

Abstract
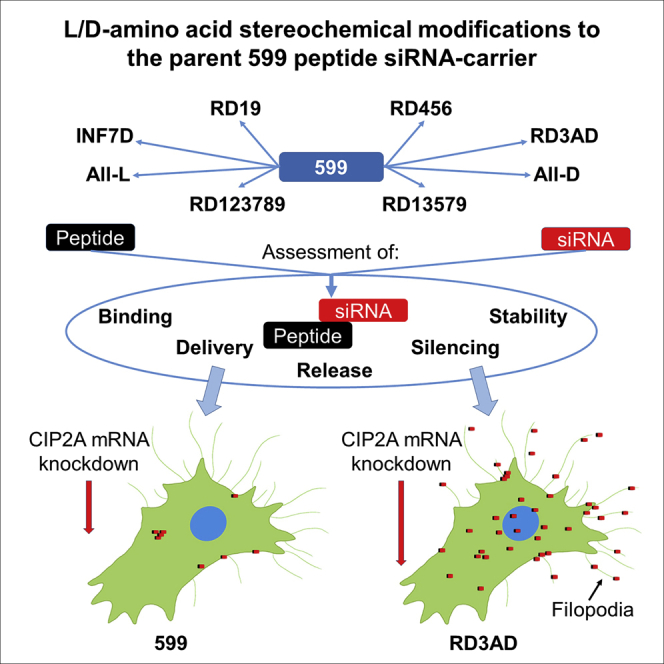
The 599 peptide has been previously shown to effectively deliver small interfering RNAs (siRNAs) to cancer cells, inducing targeted-oncogene silencing, with a consequent inhibition of tumor growth. Although effective, this study was undertaken to advance the 599 peptide siRNA-carrier design through L/D-amino acid stereochemical modifications. Consequently, 599 was modified to generate eight different peptide variants, incorporating either different stereochemical patterns of L/D-amino acids or a specific D-amino acid substitution. Upon analysis of the variants, it was observed that these modifications could, in some instances, increase/decrease the binding, nuclease/serum stability, and complex release of siRNAs, as well as influence the gene-silencing efficiencies of the complex. These modifications were also found to affect cellular uptake and intracellular localization patterns of siRNA cargo, with one particular variant capable of mediating binding of siRNAs to specific cellular projections, identified as filopodia. Interestingly, this variant also exhibited the most enhanced gene silencing in comparison to the parent 599 peptide, thus suggesting a possible connection between filopodia binding and enhanced gene silencing. Together, these data demonstrate the utility of peptide stereochemistry, as well as the importance of a key D-amino acid modification, in advancing the 599 carrier design for the enhancement of gene silencing in cancer cells.
Keywords: peptide, stereochemistry, cancer, siRNA, RNAi, CIP2A, filopodia, L/D-amino acid, chirality
Graphical abstract
In the present study, Jakymiw and colleagues demonstrate the utility of specific L/D-amino acid stereochemical modifications in advancing a peptide siRNA-carrier design for the enhancement of gene silencing in cancer cells, with a possible connection to a more efficient mode of cell entry mediated by filopodia.
Introduction
One of the more recent promising avenues in cancer treatment has been in the use of RNA interference (RNAi), a highly conserved post-transcriptional gene-silencing mechanism that involves the specific targeting of mRNA for degradation by small noncoding double-stranded RNA molecules.1,2 In fact, the discovery that the introduction of chemically synthesized small interfering RNAs (siRNAs) into cultured mammalian cells could efficiently induce sequence-specific gene silencing3 made evident the therapeutic potential of RNAi as a means to specifically target and silence disease-causing genes.4 Since then, many studies have shown the therapeutic potential of RNAi, including various clinical trials, culminating in the world’s first US Food and Drug Administration (FDA)-approved siRNA drug in 2018.4, 5, 6, 7 Inclusive of these studies were preclinical and early-phase clinical trials of human cancer patients that have demonstrated RNAi as a promising therapeutic tool for the treatment of cancer.4, 5, 6, 7
To maximize gene-silencing efficiency, the therapeutic administration of siRNA requires a delivery platform that can overcome numerous challenges typically associated with this form of therapy, including rapid renal excretion, degradation by RNases, homing to specific cell types, intracellular uptake, endosomal entrapment, and release of the siRNA cargo from the delivery platform.4,8,9 Currently, there are various types of siRNA delivery platforms, ranging from viral to non-viral vectors, each having their pros and cons when it comes to mitigating the obstacles described above, with the non-viral vectors encompassing technologies such as lipids, polymers, aptamers, and peptides.4,10,11 In particular, peptides have been found to be a highly favorable drug carrier, due to the facile ability to control their design, which allows for the adjustment of function and physiochemical properties.4 In fact, when peptide delivery systems are designed properly, peptide-bound siRNA complexes are able to overcome many of the delivery challenges faced by naked siRNAs. One such peptide that we have previously reported on is the chimeric peptide 599, which was specifically designed to overcome challenges associated with siRNA cell uptake and endosomal entrapment through its cationic nona-arginine C terminus and fusogenic INF7 N terminus, respectively.12 Together, these features led to the ability of 599 to enhance the intracellular delivery and bioavailability of therapeutic siRNAs designed to target the CIP2A oncogene (siCIP2A) into cancer cells in vitro and upon intratumoral injection in vivo induce CIP2A silencing, resulting in the significant inhibition of tumor growth.12,13 In subsequent work, using a noncovalent multifunctional peptide complex approach, co-complexation of the 599 peptide with a cancer-cell-targeting peptide was found to synergistically mediate the effective targeting/delivery of siCIP2As to xenograft oral cancer tumors upon systemic administration and significantly enhance CIP2A silencing.14
A common concern, however, for the therapeutic application of peptide carriers in vivo is their premature degradation prior to reaching their cell target for delivery of the drug cargo.15,16 Therefore, a common strategy to combat this issue has been to modulate the stereochemistry of peptides, such that they comprise D-amino acids, thereby rendering them more protease resistant than their L-amino acid counterparts,16, 17, 18, 19 with increased stability not limited to peptides composed entirely of D-amino acids, but also observed for peptides with partial D-amino acid substitutions.16,19,20 In fact, the nona-arginine tract in 599 was designed to contain D-arginines for this particular reason and shown to be able to protect siRNAs from degradation by serum and ribonucleases in vitro and upon intratumoral injection in vivo.12,13 Although the inclusion of D-arginines within the design of peptide carriers may confer protease resistance, the inclusion of L-arginines also appears to have its advantages. Interestingly, in assessing the effects of attaching cell-binding ligands to arginine-rich cell-penetrating peptides (CPPs), Zeller et al.21 found that ligands attached to L-arginine-rich CPPs, but not D-arginine-rich CPPs, could prolong the dynamics of gene silencing due to the enhanced release of siRNAs from late endosomes to the cytsol. This effect was demonstrated to be dependent on endosomal proteolytic activity, which implied that partial degradation of the arginine-rich CPP was necessary for endosome-to-cytosol translocation of the siRNAs. Moreover, this effect could also explain why another study using an analogous approach connecting a tumor-targeting peptide with a D-arginine-rich tract for delivery of siRNAs showed no knockdown of the targeted gene, even in the presence of an endosomolytic peptide.22 Only when a selected arginine was replaced with an alanine, thought in part to reduce the electrostatic interactions between the peptide and siRNA, was gene-silencing induced, suggesting the need again for effective release of siRNAs from the peptides.22
Interestingly, for many years the accepted paradigm was that stereochemical modifications to peptide carriers had no effect on their cellular uptake; however, a study by Verdurmen et al.16 reported that peptide stereochemistry, conversely, could indeed affect their cell uptake and the capacity of endocytic uptake in a cell-type dependent manner. Moreover, in a subsequent study, Favretto et al.23 reported that peptide stereochemistry involving single amino acid stereochemistry designs could also affect the cell uptake efficiency of peptide-based antisense oligonucleotide polyplexes. Taken together, these studies made evident that modulation of peptide stereochemistry could undoubtedly affect the properties/functions of the peptide carrier beyond the prototypical protease resistant function. Nonetheless, no studies to this point have characterized whether more precise alterations to the stereochemical patterns of L/D-amino acids and/or specific D-amino acid substitutions within a peptide carrier design could impact the uptake efficiency of complexed cargo, in particular siRNAs. Thus, to further advance our understanding of stereochemistry in peptide carrier design/function for siRNA-based cellular delivery, eight 599 peptide variants were designed, incorporating either different stereochemical patterns of L/D-amino acids or a specific D-amino acid substitution, and then characterized in their ability to bind, deliver, stabilize, and release siRNAs, as well as induce effective gene silencing. Our results demonstrated that incorporation of different stereochemical patterns of L/D-amino acids or a specific D-amino acid substitution within the 599 peptide design could in some instances increase/decrease the binding, nuclease/serum stability, and complex release of siRNAs, as well as influence the gene-silencing efficiencies of the complex, in comparison to the native 599 peptide. Moreover, these modifications in 599 peptide design were also found to affect cellular uptake and intracellular localization patterns of siRNA cargo to various degrees, with one particular 599 peptide variant that contained the specific D-amino acid substitution capable of mediating a more ordered binding of siRNAs to specific cellular projections, identified as filopodia. Interestingly, this specific variant also exhibited the most enhanced intracellular siRNA delivery and gene silencing in comparison to the native 599 peptide, thus implying that its peptide design modification could be responsible for directing a more efficient mode of siRNA drug delivery, resulting in the enhancement of gene silencing.
