

Abstract
Middle east respiratory syndrome coronavirus (MERS-CoV) is a fatal pathogen that poses a serious health risk worldwide and especially in the middle east countries. Targeting the MERS-CoV 3-chymotrypsin-like cysteine protease (3CLpro) with small covalent inhibitors is a significant approach to inhibit replication of the virus. The present work includes generating a pharmacophore model based on the X-ray crystal structures of MERS-CoV 3CLpro in complex with two covalently bound inhibitors. In silico screening of covalent chemical database having 31,642 compounds led to the identification of 378 compounds that fulfils the pharmacophore queries. Lipinski rules of five were then applied to select only compounds with the best physiochemical properties for orally bioavailable drugs. 260 compounds were obtained and subjected to covalent docking-based virtual screening to determine their binding energy scores. The top three candidate compounds, which were shown to adapt similar binding modes as the reported covalent ligands were selected. The mechanism and stability of binding of these compounds were confirmed by 100 ns molecular dynamic simulation followed by MM/PBSA binding free energy calculation. The identified compounds can facilitate the rational design of novel covalent inhibitors of MERS-CoV 3CLpro enzyme as anti-MERS CoV drugs.
Keywords: 3CLpro, Covalent docking, Covalent inhibitors, MERS-CoV, Molecular dynamics simulation, Pharmacophore modeling
Graphical abstract
1. Introduction
Coronaviruses infect most vertebrate species, including humans. In humans, they generally cause mild-to-moderate respiratory tract infections [1]. However, viruses belonging to this group namely Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV), MERS-CoV, and recently identified Severe Acute Respiratory Syndrome-related Coronavirus (SARS-CoV-2, which causes COVID-19 disease) have been shown to cause fatality in humans [2,3]. MERS-CoV was discovered in 2012 in the Middle East [4,5]. It is transmitted from camels to human and human-to-human transmission has been reported [6,7]. As of January 2021, a total of 2566 confirmed cases of MERS, including 882 related deaths (fatality rate: 34.4%), have been reported. The majority of the cases were reported from Saudi Arabia (2121 cases), including 788 deaths (fatality rate: 37.1%) [8,9]. MERS-CoV causes severe pneumonia in most cases of infection, quite similar to SARS. The patients are put on respiratory support and, unlike SARS, also monitored for renal failure [5,10].
MERS-CoV belongs to the genus β-coronavirus (Subfamily: Coronavirinae; Family: Coronaviridae; Order: Nidovirales), and it is closely related to bat coronavirus [5,11]. MERS-CoV is a single-stranded RNA genome of positive polarity and features the largest RNA genome known to date (~30 kb) [12]. The entry of MERS-CoV begins with the binding of its spike protein (S) on the dipeptidyl peptidase 4 (DPP4) receptor on the host cell surface [5,11]. Upon entry of the virus particle, it is decoded and generate large polyproteins pp1a (4382 amino acids) and pp1ab (7073 amino acids) via translation of open reading frame (ORF 1a & 1b) [11,12]. These polyproteins are then proteolytically cleaved by two viral proteases; 3-Chymotrypsin-like cysteine protease (3CLpro) and papain-like protease (PLpro), into at least 15 non-structural proteins (NSPs) [13]. 3CLpro cleaves the polyprotein at 11 distinct sites to generate many of the non-structural proteins, which are important in viral replication [13]. Thus, this protease plays a critical role in replication of virus [[14], [15], [16]]. The interruption of any of these replication processes would become a potential molecular target for antiviral drug development. 3CLpro shows weak dimerization and is believed to be dimerized in the presence of its substrate [17]. Structure-based activity studies and various high throughput studies have identified distinct inhibitors of SARS-CoV 3CLpro [16,[18], [19], [20], [21], [22]]. Many of these inhibitors have been tested against MERS-CoV. However, there are no studies that show that SARS-CoV inhibitors have a distinct effect against MERS-CoV [23]. Thus, it is essential to identify novel inhibitors of MERS-CoV 3CLpro.
Some peptidomimetic compounds could inhibit the dimerization of MERS-CoV 3CLpro [13,24,25]. The inhibitors reported to date comprise: N3 (IC50 0.288 μmol/l, EC50 ~ 0.3 μM) [26], 5-chloropyridyl esters GRL-001 [27], benzotriazole derivatives [17], dipeptidyl derivative GC376 ((EC50 1.6 μM) [28], pyrazolone derivatives (EC50 5.8 μM to 7.5 μM) [25], pyrrolidinone derivatives (EC50 1.7 μM to 4.3 μM) [29], pyrrolidinone-based peptide GC813 (EC50 0.5 μM to 0.8 μM) and piperidine embodied peptidomimetics [24]. These inhibitors validated the scope of targeting this enzyme with covalent compounds with novel chemical scaffolds for strong anti-MERS-CoV activity. Moreover, 3CLpro is highly conserved across coronaviruses, therefore, there is a possibility for the identification of compounds that could have broad spectrum anti-viral activity [[30], [31], [32], [33]].
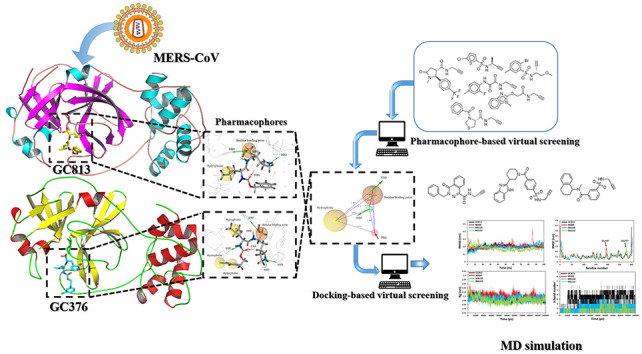
Covalent inhibitors contain electrophilic warheads including epoxide, aziridine, ester, ketone, α, β-unsaturated carbonyl, acetylene, nitrile, sulfur tethers, etc. that react and form covalent bond with nucleophilic residues like serine, cysteine etc. in the active site of the enzymes/proteins [32,34]. They have strong target affinity and if the reactivity of the electrophilic warhead is controlled, these inhibitors can provide better therapeutic options due to their outstanding pharmacodynamic properties [32,35]. The small molecule database could be utilized in either ligand-based or structure-based virtual screening for an effective identification of inhibitors [30]. In this contribution, a virtual screening approach based on a pharmacophore modeling followed by a covalent docking, molecular dynamic simulations and MM/PBSA binding free energy analyses were utilized to explore potential small covalent inhibitors of MERS-CoV 3CLpro enzyme as anti-MERS-CoV drugs. The workflow illustrating the methodology for the identification of novel MERS-CoV 3CLpro enzyme inhibitor is shown in Fig. 1 .
