

Abstract
The gene KCNT1 encodes the sodium-activated potassium channel KNa1.1 (Slack, Slo2.2). Variants in the KCNT1 gene induce a gain-of-function (GoF) phenotype in ionic currents and cause a spectrum of intractable neurological disorders in infants and children, including epilepsy of infancy with migrating focal seizures (EIMFS) and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE). Effective treatment options for KCNT1-related disease are absent, and novel therapies are urgently required. We describe the development of a novel class of oxadiazole KNa1.1 inhibitors, leading to the discovery of compound 31 that reduced seizures and interictal spikes in a mouse model of KCNT1 GoF.
Keywords: Small molecule inhibitor, KCNT1 GoF mutations, HTS, oxadiazole series, SAR, DMPK, KNa1.1
KCNT1 encodes the neuronal potassium channel KNa1.1 (Slack, Slo2.2) which is highly expressed throughout the central nervous system.1 Like other voltage-gated potassium channels, functional KNa1.1 channels are tetramers composed of four subunits. Each subunit contains a voltage sensing domain (S1–S4) and a pore-forming (S5-pore loop-S6) domain. However, KNa1.1 is only weakly gated by voltage and is activated by alterations in cytoplasmic signaling cascades, energy state (ATP, NAD+), and increases in intracellular sodium.1 These features allow the channel to open in response to short-term increases in neuronal activity whereby increased potassium efflux would be thought to reduce neuronal activity. Paradoxically, disease-causing variants in KCNT1 have invariably been found to increase the activity of the channel in a gain-of-function (GoF) manner. To date, orally active, selective inhibitors of KNa1.1 have not been reported. Therefore, novel orally active inhibitors of KNa1.1 would be important as potential therapeutics as well as tools to advance the understanding of the role of KNa1.1 in neurophysiology.
The KNa gene family contains both KNa1.1 (Slack, Slo2.2; encoded by KCNT1) and KNa1.2 (Slick, Slo2.1; encoded by KCNT2).1 KNa1.1 is thought to conduct a sodium-activated potassium current in various central and peripheral nervous system (CNS and PNS) networks, including both inhibitory and excitatory CNS networks. KNa1.2 is thought to conduct the sodium-activated potassium current in the heart.2 KNa1.1 and KNa1.2 have been suggested to form heterotetramers in cells that coexpress both genes.1 However, association of KNa1.1 and KNa1.2 in vivo has not been demonstrated.
KNa1.1 is thought to shape neural excitability on a subsecond time scale due to acute sensing of intracellular ionic and energy states.3 KNa channels have a large C-terminal region, which contains two regulators of K+ conductance (RCK), which is where Na+ binding is thought to occur.1,4 Increases in intracellular Na+ concentration associated with neuronal activity enhances KNa1.1 channel activity and has been proposed to provide a vital feedback mechanism for controlling neural activity.5 Previous work has also linked activation of KNa1.1 to the presence of subthreshold INa (persistent INa) and enhancements in INa are well-known to cause neuronal hyperexcitability.6 How variants in KNa1.1 disrupt this regulation to produce a GoF phenotype is currently not well understood.7,8 However, newly reported mouse models of KCNT1 GoF will invariably provide new insights.7,9,10
KCNT1 channel mutants are located around three functional areas: the NAD+ binding region situated in the C-terminus, the pore-forming region (between the S5 and S6 loops), and the RCK domains. All disease-causing variants are thought to be GoF and are associated with drug-resistant forms of infantile epilepsy such as the devastating epilepsy of infancy with migrating focal seizures (EIMFS)3,11−13 or the less severe autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE).14−16 Less common epileptic disorders include West syndrome,17 infantile spasm,18 and Ohtahara syndrome.19 Additional comorbidities are often present, including hypotonia, severe developmental delay, and movement disorder.20
Treatment options for KCNT1-related disease are extremely limited, and the seizures and comorbidities are intractable to conventional antiepileptics. Bepridil,21,22 clofilium,23 and quinidine1,22−25 have been proposed as treatment options. However, because of nonspecificity in their mode of action1,22,26 and recent questions around efficacy, their use is limited.21,24,27 Currently, several groups have investigated agents that specifically inhibit KNa1.1. Spitznagel et al. recently reported the discovery of VU0606170, a small molecule KNa1.1 inhibitor with low micromolar potency.5 VU0606170 was used to probe KNa1.1 inhibition in neuronal cultures and proved useful at significantly reducing hyperexcitability in spontaneously firing cortical neuron cultures. Among other groups working on KNa1.1 inhibitors, Cole et al. recently used the chicken KNa1.1 cryo-EM structure to perform a virtual screen of 100,000 commercial compounds to find those that were predicted to bind in the channel pore.22 A set of 17 compounds was selected from this exercise, of which 6 molecules were found to inhibit the KNa1.1 channel with micromolar potency. Herein we describe our efforts in this area and describe for the first time in vivo activity of a small molecule KNa1.1 inhibitor in a mouse model of KCNT1 GoF.