Results
L/D-amino acid stereochemical modifications to the 599 peptide carriers can either increase or decrease their ability to bind, deliver, protect, and release siRNAs
In advancing the 599 peptide carrier design for siRNA-based therapeutics, for the purposes of this study, we focused on the role of amino acid stereochemistry. Previously, it had been demonstrated that modulation of peptide stereochemistry could affect the cell uptake efficiency of both free arginine-rich-containing CPPs and that of antisense oligonucleotide-bound CPP polyplexes.16,23 Accordingly, to examine the effects of peptide stereochemistry on the delivery of siRNA cargo, we designed seven 599 peptide variants comprising different patterns of L/D-amino acids (see Table 1 for peptide sequences), with several designs based on the polyarginine stereopeptide sequences used in the Verdurmen et al.16 study. Additionally, we designed an eighth 599 peptide variant comprising a D-arginine substitution for D-alanine (RD3AD), based on evidence that a similar modification within a receptor-targeting CPP could lead to improved siRNA release from a multifunctional peptide complex.22
Table 1.
The amino acid sequences of 599 peptide and its L/D-amino acid stereochemically modified variants
| Peptide name | Peptide sequencea,b |
|---|---|
| 599 | GLFEAIEGFIENGWEGMIDGWYGGGGrrrrrrrrrK-biotin |
| RD3AD | GLFEAIEGFIENGWEGMIDGWYGGGGrrarrrrrrK-biotin |
| RD456 | GLFEAIEGFIENGWEGMIDGWYGGGGRRRrrrRRRK-biotin |
| All-L | GLFEAIEGFIENGWEGMIDGWYGGGGRRRRRRRRRK-biotin |
| RD19 | GLFEAIEGFIENGWEGMIDGWYGGGGrRRRRRRRrK-biotin |
| RD123789 | GLFEAIEGFIENGWEGMIDGWYGGGGrrrRRRrrrK-biotin |
| RD13579 | GLFEAIEGFIENGWEGMIDGWYGGGGrRrRrRrRrK-biotin |
| All-D | GlfeaieGfienGweGmidGwyGGGGrrrrrrrrrk-biotin |
| INF7D | GlfeaieGfienGweGmidGwyGGGGRRRRRRRRRK-biotin |
D-amino acids are indicated by lowercase letters.
Substituted D-arginine residue for D-alanine in RD3AD is underlined.
Upon chemically synthesizing the peptides, gel-shift assays were performed to determine first if L/D-amino acid stereochemical modifications to the 599 peptide could alter its ability to bind free siRNAs through electrostatic interactions between the anionic siRNAs and the C-terminal cationic polyarginine sequence found within the peptide. Using siCIP2A, an siRNA designed to target the CIP2A oncogene, it was found that complexation of this siRNA with increasing amounts of 599 peptide or its peptide variants, ranging from 1- to 50-fold molar excess of the siRNA, could significantly affect the binding properties of the 599 peptide to siRNAs (Figure 1; Table S1). In particular, at peptide-to-siRNA (Peptide:siRNA) molar ratios of 1:1 through 20:1 (equivalent to nitrogen:phosphate [N/P] ratios of 0.25 through 5 for all peptides except RD3AD, whose N/P ratios were slightly less due to the D-arginine substitution for D-alanine; please see Materials and methods section for the specific N/P ratio values for all peptides with respect to the Peptide:siRNA molar ratios), most 599 peptide variants were found to bind significantly better to the siRNA compared to the native 599 peptide, with peptides RD456 and RD19 showing the most consistent enhancement in binding. Conversely, peptides RD123789 and All-L (comprising only all L-amino acids), exhibited largely no significant changes in siRNA binding relative to 599. At Peptide:siRNA molar ratios of 30:1 (N/P ratios of 7.5) and greater, however, all peptide variants bound equally to ~100% of the siRNAs, with no significant differences observed in comparison to 599. Thus, these data indicated that siRNA-binding capacities to peptide carriers could be influenced by sequence-specific L/D-amino acid stereochemical changes, but only at Peptide:siRNA molar (N/P) ratios below a defined threshold.
Figure 1.

siRNA binding analysis of 599 and its peptide variants
Densitometric quantitation of siCIP2A levels complexed to 599 or its peptide variants at increasing Peptide:siRNA molar (nitrogen:phosphate [N/P]) ratios. Data are mean ± SEM of three independent samples, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and p ≥ 0.05 is not significant (ns) compared to 599 (2-way ANOVA).
To further examine the effects of the L/D-amino acid stereochemical modifications to the 599 peptide in its ability to deliver siRNAs into cells, we next proceeded to perform a series of quantitative uptake assays. Previously, it was determined that increasing the Peptide:siRNA molar ratio for the 599 peptide enhanced the delivery of the complexed siRNAs into cells with the 50:1 Peptide:siRNA molar (12.5 N/P) ratio being found to be optimal for the binding and intracellular delivery of siRNAs.12 Nevertheless, in this study, because ~100% of siRNAs were found to bind to the 599 peptide and its variants at the 30:1 Peptide:siRNA molar (7.5 N/P) ratio and greater, we decided to examine whether any of these variants exhibited improved siRNA cell uptake in comparison to the parent 599 peptide at both the 30:1 and 50:1 Peptide:siRNA molar (7.5 and 12.5 N/P) ratios. In monitoring the quantitative delivery of a DY-547 fluorophore-labeled siCIP2A (DY547-siCIP2A; Figure 2A), analysis of our data revealed that at the Peptide:siRNA molar ratio of 30:1 (N/P ratio of 7.5) no 599 peptide variant differed significantly from 599 in its ability to deliver siRNAs into CAL 27 cancer cells 2 h post-treatment, with the exception of INF7D, which had a dramatic and significant reduction in siRNA delivery into cells. However, at Peptide:siRNA molar ratios of 50:1 (N/P ratios of 12.5), differences in siRNA uptake became more apparent between the peptide variants, with RD19, RD123789, and RD3AD showing on average greater siRNA delivery into cells, but with only RD3AD exhibiting a significantly greater internalization, nearly 2-fold higher than 599. Conversely, RD13579, in addition to INF7D, resulted in significantly decreased levels of siRNA cell internalization. As a control, a commercially available lipid-based transfection reagent (LTR), Lipofectamine 3000 (LF3000), was also used, which was found to deliver siRNAs comparably to 599 and its variants at a Peptide:siRNA molar ratio of 30:1 (N/P ratio of 7.5) but significantly less compared to the 50:1 Peptide:siRNA molar (12.5 N/P) ratio. Visual inspection of the delivery of DY547-siCIP2As into the CAL 27 cancer cells mediated by 599 and its peptide variants (at the 50:1 Peptide:siRNA molar [12.5 N/P] ratio) using confocal fluorescence microscopy (Figure 2B) demonstrated largely a random punctate uptake pattern, similar to what was previously reported for 599,12 with the exception of RD3AD, which exhibited both random and highly ordered linear punctate uptake patterns (for visualization of the latter pattern see arrowheads in the RD3AD panel). Moreover, closer inspection of the RD3AD-treated cells revealed numerous large and small foci in comparison to cells treated with 599 or the other peptide variants, whose foci tended to be larger and fewer in number. Together, these differences in staining patterns for RD3AD were potentially indicative of an alternate mode of cell entry mediated by RD3AD that was more efficient in siRNA delivery, especially in light of the fact that higher levels of siRNA cell internalization were observed for this peptide in the quantitative uptake assays. Nevertheless, these results indicated that varying the peptide stereochemistry patterns within 599 could influence the degree of siRNA delivery into cells, but that a specific D-amino acid substitution within the 599 peptide carrier, as observed through RD3AD, was needed to enhance the cellular internalization of the complexed siRNA cargo. Of note, measurements of the physicochemical properties of the 599 peptide variants in complex with siRNAs at the 50:1 Peptide:siRNA molar (12.5 N/P) ratio, including particle size, zeta potential, and polydispersity index (Table 2), did not reveal any distinguishable characteristics between the complexes that could account for the observed differences in siRNA cellular uptake patterns or that would imply that these differences were a consequence of variances in particle aggregation properties. Nonetheless, based on the above findings, combined with the observation that none of the 599 peptide variants induced long-term cytotoxic effects at the 50:1 Peptide:siRNA molar (12.5 N/P) ratio compared to untreated cells (Figure S1), nor were they significantly different from 599 (Figure 3), this particular Peptide:siRNA molar ratio was used for subsequent experimentations.
