

Abstract
Many proteins have been implicated in synaptic and experience-dependent plasticity. However, few demonstrate the exquisite regulation of expression and breadth of functional importance as the immediate early gene product Arc. Here we review and attempt to synthesize the disparate views of Arc in neuronal function. The main conclusion garnered from this body of work is that Arc is a critical effector molecule downstream of many molecular signaling pathways and that dysregulation of Arc expression can have dire consequences for normal brain function.
Brains store and process information from the outside world and do so elegantly through synaptic connections between interconnected networks of neurons. Knowing how neuronal networks are coordinated during information processing and how proteins and genes contribute to circuit modification during experience is fundamental to understanding brain function. Since the 1960s it has been known that long-term storage of information in the brain is dependent on rapid, de novo RNA and protein synthesis1. Similar macromolecular synthesis is essential for long-term forms of synaptic plasticity such as long-term potentiation (LTP) and depression (LTD)2. These activity-dependent changes in synaptic efficacy are suggested to underlie learning and memory3 and have been intensively studied. Many labs set out to identify the gene program that mediates protein synthesis–dependent plasticity, with a plethora of hits. Among these, the immediate early gene Arc (also known as Arg3.1)4,5 has proven to be the most tightly coupled to behavioral encoding of information in neuronal circuits in vivo6.
Arc is a single copy gene that is highly conserved in vertebrates and is induced in divergent behavioral paradigms in many species. Indeed, Arc mRNA and protein induction during behavioral learning is so robust and reproducible that cellular imaging of Arc induction is a powerful methodology for detecting neural networks that underlie information processing and memory6. In vivo, Arc is coordinately induced in populations of neurons that mediate learning such as place cells of the hippocampus7 and behavior-specific neural networks in the cortex8. For example, 5 min of spatial exploration elicits transcriptional induction of Arc in ~40% of CA1 neurons6.
Unlike most other immediate early gene products, Arc protein is not a transcription factor, but is instead a cytosolic protein that acts as an effector protein downstream of multiple neuronal signaling pathways.
Arc expression is confined to the brain and testis and seems to almost exclusively be expressed in CaMKII-positive glutamatergic neurons in hippocampus and neocortex9, with little or no expression in glial cells. Arc protein is found in the postsynaptic density (PSD) and co-purifies with the NMDA receptor complex10,11, but is not found in presynaptic terminals or axons. The tight transcriptional regulation of Arc seems to be determined by multiple transcriptional enhancer sites that contain binding domains for a set of transcription factors, including SRF, MEF2 and CREB. The precise signaling cascades involved in Arc transcription are not well defined. One study showed that PKA and MAPK cascades are involved in Arc induction12. Arc transcription is also regulated by neuronal spiking and calcium influx though voltage-sensitive calcium channels (VSCCs)13 and by group 1 metabotropic glutamate receptors (mGluRs)14. The precise kinetics of transcription and translation of Arc appear to differ according to which receptors and signaling pathways are used (Fig. 1a) and this has important implications for Arc’s role in neuronal plasticity, as discussed below.
Figure 1.
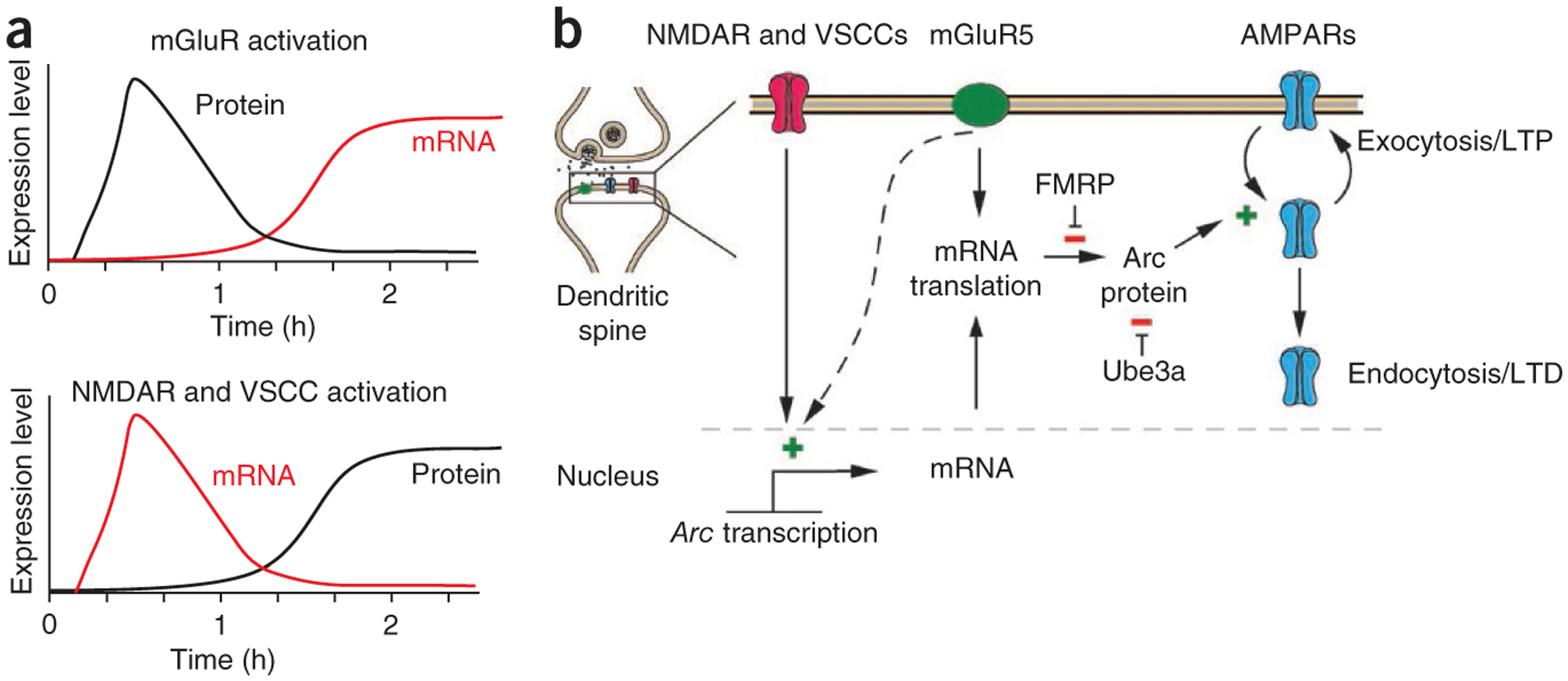