A high throughput screen (HTS) using a rubidium (86Rb) flux assay in HEK-TREX cells stably expressing the human EIMFS variant P924L (hKNa1.1-P924L) was developed. Cells were preloaded with Rb and then incubated for 10 min with test compound in the presence of elevated KCl (5.4 mM) to depolarize the resting membrane potential and activate KNa1.1 mediated Rb efflux. The amount of Rb efflux was quantitated and expressed as percent efflux. Approximately 72,000 compounds were screened using a custom-built library designed to maximize chemical diversity and obtained from several commercial suppliers. Screening was performed at 10 μM of compound and yielded an approximate 1% hit rate when defined as greater than 55% inhibition. These hits were reconfirmed for activity by screening in the same assay in a concentration response format where 270 of the hits were found to have a half maximal inhibitory concentration (IC50) of 15 μM or less. The 270 compounds were subsequently tested in an automated, SyncroPatch patch clamp assay to refine the assessment of activity directly on KNa1.1 current at physiological membrane potentials. HEK-TREX cells stably expressing human wildtype KNa1.1 (hKNa1.1-WT) were voltage clamped at −80 mV, and inhibition was measured using a voltage step to 0 mV. In this assay, KNa1.1 was activated by increasing intracellular Na+ to 70 mM. For the hKCNT1-WT and mKCNT1-WT assays, IC50s were generated using concentrations ranging from 0.001 to 30 μM in half log steps and a minimum of three cells (replicates) per concentration (Tables 1–3). For compound 31, all data represent a minimum of two (typically >3) independent experimental runs with at least five (typically >10) cells per concentration. Confirmed hits were subsequently clustered using chemoinformatic methods, and compound clusters were evaluated for their attractiveness to take forward into new analogue synthesis and development of structure–activity relationships (SARs). One cluster of interest, containing a phenyl oxadiazole scaffold, was selected for SAR studies and is described in this paper. Two such examples of oxadiazole28 HTS hits are shown in Figure 1.
Table 1. Initial SAR Exploration around HTS Hit.

Table 3. Effects of Pyridine Substitution on Activity and Compound Properties.


Figure 1.

Oxadiazole HTS hits.
Compound 2 was of particular interest, and initial development of the scaffold sought to understand the basic SAR, as shown in Table 1. Compound 2 is a racemic mixture, and the individual enantiomers (compounds 3 and 4) were synthesized in a chiral fashion according to the scheme outlined in Scheme 1. Coupling of N′-hydroxy-3-methylbenzimidamide with the corresponding protected amino acid gave the desired chiral oxadiazole intermediate, which was then deprotected and coupled to the pyrazole carboxylic acid to afford compound 3. All subsequent analogues were synthesized according to this general scheme.
Scheme 1. Synthesis of Compound 3.

Comparing the activity of compounds 3 and 4, it was apparent that the stereochemistry of the methyl was important for inhibition at KNa1.1, with the (S) isomer being the more active enantiomer. Not only is the stereochemistry important, but removal of the chiral methyl group led to greater than a 10-fold loss in activity (data not shown).