Figure 2.

Intracellular delivery of siRNAs in complex with 599 or its peptide variants
(A) Quantitative siRNA uptake into CAL 27 cancer cells 2 h post-treatment with DY547-siCIP2A in complex with 599 or its peptide variants at either the 30:1 or 50:1 Peptide:siRNA molar (7.5 or 12.5 N/P) ratios. For comparison, the cells were also transfected using the commercial lipid-based transfection reagent (LTR), Lipofectamine 3000 (LF3000). The amount of siRNA delivered into cells in pmol per mg of protein is reported with each treatment normalized to DY547-siCIP2A alone. Data are mean ± SEM of three independent samples, where ∗p < 0.05, ∗∗p < 0.01, and p ≥ 0.05 is not significant (ns) compared to 599 (2-way ANOVA). (B) Confocal fluorescence microscopy analysis of CAL 27 cancer cells 2 h post-treatment with DY547-CIP2A (red) in complex with 599 or its peptide variants at 50:1 Peptide:siRNA molar (12.5 N/P) ratios. Nuclei (blue) were counterstained with DAPI. The arrowheads indicate highly ordered linear punctate uptake patterns of siRNAs in the RD3AD panel. Scale bar: 35 μm.
Table 2.
Physicochemical properties of 599 peptide and its L/D-amino acid stereochemically modified variants
| Peptide name | Particle size (nm)a | Zeta potential (mV)b | PDI |
|---|---|---|---|
| 599 | 172.2 ± 81.6 | 30.7 ± 10.6 | 0.349 |
| RD3AD | 213.3 ± 119.3 | 35.3 ± 8.9 | 0.336 |
| RD456 | 200.5 ± 98.5 | 39.2 ± 11.2 | 0.344 |
| All-L | 190.2 ± 82.4 | 39.4 ± 7.3 | 0.327 |
| RD19 | 211.4 ± 98.0 | 31.4 ± 6.7 | 0.381 |
| RD123789 | 224.1 ± 118.4 | 34.1 ± 6.5 | 0.342 |
| RD13579 | 201.2 ± 92.8 | 35.3 ± 7.2 | 0.325 |
| All-D | 185.9 ± 72.0 | 28.3 ± 7.7 | 0.366 |
| INF7D | 190.4 ± 94.6 | 28.2 ± 10.2 | 0.296 |
PDI, polydispersity index.
Mean hydrodynamic size based on dynamic light scattering measurements. Errors indicate SD from three independent experiments.
Mean zeta potential measurements. Errors indicate SD from three independent experiments.
Figure 3.

Assessment of cytotoxicity after treatment of cancer cells with 599 or its peptide variants in complex with siRNA
The long-term cellular toxicity (as measured by a cell proliferation assay) of CAL 27 cancer cells was assessed 48 h post-treatment with 599 or its peptide variants in complex with a non-targeting siRNA (siNT) at 50:1 Peptide:siRNA molar (12.5 N/P) ratios. Data are mean ± SEM of three independent samples, where p ≥ 0.05 is not significant (ns) compared to 599 (1-way ANOVA).
siRNAs delivered in vivo encounter RNases in the physiological environment (e.g., serum) that can degrade them before they reach their therapeutic target.9,13 Consequently, siRNA-delivery systems must be able to protect the small RNAs from degradation in order for them to maintain their gene-silencing function. Previously, 599 had been shown to protect siRNAs from degradation in the presence of RNase A and human serum.13 Therefore, to further assess whether L/D-amino acid stereochemical modifications to a peptide carrier could alter its siRNA protective ability, the resistance of 599 and its variants against RNases was assayed by complexing the peptides to siCIP2A and incubating either in the presence of RNase A or human serum for 1 h (Figure 4). Data from the assays found that in the presence of RNase A, only RD456 had a significant inability to protect siRNAs, resulting in ~25% of the complexed siRNAs being degraded. Alternatively, in the presence of human serum, several peptides, including, All-L, RD19, RD123789, and INF7D, were significantly less able to protect their complexed siRNAs, with degradation ranging between ~20% and 60%. All other 599 peptide variants, including RD3AD, RD13579, and All-D, did not differ from 599 in their ability to protect siRNAs in the presence of either RNase A or serum. Thus, together, these results implied that the stereochemistry of peptide carrier designs does play a critical role in the preservation of complexed siRNAs, with the L/D-arginines within the polyarginine tract of the 599 design, in particular, appearing to be the determinant factors in the sensitivity of the complexed siRNAs to RNase/serum-mediated degradation.
Figure 4.

Analyses of 599 and its peptide variants in their ability to protect siRNAs from RNase and serum-mediated degradation
Naked and peptide-complexed siCIP2As at a 0:1 and 50:1 Peptide:siRNA molar (0 and 12.5 N/P) ratio, respectively, were incubated in the absence or presence of either RNase A or 50% human serum for 1 h at 37°C, after which densitometric quantitation was performed to determine the levels of protected siRNAs. Data are mean ± SEM of three independent samples, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and p ≥ 0.05 is not significant compared to 599 (Student’s t test).
Gene-silencing efficiency is dependent on the release of siRNAs from the delivery complex upon entry into the cell cytoplasm to allow for incorporation of the siRNAs into the RNA-induced silencing complex (RISC).22,24 Thus, in an effort to explore whether specific L/D-amino acid stereochemical modifications could affect siRNA release dynamics, we assessed the dissociation of siRNAs from 599 and its peptide variants by performing quantitative heparin competition assays (Figure 5; Table S2). Results from these experiments demonstrated that the siRNAs began to dissociate from all the peptide complexes to varying degrees upon addition of ~6 pmol (0.1 μg) of heparin, with 599 capable of releasing approximately 100% of the complexed siRNAs upon increasing competitive amounts of the negatively charged reagent. No statistical differences in siRNA release were observed for RD456, RD19, RD123789, and INF7D, in comparison to 599. Conversely, RD3AD and All-L/All-D/RD13579 showed significant differences in terms of siRNA dissociation compared to 599, where ~90% and ~70% of their complexed siRNAs were maximally released, respectively, at 4-fold excess levels and higher (≥2 μg; ≥120 pmol) of heparin. Intriguingly, two out of the three peptide variants which exhibited the greatest impairment in siRNA release capability, were the carriers that comprised single amino acid stereochemistries (All-L or All-D). Moreover, although still very effective at releasing siRNAs, substitution of the third D-arginine for D-alanine within the polyarginine tract of RD3AD did not appear to be universal in conferring enhanced siRNA release from a peptide complex (at least compared to the parent peptide), as was reported by Jun et al.22 in their studies of a multifunctional peptide complex for delivery of siRNAs. Nonetheless, the fact that 6 out of 7 peptides that comprised both L- and D-amino acid sequence patterns showed ≥90% siRNA release, suggested that the incorporation of alternating amino acid stereochemistries within peptide carrier designs appeared to be an important factor in ensuring efficient siRNA release from the complex.
Figure 5.

Analysis of siRNA release from 599 and its peptide variant complexes
Densitometric quantitation of released siRNAs from 599 and its peptide variants complexed at the 50:1 Peptide:siRNA molar (12.5 N/P) ratio upon addition of increasing amounts of heparin (0–10 μg). Data are mean ± SEM of three independent samples, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and p ≥ 0.05 is not significant (ns) compared to 599 (2-way ANOVA).
