

Summary.
Background:
Fibrinolysis may exacerbate bleeding in patients with hemophilia A (HA). Accordingly, antifibrinolytics have been used to help maintain hemostatic control. Although antifibrinolytic drugs have been proven to be effective in the treatment of mucosal bleeds in the oral cavity, their efficacy in non-mucosal tissues remain an open question of significant clinical interest.
Objective:
To determine whether inhibiting fibrinolysis improves the outcome in non-mucosal hemophilic tail vein transection (TVT) bleeding models, and to determine whether a standard ex vivo clotting/fibrinolysis assay can be used as a predictive surrogate for in vivo efficacy.
Methods:
A highly sensitive TVT model was employed in hemophilic rodents with a suppressed fibrinolytic system to examine the effect of inhibiting fibrinolysis on bleeding in non-mucosal tissue. In mice, induced and congenital hemophilia models were combined with fibrinolytic attenuation achieved either genetically or pharmacologically (tranexamic acid [TXA]). In hemophilic rats, tail bleeding was followed by whole blood rotational thromboelastometry evaluation of the same animals to gauge the predictive value of such assays.
Results:
The beneficial effect of systemic TXA therapy observed ex vivo could not be confirmed in vivo in hemophilic rats. Furthermore, neither intravenously administered TXA nor congenital knockout of the fibrinolytic genes encoding plasminogen or tissue-type plasminogen activator markedly improved the TVT bleeding phenotype or response to factor therapy in hemophilic mice.
Conclusions:
The findings here suggest that inhibition of fibrinolysis is not effective in limiting the TVT bleeding phenotype of HA rodents in non-mucosal tissues.
Keywords: blood coagulation factors, fibrinolysis, hemophilia A, tail, tranexamic acid
Introduction
Hemophilia A (HA) is a bleeding disorder caused by coagulation factor VIII (FVIII) deficiency. In the absence of adequate levels of circulating FVIII, thrombin generation is compromised, leading to impaired blood coagulation [1]. Accordingly, individuals with HA form unstable blood clots with an abnormal structure [2,3]. Moreover, insufficient thrombin generation is hypothesized to lead to inadequate downregulation of the fibrinolytic system, owing to diminished activation of thrombin-activatable fibrinolysis inhibitor (TAFI; also known as carboxypeptidase B2) [4]. Consequently, clots are degraded prematurely, potentially causing rebleeding. To correct the bleeding phenotype, current HA treatments aim to restore thrombin generation by supplying exogenous clotting factors [5]. Such therapies include administration of FVIII or bypassing agents, such as recombinant activated FVII (rFVIIa), in patients who have formed inhibitory antibodies against FVIII.
An alternative treatment strategy is to limit premature clot degradation in HA patients by applying pharmaceutical agents to suppress the fibrinolytic pathway. The beneficial effect of antifibrinolytic drugs as adjunctive HA therapy for mucosal bleeds, and especially oral surgical procedures, is widely recognized [6–8]. However, the efficacy of antifibrinolytic drugs in treating non-mucosal bleeding in HA patients lacks formal clinical and preclinical validation.
Two studies have suggested a potential beneficial effect of antifibrinolytic drugs for bleeding in non-mucosal tissues [9,10]. However, these studies did not provide definitive conclusions, and have, to our knowledge, never been followed up by larger preclinical or clinical studies. Whole blood assays [11–13] indicate that antifibrinolytics can markedly improve clot stability in HA patients, but whether such ex vivo and in vitro analyses are indicative of hemophilic bleeding pathology in vivo has not been confirmed by preclinical or clinical studies. Therefore, we directly evaluated the hypothesis that inhibition of fibrinolysis increases clot stability ex vivo, and improves the bleeding phenotype and/or response to rFVIIa/recombinant FVIII (rFVIII) treatment in vivo in hemophilic rodents. To test this hypothesis, the effect of inhibiting fibrinolysis in non-mucosal tissue was determined by studying such animals, with an inhibited fibrinolytic system, in a tail-bleeding model. Furthermore, the predictive value of a standard ex vivo whole blood clotting assay was assessed in rats subjected to the in vivo bleeding model.