The pyrazole substitution pattern was found to be critical to activity. Comparing compound 5 to 3, we found that moving the methyl to the alternative pyrazole nitrogen abolished activity. Removal of the methyl on the left-hand side aromatic ring (compound 6) showed a significant loss of activity, although compound 11 demonstrates that fluorine is a suitable replacement for methyl (trifluoromethyl pyrazole has similar activity to cyclopropyl pyrazole). In an effort to establish if the oxadiazole is simply a spacer to correctly orientate the other features of the molecules, we synthesized compounds 8–10 where alternative, readily accessible heterocycles were incorporated as replacements for the oxadiazole. All changes to the oxadiazole caused a reduction in activity, suggesting that electronic effects of the heterocycle may play a part in determining activity. Finally, compound 12 showed that N-methylation of the amide is not tolerated and compound 13 demonstrates that a sulfonamide cannot be used to replace the amide.
Our initial SAR scoping yielded molecules with high lipophilicity (measured logD7.4 of 4 and higher), and we wished to establish if decreasing the lipophilicity was compatible with potent KNa1.1 inhibition. We probed the introduction of various polar groups in the scaffold, with one such example being the use of heterocyclic rings in the left region as replacements for the existing substituted aromatic rings. The compounds in Table 2 describe the introduction of left-hand side heterocycles and their effect on KNa1.1 inhibition. Compounds 18–20 contain unsubstituted pyridyl rings, and they were found to be poorly active, with the 4-pyridyl analogue, compound 19, being the most active. However, in some cases, substitution on the pyridine ring did improve activity, with the 3-methyl substitution of the 4-pyridine (compound 17) showing very promising KNa1.1 inhibition with an IC50 of 165 nM. The same was not true for the 2-pyridines and 3-pyridine analogues which were significantly less active (compare compound 17 to 14–16). Ring systems with additional heteroatoms such as compounds 21 and 22 were found to be weakly active.
Table 2. Left Hand Side Heterocycle SAR.


The SAR of compound 17 was further probed by varying the substituents on the pyridine ring as shown in Table 3. Compound 17 showed good aqueous kinetic solubility and in vitro stability in human liver microsomes. Changing the methyl group of compound 17 to the o-methoxy group in compound 23 enhanced the activity and maintained good in vitro metabolic stability. Tertiary amines at the meta position have weaker activity and poorer metabolic stability as seen with compounds 24 and 25. The secondary amine 30 did show comparable activity to compound 17 but was weaker than the methoxy analogue compound 23 as well as less metabolically stable (HCLint of 28 μL/min/mg compared to <9.6 μL/min/mg).
Placing a pendent methyl ether at the meta position, as shown by compound 28, caused a considerable loss in activity, which was also found with the substituted ether compounds 32–34. Addition of a second substituent to the aromatic ring caused a greater than 10-fold drop in activity (compare compound 26 to 23), and the cyclopropyl analogue 27 was found to be the most potent analogue in the series.
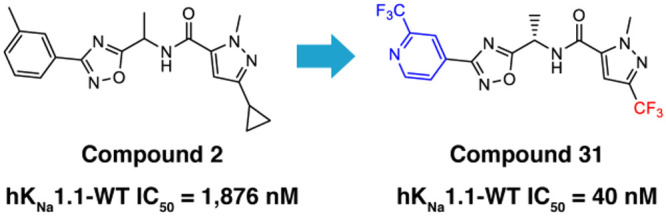
Where measured, it should be noted that all compounds showed a significant loss of activity at the mouse wildtype KNa1.1 (mKNa1.1-WT) channel compared to hKNa1.1-WT (see Table 3). For example, our initial pyridine hit, compound 17, had an IC50 of 165 nM at hKNa1.1-WT but an IC50 of >14,000 nM at mKNa1.1-WT. Given that our preclinical in vivo testing was performed in a mouse model of KCNT1 GoF, an in vivo assessment of the series was technically challenging due to a drop-off with mouse activity. The reason for the marked difference in potency between human and mouse wildtype channels is not well understood. Fortunately, the trifluoromethyl pyridyl analogue 31, although less active than some analogues at hKNa1.1-WT (IC50 40 nM), was found to be the most potent compound at mKNa1.1-WT (IC50 622 nM) with a modest 15-fold shift between the orthologues (Figure 2). Moreover, activity was retained at mKNa1.1-P905L (25-fold shift, Figure 2), which represents the mouse orthologue containing the human P924L EIMFS variant. This retained activity facilitated in vivo testing. Based on the described SAR findings, compound 31 was selected as a candidate to characterize further in our in vitro and in vivo assays. Additional patch clamp based, primary pharmacology revealed IC50’s of 49 nM for cynomolgus monkey KNa1.1-WT and 545 nM for rat KNa1.1-WT.