Regulated Arc expression modulates trafficking of AMPA receptors and stabilization of LTP and LTD. (a) Arc transcription and translation are differentially regulated by glutamate receptors and voltage-sensitive calcium channels (VSCCs). Top, group 1 mGluR activation results in rapid and local translation of Arc mRNA that pre-exists in dendrites. Arc transcription in response to mGluR activation lags behind, peaking 1–2 h after activation14. Bottom, NMDA or VSCC activation results in rapid Arc transcription and a delayed increase in Arc protein through conventional translation at the cell body. (b) Arc synthesis modulates AMPAR trafficking. NMDA and/or VSCC activation induces rapid synthesis of Arc mRNA, which is subsequently translated at the cell body or in response to local mGluR activation. Translation of Arc mRNA in dendrites is inhibited by FMRP and Arc protein is rapidly degraded through Ube3a binding and subsequent ubiquitination. Arc protein increases AMPAR endocytosis, which can lead to LTD. Arc may also act to stabilize the internal pool of AMPARs so that the surface levels of AMPARs remain constant after plasticity occurs, which would lead to a sustained increase or decrease in surface AMPARs depending on the direction of the initial plasticity trigger.
Arc mRNA is transported to dendrites and becomes enriched at the site of local synaptic activity, suggesting that Arc protein is locally synthesized15. In addition to regulated transport of Arc mRNA in dendrites, Arc mRNA undergoes a form of nonsense-mediated decay in dendrites that results in limited translation of protein from a single mRNA16. This exquisite regulation of mRNA and protein localization and expression suggests that Arc is important for synaptic function and that dysregulation of Arc expression may have dire consequences for brain function.
Arc has been studied from a number of perspectives17,18, each yielding a different view of Arc’s contribution to neuronal and synaptic function. Here we briefly review recent studies and attempt to integrate them into the evolving models of Arc’s function. The main conclusion garnered from this body of work is that Arc is poised to be a master regulator of synaptic plasticity.
A view from the top: information storage
The properties of activity-dependent Arc protein and mRNA induction immediately suggested a role in memory consolidation, so it is not surprising that the first studies on Arc concentrated on its regulation and function in the hippocampus. Arc knockout (Arc−/−) mice exhibit impaired consolidation of long-term memory, without alteration of short-term memory19. Infusion of Arc antisense oligodeoxy-nucleotides (ODNs) in the rat hippocampus blocked consolidation in a spatial memory task20. Similarly, Arc antisense ODN infusion in the lateral amygdala blocked the consolidation of Pavlovian fear conditioning21. These findings suggest that Arc has a conserved role in information storage in limbic forebrain memory systems.
Of the various known forms of activity-dependent synaptic plasticity, LTP has attracted the most interest as a memory storage mechanism. Initial studies found that Arc transcription was induced by LTP-like stimuli such as high-frequency stimulation15; early studies therefore assessed the role of Arc in LTP. In vivo infusion of Arc antisense ODN in the hippocampus 1.5 h before LTP induction blocked the maintenance of LTP20. Moreover, although Arc−/− mice exhibit enhanced early phase LTP, they were reported to have deficient late-phase LTP both in vivo in the dentate gyrus and in the CA1 region from hippocampal slices19. Recent studies have also shown that potentiation induced by BDNF application is Arc dependent22. Surprisingly, brief infusion of Arc antisense ODNs during early-phase LTP (15 min post-induction) resulted in transient inhibition of LTP and application of antisense at 2 h, but not 4 h, post-induction resulted in a rapid reversal (within 15 min) of LTP22. It is unclear how antisense could mediate such a rapid and dramatic effect, as a decrease in protein expression was only observed an hour after antisense infusion at the earliest and other studies have only found a decrease in Arc protein 3–6 h after antisense application. Nevertheless, these results suggest that maintenance of LTP requires sustained Arc synthesis during a specific time window.