Fig. 1.

Workflow illustrating the methodology for the identification of novel MERS-CoV 3CLpro enzyme inhibitors.
2. Materials and methods
2.1. Establishment of a structure-based pharmacophore model
LigandScout 4.3 program [36] was utilized to construct a pharmacophore model based on two X-ray structures of MERS-CoV 3CLpro in complex with covalent inhibitors. The PDB files of both structures, 5WKK and 5WKJ, with X-ray resolution of 1.55 and 2.05 Å, respectively, were obtained from protein data bank. Two independent pharmacophore hypotheses were initially generated from the 5WKK and 5WKJ of MERS-CoV 3CLpro structures in complex with GC813 and GC376 inhibitors, respectively, using a default setting [24]. The shared pharmacophore features along with exclusion volumes were then excreted to construct the final pharmacophore model that was used as 3D query for pharmacophore-based virtual screening of covalent chemical database. The developed 3D pharmacophore model was qualitatively validated using Receiver Operating Characteristic (ROC) method implemented in LigandScout software. The analysis was carried out by screening the model against two sets of compounds: 9 known actives and 512 decoys. The decoy set was generated from the active molecules using DUDE decoy online generator [37].
2.2. Preparation of covalent chemical database
The virtual screening was performed with three covalent focused chemical databases consisting of a total 31,642 compounds. The libraries include Enamine covalent library (ww.enamine.net), ChemDiv covalent inhibitor library (http://www.chemdiv.com) and Life chemicals cysteine focused covalent inhibitor library (www.lifechemicals.com), composed of 21,969, 8293 and 1383 compounds, respectively. Discovery Studio Visualizer was used to combine all three databases in one SDF file [38]. The duplicates in the chemical database were removed by using OpenBabel (ver. 3.0.0) [39]. As a result, a covalent library of 28,790 compounds was prepared.
2.3. Virtual screening
2.3.1. Structure-based pharmacophore dependent virtual screening
A structure-based pharmacophore virtual screening was performed using LigandScout 4.3. The hits were ranked based on the pharmacophore fit scores and the only compounds that meet the all-pharmacophore query were considered for further investigation. To reduce the number of candidate compounds, Lipinski rule of five [40] in LigandScout 4.3 program tool were applied. Compound with the following parameters is considered for docking-based virtual screening; molecule weight < 500 g/mol, LogP <5, Hydrogen bond donors <5, hydrogen bond acceptors <10 and rotatable bonds <10.
2.3.2. Covalent docking based virtual screening
To filter the candidate compounds further, the obtained 260 candidate compounds were subjected to covalent docking based virtual screening (CovDock-VS) against the structure of MERS-CoV 3CLpro (PDB: 5WKK). Molecular docking studies were carried out by using Maestro program, version-10.5 (GUI of Schrodinger 2017). X-ray structures of MERS-CoV 3CLpro having PDB ID of 5WKK with resolution of 1.55 Å was chosen for covalent docking [24,41]. Protein Preparation Wizard of Maestro was used for the preparation and energy minimization of the crystal structure. Hydrogen atoms were added, water molecules were deleted, and the tautomeric and protonation states of Asp, Glu, Arg, Lys and His amino acids were adjusted and finally energy of the crystal structure was minimized by OPLS2005 force field. The ligands were prepared by LigPrep facility in Schrodinger. Various stereoisomers, tautomers, and the protonation and ionization states of ligands were generated at pH 7.4 using ionizer, as a result of which a library of 435 ligands were constructed. Finally, energy of the ligands was minimized by OPLS2005 force field. Before molecular docking, the reactive amino acid residue, Cys148 was mutated to alanine, to dock the pre-reactive forms of ligands. The ligands generated by LigPrep were docked into the receptor grid in the standard Glide XP mode. The reactive functional groups of the ligands were constrained within 5 Å of the Cβ atom of the reactive amino acid residue. Post-docking minimization was performed and up to three best poses per ligand were written out. All poses were examined manually, and the pose of the best docking score was retained unless noted otherwise. Then, the amino acid was mutated back to Cys148 and sampled using Prime VSGB2.0 with OPLS2005 force field. Covalent docking was performed using the CovDock application with Cys148 as the nucleophilic residue, which underwent a conjugate addition to an alkyne (carbonyl-activated) as preset by CovDock. The covalent bond was formed for ligand poses having reactive functional groups within 5 Å according to the reaction specified. The selection and ranking of the ligands were done on the basis of the Glide Scores of the binding mode of pre-reactive complexes.
2.4. Drug-likeness and ADME-T profiles analysis
The available QikProp tool in Schrodinger was used for finding the drug-likeness properties. Based on Lipinski's rule of five, the properties that have been considered were molecular weight (MW), hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), lipophilicity (log P), rotatable bonds and aqueous solubility (QP log S). The ADME-T (absorption, distribution, metabolism, and excretion - toxicity) profile is very essential for predicting the pharmacokinetics properties of compounds [42]. ADME-T properties of hit compounds were determined using QikProp module of Maestro program, version-10.5 (GUI of Schrodinger 2017).