Figure 2.

In vitro profile of compound 31 on human and mouse KNa1.1.
KNa1.1 inhibition data described so far were determined using the human or mouse wildtype channels. Given that we aim to treat individuals with GoF KNa1.1 variants, it is important to profile compounds of interest for activity across several variants due to their disparate locations in the channel protein. Cell lines stably expressing individual human KCNT1 GoF variants were created to represent recurrent variants located across the channel structure: transmembrane domains, RCK1, and RCK2. The activity of compound 31 at these variants was assessed (Figure 3), and all variants were less active when compared to hKNa1.1-WT with activity at the human variants ranging from 221 nM (G288S) to 1768 nM (F346L). Such a range of activities is a feature we have seen for several other members of the oxadiazole series (data not shown).
Figure 3.
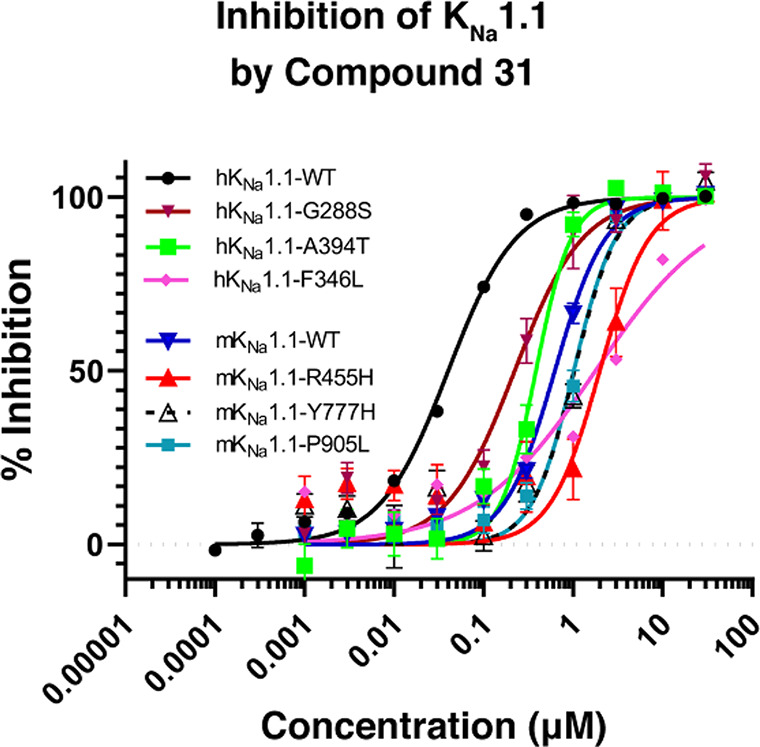
Effect of compound 31 on human and mouse KNa1.1 variants.
Further characterization of compound 31 included an assessment of potential secondary activity across a panel of 80 targets at 10 μM compound concentration, using binding displacement assays. Only two hits showed >50% activity: translocator protein (TSPO) (63% displacement) and GABAA Cl– channel (74% displacement). Furthermore, using patch clamp analysis in a panel of ion channels, compound 31 showed no significant inhibition at hERG (IC50 11.9 μM), hNaV1.5 (IC50 42 μM), CaV1.2 (IC50 10.7 μM), or lKs (IC50 18.5 μM). Finally, additional patch clamp based assessment of compound 31 at the potassium channels BK (IC50 13.2 μM) and KNa1.2/Slick/KCNT2 (IC50 16.4 μM) showed weak activity. This package of data indicates that compound 31 shows good overall selectivity for the KNa1.1 channel.