Arc has also been implicated in forms of hippocampal LTD14,19,23. Arc−/− mice exhibit very little LTD mediated through mGluRs14. Although NMDA receptor–dependent LTD was shown to be intact in one study14, an earlier report found that NMDAR-dependent LTD was also affected in Arc−/− mice19. Recent findings suggest that Arc’s role in protein synthesis–dependent LTD is conserved in other brain regions, as Arc is also required for late-phase LTD in cerebellar Purkinje neurons24. From the point of view of hippocampal LTP and LTD, one might therefore conclude that Arc induction is critical for protein synthesis–dependent consolidation of synaptic modifications, regardless of the polarity of the change. According to this view, local post-translational modifications change the strength of the synapse on the basis of local information, for example, the amount and duration of NMDAR activation. The timely induction of Arc ensures that these labile modifications are made permanent (Fig. 1b). This model is consistent with the idea that synapses are ‘tagged’ by plasticity-inducing stimuli and capture proteins, possibly including Arc, that are required for stabilization of LTP and LTD25.
A view from the bottom: synaptic function
The first clues to a molecular basis for Arc’s synaptic function came from a yeast two-hybrid screen that pulled out two interacting proteins, dynamin and endophilin, which are essential proteins required for regulated endocytosis. A series of studies went on to show that Arc affects the trafficking of AMPA-type glutamate receptors (AMPARs) by directly interacting with the endocytic machinery26. AMPARs are highly dynamic and undergo rapid shuttling between the plasma membrane and internal recycling pools. Canonical LTD and LTP involve modulation of endocytosis and exocytosis of AMPA receptors, respectively27. Arc overexpression in hippocampal culture and slices results in downregulation and accelerated endocytosis of surface AMPARs. In contrast, Arc−/− neurons have abnormally high basal levels of surface GluR1 and slower receptor endocytosis. The precise molecular mechanisms underlying Arc’s role in endocytosis of AMPA receptors remains unknown. Neither Arc nor endophilin and dynamin interact directly with AMPA receptors. Nor is it clear how Arc affects the endocytic function of the endophilin and dynamin complex, although it seems likely that catalysis of endosome formation occurs. It is notable that, although endophilin alone associates with vesicles in neurons that include AMPARs, it is only when Arc is expressed that a reduction in surface AMPA receptors is detected. This suggests that Arc acts as an adaptor to specifically localize or concentrate endocytic proteins at sites at which AMPAR endocytosis can occur. Arc may also increase diffusion of AMPARs out of the PSD through yet to be identified adaptor proteins that interact with Arc and AMPARs. The major scaffolding protein PSD-95 is a likely candidate, as an interaction with Arc has been reported11 and PSD-95 is a major regulator of AMPAR function28.
These findings support another view, that activity-dependent Arc induction is involved in neuronal homeostasis (Fig. 2a). According to this idea, persistent increases in activity are compensated by scaling down AMPARs as a result of an increase in Arc protein and persistent decreases in activity are compensated by scaling up AMPARs as a result of a lack of Arc protein. It is now well established in neuronal culture systems that this type of homeostatic plasticity occurs; neurons scale up AMPARs under conditions of low network activity and scale down AMPARs under conditions of increased network excitability29. This process is thought to be important for normal network function, as it can normalize neuronal output without changing the relative strength of individual synapses29. The properties of Arc induction make it an ideal molecular register of the history of recent synaptic and cellular activity and synaptic scaling could occur simply by ongoing, global adjustments of the rate of AMPAR endocytosis by Arc. This view, that activity-dependent expression of Arc is sine qua non for homeostatic synaptic scaling, is supported by the finding that Arc−/− neurons in culture exhibit severe deficits in AMPAR scaling27. Further in vivo evidence that Arc has a role in synaptic scaling comes from recent studies in visual cortex (see below).
Figure 2.