Methods
Animals
FVIII−/− mice with a mixed 129SV/C57BL/6 background [14] and C57Bl/6 mice were obtained from Taconic (Ry, Denmark), and FVIII−/− rats were bred at Novo Nordisk A/S (Maaloev, Denmark) [15]. FVIII−/− rats and mice are animal models of congenital HA. Tissue-type plasminogen activator (t-PA) wild-type (WT), t-PA−/− [16], plasminogen WT and plasminogen−/− mice [17], all with a C57Bl/6 background, were bred at the Cincinnati Children’s Hospital Research Foundation (Cincinnati, OH, USA).
The animals were group-housed under standardized conditions; both sexes were used; and mice were aged 8–16 weeks and rats were aged 8–23 weeks when they were included in the studies. Experiments were approved and performed according to guidelines of the Danish Animal Experiments Council, the Novo Nordisk Ethical Review Council, and the Cincinnati Children’s Hospital Research Foundation’s Institutional Animal Care and Use Committee.
Proteins and reagents
Monoclonal anti-FVIII antibody 4F30 was produced in-house (Novo Nordisk A/S, Bagsværd, Denmark). rFVIIa (NovoSeven) (Novo Nordisk A/S, Bagsværd, Denmark) was reconstituted in sterile water, and diluted to concentrations corresponding to intended dosages in 10 mM glycyl-glycine, 165 mM mannitol, 50 mM NaCl, 10 mM CaCl2, and 0.01% Tween-80 (pH 6.0) (vehicle buffer). rFVIII (Advate) was purchased from Baxter Bioscience (Vienna, Austria), reconstituted with its diluent, and further diluted to various concentrations with 12 mM l-histidine, 209 mM mannitol, 1.9 mM CaCl2 (dihydrate), 108 mM NaCl, 12 mM Tris, 0.33 mM glutathione, 26.4 mM trehalose (dihydrate), and 0.015% polysorbate 80 (pH 7.4) (vehicle buffer). Tranexamic acid (TXA) (Cyklokapron, 100 mg mL−1) was purchased from Pfizer (New York, NY, USA), and was stored at room temperature or at 4 °C, and the other compounds were stored at −80 °C.
Induced HA model
A condition similar to acquired HA was induced in mice by intravenous injections of 143.8 mg kg−1 4F30 into the lateral tail vein 5 min prior to any other test compound (Data S1, Figure S1).
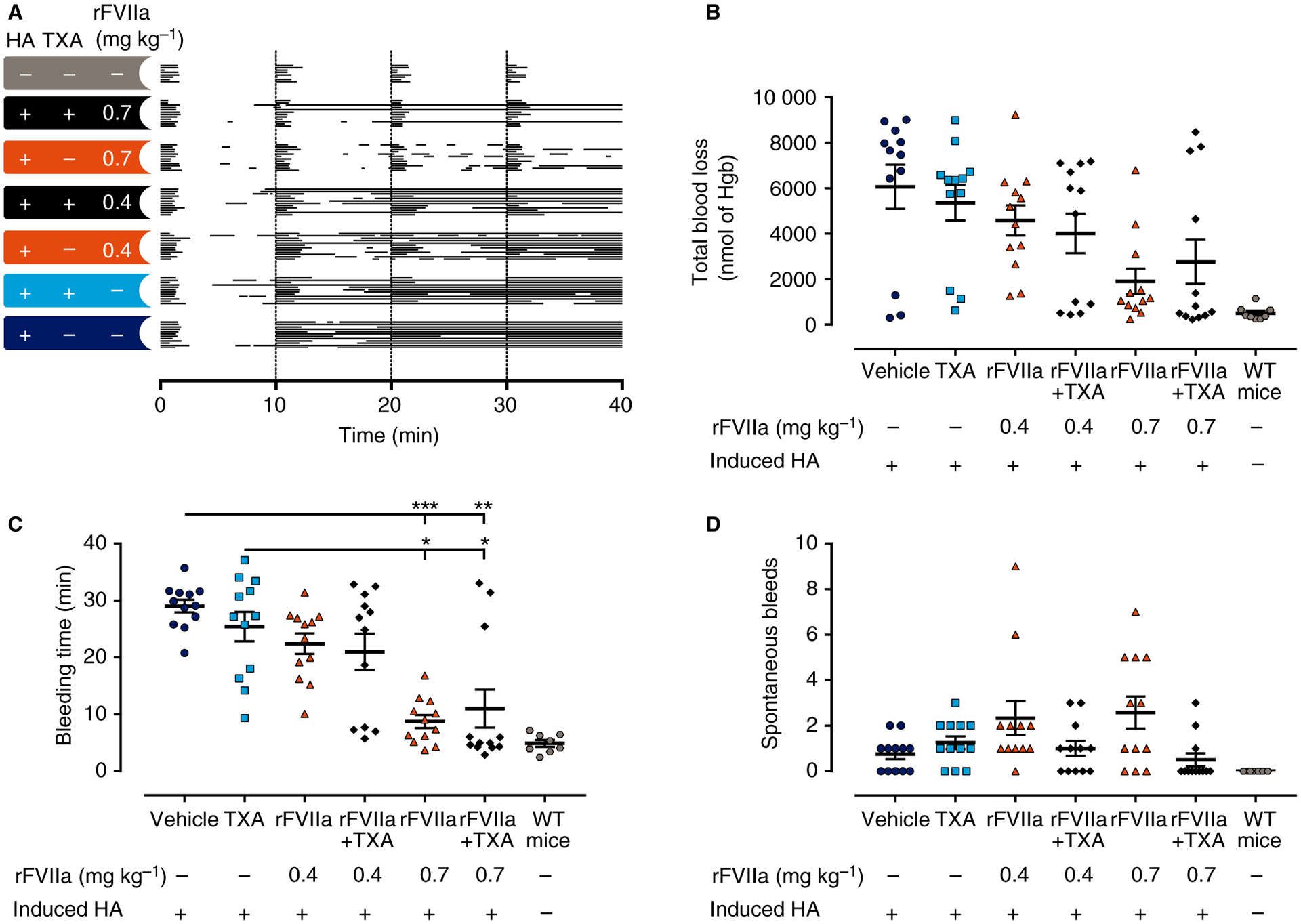
Tail vein transection (TVT) bleeding model