2.5. Molecular dynamic simulation
A 100 ns molecular dynamic (MD) simulation on covalently docked structures was performed using GROMACS 2018.1 package [43] with the OPLS-AA all-atom force fields [44]. MD simulations were carried out in the presence of ligands covalently bound to Cys148. The topology parameters of ligands were generated using SwissParam [45] and LigParGen [46] web servers. Each of docked-ligand and protein complex was then solvated in a triclinic box with TIP3P water molecules [47] with at least 1 nm distance from the docked-ligand and protein system. The system was further neutralized by adding appropriate numbers of counter ions. Periodic boundary conditions were applied during the MD simulation. A steepest decent algorithm of a maximum step size 0.01 nm with a tolerance of 239 kcal/mol/nm was used for energy minimization. A LINear Constraint Solver (LINCS) algorithm was used for bond lengths constrained. Electrostatic calculations were achieved using particle mesh Ewald method. After, energy minimization, the system was equilibration using canonical ensembles NVT followed by isothermal-isobaric ensemble (NPT) for 100 ps. All the simulations were carried out at constant temperature (300K) and pressure (1 atm). The production run was 100 ns, with trajectories generated every 2 fs and saved every 2 ps. All the preliminary analyses such as root mean square deviation (RMSD), root mean square fluctuations (RMSF), radius of gyration (Rg), and hydrogen bond analysis were carried out by GROMACS analysis programs.
2.6. MM/PBSA binding free energy calculation
MM/PBSA binding free energy analysis was performed using G_mmpbsa module of GROMACS v.5.1.4 [48]. Briefly, Total 5000 snapshots from the last 20 ns stable simulation trajectories of every system were extracted with the interval of 2 ps for the calculation of MM/PBSA. Following equations have been used to calculate binding free energy:
Representations in the above equations are: ΔGbind = binding energy; -TΔS = entropic energy; ΔEMM = molecular mechanics potential energy [van der Waals (ΔEvdw) energy + electrostatic (ΔEelec)]; ΔGsol = solvation free energy [polar solvation energy (ΔEpolar) + non-polar solvation energy (ΔESASA). MM/PBSA analysis has been widely used in binding free energy calculations for anti-viral inhibitors [32,[49], [50], [51], [52], [53], [54], [55]].
3. Results
MERS-CoV infection is recognised as a global health issue. Currently, there is no treatment or vaccine to combat or prevent MERS-CoV infection. MERS-CoV 3CLpro has emerged as an attractive target to inhibit replication of this virus [5,56]. Crystal structures of this protease in complex with dipeptidyl and tetrapeptidyl peptidomimetic inhibitors, namely GC813 and GC376, respectively, showed that both compounds formed a tetrahedral hemithioacetal upon reaction with the catalytic Cys148 residue in the active site [24]. This shows the viability of targeting the active site Cys148 with covalent inhibitors. In fact, targeting protease enzymes with covalent inhibitors is an effective general strategy in antiviral drug discovery [32,34]. In this context, the main objectives of this study were to use a pharmacoinformatic approach and molecular dynamic simulation to explore novel covalent inhibitors of MERS-CoV 3CLpro. This approach has been successfully used to discover new drugs in a time- and cost-effective manners [55,57,58].
3.1. Construction of the pharmacophore model
Pharmacophore describes the three-dimensional arrangement of essential steric and electronic features for optimal binding of a ligand to a macromolecule. The design of pharmacophore model could be either structure-based or ligand-based depending on the available target or known ligands information, respectively [57,59,60]. In this study, the structure-based pharmacophore model was constructed from the shared features of two independent pharmacophore models that was designed from two X-ray structures of MERS-CoV 3CLpro in complex with two potent and covalent inhibitors namely GC813 and GC376. GC813 and GC376 inhibit the MERS-CoV 3CLpro enzyme at 0.5 and 0.9 μM, respectively (Fig. 2A) [24]. Both compounds form a covalent tetrahedral adduct with the cysteine 148 (Cys148) of the catalytic dyad His41-Cys148 (Fig. 2B & C). Overlapping both ligands suggested that they both adapt the same binding mode within the active site of MERS-CoV 3CLpro enzyme (Fig. 2D). Using crystal structures of ligands in complex with their targets for generating the structure-based pharmacophore model may post the efficiency of the designed pharmacophore model [61].
Fig. 2.

(A) Chemical structures of GC813 and GC376. A ribbon representation of the active site of MERS-CoV 3CLpro protease enzyme in complex with (B) GC813 (red) and (C) GC376 (cyan). (D) The binding mode of GC813 (red) and GC376 (cyan) to MERS-CoV 3CLpro enzyme. The catalytic dyad His41-Cys148 are highlighted in red color.
The first pharmacophore model was designed from the interaction of GC813 with MERS-CoV 3CLpro enzyme. The pharmacophore was composed of one residue binding point which is the site for covalent binding group, one hydrophobic, three hydrogen bond donors and one hydrogen bond acceptor site (Fig. 3A). The second pharmacophore model was generated from the binding of GC376 to MERS-CoV 3CLpro. This model was consisted of one residue binding point, three hydrophobic, five hydrogen bond donors and one hydrogen bond acceptor group (Fig. 3B). Using the LigandScout 4.3 program, the shared features along with the exclusion volumes, which are essential features for the determination of the binding pocket overall shape, were extracted to result in the final pharmacophore model. This pharmacophore model was composed of one residue binding point, one hydrophobic, two hydrogen bond donors and one hydrogen bond acceptor group (Fig. 3C).
Fig. 3.

Structure-based pharmacophore models derived from X-ray structures of MERS-CoV 3CLpro enzyme in complex with (A) GC813 (PDB: 5WKK) and (B) GC376 (5WKJ). (C) A model of shared features pharmacophore of A and B. The pharmacophore features were represented in LigandScout by color codes in which, residue binding point, hydrophobic, hydrogen bind donor and hydrogen bond acceptor are depicted as orange sphere, yellow sphere, green arrow and red arrow, respectively. HBD and HBA stand for hydrogen bond donor and hydrogen bond acceptor, respectively. (D) Receiver operating characteristic (ROC) curve validation of the 3D structure-based pharmacophore model.
