fully activating these channels with their respective agonists, 10 μM of compound 20 was perfused together with the agonists. At this saturating concentration (10 μM), compound 20 failed to inhibit hTRPA1, rTRPV1, rTRPV4, and hTRPM8 currents (Figure 4a and 4b). On the other hand, compound 20 inhibited approximately 39 ± 5% of hTRPC6 and 26 ± 1% of hTRPC7 (Figure 4a and 4b), which are closely related to TRPC3.
Figure 4.

Compound 20 is a selective inhibitor of the TRPC subfamily. (a) Representative whole-cell recordings of HEK293 cells overexpressing hTRPA1, rTRPV1, rTRPV4, hTRPC3, mmTRPC6, hTRPC7, and hTRPM8. Currents were evoked with high concentrations of the agonist (red) and challenged with 20 (10 μM) in the presence of the respective agonist (blue). (b) Percentage of peak current blocked by 20 (10 μM) in the presence of each agonist. AITC, Allyl isothiocyanate; Cap, capsaicin; GSK101, GSK1016790A. Bars are mean ± SD. n is denoted above the x-axis.
Several studies found that Pyr3 inhibits Ca2+ release-activated Ca2+ channel 1 (Orai1) with comparable blocking potency.23 Similarly, we found that compound 20 inhibits STIM/Orai mediated Ca2+ influx as Pyr3 when activated with thapsigargin (Figure S3). Future modifications could be introduced to compound 20 to discriminate between TRPC3 and STIM/Orai channels. Despite the side effect of pyrazoles at STIM/Orai channels, our results demonstrate that compound 20 is a potent and selective inhibitor of hTRPC3 against other members of the TRP channel superfamily.
To preliminarily confirm the direct on-target engagement of compound 20 with hTRPC3, we performed LC-MS analyses. After incubation of compound 20 with the hTRPC3 protein2 for 1 h, we performed repeated size-exclusion chromatography to completely remove free compound 20 from the protein-bound fraction. Methanol was then added to this complex to denature the protein and extract the bound compound 20. As shown in Figure 5, LC-MS detected the presence of compound 20 in the extract solution while no peaks were detected in the control. Since unbound compound 20 was washed out during the complex preparation, this result strongly suggests the direct binding of compound 20 to hTRPC3. Future structural and functional studies with Cryo-EM will reveal the molecular interactions of the complex.
Figure 5.

LC/MS analyses of the purified 20–TRPC3 complexes (right) together with the prepared control (left) support the direct binding of 20 to TRPC3 proteins. Spectra of MS data collected from 2.5 min to avoid buffer salts to MS.
Having confirmed the selective hTRPC3 inhibition and direct molecular interactions, we next evaluated the metabolic stability and safety profile of compound 20. As expected, the half-life was extended substantially from less than 15 min for Pyr3 to more than 4 h for compound 20 in mouse, rat, and human liver microsomes, consistent with our design to replace the metabolically labile ester headgroup in Pyr3. A preliminary in vivo toxicity study was subsequently performed in mice. Because Pyr3 undergoes fast hydrolysis to its inactive metabolite Pyr8 and both showed rapid blood clearance in mice,21 compound 27, bearing the same trichloroacrylic amide group as Pyr3 but with a stable amide bond, was used together with Pyr3 as the comparison treatments for the purpose of this toxicity study. All mice (n = 5) were treated daily with compounds 20, 27, or Pyr3 at 200 mg/kg by intraperitoneal injection for 5 consecutive days, and their survival was monitored. Compound 27 resulted in 100% mortality within 48 h in treated mice. The mice in the Pyr3-treated group with 80% survival dropped by roughly an average of 15% body weight compared to their weights at the start of the experiment, indicating the potential toxicity of Pyr3. In contrast, compound 20 displayed neither mortality nor significant weight loss (Figure 6). Since the only difference between compounds 20 and 27 in structure is their tail moiety, this study provided preliminary evidence that our rational structural modification can significantly decrease the safety liability associated with the trichloroacrylic amide group in Pyr3. It should be noted that since Pyr3 rapidly hydrolyzed in vivo to its much polar acid derivative Pyr8 which may have a much faster in vivo clearance, the appearance of a lower mortality in Pyr3 treatment group than its metabolically stable analogue 27 is likely due to the fast metabolism and elimination of Pyr3.
Figure 6.
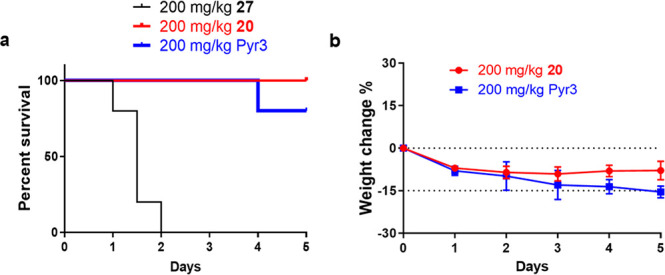
Compound 20 shows a good safety profile in mice. (a) Mouse survival curves. (b) Mean percent change in mouse body weight ± SD relative to body weight at the time of initiating drug treatment. All mice received a daily dose at 200 mg/kg body weight of compounds 20, 27, or Pyr3 (n = 5) for 5 consecutive days. Dashed lines indicate mouse weight relative to its respective baseline weight at day 0 and 15% weight loss.