The primary activity of compound 31 was further assessed in a native tissue brain slice preparation. Whole-cell patch clamp recordings from CA1 pyramidal neurons in acute mouse brain slices were performed. The effects of compound 31 on neuronal firing were evaluated in brain slices from WT mice and mice homozygous for mKcnt1-P905L (Kcnt1L/L). The Kcnt1L/L mouse line recapitulates many key features of the human disease, including spontaneous seizures, high interictal spike frequency, and reduced survival.10 In brain slices prepared from p16–30 day old WT or Kcnt1L/L mice, a current injection protocol was used to determine action potential firing at baseline and in the presence of 1 μM or 10 μM of compound 31. In neurons from WT mice, there was a nonsignificant trend toward a decrease in the total number of action potentials fired in the presence of 1 μM (p = 0.054, baseline 213.5 ± 15.21, compound 31 188.5 ± 21.09, n = 11 paired cells) and 10 μM compound 31 (p = 0.897, baseline 216.8 ± 10.65, compound 31 218.1 ± 9.43, n = 9 paired cells) (Figure 4A–F). In neurons from Kcnt1L/L mice, although 1 μM compound 31 did not alter firing (p = 0.075, baseline 201.2 ± 13.00, compound 31 219.5 ± 16.5, n = 6 paired cells), application of 10 μM compound 31 resulted in a significant reduction in firing (p = 0.0015, baseline 284.4 ± 31.10, compound 31 223.4 ± 28.01, n = 7 paired cells) (Figure 4G–L).
Figure 4.

Effect of comnpound 31 on CA1 pyramidal neurons from wildtype and Kcnt1L/L mice. (A,D,G,J) Represenative traces of baseline (black) and in the presence of 1 μM (orange) and 10 μM (red) compound 31. Quantification of (B,E,H,K) input-frequency relations and (C,F,I,L) total number of action potentials fired. Data presented as mean ± SEM and paired individual data points. **p < 0.01.
Pharmacokinetics of compound 31 in CD-1 mice showed a compound with good brain penetration and low clearance, suitable for in vivo pharmacological testing. A 0.5 mg/kg IV dose (10% DMSO, 10% Solutol, and 80% water vehicle) had a clearance of 2.0 mL/min/kg, a Vdss of 1.88 L/kg, and a half-life of over 20 h. Upon oral dosing, compound 31 was fully bioavailable and, with an oral dose of 30 mg/kg, slightly more compound was present in the brain compared to plasma, with 98.3% compound binding to mouse plasma protein and 97.8% compound binding to mouse brain tissue.
Compound 31 was assessed in vivo in the Kcnt1L/L mouse model.10 Kcnt1L/L mice (P32–40) were implanted with EEG electrodes to allow monitoring of electrographic seizure and interictal spike frequency. After collecting a 24 h recording to establish baseline interictal spike and seizure frequency, Kcnt1L/L mice were dosed with vehicle (10% DMSO, 10% Solutol, and 80% water vehicle) or compound 31 (30 mg/kg, SC) and EEG recordings collected for a further 24 h.
Figure 5A depicts mean normalized interictal spike frequency over time. After dosing with vehicle, mice continue to have rising interictal spike frequency, whereas after dosing with compound 31 (30 mg/kg, SC) there is a robust reduction in interictal spike frequency that persists over the 24 h period. A subset of Kcnt1L/L mice (n = 12) exhibited multiple seizures (average 8.8 ± 2.0 seizures/24 h) during the 24 h baseline period (Figure 5B). Not a single seizure was observed in the 24 h period after dosing with compound 31 (30 mg/kg, SC). At this dose of compound 31 (30 mg/kg, SC), the maximum free brain concentration is approximately 270 nM, which is in the region of a quarter of the in vitro IC50 (1012 nM) at mKNa1.1-P905L. For ion channel modulators, achieving in vivo activity at free concentrations in the brain which are well below the IC50 determined using in vitro assays is not unprecedented. Indeed the sodium channel blocker carbamazepine shows anticonvulsant efficacy in mice with a free brain EC50 of ∼5.6 μM despite its potency for blocking peak sodium currents in vitro (IC50) being 10–100-fold29,30 higher (unpublished data). Similarly, the Kv7 potassium channel positive modulator retigabine is efficacious at free brain concentrations of around one-third of the in vitro EC50.30,31 To determine whether compound 31 had any nonspecific sedative effects, the molecule was assessed for effects on spontaneous locomotor activity in CD-1 mice. Compound 31 was devoid of any effects on distance moved over the dose range evaluated (10–150 mg/kg; Figure 5C). Together these in vivo data suggest that compound 31 ameliorates the phenotype of Kcnt1L/Lmice and may have an acceptable therapeutic window between efficacy (free brain concentration of 270 nM) and tolerability (maximum brain concentration 1204 nM at 150 mg/kg).