L/D-amino acid stereochemical modifications to the 599 peptide siRNA-carrier affect the gene-silencing properties of the complex
599 has previously been shown to be an effective carrier in the delivery of siRNAs that could induce both in vitro and in vivo gene silencing.12,13 Nonetheless, the above findings that specific L/D-amino acid stereochemical changes could affect the binding, cell internalization, protection, and release of siRNAs mediated by 599 made evident the need to further assess the gene-silencing functionality of the 599 peptide variants in complex with siRNAs. By doing so, this would help ascertain whether these properties were related to enhancements in gene silencing and whether there was potential for the advancement in the peptide carrier design. Consequently, to assess gene silencing efficiencies mediated by 599 and its peptide variants, CAL 27 cancer cells were treated with 599 or its peptide variants complexed with either a control non-targeting siRNA (siNT) or siCIP2A, after which the CIP2A mRNA levels were assessed 48 h post-treatment by real-time PCR. Quantitation of the results demonstrated significant knockdowns in CIP2A mRNA levels for all the peptides tested, ranging between ~50% and 80%, with RD3AD having the highest observed reduction in CIP2A mRNA levels at approximately 80% (Figure 6A). However, in comparison to 599, only two peptide variants, RD3AD and All-L, were found to exhibit significant enhancements in gene silencing, with RD3AD showing the greatest improvement at ~30% (Figure 6B). To further corroborate the real-time PCR results, western blot analyses were performed, which confirmed the ability of the 599 peptide variants to deliver functional siRNAs by demonstrating the suppression of CIP2A protein levels 48 h post-treatment (Figure 6C). Intriguingly, the data also appeared to support RD3AD as having the highest observed reduction in CIP2A protein levels among the peptide variants in comparison to 599. Thus, these findings demonstrated that the gene-silencing functionality of the 599-siRNA complex could be influenced by varying the stereochemical patterns within the peptide carrier design, but that a specific D-amino acid substitution, as observed through RD3AD, was particularly significant because it revealed its potential in advancing the 599 peptide carrier design for siRNA-based therapeutics.
Figure 6.

CIP2A gene silencing mediated by 599 and its peptide variants in complex with siCIP2A
(A) Real-time PCR analysis of CIP2A mRNA levels in CAL 27 cancer cells 48 h post-treatment with siNT or siCIP2A in complex with either 599 or its peptide variants at 50:1 Peptide:siRNA molar (12.5 N/P) ratios. The CIP2A mRNA levels were normalized to endogenous 18S rRNA. Data are mean ± SEM of three independent samples, where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and p ≥ 0.05 is not significant compared to 599 (Student’s t test). (B) Fold change in CIP2A silencing mediated by the 599 peptide variants normalized to 599. Data are mean ± SEM of three independent samples, where ∗∗p < 0.01, ∗∗∗p < 0.001, and p ≥ 0.05 is not significant (ns) compared to 599 (Student’s t test). (C) Western blot analyses of CIP2A protein expression levels in CAL 27 cancer cells 48 h post-treatment with siNT or siCIP2A in complex with either 599 or its peptide variants at 50:1 Peptide:siRNA molar (12.5 N/P) ratios. β-actin protein levels were monitored to ensure equal loading of samples.
RD3AD-siRNA complexes colocalize with filopodia on cell surfaces
Having found that RD3AD in complex with siRNAs could enhance gene silencing and that this peptide mediated increased cellular internalization of the complexed siRNA cargo with an apparent highly ordered linear accumulation of siRNA-containing foci at what appeared to be the cell periphery implied that its mode of cell entry was potentially more efficient compared to 599 and the other peptide variants. Thus, in an effort to better resolve these structures and elucidate a mechanism responsible for the improved uptake of siRNAs and, consequently, the enhanced gene silencing mediated by RD3AD, the delivery of fluorophore-labeled siRNAs into cells mediated by 599 and its peptide variants were assessed at a high magnification using confocal fluorescence microscopy (Figure 7A). Results from these experiments verified that treatment of CAL 27 cancer cells with 599 or its peptide variants in complex with DY547-siCIP2As predominantly exhibited irregular-shaped foci of varying sizes (most of which were on the larger side) that localized randomly to both the periphery of cell membranes, as well as the cytoplasm, 2 h post-treatment. Notably, the exception was RD3AD, which showed a highly ordered accumulation of uniformly shaped spherical foci that lined up along cell surface protrusions, identified as filopodia. Interestingly, filopodia are highly dynamic, elongated, and thin cellular processes that have been reported to facilitate the highly efficient cell entry of viruses, bacteria, activated receptors, lipo/polyplexes, and exosomes.25, 26, 27, 28, 29 Thus, the association of the RD3AD-siRNA complex with filopodia could explain why a greater accumulation of small siRNA-containing foci were also detected in the cytoplasm of treated cells versus the 599-siRNA complex, which conversely did not appear to localize along filopodia and only showed a limited uptake of its siRNA cargo in mostly large punctate structures (Figure 7B). Further examination into whether the staining pattern for RD3AD-mediated siRNA uptake was reproducible in another cancer cell line likewise demonstrated that treatment of SCC-90 cells with either RD3AD or 599 in complex with DY547-siCIP2As produced the same staining pattern differences observed for both peptide carriers in CAL 27 treatments (Figure 7C). More specifically, the RD3AD-siRNA complexes were found to exhibit the same dramatic “beads-on-a-string” pattern along filopodia, in addition to an accumulation of numerous small cytoplasmic foci, whereas 599-siRNA complexes were predominantly larger, irregularly shaped structures that were fewer in number and localized to both the cytoplasm and periphery of the cell membrane with no apparent associations with filopodia. The fact that the staining pattern we observed for RD3AD-mediated siRNA uptake in both cancer cell lines was very reminiscent of the patterns previously reported for cell entry of viruses, bacteria, activated receptors, lipo/polyplexes, and exosomes25, 26, 27, 28, 29 implied that this peptide carrier could potentially exploit similar cell machineries for efficient delivery of its cargo by using filopodia to gain entry into cells, thereby contributing to the enhancement of gene silencing. In effect, time-course analyses of cells treated with RD3AD in complex with DY547-siCIP2A clearly demonstrated an accumulation of siRNAs along filopodia, the cell periphery, and within the cell over a period of 4 h in comparison to cells treated with 599-siRNA complexes, with the most noticeable differences observed at the 4-h time period (Figure 8). Moreover, cross-sectional analyses of the treated cells at the 4-h time period corroborated the increased uptake of siRNAs within the cells, as evidenced by the greater degree of siRNA colocalization with cytoplasmic F-actin, which helped demarcate the cell body in addition to filopodia. Conversely, 599 treatments appeared to show the siRNAs predominantly localized on the surfaces of cell bodies with limited intracellular uptake. Thus, together, these data strengthened the notion that RD3AD appeared to mediate a more efficient mode of cell entry, possibly through its associations with filopodia.
Figure 7.

Cellular localization of siRNAs mediated by 599 and its peptide variants
(A) Confocal fluorescence microscopy analysis of CAL 27 cancer cells 2 h post-treatment with DY547-siCIP2A (red) in complex with 599 or its peptide variants at 50:1 Peptide:siRNA molar (12.5 N/P) ratios. Filopodia (green) were stained with the selective F-actin label Alexa Fluor 488 phalloidin and nuclei (blue) were counterstained with DAPI. Scale bar: 17 μm. (B) Confocal fluorescence microscopy images of 599 and RD3AD-mediated localization of DY547-siCIP2A in CAL 27 cancer cells from (A) separated into their individual fluorophore signals. The merged images are also presented. Scale bar: 17 μm. (C) Confocal fluorescence microscopy analysis of SCC-90 cancer cells 2 h post-treatment with DY547-siCIP2A (red) in complex with 599 or RD3AD at a 50:1 Peptide:siRNA molar (12.5 N/P) ratio. Filopodia (green) were stained with the selective F-actin label Alexa Fluor 488 phalloidin and nuclei (blue) were counterstained with DAPI. Scale bar: 27 μm.
Figure 8.

Time-course analyses of siRNA cellular localization mediated by RD3AD in comparison to 599
Confocal fluorescence microscopy analyses of CAL 27 cancer cells 0.5, 1, 2, and 4 h post-treatment with DY547-siCIP2A (red) in complex with 599 or RD3AD at a 50:1 Peptide:siRNA molar (12.5 N/P) ratio. Alexa Fluor 488 phalloidin was used to stain the F-actin (green) in filopodia, as well as the cytoplasm of cells, so as to help demarcate the cell bodies. Nuclei (blue) were counterstained with DAPI. The upper-half panels represent merged images of the red and blue fluorophore signals only, while the lower-half panels represent all three (red, blue, and green) fluorophore signals combined. Cross-sectional views comprising z sections along the x and y planes are also presented within each panel. Yellow fluorescence reveals the potential colocalization between siRNA and F-actin. Scale bar: 70 μm.