A 40-min version of the TVT bleeding model was used [18]. The study design, treatment and doses are shown in Tables 1 and 2. rFVIIa and rFVIII doses were selected to represent at least the median effective dose (ED50) in the TVT bleeding model (Data S2, Figure S2, S3). Two TXA doses in a similar range to what has successfully been used in rodents before [19–21] were selected to achieve TXA plasma concentrations of > 100 μg mL−1, which is an exposure level that is known to be efficacious clinically [22,23], and to fully inhibit fibrinolysis in vitro [24]. Isoflurane-anesthetized mice and rats were dosed with the test compound in the right lateral tail vein. After 5 min, the left lateral tail vein was transected by use of a template guide with a #11 scalpel at a tail diameter of 2.5 mm (mice) or with a #21 scalpel at a tail diameter of 6.4 mm (rats). The tail was submerged in 37 °C saline, and all tail-bleeding episodes were monitored and recorded for 40 min, while the blood was collected. If no bleeding was observed at 10, 20 or 30 min after injury, the clot was gently challenged by being wiped twice with wetted saline gauze. The bleeds observed at 0, 10, 20 and 30 min were considered to be provoked, whereas any additional bleeds were deemed to be spontaneous, as these developed without intervention by the experimenter. After 40 min, blood was sampled by cardiac puncture in all mice and half of the rats. The remaining rats had a catheter placed in the carotid artery at t = 40 min, from which blood was collected for rotational thromboelastometry (ROTEM) analyses at t = 55 min. All samples were stabilized with 3.2% trisodium citrate (1 : 10). Animals were killed while fully anesthetized. Blood loss was determined by quantification of the collected hemoglobin (Hgb) (nmol) [25], and bleeding time (min) was defined as the sum of the durations of all observed bleeding episodes.
Table 1.
Overview of the study design for the combined tail vein transection (TVT)/rotational thromboelastometry (ROTEM) rat study
| Group | Rats | Type of HA | Rats per group for TVT (ROTEM) | Treatment (dose, mg kg−1) | Design |
|---|---|---|---|---|---|
| 1 | FVIII−/− | Congenital | 10 (5) | Vehicle | S |
| 2 | FVIII−/− | Congenital | 10 (5) | TXA (350) | S and B |
| 3 | FVIII−/− | Congenital | 10 (5) | rFVIIa (0.5 ~ ED50) | S and B |
| 4 | FVIII−/− | Congenital | 10 (5) | rFVIIa (0.5 ~ ED50) + TXA (350) | S and B |
| 5 | WT | - | 11 (5) | Vehicle | S |
B, blinded; ED50, median effective dose; HA, hemophilia A; rFVIIa, activated recombinant factor VII; S, stratified; TXA, tranexamic acid; WT, wild type. All rats were subjected to the TVT bleeding assay. Following the TVT bleeding study, half of the rats had blood drawn from a carotid artery catheter for ROTEM analyses. Data from the study are shown in Figs 1 and 2.