This pharmacophore model was then validated theoretically for its sensitivity and specificity to recognize true-active and -inactive ligands by screening a database composed of 9 known potent 3CLpro inhibitors and 512 decoy compounds using the receiver operating characteristic curve (ROC) method. A ROC curve illustrates the rate for the ability of a model to retrieve the true positive (actives) on Y-coordinate plotted against the rate for its ability to retrieve false positives (decoys) on X-coordinate. An ideal curve would increase along the Y-axis until it reaches 1, which is the maximum true positive rate, then continues horizontally to the right shown that the hit list contains only the active molecules in the dataset. Two parameters of ROC curve were calculated at different fraction of the model-ordered database (1,5;10 and 100%) including the area under the curve (AUC), which shows the capability of the model to distinguish between true-active and decoy molecules as well as the enrichment factor (EF) which represents the number of true-active compounds found by using the generated pharmacophore mode [62]. The calculated ROC parameters for the generated model were as follows: AUC1,5,10,100% 1.0, 1.0, 1.0, and 0.83, respectively; and EF1;5;10;100%, 57.9; 15.8; 15.8; and 15.8, respectively (Fig. 3D). These values complied with the requirements for AUC1,5,10,100% all to be between 0.5 and 1 and EF1;5;10;100% all to be >1 validating the ability of developed 3D model to distinguish the active molecules from decoys, therefore, its reliability to carry out the pharmacophore-based screening [63].
3.2. Pharmacophore-based virtual screening
A focused covalent chemical library containing a total of 28,790 compounds was used for virtual screening against MERS-CoV 3CLpro enzyme. The compounds possess numerous reactive groups toward cysteine residue such as α,β-unsaturated ketones, α-chloracetamides, phenylsulphonate esters, vinylsulfonamides, acrylamides, acrylonitriles, aminomethyl methyl acrylathes, methyl vinylsulfones, epoxides, activated acetylenes and sulfonyl fluorides. This chemical database was initially used to carry out the pharmacophore-based virtual screening against the generated pharmacophore model. Among the screened compounds, 378 compounds were found to meet all the pharmacophore queries. The candidate compounds were ranked based on LigandScout pharmacophore fit score, which reflects to how much extent a compound fits the pharmacophore features. The pharmacophore fit scores for the obtained candidate compounds are ranged from 57.4419 to 55.0652. To reduce the number of candidate hits, Lipinski rule of five in LigandScout 4.3 program were applied. This step is necessary to evaluate the drug-likeness based on the physicochemical properties of the molecule. Among the 378 compounds, 260 candidate compounds were obtained and considered for the docking-based virtual screening.
3.3. Covalent docking dependent virtual screening
To filter the candidate hits further, a covalent docking-based virtual screening was performed against the active site of MERS-CoV 3CLpro enzyme using CovDock-VS (Maestro ver. 11, Schrodinger). The 260 compounds were then ranked on the basis of their glide scores. The top three candidate compounds with high glide scores toward the target protein were found to be MA69 (Glide score: −9.141; PubChem CID: 45870851; chemical name: N-Methyl-3-(prop-2-ynylsulfamoyl)-N-(1,2,3,4-tetrahydronaphthalen-1-yl)benzamide), MA120 (Glide score: −9.304; PubChem CID: 42932475; chemical name: 4-[3-(1H-1,3-Benzodiazol-2-yl)piperidine-1‑carbonyl]-N-(prop-2-yn-1-yl)benzene-1-sulfonamide) and MA152 (Glide score: −8.674; PubChem CID: 16393007; chemical name: 3-Benzyl-4-oxo-N-(prop-2-yn-1-yl)-3,4-dihydrophthalazine-1-carboxamide) (Table 1 ).
Table 1.
The name, chemical structure, pharmacophore fit-score, Glide XP score, CovDock score and binding interaction of the hit compounds.
| Ligand (PubChem CID) | Chemical structure (Chemical name) | Pharmacophore fit-score | Glide score (kcal/mol) | Interacting residues |
|---|---|---|---|---|
| MA69 (45870851) |
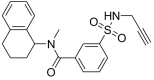 N-Methyl-3-(prop-2-ynylsulfamoyl)-N-(1,2,3,4-tetrahydronaphthalen-1-yl)benzamide |
56.72 | −9.141 | His41, Leu49, Cys145, Gly146, Cys148, His166, Gln167, Met168, Glu169, Asp190, Lys191, Gln192, Val193 |
| MA120 (42932475) |
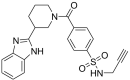 4-[3-(1H-1,3-Benzodiazol-2-yl)piperidine-1‑carbonyl]-N-(prop-2-yn-1-yl)benzene-1-sulfonamide |
56.6 | −9.304 | Met25, Thr26, Leu27, His41, Val42, Leu49, Gly146, Cys148, Gln167, Met168, Glu169, Leu170, Ala171, Asp190, Lys191, Gln192, Val193, His194 |
| MA152 (16393007) |
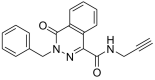 3-Benzyl-4-oxo-N-(prop-2-yn-1-yl)-3,4-dihydrophthalazine-1-carboxamide |
56.55 | −8.674 | His41, Leu49, Tyr54, Cys145, Gly146, Cys148, Gln167, Met168, Glu169, Asp190, Lys191, Gln192, |
Interestingly, these compounds possess activated acetylene groups which are involved in a covalent interaction with the Cys148 of the catalytic dyad. The active site of MERS-CoV 3CLpro enzyme has a catalytic Cys148-His41 dyad and an extended binding site [24]. Before the molecular docking, the protocol was initially validated by independent re-docking of the co-crystallized ligand, GC813 into the active binding site. The latter step was conducted to examine the ability of the docking protocol to produce the bioactive conformation. Accordingly, the docked pose with the lowest binding energy score adapted a binding mode similar to that of the co-crystallized ligand (Fig. 4A). In addition, the distance between the reactive bisulfite group and the catalytic Cys148 residue was returned. The catalytic Cys148 was 1.8 Å from the bisulfite group of the co-crystallized ligand and 3.3 Å from the bisulfite group of the docked pose.
Fig. 4.

