

Abstract
Small molecules that promote the metabolic activity of the pyruvate kinase isoform PKM2, such as TEPP-46 and DASA-58, limit tumorigenesis and inflammation. To understand how these compounds alter T cell function, we assessed their therapeutic activity in a mouse model of T cell-mediated autoimmunity that mimics multiple sclerosis (MS). TH17 cells are believed to orchestrate MS pathology in part through the production of two pro-inflammatory cytokines: interleukin-17 (IL-17) and GM-CSF. We found that both TEPP-46 and DASA-58 suppressed the development of IL-17-producing TH17 cells but increased the generation of those producing GM-CSF. This switch redirected disease pathology from the spinal cord to the brain. Additionally, we found that activation of PKM2 interfered with TGF-β1 signaling, which is necessary for the development of TH17 and regulatory T cells. Collectively, our data clarify the therapeutic potential of PKM2 activators in MS-like disease and how these agents alter T cell function.
INTRODUCTION
Multiple sclerosis is a chronic debilitating disease characterized by flares of neurologic disturbances superimposed on an insidious, progressive course of disability accumulation (1). T cells, the agents of the adaptive immune system that provide antigen-directed immune responses, are believed to play critical, if not orchestrating, roles in the MS disease process. In particular, TH17, the CD4+ T cell lineage that expresses the transcription factor RORγT (RAR-related orphan receptor gamma T), is believed to play an essential role in many pathologic aspects of disease, including blood brain barrier disruption (2, 3), peripheral immune cell chemoattraction (4), meningeal tertiary lymphoid structure organization (5), and direct cytotoxicity to glial cells (6). These functions are largely attributed to the RORγT-controlled cytokines IL-17A and GM-CSF (7, 8). RORγT expression in TH17 cells is driven by Hif-1α (hypoxia inducible factor-1 alpha) (9) and STAT3 (signal transducer and activator of transcription 3) (10). In experimental autoimmune encephalomyelitis (EAE), a mouse model for MS, genetic or pharmacologic interventions that target RORγT (7, 8, 11), Hif-1α (9, 12), STAT3 (10), IL-17A (13), or GM-CSF (14) all ameliorate disease. Thus, a great deal of evidence supports the rationale of targeting TH17 cells to treat MS.
Metabolism is essential to the pathologic functions of TH17 cells (12, 15, 16). Metabolic interventions, such as glycolytic inhibition (12), block the differentiation of TH17 cells and prevent their ability to drive EAE in adoptive transfer models. However, the targeting of metabolic pathways to defuse inflammation is limited by the anticipated consequences of global metabolic inhibition. An alternative approach is to target specific metabolic enzyme isoforms involved in inflammation.
Pyruvate kinase is the enzyme that converts phosphoenolpyruvate (PEP) to pyruvate during glycolysis. Alternative splicing of exons 9 and 10 of the Pkm transcript results in the expression of the enzyme isoforms PKM1 (which includes exon 9) and PKM2 (which includes exon 10)(17). Whereas PKM1 functions as a tetramer, PKM2 is a multifunctional protein that can exist in multiple oligomeric states. PKM2 is an active glycolytic enzyme as a tetramer or is inactive as a dimer. Numerous factors including posttranslational modifications and metabolites, such as FBP and serine, regulate the metabolic status of PKM2 (18). Apart from its enzymatic activity, both monomeric and dimeric PKM2 may function as a transcriptional coactivator for Hif-1α (19, 20) and STAT3 (21, 22). Thus, PKM2 is a multifunctional protein with enzymatic and non-enzymatic functions.
Pharmacological agents that enforce the tetramerization of PKM2, such as TEPP-46 (also known as ML-265) and DASA-58, facilitate the enzymatic ability of PKM2 to catalyze PEP to pyruvate conversion (23–25). As a result, these compounds are referred to as “PKM2 activators” (25). TEPP-46, which can be used in vivo (25), can destabilize pathogenic Hif-1α and STAT3 functions in various disease states (20, 21, 26).
Given the central role that STAT3 and Hif-1α play in the TH17 cell lineage generation, we postulated that increasing PKM2 enzymatic activity with these PKM2 activators could interfere with TH17 cell differentiation and thus curtail TH17-associated disease states. Here, we showed that PKM2 activators blocked the differentiation of IL-17A-producing T cells. This effect was not mediated by inhibition of STAT3 signaling but by blockade of the TGF-β1 pathway, because PKM2 activators also suppressed the generation of anti-inflammatory regulatory T cells (Tregs). Furthermore, we found that treatment with these compounds led to the expansion of T cell subsets capable of producing the potently encephalitogenic cytokine GM-CSF, redirecting EAE pathology from the spinal cord to the brain. Further, we generated a new mouse strain that lacked PKM2 expression in T cells, which resulted in compensatory expression of PKM1 in these cells. We discovered that PKM2 activator treatment reduced TH17 and iTreg differentiation even in the context of PKM2 deletion, perhaps due to binding to PKM1. Together, this study broadens our understanding of the mechanism of action of PKM2 activators for inflammatory disease and suggests that careful consideration of PKM activity targeting is warranted.
RESULTS
PKM2 is expressed in T cells present in areas of CNS demyelination
Our previous work revealed that PKM2 transcript is enriched in T cells isolated from the EAE spinal cord when compared to splenic T cells (27). We assessed PKM2 protein abundance in intact spinal cords from mice with EAE. Serial sections of the spinal cord were stained for myelin using luxol fast blue and PKM2, revealing dense PKM2 staining in areas of demyelination (Figure 1A). Further immunofluorescent studies revealed that PKM2 protein was present in CD3+ T cells (Figure 1B). Thus, during the immune-mediated demyelinating process in EAE, PKM2 was expressed by T cells at the protein level. Because T cells present in the CNS during EAE induced pathology include TH1 and TH17 cells, we tested PKM2 expression during in vitro differentiation of T cells. PKM2 expression was increased in TH1 and TH17 differentiated cells compared to naïve T cells (Figure 1C), supporting a functional assessment of PKM2 in the regulation of TH1/17 differentiation or function.
Fig. 1. PKM2 expression is increased in T cells of the EAE spinal cord.