Table 2.
Overview of the study designs for the mouse tail vein transection bleeding studies
| Figure | Group | Strain | Type of HA | Mice (n) | Treatment (dose, mg kg−1) | Design |
|---|---|---|---|---|---|---|
| 3A-D | 1 | FVIII−/− | Congenital | 16 | Vehicle | S and B |
| 2 | FVIII−/− | Congenital | 15 | TXA (500) | S and B | |
| 3 | FVIII−/− | Congenital | 15 | rFVIIa (0.4 ~ ED50) | S and B | |
| 4 | FVIII−/− | Congenital | 15 | rFVIIa (0.4 ~ ED50) + TXA (500) | S and B | |
| 5 | C57Bl/6 | - | 8 | Vehicle | S | |
| 3E-H | 1 | FVIII−/− | Congenital | 12 | Vehicle | S and B |
| 2 | FVIII−/− | Congenital | 12 | TXA (500) | S and B | |
| 3 | FVIII−/− | Congenital | 12 | rFVIII (1 U kg−1 ~ ED50) | S and B | |
| 4 | FVIII−/− | Congenital | 12 | rFVIII (1 U kg−1 ~ ED50) + TXA (500) | S and B | |
| 5 | C57Bl/6 | - | 8 | Vehicle | S | |
| 4A-D | 1 | C57Bl/6 | Induced | 12 | TXA (350) | S |
| 2 | C57Bl/6 | Induced | 12 | Vehicle | S and B | |
| 3 | C57Bl/6 | Induced | 12 | rFVIIa (0.4) | S and B | |
| 4 | C57Bl/6 | Induced | 12 | rFVIIa (0.4) + TXA (350) | S and B | |
| 5 | C57Bl/6 | Induced | 12 | rFVIIa (0.7 ~ ED50) | S & B | |
| 6 | C57Bl/6 | Induced | 12 | rFVIIa (0.7~ED50) + TXA (350) | S and B | |
| 7 | C57Bl/6 | - | 8 | Vehicle | S | |
| 5A-D | 1 | t-PA WT | Induced | 8 | Vehicle | S |
| t-PA−/− | Induced | 8 | ||||
| 2 | t-PA WT | Induced | 10 | rFVIIa (0.7 ~ ED50) | S | |
| t-PA−/− | Induced | 8 | ||||
| 3 | t-PA WT | Induced | 8 | rFVIIa (1.5) | S | |
| t-PA−/− | Induced | 8 | ||||
| 4 | t-PA WT | Induced | 8 | rFVIIa (2.7) | S | |
| t-PA−/− | Induced | 8 | ||||
| 5 | t-PA WT | - | 8 | Vehicle | S | |
| t-PA−/− | - | 8 | ||||
| 5E-D | 1 | Plg WT | Induced | 8 | Vehicle | S |
| Plg−/− | Induced | 11 | ||||
| 2 | Plg WT | Induced | 10 | rFVIIa (0.7 ~ ED50) | S | |
| Plg−/− | Induced | 7 | ||||
| 3 | Plg WT | Induced | 7 | rFVIIa (1.5) | S | |
| Plg−/− | Induced | 8 | ||||
| 4 | Plg WT | Induced | 8 | rFVIIa (2.7) | S | |
| Plg−/− | Induced | 9 | ||||
| 5 | Plg WT | - | 7 | Vehicle | S | |
| Plg−/− | - | 5 |
B, blinded; ED50, median effective dose; FVIII, coagulation factor VIII; HA, hemophilia A; Plg, plasminogen; rFVIIa, activated recombinant factor VII; rFVIII, recombinant factor VIII; S, stratified; t-PA, tissue-type plasminogen activator; TXA, tranexamic acid; WT, wild type. Data from the study are shown in Figs 3–5.