(A) The best docked conformation of CG813 (yellow) overlapped with co-crystal ligand (red). The distance between the Cys148 and the reactive bisulfite group of CG813 is ladled in black color. (B) Surface representation of covalent binding mode of MA69 (green), MA120 (pink) and MA152 (cyan) to MERS-CoV 3CLpro. 3D representation of the mechanism of interaction of (C) MA69, (D) MA120 and (E) MA152 with MERS-CoV 3CLpro. Compounds were shown to form covalent bonds with Cys148 (highlighted in red label) within the active site of MERS-CoV 3CLpro.
These results validate the robustness of the docking protocol. The best binding poses for MA69, MA120 and MA152 are depicted in (Fig. 4B). These candidate compounds adapted similar binding modes within the active site of MERS-CoV 3CLpro enzyme. The reactive acetylene groups within the chemical structures of these compounds formed covalent bond with the catalytic Cys148 (Fig. 4C-E). The reactive groups are oriented toward the catalytic Cys148-His41 dyad within the active site. Therefore, these compounds have high potential to inhibit the MERS-CoV 3CLpro enzyme.
3.4. Drug-likeness and ADME-T properties of hit compounds
The selected hit compounds were then evaluated for the drug-likeness and ADME-T properties by QikProp module of Schrodinger. From pervious filter, these hits follow the Lipinski's rule of five. These compounds have molecule weights (MW) < 500, log P < 5, hydrogen bond donors (HBD) <5, hydrogen bond acceptors (HBA) < 10. In addition, the number of rotatable bonds was <7, which is an essential parameter for good oral bioavailability. The value of QPlog S is an indicator of aqueous solubility of compounds with an acceptable range from −9.5 to 0.47. The lower the QP log S value, the higher the solubility and drug absorption profile. The identified hit compounds were found to perfectly obey the Lipinski's rule of five with ideal rotatable bonds numbers and QPlog S values, essential for orally active drugs [42]. Ghose et al. derived a guideline for the selection and optimization of CNS and non-CNS orally active compounds, through the analysis of 35 physicochemical and pharmacokinetic properties of 317 CNS and 626 non-CNS oral drugs [42]. Interestingly, the physicochemical and pharmacokinetic profile of compounds MA69, MA120, and MA152, was found to be within the limits as proposed and presented in the Table 2 .
Table 2.
ADME-T properties of compounds MA69, MA120, and MA152, predicted by QikProp, Schrodinger.
| S. No. | Property | Description | Range for orally active non-CNS drugs |
Compound MA69 | Compound MA120 | Compound MA152 | |||
|---|---|---|---|---|---|---|---|---|---|
| QL | PL | PU | QU | ||||||
| 1 | #stars | drug likeness penalty; the higher the value, the less drug-like the molecule | 0 | 0 | 0 | 8 | 0 | 0 | 0 |
| 2 | #amine | no. of basic amines | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| 3 | #amidine | no. of amidines groups | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | #acid | no. of carboxylic acid groups | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| 5 | #amide | no. of amides groups | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| 6 | #rotor | no. of rotatable bonds (without CX3, alkene, amide, small ring) | 0 | 2 | 6 | 17 | 6 | 5 | 5 |
| 7 | CNS | a qualitative CNS activity parameter | −2 | −2 | −1 | 1 | −1 | −2 | 0 |
| 8 | dipole | computed dipole moment | 0.96 | 3.7 | 7.7 | 12 | 3.56 | 7.46 | 6.25 |
| 9 | SASA | solvent accessible surface area | 265 | 459 | 660 | 1023 | 680.74 | 745.25 | 614 |
| 10 | FOSA | SASA on saturated carbon and attached hydrogen | 0 | 69 | 304 | 667 | 253.67 | 220.52 | 124.89 |
| 11 | FISA | SASA on N, O, and H attached to heteroatoms | 0 | 81 | 176 | 306 | 111.08 | 169.13 | 92.77 |
| 12 | PISA | π component of SASA | 0 | 0 | 138 | 371 | 314.13 | 353.73 | 396.34 |
| 13 | WPSA | weakly polar component of the SASA (halogens, P, and S) | 0 | 0 | 0 | 144 | 1.85 | 1.86 | 0 |
| 14 | volume | solvent accessible volume (Å3) | 410 | 763 | 1178 | 2082 | 1211.06 | 1306.56 | 1050.95 |
| 15 | donorHB | estimated no. of hydrogen bonds that would be donated to the solvent water | 0 | 1 | 2.5 | 5 | 1.5 | 2.5 | 1.5 |
| 16 | accptHB | estimated no. of hydrogen bonds that would be accepted from the solvent water | 0 | 4 | 8.2 | 16.1 | 7.5 | 9 | 4.5 |
| 17 | glob | a globularity descriptor (1 for a sphere) | 0.73 | 0.81 | 0.88 | 0.94 | 0.80 | 0.77 | 0.81 |
| 18 | QPpolrz | predicted polarizability (Å3) | 10 | 25 | 41 | 71 | 41.46 | 46.30 | 36.48 |
| 19 | QPlogPo/w | octanol−water logP | −2.6 | 0.76 | 4 | 7.3 | 3.29 | 2.78 | 3.73 |
| 20 | QPlogS | solubility in log(moles/l) | −9.4 | −4.9 | −2.3 | 0.47 | −4.96 | −5.72 | −4.97 |
| 21 | CIQPlogS | log of conformation-independent solubility | −9.6 | −5.1 | −2.4 | 0.14 | −4.69 | −5.11 | −4.80 |
| 22 | QPPCaco | apparent Caco-2 cell permeability | 0 | 0 | 198 | 3975 | 875.98 | 246.62 | 1306.45 |
| 23 | QPlogBB | brain/blood partition coefficient | −3.1 | −1.5 | −0.36 | 0.78 | −0.93 | −1.60 | −0.67 |
| 24 | QPPMDCK | predicted apparent MDCK cell permeability (nm/s) | 0 | 0 | 133 | 5302 | 438.88 | 111.54 | 660.44 |
| 25 | QPlogKhsa | prediction of binding to human serum albumin | −1.80 | −0.67 | 0.24 | 1.42 | 0.18 | 0.15 | 0.36 |
| 26 | HumanOralAbsorption | Human oral absorption | 1 | 3 | 3 | 3 | 3 | 3 | 3 |
| 27 | PercentHumanOralAbsorption | Percent of human oral absorption | 10 | 77 | 100 | 100 | 100 | 86 | 100 |
| 28 | PSA | van der Waals surface area of polar nitrogen and oxygen atoms | 0 | 61 | 120 | 194 | 75.12 | 106.51 | 75.20 |
| 29 | #NandO | no. of N and O atoms | 0 | 3 | 6 | 13 | 5 | 7 | 5 |
| 30 | RuleOfFive | no. of violations of Lipinski's rule of five | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| 31 | RuleOfThree | no. of violations of Jorgensen's rule of three | 0 | 0 | 0 | 2 | 0 | 1 | 0 |
| 32 | #in34 | no. of atoms in three- or four-membered rings | 0 | 0 | 0 | 4 | 0 | 0 | 0 |
| 33 | #in56 | no. of atoms in five- or six-membered rings | 0 | 9 | 16 | 24 | 16 | 21 | 16 |
| 34 | #noncon | no. of atoms not able to form conjugation in nonaromatic rings | 0 | 0 | 2 | 14 | 4 | 5 | 0 |
| 35 | #nonHatm | no. of non-H atoms | 4 | 17 | 27 | 46 | 27 | 30 | 24 |
Abbreviations: QL, qualifying lower limit; PL, preferred lower limit; QU, qualifying upper limit; PU, preferred upper limit [QL, PL, QU and PU values for non-CNS drug criteria were taken from [64]].