Discussion
The results presented here highlight the importance of peptide stereochemistry, in particular, the modification of specific L/D-amino acid sequence patterns in peptide carrier designs, with regard to siRNA complexation, intracellular delivery, RNase/serum sensitivity, complex release, and gene-silencing efficiency. Moreover, our findings notably demonstrate that a peptide carrier, through a D-amino acid substitution, can enhance siRNA delivery and consequently gene silencing via potential “surfing” along filopodia. As stated previously, conventional thought was that stereochemistry in peptide carrier designs was primarily used to confer proteolytic resistance function to the peptide-siRNA complex, making the peptide and its therapeutic cargo more stable for in vivo therapeutic applications. However, Zeller et al.21 alternatively found that the proteolytic digestion of the peptide carrier in endosomes was actually critical to prolonging the dynamics of gene silencing due to the enhanced release of siRNAs from late endosomes to the cytosol. Thus, these data made evident the need for a balance in the design of peptide carriers when modulating their L/D-amino acid stereochemistries. Additionally, in studies by Verdurmen et al.16 and Favretto et al.,23 it had been demonstrated that modulation of peptide stereochemistry could also affect the cell uptake efficiency of both free arginine-rich-containing CPPs and that of antisense oligonucleotide-bound CPP polyplexes, further implying that peptide stereochemistries could potentially confer additional functions, beyond the prototypical protease-resistant function. Accordingly, to further expand our knowledge in the application of stereochemistry in peptide carrier designs, this study sought to determine how specific L/D-amino acid stereochemical modifications within the 599 peptide carrier, which we previously designed, may impact peptide carrier properties/functions and advance the 599 peptide carrier design for the enhancement of gene silencing in cancer cells.
Because the 599 peptide was designed to electrostatically complex with siRNAs,12 we initially examined whether alteration of L/D-amino acid sequence patterns, as well as a specific D-arginine substitution for D-alanine, would alter the binding affinity of the 599 peptide carrier. Results from our studies found that these modifications to the 599 peptide could indeed alter the ability to bind free siRNAs in a dose-dependent manner, with all peptide variants binding ~100% of siRNAs at ≥30:1 Peptide:siRNA molar (≥7.5 N/P) ratios. Although there were no differences in siRNA binding at this particular Peptide:siRNA molar ratio and greater, most peptides (with the exception of All-L and RD123789) did bind significantly better than 599 between the 1:1 and 20:1 Peptide:siRNA molar (0.25 to 5 N/P) ratios, with RD456 showing the most significant improvement overall. Moreover, 6 out of the 8 peptide variants exhibited the highest differences in binding ability at the 10:1 Peptide:siRNA molar (2.5 N/P) ratio, with RD456, RD19, and INF7D showing a ~20% binding improvement relative to 599. This finding was particularly interesting because 599, which has been previously determined to function optimally at the 50:1 Peptide:siRNA molar (12.5 N/P) ratio, is typically formulated by mixing the relative molar amounts of both the peptide and siRNA in one reaction.12 Therefore, one could potentially envision improving the siRNA binding capacity of the complex, by complexing RD456, for example, by sequentially adding the peptide five times to the siRNA (with the starting siRNA amount increased by ~20%) at 10:1 Peptide:siRNA molar (2.5 N/P) ratio increments, to allow it to still function near the optimal Peptide:siRNA molar ratio.
Subsequent investigation into the intracellular delivery of siRNAs mediated by the 599 peptide variants revealed that even though all the peptides bound ~100% of the siRNAs at the 30:1 Peptide:siRNA molar (7.5 N/P) ratio, increasing the Peptide:siRNA molar ratio to 50:1 (N/P ratio of 12.5) further increased siRNA uptake into cells 2 h post-treatment in comparison to naked siRNAs for 5 out of 7 peptides, in addition to 599, which was consistent with our previously published data pertaining to 599,12 along with other reports that similarly found that increasing CPP concentrations typically resulted in increased siRNA uptake ability.30,31 Regarding the stereochemical changes to the 599 peptide design, alteration of L/D-amino acid sequence patterns was found to only adversely affect the intracellular delivery of siRNAs for two peptides, INF7D and RD13579, in comparison to 599, but otherwise these changes were largely tolerable, with no significant differences observed for the other 5 peptides in comparison to 599. These data did, however, make evident that changing the chirality of the fusogenic INF7 N-terminal region to the non-natural D-amino acids was potentially detrimental to the uptake of siRNAs, as observed through All-D and INF7D, which both showed on average lower siRNA uptake abilities in comparison to 599, suggesting that this type of modification was to be avoided, possibly due to its functional importance in membrane destabilization.32 Notably, the only peptide that showed a dramatic and significant increase in intracellular siRNA delivery, which was nearly 2-fold higher than 599, was RD3AD, which was modified with a specific D-arginine substitution for D-alanine and whose design was based on evidence that a similar modification within a receptor-targeting CPP could lead to enhanced gene silencing mediated by a multifunctional peptide complex.22 Interestingly, confocal fluorescent microscopy analyses of the cellular uptake patterns for both the stereochemically modified peptides and RD3AD in complex with fluorophore-labeled siRNAs revealed that all the peptide variants, with the exception of RD3AD, exhibited predominantly random punctate patterns whose foci tended to be larger and fewer in number, similar to 599, the difference being that RD3AD also showed highly ordered linear punctate uptake patterns, as well as both numerous large and small foci. Thus, these differences in cellular uptake patterns for RD3AD could possibly account for the higher detectable levels of siRNAs found within cells treated with RD3AD-siRNA complexes and could be indicative of an alternate and more efficient mode of cell entry compared to 599 and the other peptide variants.
In further analyzing the effects of the specific L/D-amino acid stereochemical modifications on 599 peptide properties, in terms of cytotoxicity, none of the peptide variants differed significantly in their ability to affect the long-term viability of treated cells compared to 599 or to untreated cells (with the latter comparison also true for 599, which corroborated our previous findings that 599 treatments in complex with siRNAs were non-cytotoxic12). Conversely, regarding siRNA stability, the data did, however, suggest that these modifications did play a critical role in the preservation of complexed siRNAs, with the L/D-arginines within the polyarginine tract of the 599 design, in particular, appearing to be the determinant factors in the sensitivity of the complexed siRNAs to RNase/serum-mediated degradation. Intriguingly, the central arginine residues (#4, 5, and 6) of the nona-arginine tract appeared to be key in determining the stability of the complex, as RD456 was found to be the only peptide variant whose complexed siRNA was susceptible to direct RNase treatment, resulting in ~25% siRNA degradation. Furthermore, in the presence of serum (which contains both proteases and nucleases8,18,33), the fact that peptides All-L, RD19, RD123789, and INF7D, which all had L-arginines at positions #4, 5, and 6 of the nona-arginine tract, were significantly less able to protect their complexed siRNAs compared to 599, with degradation ranging between ~20% and 60%, whereas RD456 could, indicted that the incorporation of L-amino acid chirality at these three positions was to be avoided, as it would make the complex more susceptible to proteolytic digestion, resulting in its destabilization, with a consequent exposure of the siRNAs to serum nucleases. Thus, it appeared that because the RD456 design renders the peptide susceptible to RNase-mediated degradation of its siRNA cargo and maintaining L-arginines within the central region of the polyarginine tract (All-L, RD19, RD123789, and INF7D) renders the peptide susceptible to protease degradation, these particular modifications were to be avoided in any advancement of 599 peptide designs. Additionally, the data pertaining to siRNA release also indicated that the incorporation of alternating amino acid stereochemistries within the 599 peptide carrier design appeared to be an important factor in ensuring efficient siRNA release from the complex, accounting for ≥90% siRNA release, as 2 out of 3 peptides that had the poorest release capabilities comprised single amino acid stereochemistries (All-L or All-D). Interestingly, this parity in poor siRNA release capabilities between All-L and All-D corroborated the findings from an earlier study by Favretto et al.,23 which likewise found that polyplexes comprising either all D- or all L-CPPs were less likely to fall apart and were equally stable in the presence of heparin when formulated at high peptide concentrations. Taken together, these data also potentially implied that the poor siRNA release mediated by All-L and All-D were the consequence of greater complex stability conferred by the single amino acid stereochemistry peptide designs. However, it should be noted that alternating the chirality for every other amino acid within the polyarginine tract, which is responsible for electrostatically complexing the siRNAs,12 was also found to be equally poor at releasing siRNAs from the complex, as was the case for RD13579. Surprisingly, although still very effective at releasing siRNAs, substitution of the third D-arginine for D-alanine within the polyarginine tract of RD3AD did not universally confer enhancement of siRNA release from the peptide complex (at least compared to the parent peptide), as was reported by Jun et al.22 in their studies of a multifunctional peptide complex for delivery of siRNAs. Thus, the introduction of an uncharged amino acid into the polyarginine tract, to decrease the electrostatic interactions between the siRNA and peptide carrier, does not necessarily result in greater release of the siRNA. Interestingly, the Jun et al.22 study, however, never made clear the chirality of the substituted alanine residue in their peptide carrier design, thus opening the possibility that this discrepancy in findings could be related to L/D-amino acid stereochemistry differences.