Whole blood ROTEM assay
Whole blood coagulation profiles were obtained by use of a ROTEM delta hemostasis analyzer (TEM International, Munich, Germany) and prewarmed plastic test cups. Native clot formation was assessed by adding 20 μL of the recalcification reagent star-tem (TEM International) and 300 μL of stabilized whole blood to a test cup (recalcification assay). Clot formation, after initiation of coagulation with tissue factor (TF), was assessed by adding 20 μL of star-tem, 20 μL of the TF-containing reagent r ex-tem (TEM International) and 300 μL of stabilized whole blood to a test cup (TF assay). The stability of blood clots was assessed with an adapted TF/t-PA assay, in which whole blood was spiked with t-PA prior to the ROTEM assay (Merck Life Science A/S, Hellerup, Denmark); coagulation was also initiated as in the TF assay described above. This ensured sufficient clot formation in all treatment groups, so any observed differences in the TF/t-PA assay should be attributable to differences in clot stability and resistance to t-PA-mediated fibrinolysis. t-PA was diluted in 20 nM HEPES buffer with 150 mM NaCl and 0.01% Tween-80 (pH 7.4) to obtain a final concentration of 20 nM t-PA. All ROTEMs were run for 120 min, and standard ROTEM parameters were collected as described in [26], including the amplitudes at 5, 10, 15, 20, 25, 30, 45, 60 and 120 min.
TXA plasma concentration
The TXA plasma concentration was determined by solid-phase microextraction coupled to liquid chromatography–tandem mass spectrometry (LC-MS/MS), a method that has been previously described by Gorynski et al. [27]. In the current experiments, 10 μL of plasma was used instead of 800 μL. Sample preparation steps were performed with the Concept-96 manual unit (PAS Technology, Magdala, Germany), liquid chromatography was performed with the Discovery HS F5 100 × 2.1-mm, 3-μm chromatographic column from MilliporeSigma/Supelco (Bellefonte, PA, USA), and LC-MS/MS analysis was performed with a Shimadzu LC-MS 8060 triple quadrupole mass spectrometer coupled to the Nexera UHPLC system.
Statistical analyses
Data were analyzed with GRAPHPAD PRISM 7.03, Excel 2010, and R version 3.3.2. The ROTEM measurements were analyzed with Welch’s ANOVA with the Games–Howell post hoc test, as the data were continuous but without variance homogeneity of the residuals. Continuous data with variance homogeneity of the residuals, including total blood loss, bleeding time, and number of spontaneous bleeds, were analysed with ANOVA with Bonferroni’s correction for multiple comparisons. Otherwise, a Kruskal–Wallis test with Dunn’s multiple comparisons test was used. In the cases where the impact of a different genotype was also analyzed (experiments with t-PA or plasminogen mice), the blood loss, bleeding time and spontaneous bleeds were analyzed with two-way ANOVA with Bonferroni’s correction for multiple comparisons, after Q–Q plots and residual versus fitted plots had confirmed that the assumptions of the test were not violated (P < 0.05 was considered to be significant).
Results
Antifibrinolytic therapy did not increase the rate of clot formation, but increased clot stability ex vivo
The effect of TXA was tested in the same FVIII−/− rats with both an ex vivo ROTEM assay and an in vivo bleeding model. The schematic depiction of a ROTEM trace in Fig. 1A demonstrates how to interpret whole blood coagulation profiles. To illustrate the differences in the coagulation profiles, Fig. 1B–D shows representative examples of recorded ROTEM traces from each treatment group after coagulation had been initiated with recalcification only (Fig. 1B), TF (Fig. 1C), and TF/t-PA (Fig. 1D). The impact of the treatments on whole blood coagulation was analyzed by plotting the average clot firmness for each of the measured time points. In the recalcification assay, TXA was not found to increase the rate of clot formation, as no difference in clot firmness was detected between vehicle-treated and TXA-treated rats (Fig. 2A). However, the TF/t-PA assay revealed that, in contrast to whole blood samples from vehicle-treated rats, samples from TXA-treated FVIII−/− rats could resist the t-PA-mediated clot degradation (Fig. 2B).
Fig. 1.