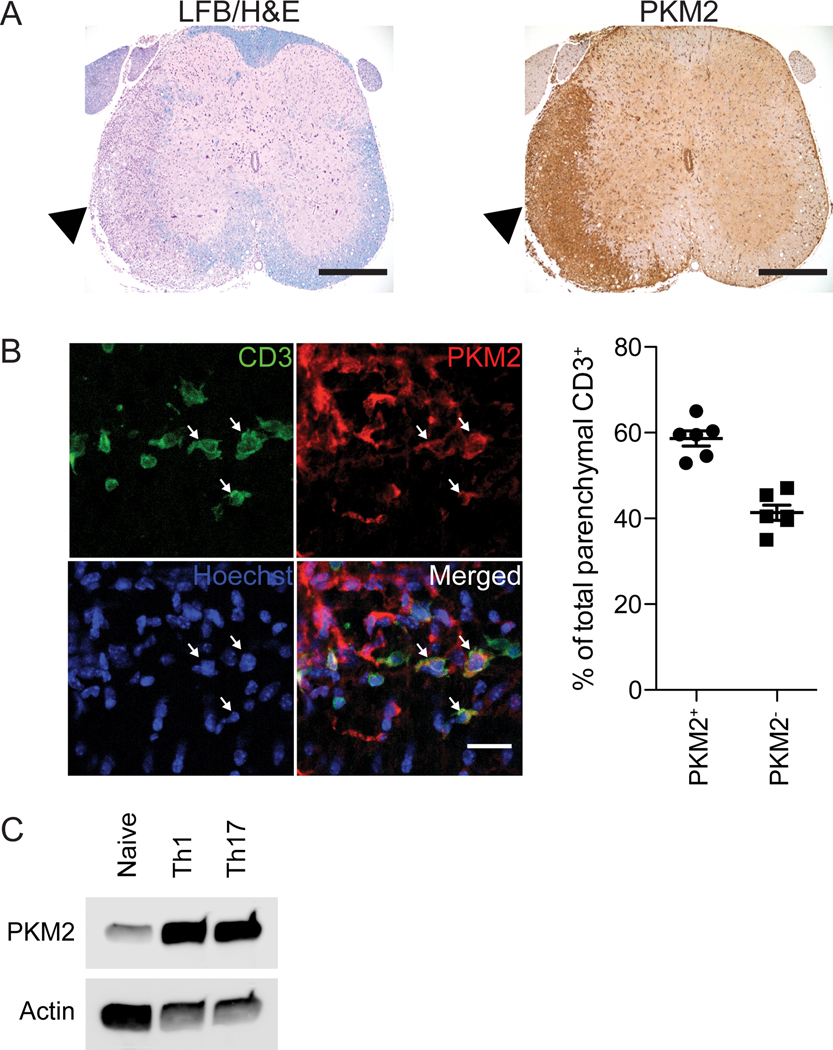
(A) Immunohistochemistry for luxol fast blue (LFB) with the hematoxylin and eosin (H&E) counterstain (left) or PKM2 (right) in adjacent spinal cord sections. Images are representative of 2 independent experiments. Scale bar, 400 μm. (B) Representative immunofluorescence (left panel) and quantification (right panel) of PKM2 and CD3 expression in the spinal cord of mice with EAE. Arrows indicate T cells with prominent PKM2 staining. Images are representative of N=3 independent experiments from n=6 total WT mice with EAE. Quantification of parenchymal CD3+ cells with PKM2 staining from each mouse is presented in the graph. Scale bar, 20 μm. (C) Immunoblot for PKM2 in naïve, TH1 and TH17 T cells. Blots are representative of N=4 independent experiments.
PKM2 modulators inhibit differentiation of TH17 cells
Nonmetabolic, transcription regulatory functions of PKM2 are critical for the coactivation of Hif-1α (19) and STAT3 (22). TEPP-46 and DASA-58 are PKM2 “activators” that promote tetramerization and glycolytic activity (Figure 2A) (23, 24). Indeed, treatment of cytoplasmic extract with TEPP-46 followed by crosslinking increased the tetrameric form of PKM2 (Figure 2B). It is currently unclear how nonmetabolic functions of PKM2 affect T cell biology and, more specifically, how they might affect the Hif-1α/STAT3-dependent acquisition of TH17 identity. Using an in vitro differentiation protocol (Figure 2C, left), we discovered that TEPP-46 and DASA-58 treatment delayed the induction of RORγT (Figure 2C, middle and right), suggesting delayed TH17 differentiation. To determine if this activity is unique to TH17 cells, we also assessed the effects of TEPP-46 on the differentiation of IFN-γ-producing TH1 cells (Figure 2C, left). Instead of inhibiting their differentiation, we found that TEPP-46 potentiated skewing of T cells into IFN-γ producing TH1 cells (Figure 2D). qRT-PCR for genes associated with TH17 cells (Figure 2E, left) showed that treatment of TH17 cells with TEPP-46 delayed and reduced the induction of STAT3-dependent genes IL-17A, RORγT, IL-21, IL-1R1, and IL-23R, but not that of the aryl hydrocarbon receptor-dependent target IL-22 (28)(Figure 2E, right). To investigate the effect of PKM2 activators on the functionality of differentiating TH17 cells, we assessed cytokine production capacity 2 days post treatment, a time point when we see robust IL-17A production (Figure 2F, left). As anticipated, PKM2 activator treatment potently blocked IL-17A production (Figure 2F, right). However, TEPP-46 and DASA-58 did not inhibit, but instead potentiated the generation of T cells capable of producing GM-CSF (Figure 2F, right). We found that TEPP-46 treatment did not correlate with sustained changes in overall metabolism after 2 days of differentiation (Figure 2G). Together, these data reveal that PKM2 activators delay and inhibit the acquisition of TH17 lineage markers and IL-17A expression, but do not block differentiation of GM-CSF-producing T cells and do not increase overall glycolysis rates in differentiated TH17 cells.
Fig. 2. PKM2 activators alter differentiation of TH17 cells in vitro.

(A) Proposed mechanism of action for the PKM2 activators TEPP-46 and DASA-58 in the oligomerization of PKM2. Cyto, cytoplasm. Nuc, nucleus. (B) Immunoblot for PKM2 oligomerization in cytoplasmic extracts of CD4+ T cells treated with TEPP-46 or vehicle. Blots are representative of N=2 independent experiments. (C) Intracellular flow cytometry for RORγT abundance in unstimulated CD4+ T cells or TH17 cells differentiated with TEPP-46, DASA-58 or vehicle (DMSO) for 24 hours. Representative histograms (left) and quantification (right) (N=2 independent experiments). (D) Intracellular flow cytometry for IFN-γ-production in TH1 cells differentiated with TEPP-46 or DMSO for 48 hours, then stimulated with PMA/ionomycin for 6 hours. Dot plots are representative of N=4 independent experiments with quantification of pooled data in the graph. (E) qRT-PCR analysis of TH17 lineage mRNA expression in TH17 cells differentiated with TEPP-46 or DMSO at indicated time points. Data are means ± SEM pooled from N=2 independent experiments on 2 mice. (F) Intracellular flow cytometry for IL-17A/GM-CSF production by TH17 cells differentiated in the presence of DMSO, TEPP-46, or DASA-58. Dot plots are representative of N=2 independent experiments on 5 mice with quantification of pooled data in right panel as mean ± SEM. (G) Seahorse analysis of extracellular acidification rates (ECAR) and oxygen consumption rates (OCR) in unstimulated CD4+ T cells (green) and TH17 cells differentiated with TEPP-46 (red) or DMSO (blue) for 48 hours. Seahorse traces and OCR/ECAR quantification are representative of N=2 independent experiments on 6 mice and presented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 by one-way ANOVA with Tukey post test (C and F), paired Student’s t-test (D), or two-way ANOVA with Sidak post test (E).
PKM2-deficient T cells utilize PKM1
To test the consequence of the loss of PKM2 function in T cells, we bred the Cd4-Cre mouse line with mice bearing the floxed exon 10 of the Pkm gene (Pkm2fl/fl) to generate mice with PKM2 deleted in T cells (referred to as PKM2-KO mice) (Figure S1A). We confirmed that PKM2 was efficiently deleted by immunoblot analysis of purified CD4+ T cells from PKM2-KO mice, though, PKM1 was robustly expressed in PKM2-KO T cells (Figure S1B). Baseline functional metabolism of splenic CD4+ T cells was comparable between WT and KO PKM2 mice (Figure S1C). We found that the frequencies of T cells and T cell precursors were not altered by PKM2 deficiency in multiple immune organs analyzed (Figure S1D–G). Further, PKM2-KO T cells proliferated to a similar extent as WT T cells and were not defective in their ability to differentiate into helper T cell subsets, including IL-17A producers (Figure S1H). Similarly, OCR and ECAR were comparable in WT and PKM2-KO T cells skewed toward the TH17 lineage (Figure S1I).