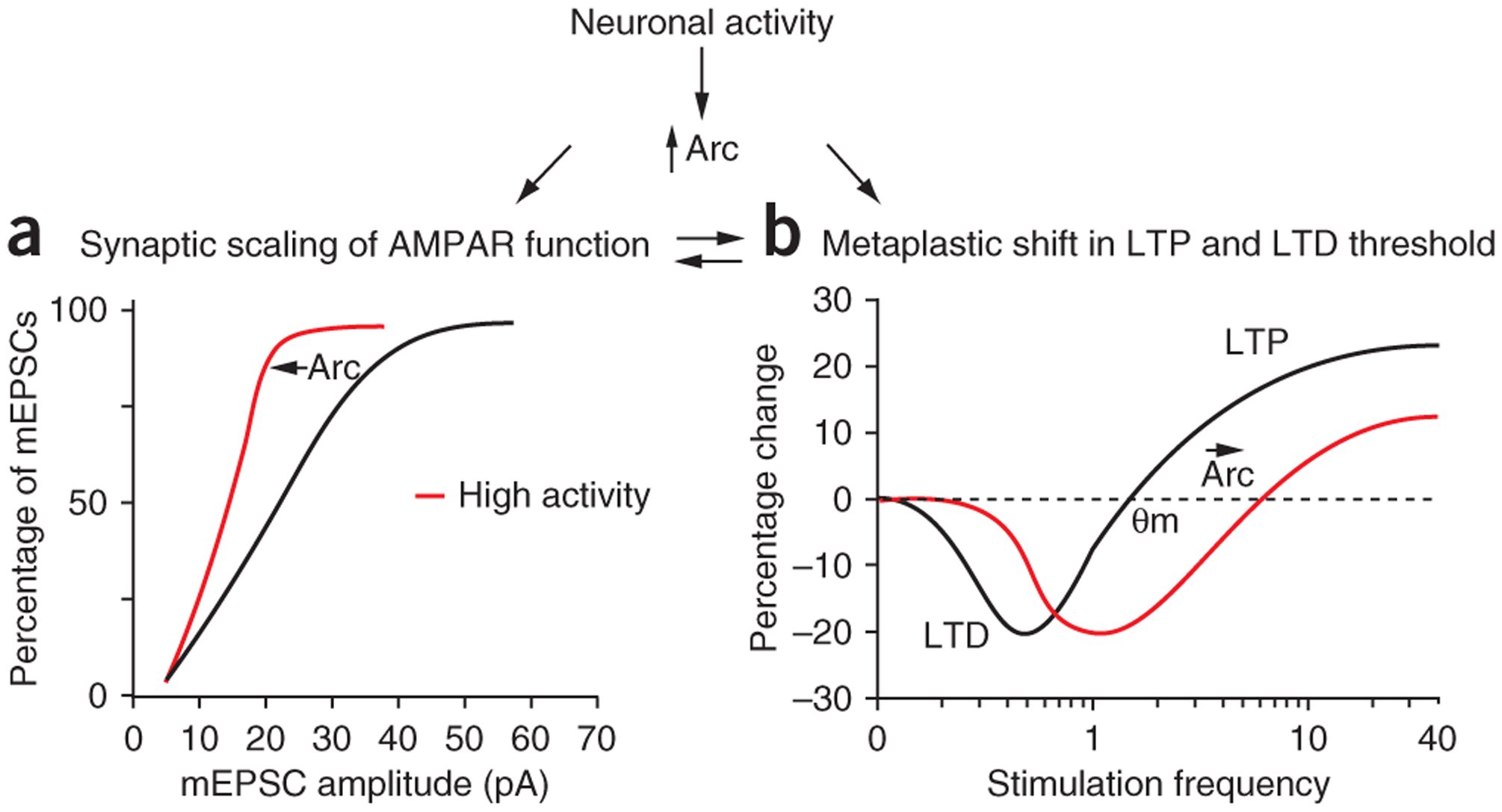
Arc regulates neural circuit homeostasis. (a,b) Neuronal activity regulates Arc protein expression, which can act as a sensor for the amount of activity a neuron experiences in an epoch of time. Chronic changes in neuronal activity result in homeostatic processes that maintain a relative constant neuronal output. Many mechanisms have been implicated in these homeostatic changes; two important mechanisms are synaptic scaling of AMPARs (a) and modification of the LTP and LTD threshold (b). Arc has been shown to be critical for synaptic scaling of AMPARs but may also be involved in setting the threshold for LTP versus LTD. These processes are not mutually exclusive and may act in concert, depending on the precise activity patterns the neuron is subjected to. θm denotes modification threshold.
Arc has also been implicated in actin remodeling of dendritic spines. LTP in the dentate gyrus of awake rats is associated with a long-lasting increase in spine F-actin content, an increase in synapse diameter and enhanced cofilin phosphorylation30, which promotes actin polymerization. Reversal of LTP after infusion of Arc antisense 2 h after high-frequency stimulation was correlated with dephosphorylation of hyperphosphorylated cofilin and a loss of F-actin and Arc antisense was unable to reverse LTP when the F-actin–stabilizing drug, jasplakinolide was used22. These results suggest that Arc synthesis promotes F-actin polymerization and spine remodeling, although the molecular mechanism of Arc’s role in actin dynamics is unclear. A direct molecular link between Arc and LTP stabilization has yet to be made, unlike Arc’s role in LTD. Therefore, it is possible that Arc is important for LTP stabilization, but in an indirect manner that may be a result of Arc’s primary role in LTD or other homeostatic processes (Fig. 2b).
A view from the visual cortex: experience-dependent plasticity
Visual cortex is the preeminent experimental model for connecting elementary molecular mechanisms of synaptic plasticity with their behaviorally relevant consequences in an intact neural network. Brief monocular deprivation sets in motion a stereotyped choreography of synaptic modifications in visual cortex that, at a minimum, engage mechanisms of excitatory synaptic depression, metaplasticity, synaptic potentiation and possibly homeostatic synaptic scaling31. The net result is visual impairment through the deprived eye and a compensatory increase in vision through the nondeprived eye. Other manipulations of experience, such as selective exposure to gratings of a particular orientation, can cause stimulus-selective response potentiation (SRP), which appears to utilize the mechanisms of LTP32. Conversely, total deprivation of light or pattern vision through both eyes can cause a change in AMPARs akin to homeostatic synaptic scaling observed in vitro33. Arc mRNA and protein expression in the superficial layers and layer 6 can be manipulated dramatically with visual deprivation and experience8. Thus, the visual cortex is an ideal place to dissect the precise role of Arc in synaptic plasticity.