Figure 5.

Compound 31 normalizes EEG phentotype in Kcnt1L/L mice without affecting spontaneous locomotor activity. Acute administration of compound 31 (30 mg/kg, SC) decreased interictal spike frequency (A) and decreased mean number of seizures (B) in Kcnt1L/L mice. Compound 31 (10–150 mg/kg) did not affect spontaneous locomotor activity in wildtype CD-1 mice (C). Data are mean ± SEM, n = 4–10; *P < 0.05 vs respective baseline; ***P < 0.0001 vs vehicle.
In conclusion, we have discovered a series of oxadiazole small molecule KNa1.1 inhibitors that successfully demonstrate in vivo activity in a mouse model of KCNT1 GoF. Future efforts will be aimed at evaluating the effects of longer-term dosing on survival and behavior in the Kcnt1L/L mouse to better understand if the series holds therapeutic potential.
Acknowledgments
We thank Zhu Bai and the WuXi AppTech chemistry team for help with the compound syntheses discussed in this article as well as Rahul Nagawade and the chemistry team at Sai Life Sciences. We also thank Melody Li and Nikola Jancovski at The Florey Institute for performing the mouse in vivo EEG studies and Cristobal Alhambra for computational chemistry support provided for the HTS analysis. We also thank Kalpana Shankar at Simpson Healthcare for assistance with preparing the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00675.
Details of the chemical synthesis and characterization for compounds 3, 4, 27, and 31; ethical statement regarding the use of experimental animals in the studies; descriptions of the patch clamp procedure and the assays used to detect excitability; information regarding in vitro pharmacology testing of compound 31 in binding assays, with associated histogram (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given final approval to the final version of the manuscript.
The authors declare no competing financial interest.
This letter published ASAP on March 9, 2021, with an error in Figure 2. The corrected version was reposted on March 29, 2021.
Supplementary Material
References
- Kaczmarek L. K. Slack, Slick and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013, 2013, 354262. 10.1155/2013/354262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. O.; Wang Y. T.; Nadtochiy S. M.; Miller J. H.; Jonas E. A.; Dirksen R. T.; Nehrke K.; Brookes P. S. Cardiac metabolic effects of K(Na)1.2 channel deletion and evidence for its mitochondrial localization. FASEB J. 2018, 32 (11), fj201800139R. 10.1096/fj.201800139R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G. E.; Kaczmarek L. K. Emerging role of the KCNT1 Slack channel in intellectual disability. Front. Cell. Neurosci. 2014, 8, 209. 10.3389/fncel.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A.; Kaczmarek L. K. For K+ channels, Na+ is the new Ca2+. Trends Neurosci. 2005, 28 (8), 422–8. 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Spitznagel B. D.; Mishra N. M.; Qunies A. M.; Prael F. J. 3rd; Du Y.; Kozek K. A.; Lazarenko R. M.; Denton J. S.; Emmitte K. A.; Weaver C. D. VU0606170, a Selective Slack Channels Inhibitor, Decreases Calcium Oscillations in Cultured Cortical Neurons. ACS Chem. Neurosci. 2020, 11 (21), 3658–3671. 10.1021/acschemneuro.0c00583. [DOI] [PubMed] [Google Scholar]