We next hypothesized that PKM1 may be compensating the loss of PKM2, possibly due to the redirected RNA splicing caused by Pkm exon 10 deletion. Indeed, qRT-PCR studies showed induction of the PKM1 transcript in PKM2 KO T helper subsets (Figure S1J). Immunoblot analysis confirmed PKM1 protein expression in PKM2 deficient cells, which was not detectable in CD4+ T cells from WT mice (Figure S1B). These data demonstrate that deletion of PKM2 may be functionally compensated by an increase in PKM1 expression.
The PKM2 activator TEPP-46 binds to PKM1
The targeting specificity of TEPP-46 and DASA-58 for PKM2 is based on the structural differences between PKM1 and PKM2 (23, 24). Thus, we did not anticipate finding that the PKM2 activators TEPP-46 and DASA-58 potently inhibited TH17 differentiation in PKM2 deficient T cells (Figure 3A). To exclude the possibility of incomplete deletion of PKM2, which would allow selective PKM2 positive T cell expansion during in vitro culture, we analyzed PKM2 protein expression in differentiated TH17 cells. However, we did not observe detectable amounts of PKM2 even after 48 hours of TH17 differentiation of PKM2-KO CD4+ T cells (Figure 3B). To next address the possibility that PKM2 activators may be target PKM1 in the absence of PKM2, we created a photoactivatable TEPP-46 derivative, modified TEPP-46 (mTEPP-46, Figure 3C). mTEPP-46 had a dose-dependent effect on TH17 cytokine production, similar to that of the parent molecule (Figure 3D-E). Because of the limited protein yields from primary T cells, we analyzed the binding of mTEPP-46 to PKM1 and PKM2 in Jurkat T cells, which express both PKM isoforms endogenously (Figure 3F, input). mTEPP-46 was added to Jurkat cells and, following photo-crosslinking, proteins bound to mTEPP-46 were identified. We found that mTEPP-46 exhibited binding to both PKM1 and PKM2 in Jurkat cells (Figure 3F–G). These data show that TEPP-46 can bind to PKM1 and PKM2 to potentially modulate TH17 cell differentiation.
Fig. 3. PKM2 activators inhibit differentiation of PKM2-KO TH17 cells and bind PKM1.

(A) Intracellular flow cytometry analysis of GM-CSF and IL-17A production by WT (upper) and PKM2-KO (lower) TH17 cells differentiated with DMSO, TEPP-46, or DASA-58 for 2 days. Data are means ± SEM pooled from N=2 independent experiments on 5 mice. (B) PKM1 and PKM2 immunoblot on cell extracts from TH17 cells from PKM2-WT or PKM2-KO mice. Blots are representative of N=2 independent experiments. (C) Chemical formula of TEPP-46 and modified TEPP-46 (mTEPP-46). (D and E) Intracellular flow cytometry analysis of GM-CSF or IL-17A production by TH17 cells differentiated with DMSO, TEPP-46, DASA-58, or mTEPP46 for 48 hours. Data are from N=1 independent experiment with 2 mice/treatment group. (F and G) Immunoblot for mTEPP-46 binding to PKM1 and PKM2. Blots (F) are representative of and quantified values (G) are from N=3 independent experiments. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 by one-way ANOVA with Tukey post test (A) or paired Student’s t-test (G).
TEPP-46 increases the enzymatic activity of PKM2 but not that PKM1 in CD4+ T cells
Because mTEPP-46 bound to PKM1, we hypothesized that an overlapping TEPP-46-sensitive function of PKM2 and PKM1 in CD4+ T cells could explain why PKM2 activators affected PKM2-KO T cells. To date, the most frequently used measure of TEPP-46 selectivity for PKM species is an in vitro assay for enzymatic activity that reports conversion of phosphoenol pyruvate (PEP) to pyruvate (25). To determine if TEPP-46 impacted the metabolic functions of PKM1-expressing CD4+ T cells, we measured the effect of TEPP-46 on PK enzymatic activity (Figure 4A). Although TEPP-46 increased PK activity in PKM2 expressing cells, it was unable to do so in PKM2-KO cells. Thus, as previously shown, TEPP-46 is indeed selective for PKM2 when analyzed strictly through the lens of enzymatic activity. We subsequently studied the effects of TEPP-46 on the functional metabolism of PKM2-WT CD4+ T cells, expecting that TEPP-46 would promote glycolysis. Instead, we found that TEPP-46 impaired maximal glycolytic flux in unstimulated CD4+ T cells (Figure 4B),but did not impact oxidative phosphorylation (Figure 4C). In contrast, TEPP-46 did not impact the metabolism of CD4+ T cells differentiated for 48 hours, suggesting that any impact of TEPP-46 on CD4+ T cells related to metabolism occurred during the initial differentiation phase (Figure 4B-C). Thus, although TEPP-46 selectively increased PK activity in PKM2-WT T cells, overall glycolytic flux was not modified.
Fig. 4: TEPP-46 impacts enzymatic activity of PKM2, but not that of PKM1, but does not increase overall glycolytic flux in T cells.

(A) Pyruvate kinase (PK) activity assay on unstimulated CD4+ T cells from PKM2-WT and PKM2-KO mice treated with DMSO or TEPP-46 (50μM). Data are pooled from N=2–3 independent experiments with T cells freshly isolated from n=8 PKM2-WT and n=5 PKM2-KO mice. Significance was determined by repeated measures two-way ANOVA with Sidak post-test. (B and C) Assessment of WT CD4+ T cell functional metabolism by glycolytic (data pooled from N=3 independent experiments on CD4+ T cells from n=6 WT mice) (B) and mitochondrial stress tests (data pooled from N=2 independent experiments with n=4 WT mice) after 45 minutes of treatment with DMSO, TEPP-46 (50μM) or 2-deoxyglucose (2-DG, 1mM) (C). Significance determined by repeated measures two-way ANOVA with Bonferroni post-test. ****p<0.0001. All data are expressed as mean ± SEM. Oligo = oligomycin; FCCP = mitochondrial uncoupler; Rot/AA = rotenone + antimycin A.
PKM2 activators block TGF-β1 signaling
TH17 cell differentiation is driven by TGF-β1 (transforming growth factor β1) and IL-6, cytokines that also govern the development of a related T cell type – the anti-inflammatory regulatory T cells (Treg) (29–31). IL-6 activation induces STAT3 phosphorylation that is considered critical for tipping the balance between TH17 and Treg cells in favor of TH17 cells (Figure 5A)(32). Mice lacking Stat3 in CD4+ T cells are impaired in the generation of IL-17-producing TH17 cells and are resistant to EAE induction (33, 34). Therefore, we posited that STAT3 phosphorylation could be perturbed in the presence of PKM2 activators. TEPP-46-induced skewing of CD4+ T cells to TH17 cells did not result in deficient STAT3 phosphorylation (Figure 5B).