Finally, to explore the potential biological effects of 20 in a TRPC3 related disease model, we evaluated its neuroprotective ability in primary cultured neurons (14 DIV prepared from E17 rats as described24). Compound 20 displayed similar protective effects as Pyr3 in preventing soluble oligomeric Aβ-induced synaptic toxicity on dendritic spine morphology as evidenced by MAP-2 staining (Figure 7).
Figure 7.

Effects on neuron dendritic morphology. (a) Compound 20 and Pyr3 protect against Aβ-induced damage to dendrites. Mature hippocampal neurons (DIV = 14) were treated with CHO control medium, or naturally secreted Aβ-containing conditioned medium (7PA2)25 or in combination with compound 20 or Pyr3 at different concentrations, for 16 h, followed by immunocytochemistry using an antibody against MAP2 (green) and DAPI (blue). Scale bar: 100 μm. (b) Lack of toxicity of the inhibitor compounds in neuronal culture. Hippocampal neurons (DIV = 14) were treated with compound 20 or Pyr3 alone at different concentrations, followed by immunocytochemistry staining with MAP2 (green) and DAPI (blue). Scale bar: 100 μm. (c) Quantification of dendritic length. Bars are mean ± SEM. **p < 0.01 (one-way ANOVA). ****p < 0.0001 (one-way ANOVA). n = 50 dendrites.
In summary, we describe here the rational design and preliminary characterization of a new metabolically stable, low-toxic, and selective hTRPC3 inhibitor compound 20. We also demonstrated the direct binding of compound 20 to hTRPC3 protein and its potential neuroprotective effects. These results indicate that further characterization and optimization of compound 20 may lead to a selective TRPC3 inhibitor as a potentially viable agent for diseases where overexpression of TRPC3 is involved.
Acknowledgments
We thank Dr. Valeria Vásquez and Dr. Lubin Lan (retired) for technical support. We thank Dr. Amanda Clarke at UTHSC for editorial assistance. We thank Dr. Salvatore Mancarella for experimental advice. This work was partially supported by NIH Grants R01GM125629 (to J.F.C.-M.) and R01AG049772 (to F.-F.L.) and funds from a UTHSC CORNET award and the UTCoP Drug Discovery Center (to W.L.). The contents of the article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Glossary
Abbreviations
- AITC
allyl isothiocyanate
- Cap
capsaicin
- GSK101
GSK1016790A
- GSK170
GSK1702934A
- NCS
N-chlorosuccinimide
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- Pyr3
pyrazole 3
- SAR
structure–activity relationship
- TRPC
transient receptor potential canonical.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00571.
Whole-cell recording, experimental procedures and methods, characterization data, and 1H and 13C NMR spectra (PDF)
Author Contributions
∇ S.Z. and L.O.R. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): W.L., Z.W., and F.L. are listed as inventors for a filed provisional patent application covering these compounds.
Supplementary Material
References
- Venkatachalam K.; Montell C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Valdez F.; Azumaya C. M.; Romero L. O.; Nakagawa T.; Cordero-Morales J. F. Structure-function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating. J. Biol. Chem. 2018, 293, 16102–16114. 10.1074/jbc.RA118.005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y.; Wakamori M.; Miyakawa T.; Hermosura M.; Hara Y.; Nishida M.; Hirose K.; Mizushima A.; Kurosaki M.; Mori E.; Gotoh K.; Okada T.; Fleig A.; Penner R.; Iino M.; Kurosaki T. Transient receptor potential 1 regulates capacitative Ca2+ entry and Ca2+ release from endoplasmic reticulum in B lymphocytes. J. Exp. Med. 2002, 195, 673–681. 10.1084/jem.20011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. L. The transient receptor potential superfamily of ion channels. J. Am. Soc. Nephrol. 2004, 15, 1690–1699. 10.1097/01.ASN.0000129115.69395.65. [DOI] [PubMed] [Google Scholar]
- Numaga-Tomita T.; Oda S.; Nishiyama K.; Tanaka T.; Nishimura A.; Nishida M. TRPC channels in exercise-mimetic therapy. Pfluegers Arch. 2019, 471, 507–517. 10.1007/s00424-018-2211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H. B.; Webb S. E.; Yue J. B.; Moreau M.; Leclerc C.; Miller A. L. TRPC3 is required for the survival, pluripotency and neural differentiation of mouse embryonic stem cells (mESCs). Sci. China: Life Sci. 2018, 61, 253–265. 10.1007/s11427-017-9222-9. [DOI] [PubMed] [Google Scholar]