In juvenile mice (~postnatal day 30, P30), closure of the contralateral eyelid for 1–3 d produces a depression of deprived eye responses without affecting responses to stimulation of the other eye. This response depression is mediated largely by reduced efficacy of excitatory thalamo-cortical synapses conveying the input from the deprived eye, at least at the level of cortical layer 4. The mechanism for deprivation-induced synaptic depression in layer 4 is endocytosis of AMPARs triggered specifically by NMDAR activation31, analogous to the well-studied model of hippocampal homosynaptic LTD. Consistent with a role for Arc in AMPAR endocytosis, deprived eye depression following 3 d of monocular deprivation is completely absent in Arc−/− mice34 (Fig. 3).
Figure 3.
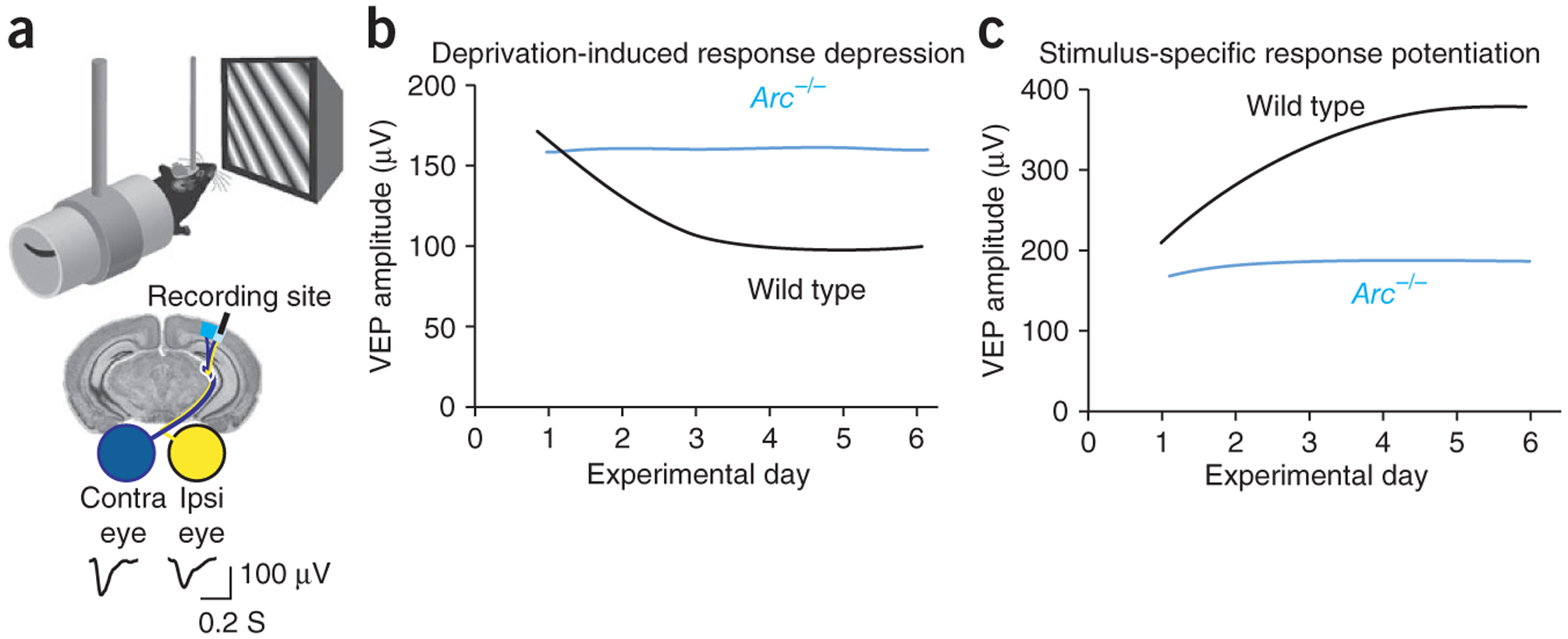
Arc is required for bidirectional experience-dependent plasticity in visual cortex in vivo. (a) Experience-dependent plasticity in the visual cortex can be measured using chronic visually evoked potential (VEP) recordings in V1 layer 4. (b) Schematic of the time course of VEP changes in visual cortex after monocular deprivation. In wild-type mice, monocular deprivation results in a substantial decrease in deprived eye VEPs (black line). However, deprivation has no effect in Arc−/− mice (blue line)34. (c) Repeated exposure to gratings of a specific orientation results in SRP of VEPs in wild-type mice (black line). Mice that lack Arc do no exhibit substantial SRP (blue line)34. Thus, the visual cortex is rendered immutable by deprivation or experience in the absence of Arc.