Fig. 5: TEPP-46 inhibits Smad2/3 activation during TH17 differentiation.

(A) Schematic showing cytokines that drive RORγT+ TH17 and FoxP3+ Treg cell differentiation. (B) TH17 cells skewed for 24 hours with TEPP-46 (50μM) or vehicle (DMSO) were lysed and cell extracts subjected to immunoblot analysis of phosphorylated STAT3 (P-STAT3) and total STAT3 (T-STAT3). Quantification is pooled densitometric analysis from n=7 mice from three independent experiments. Paired Student’s t-test. (C) Flow cytometry analysis of TGF-β1-mediated induction of FoxP3 in the presence of indicated concentration of TEPP-46 or DASA-58 or vehicle (DMSO). Quantification is of n=6–7 mice per group pooled from N=3 independent experiments. Repeated measures one-way ANOVA with Tukey post test. (D) assessment of P-SMAD2 in response to TGF-β1 treatment in Jurkat cells pretreated with DMSO or TEPP-46. Quantification of N=4, each dot pair represents one independent experiment; paired t test. (E) Cells treated with TGF-β1 in the presence of TEPP-46 (50μM) or vehicle (DMSO) for 24 hours were subjected to qRT-PCR analysis for Ccr8 and Irf8 expression. Quantification is of n=6 mice per group pooled from N=3 independent experiments. Repeated measures one-way ANOVA with Tukey post test. (F) Purified CD4+ T cells were skewed towards TH17 in the presence of increasing concentrations of TGF-β1 in the presence of TEPP-46 (50μM) or vehicle (DMSO) for 48 hours and subjected to flow cytometry analysis of IL-17A and GM-CSF production after 6 hours of PMA/ionomycin stimulation. Quantification is of n=4 mice per group pooled from N=2 independent experiments. Repeated measures two-way ANOVA with Tukey post test. **p<0.01, ****p<0.0001.
We reasoned that PKM2 activators might inhibit a lineage-driving factor common between Treg and TH17 cells. To test this hypothesis, we determined the effects of PKM2 activators on TGF-β1-dependent generation of inducible Tregs (iTregs) in vitro. After 2 days of stimulation, TGF-β1 treatment induced a large population of FoxP3-expressing T cells (up to 50%) which was significantly inhibited by TEPP-46 and DASA-58 (Figure 5C). TGF-β1 signaling is predominantly mediated by the receptor-induced activation of the transcription factors Smad2 and Smad3, which, together with Smad4, regulate gene expression (35). TEPP-46 impaired TGF-β-mediated phosphorylation of Smad2 in Jurkat T cells (Figure 5D). We thus assessed the effects of TEPP-46 on Smad2/3-dependent and independent gene targets (36). We found that TEPP-46 blocked induction of the Smad2/3-dependent target Irf8 (which encodes interferon regulatory factor 8), but did not alter expression of the Smad2/3-independent CCR8 (which encodes C-C motif chemokine receptor 8) (Figure 5E). These results are consistent with the significantly reduced expression of the Smad2/3-dependent FoxP3 (Figure 5C). To test whether increased TGF-β1 signaling alleviated the inhibitory effect of TEPP-46 on TH17 differentiation, we tested a range of TGF-β1 concentrations during TH17 skewing. Indeed, increasing amounts of TGF-β1 increased production of IL-17A, with a concomitant decrease in GM-CSF, in TH17 cells differentiated in the presence of TEPP-46 (Figure 5F). A possible mechanism by which TEPP-46 impacts TGF-β1 signaling is by impinging upon autocrine signaling by inhibiting either TGF-β1 or TβRII induction. However, we found that neither TEPP-46 nor DASA-58 impaired production of either of these two essential arms of autocrine TGF-β signaling in T cells at the mRNA level (Figure S2). These data suggest that PKM2 activators inhibit TH17 cell differentiation by reducing TGFβ1 induced Smad2/3-dependent gene expression through a cell-intrinsic process downstream of TGF-β1-TβRII ligation.
PKM2 activator-mediated enhancement of GM-CSF production requires STAT5 signaling
TGF-β1 signaling inhibits the phosphorylation of STAT5 (37), the primary transcription factor associated with GM-CSF induction (38, 39) (Figure 6A). Because we found that PKM2 activators decreased IL-17A production but increased GM-CSF production, we tested the effects of TEPP-46 on STAT5 phosphorylation in differentiated TH17 cells. TEPP-46 treatment resulted in a ~2-fold increase in the phosphorylation of STAT5 (Figure 6B). To next test if STAT5 was required for TEPP-46-mediated changes to the TH17 cytokine profile, we used the selective STAT5 inhibitor STAT5-IN-1 (40). STAT5 inhibition reversed the effects of TEPP-46 on GM-CSF and IL-17A production (Figure 6C). Collectively, these results suggest that TEPP-46 treatment shifts the T cell signaling balance in favor of STAT5 activation.
Fig. 6: TEPP-46 enhances STAT5 activation during TH17 differentiation.

(A) Schematic of P-STAT3 and P-STAT5 effects on IL-17A and GM-CSF expression. (B) TH17 cells incubated with TEPP-46 (50μM) or vehicle (DMSO) for 24 hours were subjected to immunoblot analysis for phosphorylated STAT5 (P-STAT5) or total STAT5 (T-STAT5). Quantification is of n=7 mice pooled from N=3 independent experiments. Paired Student’s t-test. (C) Purified CD4+ T cells were skewed towards TH17 in the presence of TEPP-46 (50μM), ± STAT5 inhibitor (STAT5-IN-1, 100μM) for 48 hours, and subjected to flow cytometry analysis of GM-CSF and IL-17A expression. Quantification is of n=7 mice per group pooled from N=3 independent experiments. Repeated measures two-way ANOVA with Tukey post test. Significance is as follows: **p<0.01, ****p<0.0001. Data are presented as mean ± SEM.
PKM2 activator treatment drives highly encephalitogenic EAE
We next sought to test the functional consequences of PKM2 activator treatment in EAE mouse models of MS. Because of the reported unsuitability of DASA-58 for in vivo studies (25), we used TEPP-46 for these studies. We first assessed the effect of TEPP-46 on passive EAE, which is induced by TH17 cell transfer into immunodeficient recipients (Figure 7A), with 2D2 mice, in which CD4+ T cells express a transgenic T cell receptor specific for the myelin oligodendrocyte glycoprotein (MOG)(41). We skewed 2D2 splenocytes toward the TH17 lineage in the presence or absence of TEPP-46 and confirmed that TEPP-46 treatment resulted in decreased IL-17A and increased GM-CSF production (Figure 7B). We then transferred skewed cells into immunodeficient (Rag1−/−) mice and scored EAE development. Mice receiving the vehicle treated cells developed a severe ascending paralytic syndrome that is classically associated with EAE. However, whereas some mice receiving the TEPP-46-treated cells exhibited classic EAE disease signs, many presented with disease signs commonly associated with atypical EAE (42) (Figure 7C). Flow cytometric analyses revealed that mice with TEPP-46-associated atypical EAE showed increased accumulation of immune cells in the brain and in the cerebellum and brainstem, with fewer cells in the spinal cord, when compared to mice that received vehicle treated cells (Figure 7D). Histology examination confirmed that TEPP-46 enhanced the accumulation of immune cells in the periventricular regions of the brain (Figure 7E). We noted CD45+ cell enrichment in the periventricular regions in 66% of the mice receiving TEPP-46 treated cells, compared to 14% of mice that received vehicle treated cells. These data could result from the TEPP-46-induced increase in GM-CSF, which drives atypical EAE (42). These findings collectively call into question the therapeutic potential of TEPP-46 in the treatment of multiple sclerosis.