Despite the differences in the functional properties described above, the modification of L/D-amino acid sequence patterns in the 599 peptide carrier design still resulted in effective gene silencing of the targeted CIP2A oncogene, ranging between ~50% and 70% knockdown. However, it was RD3AD, with its specific D-amino acid substitution, that showed the greatest silencing at ~80%. Moreover, although most peptide variants showed improvements, only RD3AD and All-L were found to be significantly more efficient, ~30% and ~20%, respectively, at gene silencing than 599. The observation that All-L was more efficient at gene silencing compared to 599 was not unexpected considering the Zeller et al.21 study, which reported that L-arginine-rich CPPs, but not D-arginine-rich CPPs, could prolong the dynamics of gene silencing due to the requirement for partial degradation of arginine-rich CPPs by endosomal proteases in order to enable more effective endosome-to-cytosol translocation of the complexed siRNAs. The fact that the All-L-siRNA complex was found to be unstable in serum compared to 599-siRNA complexes suggested that its design was more readily susceptible to proteolytic degradation and therefore better at translocating siRNAs from the endosome to the cytosol. The same could not be said for INF7D, however, which also comprised an all L-amino acid polyarginine tract and whose complex had the greatest instability in serum, but at the same time also had the poorest siRNA cell uptake, which most likely counteracted and contributed to it having the weakest gene-silencing effect among the peptide variants.
Finally, the findings that RD3AD exhibited the most enhanced siRNA delivery into cells, as evidenced by the quantitative uptake assays and further corroborated by 2-dimensional/3-dimensional (2D/3D) confocal fluorescence imaging, and consequently greater gene silencing, were significant because they implied that its mode of cell entry was potentially more efficient compared to 599 and the other peptide variants. In fact, the discovery that RD3AD-siRNA complexes could localize along filopodia on cell surfaces supported this notion, especially in light of the fact that viruses, bacteria, activated receptors, lipo/polyplexes, and exosomes have been reported to bind to filopodia and induce a rapid, but directed, retrograde surfing movement toward the cell body for entry into cells.25, 26, 27, 28, 29 Particularly noteworthy is that at the filopodial base are endocytic hotspots, which have been described as active areas of actin remodeling that potentially allow for easier cell entry, in comparison to other sites along the cell membrane that are more difficult to penetrate due to the dense cortical actin cytoskeleton.26 Interestingly, in the case of exosomes, they have been found to sort into endosomal trafficking circuits at the base of filopodia that are targeted to the endoplasmic reticulum (ER) as a possible site of cargo release.25 Coincidentally, the ER has also been identified as the central nucleation site of siRNA-mediated silencing.34 Thus, this directed transport along filopodia to endocytic hotspots, followed by endocytosis and trafficking to the ER, could potentially allow for the efficient entry of siRNA cargo to cellular RNAi machinery that could be exploited by peptide carriers.
In conclusion, our data collectively demonstrate the utility of peptide stereochemistry, as well as the importance of a key D-amino acid modification, in advancing the 599 carrier design for the enhancement of gene silencing in cancer cells. Moreover, this study makes evident the need for a balance in the design of peptide carriers when modulating their stereochemistry, but also demonstrates the potential in fine-tuning peptide carrier properties/functions by altering their L/D-amino acid sequence patterns. It will be interesting in future studies to further delineate the mechanisms of cell uptake and expand upon how peptide stereochemistry and/or the specific D-alanine substitution in the 599 peptide carrier design might exploit host cell machineries, like filopodia, to gain more efficient siRNA drug entry into cells. Additionally, future studies will need to confirm the direct cell uptake action of filopodia in mediating the intracellular delivery of RD3AD-siRNA complexes, which could be tested through the use of several chemical inhibitors, such as cytochalasin D and SMIFH2, which are known inhibitors of filopodia-mediated retrograde trafficking and filopodia structures, respectively.25,35, 36, 37 Furthermore, because lipo/polyplexes have been reported to use syndecan-dependent transport mechanisms in filopodia to reach the cell surface, one could also envision testing whether heparinase and sodium chlorate, which are known disruptors of syndecans,28 can likewise impair filopodia-mediated cell uptake of RD3AD-siRNA complexes. Nonetheless, the future study of these modifications in peptide carrier-siRNA cargo complex formation/disassembly and function from both a mechanobiology and 3D structural perspective are just as enticing, which could lead to further advancement of the 599 peptide carrier design and its prospective translation to the clinic as a delivery vehicle for siRNA-based human cancer therapies.
Materials and methods
Peptide synthesis
The peptides listed in Table 1 were synthesized by the solid-phase peptide synthesis process and purified (>95% purity) by high-performance liquid chromatography (HPLC) at GenScript (Piscataway, NJ, USA). D-amino acids were used directly during the synthesis process, and hydroxyl benzotriazole (HBOt) was added to suppress racemization. All peptides were also biotinylated on the C-terminal lysine residue, similar to 599,12 which was originally added to the design to serve as a biological tag. Electrospray ionization mass spectrometry and HPLC analyses of the purified peptides did not reveal any instances of amino acid racemization.
siRNAs
siCIP2A, its DY-547 fluorophore-labeled derivative, DY547-siCIP2A, and the negative control siGENOME Non-Targeting siRNA #5, siNT, were previously described12, 13, 14,38 and synthesized by Horizon Discovery Dharmacon (Lafayette, CO, USA).
Cell culture
Human tongue squamous cell carcinoma (SCC) cell lines CAL 27 and UPCI:SCC090 (SCC-90) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines were cultured in ATCC-specified complete growth medium in a 37°C incubator with 5% CO2. All experiments using these two cell lines were performed at passages less than 20.
siRNA binding assay
30 pmol of siCIP2A was complexed with 599 peptide (or the peptide variants) at 1:1, 5:1, 10:1, 20:1, 30:1, 40:1, and 50:1 Peptide:siRNA molar ratios (equivalent to 0.25, 1.25, 2.5, 5, 7.5, 10, and 12.5 N/P ratios, respectively, with the exception of RD3AD, whose N/P ratios were instead 0.225, 1.125, 2.25, 4.5, 6.75, 9, and 11.25, respectively) at room temperature (RT) for 25 min. Afterward, the samples were electrophoresed on a 4% agarose gel and stained with ethidium bromide. Free siCIP2A was used as a normalizing control. An Ultra-Low Range DNA Ladder (Thermo Fisher Scientific, Waltham, MA, USA) was used as a molecular weight marker. Resulting siCIP2A bands were imaged and quantified using a G:Box Chemi XX6 and GeneSys image capture software (Syngene, Frederick, MD, USA), respectively.