Monocular deprivation for longer periods (>5 d) has the additional consequence of causing potentiation of the nondeprived eye responses (open-eye potentiation, OEP). It has been suggested that OEP is a manifestation of homeostatic scaling35. However, unlike scaling, OEP is prevented by blockade of NMDARs after induction of deprived-eye depression36. An alternative basis for OEP is experience-dependent potentiation of nondeprived eye inputs following metaplastic reduction of the LTP threshold31. This view, inspired by the Bienenstock-Cooper-Munro (BCM) theory37 and well supported by experimental data31, posits that the amount of NMDAR activation required to transition from LTD to LTP is variable, and depends on the recent history of cellular activity. As a molecular archivist of cellular activity, adjustments in the levels of Arc could set the level of the modification threshold. Consistent with this, no OEP is observed following 7-d monocular deprivation in Arc−/− mice34.
The case that naturally occurring synaptic plasticity in visual cortex employs the mechanisms of LTP is stronger for the phenomenon of SRP. In response to brief exposure to gratings of a single orientation, responses to this stimulus grow over the course of several days. Similar to LTP, this ‘input-specific’ change persists for many days and requires cortical NMDAR activation, synaptic delivery of GluR1-containing AMPARs32 and protein synthesis (J.D.S. and M.F.B., unpublished data). No SRP occurs in the visual cortex of Arc−/− mice34 (Fig. 3c).
The view that emerges from these studies of visual cortex is that, by adolescence, excitatory synapses are rendered essentially immutable by experience or deprivation if Arc is not expressed in their postsynaptic target. Despite this profound defect in acquired properties, the innate organization and levels of visual responsiveness appear to be normal in Arc−/− mice. It appears that a requirement for Arc paints a bright line that separates the contributions of ‘nurture’ (those dependent on the quality of sensory experience) from ‘nature’ (those dependent on genetic instructions alone) on the development of glutamatergic synaptic connections in the cortex. Viewed from this perspective, Arc is more than an LTP or LTD molecule or a simple regulator of cell-wide AMPAR endocytosis rate. Arc may be a gain control or stabilizer for excitatory synaptic plasticity regardless of how this plasticity is induced or manifested. The obvious question then arises of whether the well-known developmental decline in visual cortical plasticity is possibly caused by reduced expression of Arc.
Of course, one must be cautious extrapolating from the effects of a knockout to establish the normal role of the affected gene and protein. The loss of multiple forms of plasticity could be an indirect consequence of disrupted synaptic development in the absence of Arc. This caveat is underscored by a recent study of synaptic scaling defects in the Arc−/− mouse38. Normally, at P21, total light deprivation causes AMPAR currents to scale up and subsequent light exposure reverses this change. In Arc−/− mice, neither up nor down scaling occurs38. However, AMPAR currents are increased in Arc−/− mice even in a normally lighted environment. A simple interpretation of these results is that the absence of Arc mimics light deprivation, causing upward scaling of AMPARs. No downward scaling is possible because there is no light-induced increase in Arc. If this interpretation is correct, all other defects in synaptic plasticity in vivo could be a result of synapses already saturated with a full complement of AMPARs. Inconsistent with the hypothesis of global upward scaling of AMPARs, evoked responses in visual cortex of Arc−/− mice are not substantially larger than wild-type responses34. However, it must be noted that the scaling results apply to layer 2/3 at the age of P21, whereas the in vivo plasticity results apply to layer 4 at the age of P30.
The Goldilocks protein: synaptic dysfunction
Arc expression is exquisitely regulated at many different levels, including transcription, mRNA degradation, translation and protein degradation. This suggests that disruption of Arc expression could have profound effects on Arc-dependent plasticity. Indeed, dysregulation of Arc expression has been implicated in Fragile X syndrome (FXS)14 and Alzheimer’s disease39,40. In the case of FXS, it has been proposed that fragile X mental retardation protein (FMRP), the protein that is mutated in FXS, is a negative regulator of Arc translation14. Thus, Fmr1−/− (Fmrp) mice, a mouse model of FXS, have disrupted Arc translation14. LTD is enhanced in Fmr1−/− mice and this enhancement is abolished by removing Arc protein from Fmr1−/− mice14, suggesting that some of the plasticity and cognitive phenotypes observed in FXS are possibly the results of disruption of Arc expression.
In very recent findings, Arc was found to be a direct target of the ubiquitin ligase Ube3a41. Ube3a is a gene that, when mutated, causes a debilitating neurological disorder called Angelman syndrome42 and duplication of Ube3a has also recently been implicated in autism spectrum disorders. Loss of Ube3a activity causes an increase in Arc and a concomitant decrease in synaptic AMPARs that is Arc dependent41. Notably, Ube3a mutant mice have severe deficits in experience-dependent and synaptic plasticity in V1 (ref. 43) that are markedly similar to the phenotypes found in Arc−/− mice. These deficits are reversed when mutant mice are visually deprived, suggesting that the deficits are caused by activity-dependent overexpression of protein(s), possibly including Arc. Thus, high levels of Arc may also be deleterious to normal synaptic plasticity. In the case of Ube3a duplication, one can hypothesize that Arc levels would be lower than normal, possibly contributing to the autism phenotype.