Fig. 7: PKM2 activator treatment redirects TH17 cells to adopt an encephalitogenic program.

(A) Schematic showing the stimulation of splenocytes from 2D2 mice and passive EAE induction. (B) Flow cytometry analysis of IL-17A and GM-CSF production by TCRβ+CD4+ cells on the day of splenocyte transfer. *p<0.05 by repeated measures one-way ANOVA with Tukey post test. (C) EAE phenotypes observed at day 16 post transfer of splenocytes treated with TEPP-46 (50μM) or vehicle (DMSO). Data shown are from n=19 DMSO- and n=24 TEPP-46-treated Rag1−/− mice that received 2D2 cells, pooled from four independent experiments. (D) Flow cytometry analysis of MHC-II and Ly6G expression on myeloid cells (defined as live, singlet, CD45hiCD11b+ cells) isolated from the indicated parts of the CNS of Rag1−/− mice receiving DMSO- or TEPP-46-treated cells. Data are expressed as mean ± SEM from n=10–12 mice per group pooled from N=2 independent experiments. Two-tailed Welch’s t-test. *p<0.05. (E) Representative images (left panels) of anti-CD45 staining in the periventricular brain regions from recipients of DMSO- or TEPP-46-treated splenocytes and quantification (right panel) or mice presenting with perivascular infiltrates. Data are compiled from two independent experiments with a total of 6 mice. Scale bar is 400 μm.
DISCUSSION
Immune cell activation is linked to a metabolic adaption characterized by a transition from oxidative phosphorylation to increased reliance on aerobic glycolysis, also known as the Warburg effect (43, 44). The purported goal of this metabolic shift is to maintain the balance between energy production and the generation of the building blocks for cell proliferation and protein expression. This process was initially identified in cancer cells and has been proposed as an exciting therapeutic target to block excessive inflammation in diseases with prominent inflammatory components, like MS (45–47).
PK is the ultimate rate limiting enzyme of glycolysis and is an attractive target for the treatment of inflammatory diseases such as MS. However, global inhibition of glycolysis is not a viable therapeutic strategy for all but the most acutely terminal diseases. The exclusive expression of a specific isoform of the pyruvate kinase enzyme PKM2 in immune cells (27, 48) and its targetable non-metabolic functions that could contribute to inflammation has provided us an exciting avenue to destabilize a piece of metabolic machinery for anti-inflammatory purposes. Thus, the goal of our study was to explore the therapeutic potential of targeting the non-metabolic functions of PKM2 in T cells in the context of MS.
Our data presented here highlight several new aspects of PKM regulation of T cell functions (Figure 8). First, we showed that the PKM2 “activators” TEPP-46 and DASA-58, which drive PKM2 enzymatic activity (25), blocked the differentiation of IL-17-producing TH17 cells, critical drivers of MS pathology (49). In contrast to the activity of PKM2 in macrophages (22), treatment of T cells with PKM2 activators did not alter STAT3 activity, a master regulator of TH17 development (33). Second, though TH17 differentiation is inversely linked with the development of anti-inflammatory regulatory T cells, Treg (50), we found that PKM2 activators also blocked Treg differentiation. This finding suggests that PKM2 activators may not interfere with Hif-1α activity (19, 20), because inhibition of Hif-1α would be expected to decrease TH17 differentiation but increase FoxP3-expressing Tregs (9). Third, we demonstrated that TEPP-46 impaired signaling pathways elicited by TGF-β1, thereby explaining the concurrent inhibition of TH17 and Treg differentiation. Fourth, the effect of TEPP-46 and DASA-58 on TH17 cells was two-fold. Inhibition of IL-17A production coincided with the generation of T cells capable of producing GM-CSF; GM-CSF-competent Th cells are being increasingly recognized as contributors to MS pathology (39, 51, 52). As found in (53), we also determined that production of GM-CSF mRNA is hampered during TH17 differentiation in the presence of PKM2-tetramerizing compounds. However, we showed that functionally in vitro and in vivo, TEPP-46 treatment yielded a T cell population that could produce GM-CSF and generate GM-CSF-associated phenotypes when encountering re-activating stimuli. We showed that the mechanism underlying these observations likely involved TGF-β1 signaling, although it could be related to effects of TEPP-46 on epigenetic or posttranslational regulation of GM-CSF production.
Fig. 8: Proposed mechanism of action of TEPP-46 on TH17 cells.

TEPP-46 is an inhibitor of TGF-β1, a function that may be mediated by PKM1 or PKM2. Inhibition of TGF-β1 blocks the differentiation of IL-17 producing TH17 cells while maintaining GM-CSF production.
We found that increased GM-CSF production in the presence of PKM2 activators relied on the activation of STAT5 signaling. Because TGF-β1 directly inhibits STAT5 phosphorylation (37), future experiments will investigate the link between STAT5, TGF-β1, and metabolic flux in the context of PKM activator treatment. Although our data implicate TGF-β signaling as a pathway potentially affected by TEPP-46, there are other possible mechanisms by which this compound may impact T cell functions. For example, PKM2 activation by TEPP-46 regulates p53, which is critical in regulating the balance between TH17 and Treg development (54, 55). Further studies will be needed to understand the integral role of PKM2 in T cell biology.
Ultimately, in contrast to previously published data (53), the observed effects of PKM2 activators on T cell cytokine production presented here do not support the use of PKM2 activators in MS. Not only did PKM2 activators block the induction of Tregs, a finding that should be unnerving for a therapy aimed at a population of patients already predisposed to the generation of autoinflammatory conditions, but it also promoted the generation of T cells capable of producing both IFN-γ and GM-CSF. It is unclear as to why the data presented here diverge from previous observations (53). Differences in cell isolation strategy could account for this discrepancy. Because we assessed the function of TEPP-46 treatment on bulk isolated CD4+ T cells, it would be informative but less clinically applicable to study the effect of TEPP-46 on strictly naive CD4+ T cells. In addition, differences in cell skewing strategy can impact T cell cytokine production, and our conflicting results may reflect variability in the therapeutic potential of TEPP-46 based on nuances in the different skewing strategies employed. Nuances surrounding PKM2, the drugs that target it, and their function in T cells are worth exploring in the effort to further validate these compounds for clinical use.
With our passive EAE studies, we found that T cells that encountered TEPP-46 during activation and differentiation were more likely to home to and drive pathology in the brain. This brain-directed inflammation is a major hallmark of MS that is not represented in many EAE models that, for unclear reasons, more commonly target the spinal cord. Collectively, although our work does not support the use of PKM activators as a treatment of MS, our discovery that PKM activators block TGF-β1 signaling could support their potential therapeutic use in fibrotic diseases, which are driven by increased TGF-β1 function (56).