3.5. MD simulation
To confirm the screening and docking results, the complex of the three hit compounds with MERS-CoV 3CLpro were then subjected to 100 ns MD simulations to study their binding stability. The advantage of MD simulation is that the ligand-protein complex is studied under an environmental condition similar to that of human cell condition with respect to temperature, pressure, solvent and ions. Therefore, the data obtained from MD simulation could provide deep insight into the mechanism, dynamic and natural of ligand-protein interaction [65]. Over the course of 100 ns time, the ligand-protein complexes were analysed based on their root mean square deviation (RMSD), root mean square fluctuations (RMSF), radius of gyration (Rg), and hydrogen bond analysis.
3.5.1. Root mean square deviation (RMSD) and root mean square fluctuations (RMSF)
To obtain the equilibrium time of each simulated protein-ligand complex during MD simulation, the backbone's root mean square deviation (RMSD) was calculated. RMSD plot is usually used to evaluate the time required for a system to reach structural equilibrium and to estimate the duration of running a simulation. The RMSD is a valuable parameter to estimate shifts or changes in molecular conformation. The RMSD values of simulated complexes including the reference were suddenly raised due to the sudden change in the structure condition, which is related to the protein crystallization method. The latter effect was expected, as in the crystal structure the protein is rigid and when it gets solvated in water box it returns back its dynamic motion [66]. The backbone RMSD values of the simulated systems revealed that an equilibration was achieved after ~20 ns for all complexes with respect to the reference; 3CLpro-GC813 complex (Fig. 5A). Unlike other complexes, there was an observed increase in the RMSD of compound MA69-3CLpro complex up to 0.45 Å at 84 ns (Fig. 5A). This could be a result of adapting a new conformation within the active site by the ligand. The averaged RMSD values of GC813, MA69, MA120 and MA152 complexes, for the last 80 ns were 0.21 ± 0.02-, 0.21 ± 0.03-, 0.20 ± 0.02- and 0.17 ± 0.02 nm, respectively. The RMSF was calculated to assess the contribution of each residue to the complex fluctuation. The RMSF of each residue within the MERS-CoV 3CLpro structure were calculated. Residues with high fluctuation pattern such as Glu197 and Glu277 are located in the loop regions away from the active-catalytic site. The RMSF values suggested normal backbone fluctuations behaviour indicating stable hits binding (Fig. 5B).
Fig. 5.

(A) Backbone RMSD and (B) RMSF values of MERS-CoV 3CLpro inhibitors complexes in the 100 ns period of MD simulations.
3.5.2. Radius of gyration (Rg) and hydrogen bond analysis
The radius of gyration (Rg) which is an indicator of the protein compactness suggested normal behaviour in the protein structure with average values of 2.18 ± 0.01-, 2.20 ± 0.01-, 2.19 ± 0.01- and 2.18 ± 0.01 nm for GC813, MA69, MA120 and MA152 complexes, respectively (Fig. 6A). The hydrogen bond is a major factor responsible for stable conformation's maintenance of the protein and ligands [67]. H-bond analysis was performed to examine the H-bonds for ligands complexes over the 100 ns of the simulation time and presented in Fig. 6B. The Ligands' complexes showed up to two MA69, up to four MA120 and up to three MA152, during 100 ns of the simulation with respect to GC813 which showed up to 7H-bonds.
Fig. 6.

(A) Rg trajectory of MERS-CoV 3CLpro inhibitors complexes obtained after 100 ns MD simulation. (B) Hydrogen bond interactions over the time of 100 ns MD simulation.
3.6. Binding free energy calculation
The Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) is an effective and dependable approach to calculate binding free energies of small inhibitors to their protein targets [32,68]. The binding free energies and energy components of all 3 systems are listed in Table 3 . The calculated bind free energy of complex MA69 is −27.75 kcal/mol, MA120 is −19.33 kcal/mol and of complex MA152 is −25.21 kcal/mol. For all the 3 complexes, ΔEvdw and ΔEelec were favourable and showed stable interaction between ligands-protein. The 100 ns molecular dynamic simulation study reveals that the binding of these compounds to the MERS-CoV 3CLpro were stable throughout the simulation course. Collectively, the identified compounds may have high potential of inhibition of this critical enzyme for replication of MERS-CoV.
Table 3.