- Pryce K. D.; Powell R.; Agwa D.; Evely K. M.; Sheehan G. D.; Nip A.; Tomasello D. L.; Gururaj S.; Bhattacharjee A. Magi-1 scaffolds Na(V)1.8 and Slack K(Na) channels in dorsal root ganglion neurons regulating excitability and pain. FASEB J. 2019, 33 (6), 7315–7330. 10.1096/fj.201802454RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore A. N.; Colombo S.; Tobin W. F.; Petri S.; Cullen E. R.; Dominguez S.; Bostick C. D.; Beaumont M. A.; Williams D.; Khodagholy D.; Yang M.; Lutz C. M.; Peng Y.; Gelinas J. N.; Goldstein D. B.; Boland M. J.; Frankel W. N.; Weston M. C. Reduced GABAergic Neuron Excitability, Altered Synaptic Connectivity, and Seizures in a KCNT1 Gain-of-Function Mouse Model of Childhood Epilepsy. Cell Rep. 2020, 33 (4), 108303. 10.1016/j.celrep.2020.108303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quraishi I. H.; Stern S.; Mangan K. P.; Zhang Y.; Ali S. R.; Mercier M. R.; Marchetto M. C.; McLachlan M. J.; Jones E. M.; Gage F. H.; Kaczmarek L. K. An Epilepsy-Associated KCNT1Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack K(Na) Currents. J. Neurosci. 2019, 39 (37), 7438–7449. 10.1523/JNEUROSCI.1628-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quraishi I. H.; Mercier M. R.; McClure H.; Couture R. L.; Schwartz M. L.; Lukowski R.; Ruth P.; Kaczmarek L. K. Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding Slack (K(Na)1.1) Na(+)-activated K(+) channels. Sci. Rep. 2020, 10 (1), 3213. 10.1038/s41598-020-60028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbano L. E.; Li M.; Jancovski N.; Jafar-Nejad P.; Richards K.; Sedo A.; Soriano A.; Rollo B.; Jia L.; Gazina E.; Piltz S.; Adikusuma F.; Thomas P. Q.; Rigo F.; Reid C. A.; Maljevic S.; Petrou S.. Antisense oligonucleotide therapy for KCNT1 encephalopathy. bioRxiv, November 14, 2020, ver. 1. 10.1101/2020.11.12.379164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia G.; Fleming M. R.; Deligniere A.; Gazula V. R.; Brown M. R.; Langouet M.; Chen H.; Kronengold J.; Abhyankar A.; Cilio R.; Nitschke P.; Kaminska A.; Boddaert N.; Casanova J. L.; Desguerre I.; Munnich A.; Dulac O.; Kaczmarek L. K.; Colleaux L.; Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 2012, 44 (11), 1255–9. 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo F.; Ambrosino P.; Guacci A.; Chetta M.; Marchese G.; Rocco T.; Soldovieri M. V.; Manocchio L.; Mosca I.; Casara G.; Vecchi M.; Taglialatela M.; Coppola G.; Weisz A. Characterization of two de novoKCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell. Neurosci. 2016, 72, 54–63. 10.1016/j.mcn.2016.01.004. [DOI] [PubMed] [Google Scholar]
- McTague A.; Appleton R.; Avula S.; Cross J. H.; King M. D.; Jacques T. S.; Bhate S.; Cronin A.; Curran A.; Desurkar A.; Farrell M. A.; Hughes E.; Jefferson R.; Lascelles K.; Livingston J.; Meyer E.; McLellan A.; Poduri A.; Scheffer I. E.; Spinty S.; Kurian M. A.; Kneen R. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain 2013, 136 (5), 1578–1591. 10.1093/brain/awt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry C. P.; Heron S. E.; Phillips F.; Howell S.; MacMahon J.; Phillips H. A.; Duncan J. S.; Mulley J. C.; Berkovic S. F.; Scheffer I. E. Severe autosomal dominant nocturnal frontal lobe epilepsy associated with psychiatric disorders and intellectual disability. Epilepsia 2008, 49 (12), 2125–9. 10.1111/j.1528-1167.2008.01652.x. [DOI] [PubMed] [Google Scholar]
- Lim C. X.; Ricos M. G.; Dibbens L. M.; Heron S. E. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J. Med. Genet. 2016, 53 (4), 217–25. 10.1136/jmedgenet-2015-103508. [DOI] [PubMed] [Google Scholar]
- Heron S. E.; Smith K. R.; Bahlo M.; Nobili L.; Kahana E.; Licchetta L.; Oliver K. L.; Mazarib A.; Afawi Z.; Korczyn A.; Plazzi G.; Petrou S.; Berkovic S. F.; Scheffer I. E.; Dibbens L. M. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 2012, 44 (11), 1188–90. 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- Pavone P.; Polizzi A.; Marino S. D.; Corsello G.; Falsaperla R.; Marino S.; Ruggieri M. West syndrome: a comprehensive review. Neurol. Sci. 2020, 41, 3547. 10.1007/s10072-020-04600-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrachovy R. A.; Frost J. D. Jr. Infantile spasms. Handb Clin Neurol 2013, 111, 611–8. 10.1016/B978-0-444-52891-9.00063-4. [DOI] [PubMed] [Google Scholar]