Our studies presented here demonstrated that PKM2 protein may not be required for at least some of the effects of TEPP-46 and DASA-58 on T cells. Given that TEPP-46 and DASA-58 are two structurally distinct compounds exhibiting similar “off-target” effects, it is logical to postulate that they could act on PKM1. This hypothesis is strengthened by our TEPP-46 binding study, which demonstrated an association with PKM1. We predict that PKM2 activators bind to and disrupt the nonmetabolic activities of compensatory PKM1 in T cells that simply cannot be ascertained through the study of activity assays alone. Another alternative hypothesis is that TEPP-46 can bind to other molecule(s) to impact T cells function and differentiation. More work is needed to determine the complete interactome of TEPP-46.
The exclusive expression of PKM2 in primary murine T cells is interesting, particularly given the reported protein expression of both PKM1 and PKM2 in macrophages (20). The aspects of T cell function that would benefit from the selective expression of PKM2 are currently unclear, because we showed that PKM2-specific functions may be redundantly performed by PKM1. Collectively, our data suggest that there is a critical need to develop the next generation PKM2 inhibitors or an improved animal model of PKM2 deficiency to further address the unique function of PKM2 in T cells.
MATERIALS AND METHODS:
Animals
Mice were housed in accordance with the guidelines of the University of Virginia Animal Care and Use Committee (protocol #1918). PKM2-KO mice were developed by obtaining and crossing two commercially available strains from Jackson Laboratories: Cd4-Cre (#022071) and Pkm2fl/fl (#024048). 2D2 (#006912) and Rag1−/− (#002216) mice were also obtained from Jackson Laboratories. All studies were performed in age- and sex-matched mice, with both male and female mice used, at 8–12 weeks of age.
Tissue Staining
Mouse tissues for immunohistochemistry (IHC) were prepared by the Research Histology Core at the University of Virginia School of Medicine. For staining, 10 μm sections were deparaffinized in xylene and ethanol gradients and stained with Luxol Fast Blue, as previously described (57). PKM2 staining was performed using the Cell Signaling Technologies antibody against PKM2 (D784) and the Vector Labs ImmPRESS Excel Rabbit IgG Peroxidase Staining Kit (#MP-7601). Anti-CD45 IHC was performed by the UVA Biorepository and Tissue Research Facility (BTRF). For immunofluorescent (IF) studies, frozen tissue sections (30 μm) were stained with anti-PKM2 (D784) followed by Donkey anti-Rabbit Cy3, and anti-CD3-e660 (eBioscience #50–0032-82). Isotype controls were used as negative controls. IHC images were captured using the EVOS FL Auto and manufacturer-provided software. Immunofluorescence images were captured using the SP8 Leica Confocal Microscope and were analyzed with Image J.
CD4+ T cell isolation and differentiation
CD4+ T cells were isolated from the spleen and lymph node homogenates using the EasySep™ mouse CD4+ T cell isolation Kit (StemCell Technologies #19852). For in vitro differentiation of sorted CD4+ T cells, 2×106 cells were plated per well of a 24-well tissue culture plate pre-coated with anti-CD3 (2C11, 1μg/mL) and anti-CD28 (PV1, 2μg/mL). TH17 differentiation was performed in Iscove’s Modified Dubelcco’s Medium (IMDM, Gibco #12440–053) supplemented with 10% FBS, non-essential amino acids (Gibco #11140–050), 1 mM sodium pyruvate (Gibco #11360–070), HEPES (25 mM), β-mercaptoethanol (50μM, Fisher Scientific #O3446I-100), and 100 U/mL penicillin/streptomycin (Gibco #15140122). TH17 skewing cocktail was as follows: murine IL-6 (20 ng/mL), IL-23 (10 ng/mL), TGF-β1 (0.3 ng/mL), anti-IFN-γ (10 μg/mL, XMG1.2) and anti-IL-4 (10 μg/mL, 11B11). TH1 skewing was performed in RPMI-1640 (Gibco #11875–093) supplemented as IMDM, except with 10 mM HEPES (Gibco #15630–080), human IL-2 (100 U/mL), murine IL-12 (10 ng/mL), and anti-IL-4 (10 μg/mL). Blocking antibodies were purchased from BioXCell and cytokines were purchased from Thermo Fisher Scientific. Treg differentiation was achieved by culture with TGF-β1 (5 ng/mL) in XVIVO-10 (Lonza 04–743Q) supplemented with penicillin/streptomycin. TEPP-46 (50 μM) and DASA-58 (25 μM) were initially diluted in dimethylsulfoxide (DMSO) and then further diluted using the culture medium (Cayman Chemical Company or MedChemExpress). Effects of STAT5 inhibition were assessed using STAT5-IN-1 obtained from MedChemExpress (#HY-101853) and used at 100 μM.
Seahorse extracellular flux analysis
Functional metabolic assays were performed using the Seahorse Extracellular Flux XFp analysis system. CD4+ T cells were isolated from lymph nodes and spleens and plated at 4×105 cells per well to assess metabolic function in unstimulated cells. To assess metabolism after stimulation, cells were skewed toward specific Th lineages for at least 2 days and subsequently assayed at 1×105 cells per well. Glycolysis and mitochondrial stress tests were performed according to manufacturer’s instructions with reagents as follows: glucose (Gibco #A24940–01), Sodium Pyruvate (Gibco #11360–070), L-glutamine (Gibco #25030081), Oligomycin (Sigma-Aldrich #04876), FCCP (Cayman Chemical Company #15218), Rotenone (MP Biomedicals #0215015401), Antimycin A (Sigma-Aldrich #A8674), 2-deoxyglucose (Acros Organics #111980050)
Experimental Autoimmune Encephalomyelitis (EAE)
Transfer EAE studies were performed by harvesting splenocytes from 8-week old 2D2 mice and skewing them toward the TH17 lineage in the presence of 50 μg/mL MOG35–55 and TEPP-46 for 2 days. TH17 skewed cells were transferred into immunodeficient Rag1−/− recipients of the same sex. On day 0 and day 2 post transfer, recipient mice were given 200 and 400 ng of pertussis toxin (List Biologicals), respectively. Mice were scored according to the previously established criteria (27) by an investigator blinded to the mouse genotype/treatment. Briefly, disease severity was measured as follows: 0, no disease; 1, tail paralysis; 2, gait abnormality on grate walk; 3, hindlimb weakness on grate flip; 4, bilateral hindlimb paralysis or failure of plantar placement; 5, moribund.
Cell harvest from central nervous system tissues
To determine immune invasion of the CNS by flow cytometry, mice were euthanized by CO2 asphyxiation and then perfused with sterile saline with 5 U/mL heparin. Brain, spinal cord, and cerebellum were harvested in HBSS with Ca2+/Mg2+ (Thermo Fisher Scientific #14025126) and digested in 4 mg/mL collagenase IV (Worthington LS004188) with 50 U/mL DNase (Worthington LS002139). Digestion was achieved by shaking tissues at 180 RPM at 37°C for 45 minutes. To assist with digestion, CNS tissues were gently triturated through a 5 mL pipette every 15 min. Complete RPMI was used to stop digestion. Cells were filtered using a 70μM strainer, and samples were pelleted. Debris generated during the digestion was removed by Percoll density gradient (GE Healthcare #17–0891-01). Isolated cells were subsequently washed in culture medium and used for downstream studies.