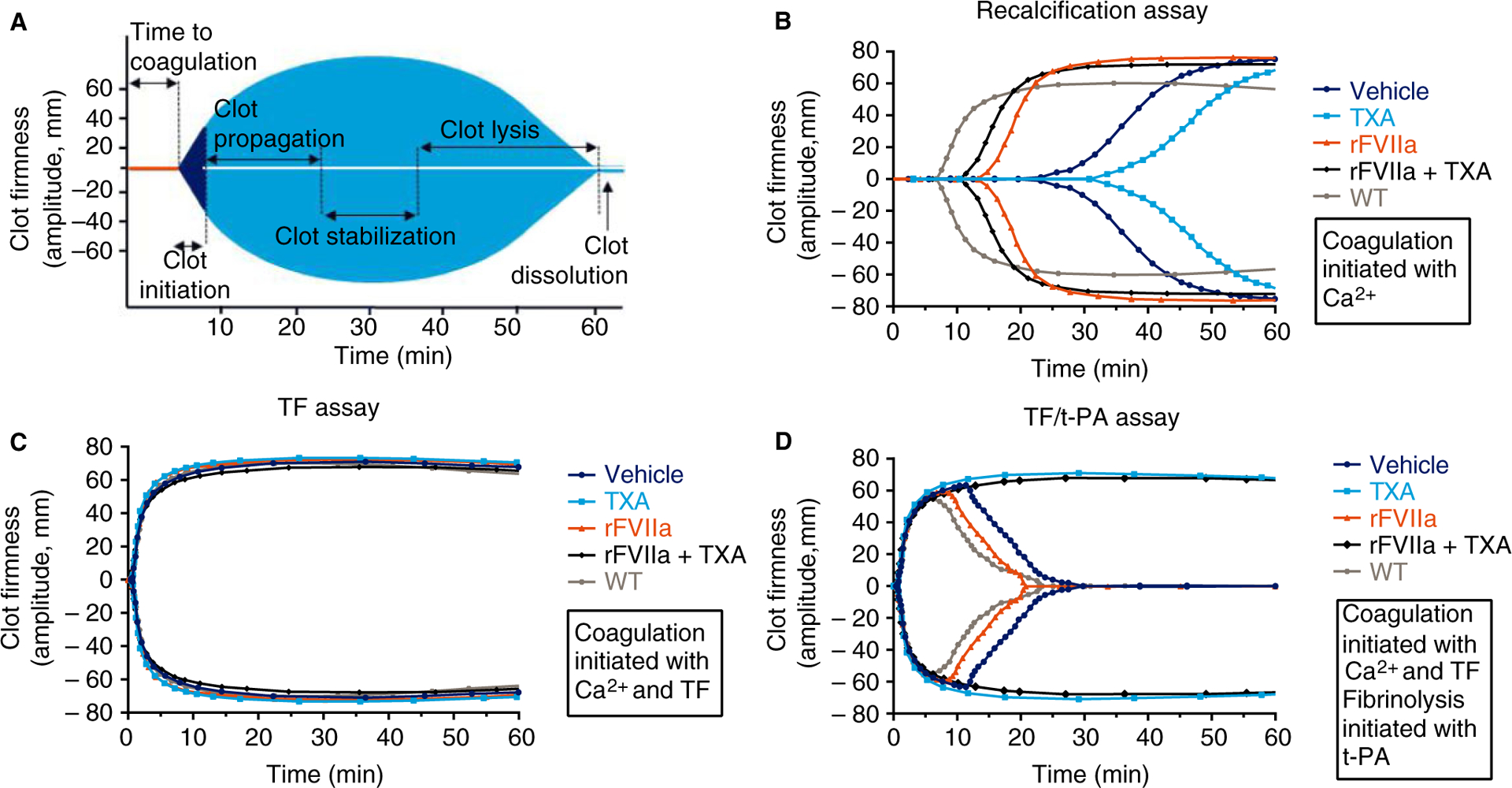
Schematic depiction and representative examples of rotational thromboelastometry (ROTEM) traces recorded during recalcification, tissue factor (TF) and TF/tissue-type plasminogen activator (t-PA) ROTEM assays with whole blood from treated FVIII−/− rats. (A) Schematic depiction of a ROTEM curve that demonstrates where clot initiation, propagation, stabilization, lysis and dissolution of a formed thrombus can be detected. (B) Representative examples of recorded ROTEM traces from the recalcified whole blood assay, which assesses global clot formation. (C) Representative examples of recorded ROTEM traces for the TF assay, which assesses global clot formation following activation of coagulation with TF. (D) Representative examples of ROTEM traces for the TF/t-PA assay, which assesses the formation and stability of clots following addition of t-PA to all blood samples. Each trace in (B), (C) and (D) shows the rat whole blood sample that best represented its treatment group on the basis of mean clotting time and maximum clot firmness results. rFVIIa, recombinant activated factor VII; TXA, tranexamic acid; WT, wild-type.
Fig. 2.