- Ohtahara S.; Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res. 2006, 70 (Suppl 1), S58–67. 10.1016/j.eplepsyres.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Gertler T.; Bearden D.; Bhattacharjee A.; Carvill G.. KCNT1-Related Epilepsy. In GeneReviews; Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., Amemiya A., Eds.; University of Washington: Seattle, WA, 1993. [PubMed] [Google Scholar]
- Gertler T. S.; Thompson C. H.; Vanoye C. G.; Millichap J. J.; George A. L. Jr. Functional consequences of a KCNT1 variant associated with status dystonicus and early-onset infantile encephalopathy. Ann. Clin. Transl. Neurol. 2019, 6 (9), 1606–1615. 10.1002/acn3.50847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole B. A.; Johnson R. M.; Dejakaisaya H.; Pilati N.; Fishwick C. W. G.; Muench S. P.; Lippiat J. D. Structure-Based Identification and Characterization of Inhibitors of the Epilepsy-Associated K(Na)1.1 (KCNT1) Potassium Channel. iScience 2020, 23 (5), 101100. 10.1016/j.isci.2020.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Los Angeles Tejada M.; Stolpe K.; Meinild A. K.; Klaerke D. A. Clofilium inhibits Slick and Slack potassium channels. Biologics 2012, 6, 465–70. 10.2147/BTT.S33827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden D.; Strong A.; Ehnot J.; DiGiovine M.; Dlugos D.; Goldberg E. M. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol. 2014, 76 (3), 457–61. 10.1002/ana.24229. [DOI] [PubMed] [Google Scholar]
- Yang B.; Gribkoff V. K.; Pan J.; Damagnez V.; Dworetzky S. I.; Boissard C. G.; Bhattacharjee A.; Yan Y.; Sigworth F. J.; Kaczmarek L. K. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology 2006, 51 (4), 896–906. 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Mullen S. A.; Carney P. W.; Roten A.; Ching M.; Lightfoot P. A.; Churilov L.; Nair U.; Li M.; Berkovic S. F.; Petrou S.; Scheffer I. E. Precision therapy for epilepsy due to KCNT1 mutations. A randomized trial of oral quinidine. Neurology 2018, 90 (1), e67–e72. 10.1212/WNL.0000000000004769. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M. P.; Fiannacca M.; Smith D. M.; Gertler T. S.; Gunning B.; Syrbe S.; Verbeek N.; Stamberger H.; Weckhuysen S.; Ceulemans B.; Schoonjans A.-S.; Rossi M.; Demarquay G.; Lesca G.; Olofsson K.; Koolen D. A.; Hornemann F.; Baulac S.; Rubboli G.; Minks K. Q.; Lee B.; Helbig I.; Dlugos D.; Møller R. S.; Bearden D. Treatment Responsiveness in KCNT1-Related Epilepsy. Neurotherapeutics 2019, 16 (3), 848–857. 10.1007/s13311-019-00739-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez B. G.; Griffin A.; Charifson P.; Reddy K.; Kahlig M. K.; Marron B., Kcnt1 Inhibitors And Methods Of Use. Patent WO/2020/227101, November 12, 2020.
- Sheets P. L.; Heers C.; Stoehr T.; Cummins T. R. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J. Pharmacol. Exp. Ther. 2008, 326 (1), 89–99. 10.1124/jpet.107.133413. [DOI] [PubMed] [Google Scholar]
- Willow M.; Gonoi T.; Catterall W. A. Voltage clamp analysis of the inhibitory actions of diphenylhydantoin and carbamazepine on voltage-sensitive sodium channels in enrupblastoma cells. Mol. Pharmacol. 1985, 27 (5), 549–558. [PubMed] [Google Scholar]
- Rogawski M. A.; Löscher W.; Rho J. M. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harbor Perspect. Med. 2016, 6 (5), a022780. 10.1101/cshperspect.a022780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.