Flow Cytometry
Antibodies used are as follows: TCRβ (H57-597), CD4 (RM4-5), CD8 (53–6.7), CD19 (eBio1D3), CD11b (M1/70), MHC-II (M5/114.15.2), CD45 (30-F11), CD25 (PC61.5), FoxP3 (FJK-16s), RORγT (B2D), T-bet (eBio4B10), IL-17A (TC11–18H10.1), IL-2 (JES6–5H4), IFN-γ (XMG1.2), GM-CSF (MP1–22E9), and CD16/32 Fc Block (93). All antibodies were purchased from eBioscience/Thermo Fisher Scientific, except IL-17A (BioLegend clone# TC11-18H10.1). Live/Dead discrimination was performed using Ghost Dye™ Violet 510 from (Tonbo Biosciences, #13–0870) or Zombie Aqua Fixable Viability kit (BioLegend #423101). For macrophage/myeloid stains, cells were used for compensation controls. For T cell stains, OneComp eBeads (Thermo Fisher Scientific 01-1111-42) were used. Staining was performed in PBS with 2% FBS and 2 mM EDTA. For intracellular cytokine staining, the eBioscience™ Intracellular Fixation and Permeabilization Kit (#88-8824-00) was used. For intranuclear staining, the eBioscience™ FoxP3/Transcription Factor Staining kit (#00-5523-00) was used. Staining was performed according to the manufacturer’s recommendations. Flow cytometry was performed using a Beckman Coulter Gallios flow cytometer and data were analyzed with FlowJo software (Tree Star, Inc.).
DSS Cross-Linking
Cytoplasmic extracts were prepared from Jurkat cells by resuspending pelleted cells in 10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1mM DTT and protease inhibitors (MedChemExpress HY-K0010) with a gauge 27 needle. After centrifugation to remove intact nuclei and membranes, extracts were supplemented with 10X PBS to adjust salt concentration to 1X PBS final and 100 μM TEPP46 (or vehicle) and kept at 37°C for 15 min. DSS crosslinker (2mM) was added for 5 min and the reaction was stopped with 20mM Tris pH8.
RNA isolation and qRT-PCR
For gene expression studies, RNA was isolated using the Bioline Isolate II RNA Mini Kit (BIO-52073) and quantified using the BioTek Epoch Microplate Spectrophotometer. Equal amounts of RNA were then converted to cDNA using the Bioline SensiFAST™ cDNA synthesis kit (BIO-65054). qRT-PCR was performed using SYBR Green system and the Thermo Fisher Scientific Pikoreal96 or BioRad CFX384 Touch™ systems. Primer sequences are in Supplementary Table 1.
Modified TEPP46
TEPP-46 (0.107 mmol) was dissolved in 5 mL of dimethylformamide in the presence of 0.161 mmol of the photolinker and 0.428 mmol of Di-isopropylethylamine. The solution was stirred under nitrogen for 1 day at 40 C. mTEPP46 was purified by gravity column (4% MeOH in dichloromethane). mTEPP46 was further characterized by NMR to confirm purity and resuspended in DMSO before use. Jurkat cells were plated 2 days before the experiment at 1×106 per ml. mTEPP46 (100μM) or vehicle was added to serum-free media and cells were incubated for 3h in 37°C. Next, photo crosslinking was induced by irradiating cells with UV light (10 min, 350nm). Cells were washed twice with PBS and protein extracts were prepared in RIPA containing protease inhibitors (MedChemExpress HY-K0010). Equal amounts of proteins (2.5 mg in 500 μl) were treated with 10 μl of 1,25 mM desthiobiotin, 10 μl of 50 mM TCEP (Tris(2-carboxyethyl)phosphine), 30μl of 1,7mM TBTA (Tris-(Benzyltriazolylmethyl)amine), 10 μl of 50 mM CuSO4 for 1h. After incubation, proteins were precipitated by methanol-chloroform extraction (58). After centrifugation, pellets were resuspended in 200 μl of RIPA buffer and incubated with neutravidin beads [Thermo High Capacity Neutravidin Agarose Resin] overnight at 4 C. The beads were washed 4x with ice-cold RIPA buffer containing protease inhibitors and resuspended with Laemmli buffer containing 50mM 1,4-Dithiothreitol (DTT) (Fisher Scientific # BP172–5).
PK Activity Assay
For PK activity assays, CD4+ T cells were freshly isolated and frozen in 1×106 cells aliquots at −80C. Cell pellets were lysed and subjected to the PK activity assay as per the manufacturer’s recommendations (Millipore Sigma # MAK072–1KT). To determine the impact of TEPP-46 on PK activity, cell lysates were treated with either DMSO or TEPP-46 (50μM) immediately prior to assaying for enzyme activity.
Western Blotting
For assessment of protein amounts by Western Blot, cells were lysed in ice-cold RIPA containing protease inhibitors (MedChemExpress HY-K0010) and 1 mM sodium orthovanadate (Sigma Aldrich #450243). Protein concentration was determined using the Pierce™ BCA assay (Thermo Fisher Scientific #23225). Lysates were denatured in SDS with 100 μM DTT and electrophoresed on BioRad Mini-PROTEAN® TGX™ Any KD™ precast gels. The BioRad TransBlot® Turbo™ system was used to transfer protein onto BioRad Immun-Blot PVDF membranes. Antibodies used are as follows: Phospho-Tyr694 STAT5 (C11C5), Phospho-Tyr705 STAT3 (D3A7), Phospho-Ser465/Ser467 SMAD2 (E8F3R), STAT5 (#9363S), STAT3 (124H6), SMAD2 (D43B4), PKM1 (D30G6), PKM2 (D784), pan PKM (#3106), Lamin B1 (D4Q4Z), Actin (AC-74). All Western Blotting antibodies were purchased from Cell Signaling Technologies except anti-Actin (Sigma Aldrich). Secondary antibodies used were as follows: anti-Mouse IgG HRP-linked whole Ab (NXA931) and anti-Rabbit IgG HRP-linked whole Ab (NA934) were purchased from Sigma Aldrich. Chemiluminescence was achieved using the Perkin Elmer LLC Western Lighting Plus-ECL, Enhanced Chemiluminescent Substrate (Thermo Fisher Scientific #50–904-9325) and captured using the GE Healthcare ImageQuant LAS4000mini. For subcellular fractionation studies, the NE-PER™ Nuclear and Cytoplasmic Extraction Kit was used (Thermo Fisher Scientific #78833).
Statistics
All statistical analyses were performed with Prism 7 (GraphPad software). Specific tests used to determine significance are described in the figure legends.
Supplementary Material
Fig. S1. Characterization of T cells from Pkm2fl/fl/Cd4-Cre mice.
Fig S2. Effects of TEPP-46 are not mediated by decreased expression of TGF-β1 or TβRII.
Table S1: Primers used in the study.
Acknowledgments:
The authors wish to thank Dr. Sanja Arandjelovic for critical reading.
Funding: This work was supported by grants from the National Institutes of Health (R01 NS083542 to A.G., R33 MH108156 to A.G., T32 GM008328 to S.M.S, A.F.C, R.M.B, T32 GM007055 to D.A.R, V.S., T32 GM007267 to S.M.S, F31 NS103327 to S.M.S., NSF2018255830 to R.M) and the Owens Family Foundation (to A.G.).