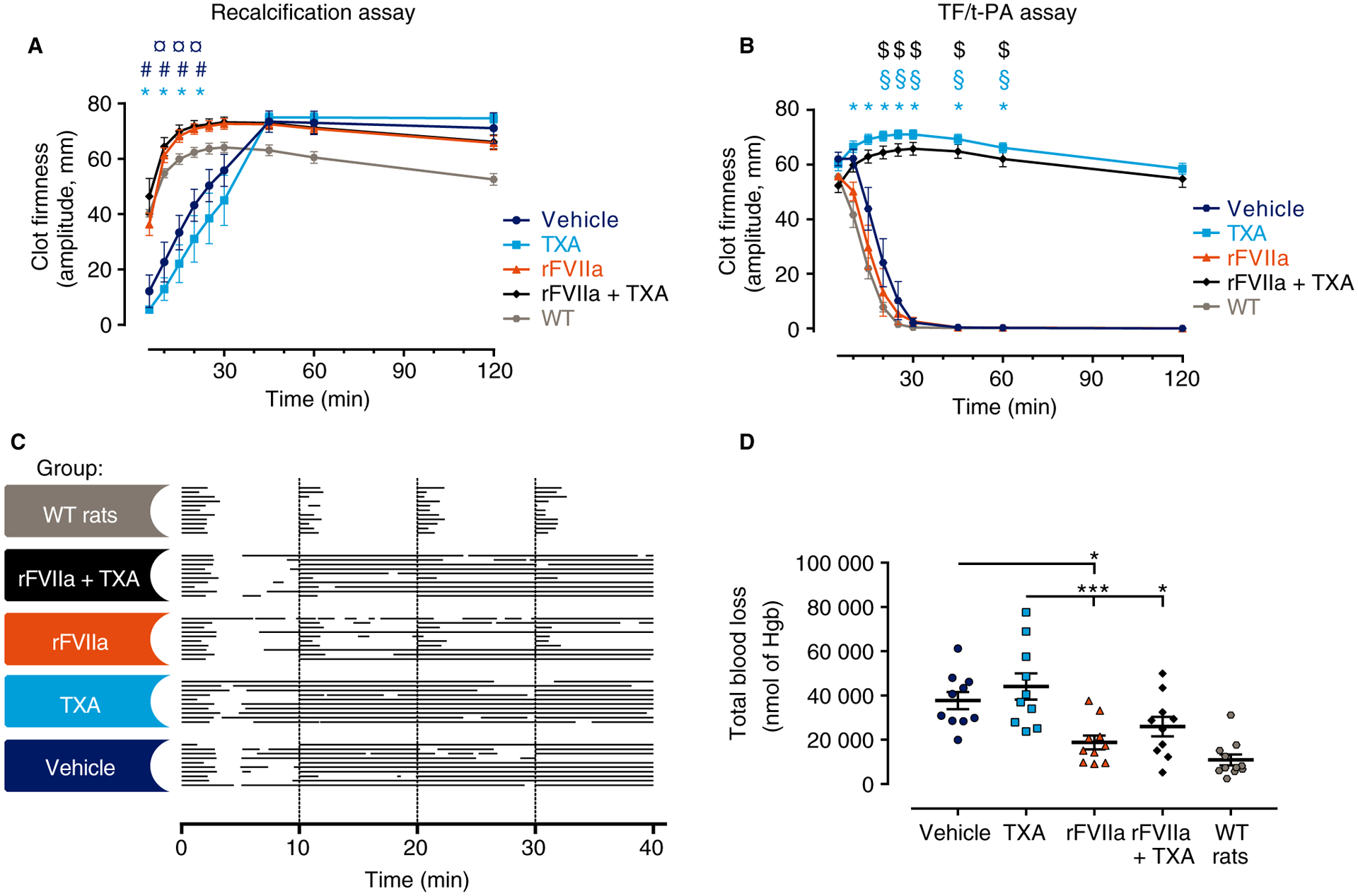
Antifibrinolytic therapy increased clot stability ex vivo, but did not improve the bleeding phenotype or the response to rFVIIa in vivo in rats with congenital hemophilia A. (A) Native clot formation in recalcified whole blood as measured by changes in amplitude over time. Clot formation was significantly reduced in whole blood from tranexamic acid (TXA)-treated and vehicle-treated rats. ¤P ≤ 0.05 for vehicle versus recombinant activated factor VII (rFVIIa); #P ≤ 0.05 for vehicle versus rFVIIa + TXA; *P ≤ 0.05 for TXA versus rFVIIa and TXA versus rFVIIa + TXA. (B) Whole blood clot formation and stability as measured by changes in amplitude over time in the tissue factor (TF)/tissue-type plasminogen activator (t-PA) assay. TXA had a significant clot-stabilizing effect ex vivo, as rats treated with TXA or a with combination of rFVIIa and TXA were almost completely resistant to a fibrinolytic challenge. $P ≤ 0.05 for rFVIIa + TXA versus vehicle and rFVIIa + TXA versus rFVIIa; §P ≤ 0.05 for TXA versus vehicle; *P ≤ 0.05 for TXA versus rFVIIa. In both (A) and (B), n = 5, with two technical replicates for each rat (mean ± standard error of the mean [SEM]). Recalcification and TF/t-PA data were analyzed with Welch’s ANOVA with the Games–Howell post hoc test. (C) Individual bleeding profiles of treated rats: a graphical representation of bleeding profiles, in which horizontal lines represent the entire bleeding profile of a single rat, and each bar in a line represents a single bleeding episode for that rat. (D) The total blood loss of FVIII−/− rats subjected to the tail vein transection (TVT) bleeding model shown as individual observations; horizontal and error bars indicate mean ± SEM. In the TVT bleeding model, no significant differences for TXA alone versus vehicle or rFVIIa alone versus rFVIIa and TXA were detected. However, blood loss was significantly reduced in rFVIIa-treated and rFVIIa + TXA-treated rats. Blood loss was analyzed with an ordinary ANOVA with Bonferroni’s correction for multiple comparisons, except for wild-type (WT) rats. *P < 0.05; ***P < 0.001. FVIII−/− rats were used, unless noted otherwise. Hgb, hemoglobin.