β-amyloid (Aβ) peptide is the major component of neuronal plaques in Alzheimer’s disease and is produced by sequential proteolytic processing of amyloid precursor protein (APP) by β-secretase (BACE) and γ-secretase. It is becoming clear that oligomeric species of Aβ can modulate synaptic transmission and plasticity and may even have a normal role in homeostatic regulation of glutamate transmission44. A number of compelling findings suggest that Arc may contribute to the cognitive and Aβ-dependent synaptic dysfunction observed in Alzheimer’s disease. Aβ depresses AMPA receptor currents in slices and induces AMPAR endocytosis via a process similar to mGluR LTD45. Oligomeric forms of Aβ have been shown to induce Arc expression46. Arc expression is severely disrupted in Alzheimer’s disease mouse models; in some cases extremely high levels of Arc have been observed as a result of seizure activity47 and in others there is a lack of normal Arc induction after experience40. Recent studies suggest that Aβ blocks BDNF-induced Arc expression, perhaps by inhibiting the PI3-Akt-mTOR pathway48. However, the role of the mTOR pathway in Arc expression is somewhat controversial, as another study found no role for mTOR in Arc induction or Arc-dependent plasticity in vivo49. These studies highlight the need to understand precisely how Arc expression is affected in Alzheimer’s disease.
Taken together, these findings suggest that overexpression or dysregulation of Arc protein levels is potentially a causative factor in a number of neurological disorders. Because Arc is a critical effector molecule, downstream of many signaling pathways, dysfunction of Arc could be a nexus point for synaptic dysfunction in diseases of cognition.
Toward an integrated view
In excitatory neurons, Arc mRNA expression is a faithful molecular read-out of the recent history of neuronal activity and myriad forms of synaptic modification are disabled by the absence of Arc. Two views of Arc function currently prevail. One is related to the synaptic homeostasis that must occur to maintain neuronal output activity in an optimal dynamic range. According to this model, increasing Arc levels raises the LTP threshold (and thereby promotes LTD) and/or directly increases the rate of AMPAR endocytosis (thereby causing downward scaling of the strength of all excitatory synapses). The second view is that Arc functions as the ‘volume knob’ on the occurrence or persistence of activity-dependent synaptic plasticity. According to this model, plasticity of all stripes is enhanced or stabilized when Arc is expressed and lost when Arc is absent.
These views are not mutually exclusive if one considers the different types of information encoded by Arc transcription (increased cellular activity) and translation (local synaptic activity). One epoch of activity could induce transcription of Arc mRNA and subsequent transport of message to activated dendrites. However, the message is dormant until a second epoch of activity occurs, which leads to rapid and local translation of Arc. Thus, the first activity epoch or experience primes the neuron with Arc mRNA. The precise amount and spatial location of synaptic activity will dictate where and when Arc protein is expressed and how it modifies synaptic function.
A number of important questions still remain as to Arc’s role in neuronal function. When and where exactly is Arc protein expressed in vivo and how do the dynamics of Arc protein differ during experience and learning and different forms of neuronal activity? Most of what is known about Arc expression is derived through static techniques that only offer a snapshot of protein expression. These methods cannot distinguish between protein made at the cell body and protein synthesized locally at dendrites. Is there something inherently different about Arc protein synthesized locally? One would predict that the composition of Arc interacting partners might be very different in dendrites versus the cell body. For example, does Arc undergo specific post-translational modifications such as phosphorylation in dendrites? Where does the locally made protein go? Does it stay in activated dendrites and spines only? Arc is also found abundantly in the cell nucleus, but is not a transcription factor and its role in the nucleus is unclear. One intriguing idea is that Arc protein made in dendrites may traffic back to the nucleus, transporting signaling molecules in a signaling endosomal compartment. Although it has been known that signaling endosomes occur in axons, it has only been recently shown that signaling endosomes also occur in dendrites50.
At the molecular level, how does Arc stabilize plasticity? Although Arc’s role in LTD and AMPAR endocytosis is compelling, there are still some important steps missing. How does Arc specifically regulate AMPARs and not other glutamate receptors? There are no direct interactions between Arc or its endocytic interactors and the AMPARs. How can Arc regulate both LTD and LTP? Does Arc stabilize the internal AMPAR pool after plasticity induction?
Arc expression is very tightly regulated, even slight changes in the timing or location of synthesis may have marked effects on Arc-dependent processes. As dysregulation of Arc expression is found in many cognitive disorders, it will be critical to assess the role of Arc in these disorders. Although Arc dysregulation may be a manifestation of the disease pathogenesis and not a causative agent, correcting Arc expression may still be a very relevant target for alleviating disease symptoms. It will be important to know precisely why and how Arc expression is disrupted in these disorders.
Finally, can Arc shed light on memory consolidation? Mice that lack Arc show a very clear and dramatic deficit in memory consolidation in multiple behavioral paradigms, but which aspect of Arc’s molecular and cellular function is critical for memory stabilization? Does Arc affect memory consolidation at the network or single cell level?