Footnotes
Competing Interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Compston A, Coles A, Multiple sclerosis. Lancet 372, 1502–1517 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, McCandless EE, Patel JR, Luker GD, Littman DR, Russell JH, Klein RS, CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med 208, 327–339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A, Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 13, 1173–1175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simmons SB, Liggitt D, Goverman JM, Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J Immunol 193, 555–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, Wucherpfennig K, Turley S, Carroll MC, Sobel RA, Bettelli E, Kuchroo VK, Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 35, 986–996 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, Li X, Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci 16, 1401–1408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR, The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B, RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12, 560–567 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F, Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG, Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179, 4313–4317 (2007). [DOI] [PubMed] [Google Scholar]
- 11.He Z, Ma J, Wang R, Zhang J, Huang Z, Wang F, Sen S, Rothenberg EV, Sun Z, A two-amino-acid substitution in the transcription factor RORgammat disrupts its function in TH17 differentiation but not in thymocyte development. Nat Immunol 18, 1128–1138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H, HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208, 1367–1376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierson ER, Goverman JM, GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight 2, e92362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, Becher B, The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 43, 502–514 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, Wendling JM, Morgan RW, Aicher TD, Carter LL, Toogood PL, Glick GD, Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol 11, 141–147 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Franchi L, Monteleone I, Hao LY, Spahr MA, Zhao W, Liu X, Demock K, Kulkarni A, Lesch CA, Sanchez B, Carter L, Marafini I, Hu X, Mashadova O, Yuan M, Asara JM, Singh H, Lyssiotis CA, Monteleone G, Opipari AW, Glick GD, Inhibiting Oxidative Phosphorylation In Vivo Restrains Th17 Effector Responses and Ameliorates Murine Colitis. J Immunol 198, 2735–2746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta V, Bamezai RN, Human pyruvate kinase M2: a multifunctional protein. Protein science : a publication of the Protein Society 19, 2031–2044 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dayton TL, Jacks T, Vander Heiden MG, PKM2, cancer metabolism, and the road ahead. EMBO reports 17, 1721–1730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL, Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O’Neill LA, Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell metabolism 21, 65–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, Goronzy JJ, Weyand CM, The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med 213, 337–354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao X, Wang H, Yang JJ, Liu X, Liu ZR, Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell 45, 598–609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boxer MB, Jiang JK, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ, Evaluation of substituted N,N’-diarylsulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. J Med Chem 53, 1048–1055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang JK, Boxer MB, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ, Evaluation of thieno[3,2-b]pyrrole[3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg Med Chem Lett 20, 3387–3393 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ, Vander Heiden MG, Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8, 839–847 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, Yorek MA, Wu IH, Lockhart S, Coppey LJ, Pfenninger A, Liew CW, Qiang G, Burkart AM, Hastings S, Pober D, Cahill C, Niewczas MA, Israelsen WJ, Tinsley L, Stillman IE, Amenta PS, Feener EP, Vander Heiden MG, Stanton RC, King GL, Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med 23, 753–762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seki SM, Stevenson M, Rosen AM, Arandjelovic S, Gemta L, Bullock TNJ, Gaultier A, Lineage-Specific Metabolic Properties and Vulnerabilities of T Cells in the Demyelinating Central Nervous System. J Immunol 198, 4607–4617 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B, The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Manel N, Unutmaz D, Littman DR, The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol 9, 641–649 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li MO, Wan YY, Flavell RA, T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM, Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198, 1875–1886 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK, Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C, STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282, 9358–9363 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Lee YS, Yu CR, Egwuagu CE, Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol 180, 6070–6076 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, ten Dijke P, TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 16, 5353–5362 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Saika S, Hara T, Nomura M, Yoshimura A, Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol 185, 842–855 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Cocolakis E, Dai M, Drevet L, Ho J, Haines E, Ali S, Lebrun JJ, Smad signaling antagonizes STAT5-mediated gene transcription and mammary epithelial cell differentiation. J Biol Chem 283, 1293–1307 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, Gran B, Piehl F, Olsson T, Codarri L, Becher B, Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nature communications 5, 5056 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, Tan P, Moh A, Kaplan MH, Zhang Y, Fu XY, STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res 24, 1387–1402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Betts BC, Veerapathran A, Pidala J, Yu XZ, Anasetti C, STAT5 polarization promotes iTregs and suppresses human T-cell alloresponses while preserving CTL capacity. J Leukoc Biol 95, 205–213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK, Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197, 1073–1081 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, Becher B, Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity 46, 245–260 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Almeida L, Lochner M, Berod L, Sparwasser T, Metabolic pathways in T cell activation and lineage differentiation. Seminars in immunology 28, 514–524 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Warburg O, The Metabolism of Carcinoma Cells. Cancer research 9, (1935). [Google Scholar]
- 45.MacIver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC, Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. Journal of Leukocyte Biology 84, 949–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC, Glucose uptake is limiting in T cell activation and requires CD28-mediated akt-dependent and independent pathways. Journal of Immunology 180, 4476–4486 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC, Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4(+) T Cell Subsets. Journal of Immunology 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T, Vander Heiden MG, Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes & development 30, 1020–1033 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dos Passos GR, Sato DK, Becker J, Fujihara K, Th17 Cells Pathways in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders: Pathophysiological and Therapeutic Implications. Mediators Inflamm 2016, 5314541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohkura N, Kitagawa Y, Sakaguchi S, Development and maintenance of regulatory T cells. Immunity 38, 414–423 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Restorick SM, Durant L, Kalra S, Hassan-Smith G, Rathbone E, Douglas MR, Curnow SJ, CCR6(+) Th cells in the cerebrospinal fluid of persons with multiple sclerosis are dominated by pathogenic non-classic Th1 cells and GM-CSF-only-secreting Th cells. Brain Behav Immun 64, 71–79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, Chang HD, Radbruch A, Zielinski CE, IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med 6, 241ra280 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, Kane H, Papadopoulou G, Pearce EL, Mills KHG, O’Neill LAJ, Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4(+) T Cell Pathogenicity and Suppresses Autoimmunity. Cell metabolism, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takatori H, Kawashima H, Suzuki K, Nakajima H, Role of p53 in systemic autoimmune diseases. Crit Rev Immunol 34, 509–516 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Saleme B, Gurtu V, Zhang Y, Kinnaird A, Boukouris AE, Gopal K, Ussher JR, Sutendra G, Tissue-specific regulation of p53 by PKM2 is redox dependent and provides a therapeutic target for anthracycline-induced cardiotoxicity. Sci Transl Med 11, (2019). [DOI] [PubMed] [Google Scholar]
- 56.Meng XM, Nikolic-Paterson DJ, Lan HY, TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 12, 325–338 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Chuang TY, Guo Y, Seki SM, Rosen AM, Johanson DM, Mandell JW, Lucchinetti CF, Gaultier A, LRP1 expression in microglia is protective during CNS autoimmunity. Acta neuropathologica communications 4, 68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wessel D, Flugge UI, A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138, 141–143 (1984). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of T cells from Pkm2fl/fl/Cd4-Cre mice.
Fig S2. Effects of TEPP-46 are not mediated by decreased expression of TGF-β1 or TβRII.
Table S1: Primers used in the study.