Quantitative siRNA cell uptake assay
CAL 27 cells, grown to a confluency of 60% on a 24-well plate, were rinsed three times with Opti-MEM I Reduced Serum medium (Opti-MEM; Thermo Fisher Scientific). Meanwhile, 60 pmol of DY547-siCIP2A was incubated either alone or with the 599 peptide (or the peptide variants) at 30:1 and 50:1 Peptide:siRNA molar ratios at RT for 25 min to allow for complexation. After complexation, the total volume was brought to 600 μL with Opti-MEM, after which the cells were incubated with 500 μL of peptide-siCIP2A complexes for 2 h at 37°C with 5% CO2. As an additional control, the cells were also transfected with 50 pmol of DY547-siCIP2A using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s instructions. After treatment, the cells were rinsed three times with phosphate-buffered saline (PBS) before being trypsinized for 10 min at 37°C, followed by centrifugation for 5 min at 1,000 × g at 4°C. The cells were then washed with ice-cold PBS and centrifuged again for 5 min at 1,000 × g at 4°C. Next, the cells were lysed in 250 μL ice-cold 0.1 M NaOH. The cell lysates were centrifuged at 4°C for 5 min at 10,000 × g to remove cell debris. Afterward, 100 μL of each sample was transferred to a black 96-well plate to measure fluorescence at 530/590 nm using a BioTek (Winooski, VT, USA) Synergy HT plate reader. Fluorescence measurements were converted to the amount of internalized siRNAs using a standard curve that was generated per experiment using known concentrations of DY547-siCIP2A ranging from 0–100 fmol/μL. Subsequently, the amount of internalized siRNA was normalized to the amount of protein, which was quantitated using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Confocal fluorescence microscopy analysis of cell uptake and localization of siRNAs
CAL 27 (or SCC-90) cells, grown to a confluency of 60% on BioCoat Collagen Type I coated 8-chamber slides (Corning, Corning, NY, USA), were rinsed three times with Opti-MEM. Meanwhile, 20 pmol of DY547-siCIP2A was complexed with 599 peptide (or the peptide variants) at 50:1 Peptide:siRNA molar ratios for 25 min at RT in Opti-MEM. Afterward, the complexes were added to the cells for incubation at 37°C with 5% CO2 for 2 h. For the time-course experiment, the cells were incubated with the specified complexes for 0.5, 1, 2, and 4 h. Subsequently, the cells were rinsed with PBS, fixed in 4% paraformaldehyde at RT for 10 min, and then the slides were either mounted with coverslips using VECTASHIELD Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA) or permeabilized with 0.1% Triton X-100 at RT for 5 min and then incubated with Alexa Fluor 488 phalloidin (Thermo Fisher Scientific) for 20 min. Afterward, the slides containing the permeabilized cells were mounted with coverslips, as described above. Fluorescence images were obtained using a Zeiss (Thornwood, NY, USA) 880 LSM NLO (with a Fast Airyscan super-resolution detector) confocal microscope equipped with ×25 and ×63 objectives.
Physicochemical measurements
The hydrodynamic diameter, zeta potential, and polydispersity index of 599 and its peptide variants in complex with siCIP2A formulated at a 50:1 Peptide:siRNA molar (12.5 N/P) ratio in water was measured using a Malvern Panalytical (Westborough, MA, USA) Zetasizer ZS instrument.
Viability assay
Long-term cell viability was assessed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). Briefly, CAL 27 cells were grown to 60% cellular confluency on 96-well plates, after which the cells were treated in Opti-MEM with 125 nM siNT in complex with 599 peptide (or the peptide variants) at 50:1 Peptide:siRNA molar ratios for 4 h, prior to adjusting the siRNA and fetal bovine serum (FBS) concentrations to 100 nM and 10%, respectively, by adding 50% FBS/Opti-MEM media. After 48 h of treatment, the cell viability was assayed, according to the manufacturer’s instructions. Absorbance at 490 nm was measured using a BioTek Synergy HT plate reader. Untreated cells were defined as 100% viable.
RNase protection assay
30 pmol of siCIP2A was complexed with 599 peptide (or the peptide variants) at 50:1 Peptide:siRNA molar ratios at RT for 25 min. Afterwards, complexes were either treated or untreated with 0.18 μg of RNase A (Thermo Fisher Scientific) for 1 h at 37°C, followed by denaturation with 4% sodium dodecyl sulfate (SDS). Next, the samples were electrophoresed on a 4% agarose gel and resulting siCIP2A bands were imaged and quantified, as described above.
Serum protection assay
30 pmol of siCIP2A was complexed with 599 peptide (or the peptide variants) at 50:1 Peptide:siRNA molar ratios at RT for 25 min. Afterwards, complexes were either treated or untreated with 50% human serum (final concentration; VWR, Radnor, PA, USA) for 1 h at 37°C, followed by denaturation with 4% SDS. Next, the samples were electrophoresed on a 4% agarose gel, and resulting siCIP2A bands were imaged and quantified, as described above.
siRNA release assay
30 pmol of siCIP2A was complexed with 599 peptide (or the peptide variants) at 50:1 Peptide:siRNA molar ratios at RT for 25 min. Afterward complexes were incubated with either 0, 0.1, 0.5, 1.0, 2.0, 5.0, or 10 μg heparin (Millipore Sigma, St. Louis, MO, USA) for 30 min at RT. The samples were then electrophoresed on a 4% agarose gel and resulting siCIP2A bands were imaged and quantified, as described above.
Gene silencing in cells
CAL 27 cells, grown to a confluency of 60% on 24-well plates, were rinsed three times with Opti-MEM. Meanwhile, 60 pmol of either siNT or siCIP2A were complexed with 599 peptide (or the peptide variants) at 50:1 Peptide:siRNA molar ratios for 25 min at RT in Opti-MEM. Afterward, the complexes were added to the cells at a siRNA concentration of 125 nM and incubated for 4 h at 37°C with 5% CO2, prior to adjusting the siRNA and FBS concentrations to 100 nM and 10%, respectively, with the addition of 50% FBS/Opti-MEM media. 48 h post-treatment, the total RNA or protein was harvested for subsequent real-time PCR and western blot analyses.
Real-time PCR
Total RNA was harvested using the RNeasy Mini Kit (QIAGEN, Germantown, MD, USA). RNA was then reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) in a Bio-Rad (Hercules, CA, USA) T100 Thermal Cycler. Next, quantitative real-time PCR was performed in an Applied Biosystems StepOnePlus Real-Time PCR machine (Thermo Fisher Scientific) using the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) and predesigned TaqMan Gene Expression Assays (Thermo Fisher Scientific) for CIP2A (Hs00405413_m1) and 18S (4319413E), according to the manufacturer’s instructions.
Western blot analysis
Treated cells were washed twice with PBS and lysed using ice-cold RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1% NP-40) with protease inhibitor cocktail (Thermo Fisher Scientific). The protein lysates were quantified using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and then resolved by SDS-PAGE on a Mini-PROTEAN TGX stain-free, 4%–20% gradient gel (Bio-Rad). Afterward, samples were transferred to a nitrocellulose membrane using the Bio-Rad Trans-Blot Turbo Transfer System. The nitrocellulose membrane was then cut into two pieces at ~60 kDa, to generate an upper membrane (≥60 kDa) and lower membrane (<60 kDa), after which both were blocked in 5% (w/v) nonfat dried milk in Tris-HCl-buffered-saline-0.1% Tween (TBS-T; pH 7.5) for 1 h at RT. The upper membrane was then incubated with mouse monoclonal anti-CIP2A antibody (1:500, clone 2G10-3B5, Santa Cruz Biotechnology, Santa Cruz, CA, USA), and the lower membrane was incubated with mouse monoclonal anti-β-actin antibody (1:5,000, clone AC-15, Millipore Sigma) in 5% (w/v) nonfat dried milk in TBS-T overnight at 4°C. Subsequently, the membranes were washed four times, 5 min each, with TBS-T and then incubated with horseradish peroxidase-conjugated-goat anti-mouse secondary antibody (1:3,000 or 1:5,000, SouthernBiotech, Birmingham, AL, USA) in 5% (w/v) nonfat dried milk in TBS-T for 1 h at RT. Immunoreactive bands were detected using Bio-Rad Clarity or Bio-Rad Clarity Max substrate systems, according to the manufacturer’s instructions, with resulting images captured using a G:Box Chemi XX6 system (Syngene).
Statistical analysis
The results are presented as mean ± standard error of the mean (SEM) or standard deviation (SD). Statistical significance was determined using either a Student’s t test or 1-way/2-way ANOVA for comparisons between groups. A p value <0.05 was considered statistically significant. Statistical analyses for all studies were performed using GraphPad Prism 8 software (San Diego, CA, USA).
Acknowledgments
We thank Aaron Reed for his technical insights and assistance with experiments. This work was supported in part by NIDCR grants R21DE027231 (A.J.) and T32DE017551 (C.E.H.); Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, MUSC (NCI P30CA138313); Bioengineering Center for Regeneration and Formation of Tissues, Clemson University (NIGMS P30GM131959); MUSC Summer Health Professions Research Program-College of Dental Medicine (T.H. and C.W.); MUSC Summer Undergraduate Research Program-College of Graduate Studies (C.R.F.); and US Government Federal Work-Study Program (C.R.F.).