- Li H. S.; Xu X. Z. S.; Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron 1999, 24, 261–273. 10.1016/S0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- Riccio A.; Medhurst A. D.; Mattei C.; Kelsell R. E.; Calver A. R.; Randall A. D.; Benham C. D.; Pangalos M. N. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Mol. Brain Res. 2002, 109, 95–104. 10.1016/S0169-328X(02)00527-2. [DOI] [PubMed] [Google Scholar]
- Li Y.; Calfa G.; Inoue T.; Amaral M. D.; Pozzo-Miller L. Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J. Neurophysiol. 2010, 103, 2846–2856. 10.1152/jn.01140.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M. D.; Pozzo-Miller L. TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J. Neurosci. 2007, 27, 5179–5189. 10.1523/JNEUROSCI.5499-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathina S.; Das U. N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 6, 1164–1178. 10.5114/aoms.2015.56342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush E. W.; Hood D. B.; Papst P. J.; Chapo J. A.; Minobe W.; Bristow M. R.; Olson E. N.; McKinsey T. A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006, 281, 33487–33496. 10.1074/jbc.M605536200. [DOI] [PubMed] [Google Scholar]
- Cohen R.; Torres A.; Ma H. T.; Holowka D.; Baird B. Ca2+ waves initiate antigen-stimulated Ca2+ responses in mast cells. J. Immunol. 2009, 183, 6478–6488. 10.4049/jimmunol.0901615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp S.; Strauss B.; Hirnet D.; Wissenbach U.; Mery L.; Flockerzi V.; Hoth M. TRPC3 mediates T-cell receptor-dependent calcium entry in human T-lymphocytes. J. Biol. Chem. 2003, 278, 26629–26638. 10.1074/jbc.M304044200. [DOI] [PubMed] [Google Scholar]
- Oda K.; Umemura M.; Nakakaji R.; Tanaka R.; Sato I.; Nagasako A.; Oyamada C.; Baljinnyam E.; Katsumata M.; Xie L. H.; Narikawa M.; Yamaguchi Y.; Akimoto T.; Ohtake M.; Fujita T.; Yokoyama U.; Iwatsubo K.; Aihara M.; Ishikawa Y. Transient receptor potential cation 3 channel regulates melanoma proliferation and migration. J. Physiol. Sci. 2017, 67, 497–505. 10.1007/s12576-016-0480-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Qi Y. X.; Qi Z. H.; Tsang S. Y. TRPC3 regulates the proliferation and apoptosis resistance of triple negative breast cancer cells through the TRPC3/RASA4/MAPK pathway. Cancers 2019, 11, 558–573. 10.3390/cancers11040558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydar E.; Yeo S.; Djamgoz M.; Palmer C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: a potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23–34. 10.1186/1475-2867-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig S.; Schernthaner M.; Maechler H.; Kappe C. O.; Glasnov T. N.; Hoefler G.; Braune M.; Wittchow E.; Groschner K. A TRPC3 blocker, ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate (Pyr3), prevents stent-induced arterial remodeling. J. Pharmacol. Exp. Ther. 2013, 344, 33–40. 10.1124/jpet.112.196832. [DOI] [PubMed] [Google Scholar]
- Wang H. B.; Cheng X. D.; Tian J. B.; Xiao Y. L.; Tian T.; Xu F. C.; Hong X. C.; Zhu M. X. TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 2020, 209, 107497. 10.1016/j.pharmthera.2020.107497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S.; Kato K.; Nishida M.; Mio K.; Numaga T.; Sawaguchi Y.; Yoshida T.; Wakamori M.; Mori E.; Numata T.; Ishii M.; Takemoto H.; Ojida A.; Watanabe K.; Uemura A.; Kurose H.; Morii T.; Kobayashi T.; Sato Y.; Sato C.; Hamachi I.; Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5400–5405. 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagimori M.; Murakami T.; Shimizu K.; Nishida M.; Ohshima T.; Mukai T. Synthesis of radioiodinated probes to evaluate the biodistribution of a potent TRPC3 inhibitor. MedChemComm 2016, 7, 1003–1006. 10.1039/C6MD00023A. [DOI] [Google Scholar]
- Gordon-Shaag A.; Zagotta W. N.; Gordon S. E. Mechanism of Ca2+-dependent desensitization in TRP channels. Channels 2008, 2, 125–129. 10.4161/chan.2.2.6026. [DOI] [PubMed] [Google Scholar]
- Schleifer H.; Doleschal B.; Lichtenegger M.; Oppenrieder R.; Derler I.; Frischauf I.; Glasnov T. N.; Kappe C. O.; Romanin C.; Groschner K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. 10.1111/j.1476-5381.2012.02126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. M.; Wang B.; Liu D.; Li J. J.; Xue Y. Q.; Sakata K.; Zhu L. Q.; Heldt S. A.; Xu H. X.; Liao F. F. Hsp90 chaperone inhibitor 17-AAG attenuates a beta-induced synaptic toxicity and memory impairment. J. Neurosci. 2014, 34, 2464–2470. 10.1523/JNEUROSCI.0151-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Klyubin I.; Fadeeva J. V.; Cullen W. K.; Anwyl R.; Wolfe M. S.; Rowan M. J.; Selkoe D. J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.