MM/PBSA binding Free energy (kcal/mol) analysis of the hit compound's complexes.
| MM/PBSA ΔGbinding | MA69 | MA120 | MA152 |
|---|---|---|---|
| ΔEvdw | −79.55 | −83.40 | −85.04 |
| ΔEelec | −14.39 | −13.15 | −14.10 |
| ΔEMM: | −93.94 | −96.55 | −99.14 |
| -TΔS | 20.10 | 22.09 | 23.73 |
| ΔESASA | −14.78 | −16.09 | −17.99 |
| ΔEpolar | 60.87 | 71.22 | 67.89 |
| ΔGsol | 46.09 | 55.13 | 49.90 |
| ΔGbind | −27.75 | −19.33 | −25.51- |
Note: ΔEvdw: van der Waals energy; ΔEelec: electrostatic energy; ΔEMM: molecular mechanics potential energy; -TΔS: entropic energy; ΔESASA: non-polar solvation energy; ΔEpolar: polar solvation energy; ΔGsol = solvation free energy; ΔGbind: binding energy.
4. Conclusion
In this study, we identified novel irreversible inhibitors of MERS-CoV 3CLpro as potential anti-MERS-CoV drugs by integrated computational approaches including pharmacophore modeling, covalent docking, molecular dynamics simulation and binding free energy calculation analyses. These compounds carrying electrophilic acetylenes groups as reactive binding points interact covalently with the key catalytic Cys148 of the MERS-CoV 3CLpro with high binding affinity forming stable acetylene adducts. These compounds with good drug-like properties, may serve as seeds to rational development of potent irreversible MERS-CoV 3CLpro.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to thank Prince Sattam Bin Abdulaziz University, AlKharj, Saudi Arabia for providing necessary facilities to carry out this research.
References
- 1.Coleman C.M., Frieman M.B. J. Virol. 2014;88:5209. doi: 10.1128/JVI.03488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Wit E., Van Doremalen N., Falzarano D., Munster V.J. Nat. Rev. Microbiol. 2016;14:523. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.C.S.G. of the International Nat. Microbiol. 2020;5:536. [Google Scholar]
- 4.Báez-Santos Y.M., Mielech A.M., Deng X., Baker S., Mesecar A.D. J. Virol. 2014;88:12511. doi: 10.1128/JVI.01294-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tahir ul Qamar M., Saleem S., Ashfaq U.A., Bari A., Anwar F., Alqahtani S. J. Transl. Med. 2019;17:362. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azhar E.I., El-Kafrawy S.A., Farraj S.A., Hassan A.M., Al-Saeed M.S., Hashem A.M., Madani T.A. N. Engl. J. Med. 2014;370:2499. doi: 10.1056/NEJMoa1401505. [DOI] [PubMed] [Google Scholar]
- 7.Raj V.S., Osterhaus A.D., Fouchier R.A., Haagmans B.L. Curr. Opin. Virol. 2014;5:58. doi: 10.1016/j.coviro.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.W.H. Organization, http://www.emro.who.int/health-topics/mers-cov/mersoutbreaks. html (29.02. 2020) (2020).
- 9.Ramadan N., Shaib H. Germs. 2019;9:35. doi: 10.18683/germs.2019.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong Y.P., Song J.Y., Seo Y.B., Choi J.-P., Shin H.-S., Team R.R. Infect. Chemother. 2015;47:212. doi: 10.3947/ic.2015.47.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackay I.M., Arden K.E. Virol. J. 2015;12:1. doi: 10.1186/s12985-015-0439-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith E.C., Sexton N.R., Denison M.R. Ann. Rev. Virol. 2014;1:111. doi: 10.1146/annurev-virology-031413-085507. [DOI] [PubMed] [Google Scholar]
- 13.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. J. Med. Chem. 2016;59:6595. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zumla A., Chan J.F., Azhar E.I., Hui D.S., Yuen K.-Y. Nat. Rev. Drug Discov. 2016;15:327. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shirato K., Kawase M., Matsuyama S. J. Virol. 2013;87:12552. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J.Y., Kim Y.I., Park S.J., Kim I.K., Choi Y.K., Kim S.-H. Int. J. Antimicrob. Agents. 2018;52:730. doi: 10.1016/j.ijantimicag.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomar S., Johnston M.L., John S.E.S., Osswald H.L., Nyalapatla P.R., Paul L.N., Ghosh A.K., Denison M.R., Mesecar A.D. J. Biol. Chem. 2015;290:19403. doi: 10.1074/jbc.M115.651463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 19.Lee H., Mittal A., Patel K., Gatuz J.L., Truong L., Torres J., Mulhearn D.C., Johnson M.E. Bioorg. Med. Chem. 2014;22:167. doi: 10.1016/j.bmc.2013.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berry M., Fielding B.C., Gamieldien J. Viruses. 2015;7:6642. doi: 10.3390/v7122963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C.-N., Lin C.P., Huang K.-K., Chen W.-C., Hsieh H.-P., Liang P.-H., Hsu J.T.-A. Vol. 2. 2005. Evidence-Based Complementary and Alternative Medicine. [Google Scholar]
- 22.Nukoolkarn V., Lee V.S., Malaisree M., Aruksakulwong O., Hannongbua S. J. Theor. Biol. 2008;254:861. doi: 10.1016/j.jtbi.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kilianski A., Mielech A.M., Deng X., Baker S.C. J. Virol. 2013;87:11955. doi: 10.1128/JVI.02105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kankanamalage A.C.G., Kim Y., Damalanka V.C., Rathnayake A.D., Fehr A.R., Mehzabeen N., Battaile K.P., Lovell S., Lushington G.H., Perlman S. Eur. J. Med. Chem. 2018;150:334. doi: 10.1016/j.ejmech.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar V., Tan K.-P., Wang Y.-M., Lin S.-W., Liang P.-H. Bioorg. Med. Chem. 2016;24:3035. doi: 10.1016/j.bmc.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. Protein Cell. 2013;4:248. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng X., StJohn S.E., Osswald H.L., O'Brien A., Banach B.S., Sleeman K., Ghosh A.K., Mesecar A.D., Baker S.C. J. Virol. 2014;88:11886. doi: 10.1128/JVI.01528-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y., Liu H., Galasiti Kankanamalage A.C., Weerasekara S., Hua D.H., Groutas W.C., Chang K.-O., Pedersen N.C. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005531. (e1005531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar V., Shin J.S., Shie J.-J., Ku K.B., Kim C., Go Y.Y., Huang K.-F., Kim M., Liang P.-H. Antivir. Res. 2017;141:101. doi: 10.1016/j.antiviral.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McInnes C. Curr. Opin. Chem. Biol. 2007;11:494. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 31.Tahir ul Qamar M., Alqahtani S.M., Alamri M.A., Chen L.-L. J. Pharm. Anal. 2020;10:313. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alamri M.A., Tahir ul Qamar M., Mirza M.U., Bhadane R., Alqahtani S.M., Muneer I., Froeyen M., Salo-Ahen O.M. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alamri M.A., Tahir ul Qamar M., Mirza M.U., Alqahtani S.M., Froeyen M., Chen L.-L. J. Pharm. Anal. 2020;10:546. doi: 10.1016/j.jpha.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bauer R.A. Drug Discov. Today. 2015;20:1061. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh A.K., Samanta I., Mondal A., Liu W.R. ChemMedChem. 2019;14:889. doi: 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolber G., Langer T. J. Chem. Inf. Model. 2005;45:160. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- 37.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. J. Med. Chem. 2012;55:6582. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biovia D.S. Release. 2017 [Google Scholar]
- 39.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. J. Cheminformatics. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Adv. Drug Deliv. Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 41.Toledo Warshaviak D., Golan G., Borrelli K.W., Zhu K., Kalid O. J. Chem. Inf. Model. 1941;54(2014) doi: 10.1021/ci500175r. [DOI] [PubMed] [Google Scholar]
- 42.Cheng F., Li W., Zhou Y., Shen J., Wu Z., Liu G., Lee P.W., Tang Y. ACS Publ. 2012;52:3099. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 43.Hess B., Kutzner C., Van Der Spoel D., Lindahl E. J. Chem. Theory Comput. 2008;4:435. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 44.Kaminski G.A., Friesner R.A., Tirado-Rives J., Jorgensen W.L. J. Phys. Chem. B. 2001;105:6474. [Google Scholar]
- 45.Zoete V., Cuendet M.A., Grosdidier A., Michielin O. J. Comput. Chem. 2011;32:2359. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 46.Dodda L.S., Cabeza de Vaca I., Tirado-Rives J., Jorgensen W.L. Nucleic Acids Res. 2017;45:W331. doi: 10.1093/nar/gkx312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 48.Kumari R., Kumar R., Consortium O.S.D.D., Lynn A. J. Chem. Inf. Model. 2014;54:1951. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 49.Tahir ul Qamar M., Shokat Z., Muneer I., Ashfaq U.A., Javed H., Anwar F., Bari A., Zahid B., Saari N. Vaccines. 2020;8:288. doi: 10.3390/vaccines8020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou T., Wang J., Li Y., Wang W. J. Chem. Inf. Model. 2011;51:69. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Genheden S., Ryde U. Expert Opin. Drug Discovery. 2015;10:449. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muhseen Z.T., Hameed A.R., Al-Hasani H.M.H., Qamar M.T., Li G. J. Mol. Liq. 2020:114493. doi: 10.1016/j.molliq.2020.114493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mirza M.U., Ahmad S., Abdullah I., Froeyen M. Comput. Biol. Chem. 2020;89:107376. doi: 10.1016/j.compbiolchem.2020.107376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mirza M.U., Froeyen M. J. Pharm. Anal. 2020;10:320. doi: 10.1016/j.jpha.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mirza M.U., Saadabadi A., Vanmeert M., Salo-Ahen O.M., Abdullah I., Claes S., De Jonghe S., Schols D., Ahmad S., Froeyen M. Eur. J. Pharm. Sci. 2020;155:105537. doi: 10.1016/j.ejps.2020.105537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durai P., Batool M., Shah M., Choi S. Exp. Mol. Med. 2015;47 doi: 10.1038/emm.2015.76. (e181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tahir ul Qamar M., Kiran S., Ashfaq U.A., Javed M.R., Anwar F., Ali M.A., Gilani A.U.H. Int. J. Pharmacol. 2016;12:621. [Google Scholar]
- 58.Mirza M.U., Vanmeert M., Ali A., Iman K., Froeyen M., Idrees M. J. Med. Virol. 2019;91:2029. doi: 10.1002/jmv.25357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaserer T., Beck K.R., Akram M., Odermatt A., Schuster D. Molecules. 2015;20:22799. doi: 10.3390/molecules201219880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muneer I., Tusleem K., Abdul Rauf S., Hussain H.M., Siddiqi A.R. Anti-Cancer Drugs. 2019;30:363. doi: 10.1097/CAD.0000000000000727. [DOI] [PubMed] [Google Scholar]
- 61.Chen Z., Li H.-l., Zhang Q.-j., Bao X.-g., Yu K.-q., Luo X.-m., Zhu W.-l., Jiang H.-l. Acta Pharmacol. Sin. 2009;30:1694. doi: 10.1038/aps.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seidel T., Bryant S.D., Ibis G., Poli G., Langer T. Tutor. Chemoinform. 2017;281:279. [Google Scholar]
- 63.Triballeau N., Acher F., Brabet I., Pin J.-P., Bertrand H.-O. J. Med. Chem. 2005;48:2534. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- 64.Ghose A.K., Herbertz T., Hudkins R.L., Dorsey B.D., Mallamo J.P. ACS Chem. Neurosci. 2012;3:50. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang W., Donini O., Reyes C.M., Kollman P.A. Annu. Rev. Biophys. Biomol. Struct. 2001;30:211. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- 66.Kumar R., Moche M., Winblad B., Pavlov P.F. Sci. Rep. 2017;7:1. doi: 10.1038/s41598-017-14731-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamaraj B., Bogaerts A. PLoS One. 2015;10 doi: 10.1371/journal.pone.0134638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang C., Greene D.A., Xiao L., Qi R., Luo R. Front. Mol. Biosci. 2018;4:87. doi: 10.3389/fmolb.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]