Author contributions
C.E.H. conducted the confocal fluorescence microscopy and gene silencing experiments. C.R.F. performed the siRNA binding, stability, and release assays. T.H. conducted the quantitative siRNA uptake assay. G.H. performed the physicochemical measurements and N.V. aided with the data analyses and interpretation. R.D.W. performed the viability assay. P.V.P., E.B., J.C.C., and C.W. helped conduct the siRNA stability, siRNA release, gene silencing, and confocal fluorescence microscopy experiments, respectively. C.E.H. and A.J. designed the experiments, as well as analyzed and interpreted all the data. C.E.H. and A.J. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.03.013.
Supplemental information
References
- 1.Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Rana T.M. Illuminating the silence: understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 3.Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 4.Cummings J.C., Zhang H., Jakymiw A. Peptide carriers to the rescue: overcoming the barriers to siRNA delivery for cancer treatment. Transl. Res. 2019;214:92–104. doi: 10.1016/j.trsl.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobbin M.L., Rossi J.J. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu. Rev. Pharmacol. Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- 6.Das M., Musetti S., Huang L. RNA Interference-Based Cancer Drugs: The Roadblocks, and the “Delivery” of the Promise. Nucleic Acid Ther. 2019;29:61–66. doi: 10.1089/nat.2018.0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuckerman J.E., Davis M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015;14:843–856. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 8.Whitehead K.A., Langer R., Anderson D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavrilov K., Saltzman W.M. Therapeutic siRNA: principles, challenges, and strategies. Yale J. Biol. Med. 2012;85:187–200. [PMC free article] [PubMed] [Google Scholar]
- 10.Singh A., Trivedi P., Jain N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018;46:274–283. doi: 10.1080/21691401.2017.1307210. [DOI] [PubMed] [Google Scholar]
- 11.Marquez A.R., Madu C.O., Lu Y. An Overview of Various Carriers for siRNA Delivery. Oncomedicine. 2018;3:48–58. [Google Scholar]
- 12.Cantini L., Attaway C.C., Butler B., Andino L.M., Sokolosky M.L., Jakymiw A. Fusogenic-oligoarginine peptide-mediated delivery of siRNAs targeting the CIP2A oncogene into oral cancer cells. PLoS ONE. 2013;8:e73348. doi: 10.1371/journal.pone.0073348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander-Bryant A.A., Dumitriu A., Attaway C.C., Yu H., Jakymiw A. Fusogenic-oligoarginine peptide-mediated silencing of the CIP2A oncogene suppresses oral cancer tumor growth in vivo. J. Control. Release. 2015;218:72–81. doi: 10.1016/j.jconrel.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander-Bryant A.A., Zhang H., Attaway C.C., Pugh W., Eggart L., Sansevere R.M., Andino L.M., Dinh L., Cantini L.P., Jakymiw A. Dual peptide-mediated targeted delivery of bioactive siRNAs to oral cancer cells in vivo. Oral Oncol. 2017;72:123–131. doi: 10.1016/j.oraloncology.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel L.N., Zaro J.L., Shen W.C. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007;24:1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 16.Verdurmen W.P., Bovee-Geurts P.H., Wadhwani P., Ulrich A.S., Hällbrink M., van Kuppevelt T.H., Brock R. Preferential uptake of L- versus D-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011;18:1000–1010. doi: 10.1016/j.chembiol.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Elmquist A., Langel U. In vitro uptake and stability study of pVEC and its all-D analog. Biol. Chem. 2003;384:387–393. doi: 10.1515/BC.2003.044. [DOI] [PubMed] [Google Scholar]
- 18.Pujals S., Sabidó E., Tarragó T., Giralt E. all-D proline-rich cell-penetrating peptides: a preliminary in vivo internalization study. Biochem. Soc. Trans. 2007;35:794–796. doi: 10.1042/BST0350794. [DOI] [PubMed] [Google Scholar]
- 19.Youngblood D.S., Hatlevig S.A., Hassinger J.N., Iversen P.L., Moulton H.M. Stability of cell-penetrating peptide-morpholino oligomer conjugates in human serum and in cells. Bioconjug. Chem. 2007;18:50–60. doi: 10.1021/bc060138s. [DOI] [PubMed] [Google Scholar]
- 20.Tugyi R., Uray K., Iván D., Fellinger E., Perkins A., Hudecz F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA. 2005;102:413–418. doi: 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeller S., Choi C.S., Uchil P.D., Ban H.S., Siefert A., Fahmy T.M., Mothes W., Lee S.K., Kumar P. Attachment of cell-binding ligands to arginine-rich cell-penetrating peptides enables cytosolic translocation of complexed siRNA. Chem. Biol. 2015;22:50–62. doi: 10.1016/j.chembiol.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jun E., Kim S., Kim J.H., Cha K., So I.S., Son H.N., Lee B.H., Kim K., Kwon I.C., Kim S.Y., Kim I.S. Design of a multicomponent peptide-woven nanocomplex for delivery of siRNA. PLoS ONE. 2015;10:e0118310. doi: 10.1371/journal.pone.0118310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Favretto M.E., Brock R. Stereoselective uptake of cell-penetrating peptides is conserved in antisense oligonucleotide polyplexes. Small. 2015;11:1414–1417. doi: 10.1002/smll.201402101. [DOI] [PubMed] [Google Scholar]
- 24.Ren Y., Hauert S., Lo J.H., Bhatia S.N. Identification and characterization of receptor-specific peptides for siRNA delivery. ACS Nano. 2012;6:8620–8631. doi: 10.1021/nn301975s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heusermann W., Hean J., Trojer D., Steib E., von Bueren S., Graff-Meyer A., Genoud C., Martin K., Pizzato N., Voshol J. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016;213:173–184. doi: 10.1083/jcb.201506084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann M.J., Sherer N.M., Marks C.B., Pypaert M., Mothes W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005;170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lidke D.S., Lidke K.A., Rieger B., Jovin T.M., Arndt-Jovin D.J. Reaching out for signals: filopodia sense EGF and respond by directed retrograde transport of activated receptors. J. Cell Biol. 2005;170:619–626. doi: 10.1083/jcb.200503140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ur Rehman Z., Sjollema K.A., Kuipers J., Hoekstra D., Zuhorn I.S. Nonviral gene delivery vectors use syndecan-dependent transport mechanisms in filopodia to reach the cell surface. ACS Nano. 2012;6:7521–7532. doi: 10.1021/nn3028562. [DOI] [PubMed] [Google Scholar]
- 29.Romero S., Grompone G., Carayol N., Mounier J., Guadagnini S., Prevost M.C., Sansonetti P.J., Van Nhieu G.T. ATP-mediated Erk1/2 activation stimulates bacterial capture by filopodia, which precedes Shigella invasion of epithelial cells. Cell Host Microbe. 2011;9:508–519. doi: 10.1016/j.chom.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lundberg P., El-Andaloussi S., Sütlü T., Johansson H., Langel U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21:2664–2671. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- 31.Kumar P., Wu H., McBride J.L., Jung K.E., Kim M.H., Davidson B.L., Lee S.K., Shankar P., Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 32.Plank C., Oberhauser B., Mechtler K., Koch C., Wagner E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J. Biol. Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- 33.Guidotti G., Brambilla L., Rossi D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017;38:406–424. doi: 10.1016/j.tips.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Stalder L., Heusermann W., Sokol L., Trojer D., Wirz J., Hean J., Fritzsche A., Aeschimann F., Pfanzagl V., Basselet P. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013;32:1115–1127. doi: 10.1038/emboj.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isogai T., van der Kammen R., Innocenti M. SMIFH2 has effects on Formins and p53 that perturb the cell cytoskeleton. Sci. Rep. 2015;5:9802. doi: 10.1038/srep09802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barry D.J., Durkin C.H., Abella J.V., Way M. Open source software for quantification of cell migration, protrusions, and fluorescence intensities. J. Cell Biol. 2015;209:163–180. doi: 10.1083/jcb.201501081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rizvi S.A., Neidt E.M., Cui J., Feiger Z., Skau C.T., Gardel M.L., Kozmin S.A., Kovar D.R. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol. 2009;16:1158–1168. doi: 10.1016/j.chembiol.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Junttila M.R., Puustinen P., Niemelä M., Ahola R., Arnold H., Böttzauw T., Ala-aho R., Nielsen C., Ivaska J., Taya Y. CIP2A inhibits PP2A in human malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.