Arc is a fascinating but still enigmatic protein that warrants further study. The fruits of this labor will shed light not only on the question of how the nervous system stores information, but also on the synaptic dysfunction observed in a number of neurological disorders that may even lead to new therapeutic interventions.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Davis HP & Squire LR Protein synthesis and memory: a review. Psychol. Bull 96, 518–559 (1984). [PubMed] [Google Scholar]
- 2.Sutton MA & Schuman EM Dendritic protein synthesis, synaptic plasticity and memory. Cell 127, 49–58 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Whitlock JR, Heynen AJ, Shuler MG & Bear MF Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Link W et al. Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc. Natl. Acad. Sci. USA 92, 5734–5738 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lyford GL et al. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14, 433–445 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Guzowski JF et al. Mapping behaviorally relevant neural circuits with immediate-early gene expression. Curr. Opin. Neurobiol 15, 599–606 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Ramírez-Amaya V et al. Spatial exploration-induced Arc mRNA and protein expression: evidence for selective, network-specific reactivation. J. Neurosci 25, 1761–1768 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tagawa Y, Kanold PO, Majdan M & Shatz CJ Multiple periods of functional ocular dominance plasticity in mouse visual cortex. Nat. Neurosci 8, 380–388 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Vazdarjanova A et al. Spatial exploration induces ARC, a plasticity-related immediate-early gene, only in calcium/calmodulin-dependent protein kinase II-positive principal excitatory and inhibitory neurons of the rat forebrain. J. Comp. Neurol 498, 317–329 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Steward O & Worley PF Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 30, 227–240 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Husi H, Ward MA, Choudhary JS, Blackstock WP & Grant SG Proteomic analysis of NMDA receptor–adhesion protein signaling complexes. Nat. Neurosci 3, 661–669 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Waltereit R et al. Arg3.1/Arc mRNA induction by Ca2+ and cAMP requires protein kinase A and mitogen-activated protein kinase/extracellular regulated kinase activation. J. Neurosci 21, 5484–5493 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams JP, Robinson RA, Hudgins ED, Wissink EM & Dudek SM NMDA receptor–independent control of transcription factors and gene expression. Neuroreport 20, 1429–1433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park S et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steward O, Wallace CS, Lyford GL & Worley PF Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 21, 741–751 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Giorgi C et al. The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 130, 179–191 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Bramham CR, Worley PF, Moore MJ & Guzowski JF The immediate early gene arc/arg3.1: regulation, mechanisms and function. J. Neurosci 28, 11760–11767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzingounis AV & Nicoll RA Arc/Arg3.1: linking gene expression to synaptic plasticity and memory. Neuron 52, 403–407 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Plath N et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Guzowski JF et al. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci 20, 3993–4001 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ploski JE et al. The activity-regulated cytoskeletal-associated protein (Arc/Arg3.1) is required for memory consolidation of Pavlovian fear conditioning in the lateral amygdala. J. Neurosci 28, 12383–12395 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messaoudi E et al. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J. Neurosci 27, 10445–10455 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA & Huber KM Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith-Hicks C et al. SRF binding to SRE 6.9 in the Arc promoter is essential for LTD in cultured Purkinje cells. Nat. Neurosci 13, 1082–1089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frey U & Morris RG Synaptic tagging and long-term potentiation. Nature 385, 533–536 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Chowdhury S et al. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shepherd JD et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shepherd JD & Huganir RL The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol 23, 613–643 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Turrigiano GG The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukazawa Y et al. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38, 447–460 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Smith GB, Heynen AJ & Bear MF Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex. Phil. Trans. R. Soc. Lond. B 364, 357–367 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frenkel MY et al. Instructive effect of visual experience in mouse visual cortex. Neuron 51, 339–349 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Goel A & Lee HK Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J. Neurosci 27, 6692–6700 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCurry CL et al. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat. Neurosci 13, 450–457 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaneko M, Stellwagen D, Malenka RC & Stryker MP Tumor necrosis factor–alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58, 673–680 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho KK, Khibnik L, Philpot BD & Bear MF The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc. Natl. Acad. Sci. USA 106, 5377–5382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bienenstock EL, Cooper LN & Munro PW Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci 2, 32–48 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao M et al. A specific requirement of Arc/Arg3.1 for visual experience-induced homeostatic synaptic plasticity in mouse primary visual cortex. J. Neurosci 30, 7168–7178 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palop JJ et al. Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J. Neurosci 25, 9686–9693 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dickey CA et al. Amyloid suppresses induction of genes critical for memory consolidation in APP + PS1 transgenic mice. J. Neurochem 88, 434–442 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Greer PL et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kishino T, Lalande M & Wagstaff J UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet 15, 70–73 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Yashiro K et al. Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci 12, 777–783 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Venkitaramani DV et al. Beta-amyloid modulation of synaptic transmission and plasticity. J. Neurosci 27, 11832–11837 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsieh H et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lacor PN et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci 24, 10191–10200 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palop JJ et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen TJ, Wang DC & Chen SS Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor–induced Arc expression in rat cortical neurons. J. Neurosci. Res 87, 2297–2307 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Panja D et al. Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J. Biol. Chem 284, 31498–31511 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma N et al. Long-distance control of synapse assembly by target-derived NGF. Neuron 67, 422–434 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]