The potential additive contributions of TXA and rFVIIa were also determined. In the recalcification assay, the clot formation rate of whole blood samples from rFVIIa-treated FVIII−/− rats was significantly increased as compared with samples from vehicle-treated or TXA-treated FVIII−/− rats (Fig. 2A). Notably, coadministration of TXA did not further enhance the effect of rFVIIa (Fig. 2A). However, coadministration of rFVIIa and TXA substantially decreased t-PA-mediated clot degradation, which indicated that the combination therapy increased clot stability (Fig. 2B).
Antifibrinolytic therapy alone did not improve the in vivo bleeding phenotype of rats with congenital HA
To determine whether the TF/t-PA ex vivo ROTEM assay accurately predicted the effect of TXA in vivo, we analyzed the bleeding phenotype of rats following a TVT injury. All recorded bleeding episodes were first illustrated to provide a visualization of the length and number of bleeds that the rats experienced (Fig. 2C); visual examination of the bleeding profile indicated that TXA alone did not shorten the bleeding episodes (Fig. 2C). Despite the observed ex vivo reduction in fibrinolysis afforded by TXA treatment, no significant difference in blood loss was observed between vehicle-treated and TXA-treated FVIII−/− rats in vivo (Fig. 2D); mean blood losses were 37 721 ± 3898 nmol of Hgb and 44 114 ± 5914 nmol of Hgb for vehicle-treated animals and TXA-treated animals, respectively. Furthermore, TXA did not reduce the number of spontaneous bleeds (Table 3). Notably, the mean TXA plasma concentration was determined to be 228.7 μg mL−1 (Table 3), which is a plasma level that has previously been shown to be clinically efficacious [23].
Table 3.
Overview of the effect of diminishing fibrinolysis in hemophilia A (HA) animal models subjected to the tail vein transection (TVT) bleeding model as assessed by blood loss, bleeding time, and spontaneous bleeds; total blood loss, bleeding time, spontaneous bleeds and tranexamic acid (TXA) plasma concentration as observed in the TVT model
| Figure | Study | Treatment group | Blood loss (nmol of Hgb) | Bleeding time (min) | Spontaneous bleeds | TXA exposure (μg mL−1) |
|---|---|---|---|---|---|---|
| 2 | FVIII−/− rats | Vehicle | 37 721 ± 3898 | 31.5 ± 1.4 | 1.6 ± 0.4 | 0 |
| TXA | 44 114 ± 5914 | 35.2 ± 0.9 | 1.3 ± 0.26 | 228.7 ± 34 | ||
| rFVIIa | 18 821 ± 3108 | 21.6 ± 4.0 | 1.7 ± 0.87 | 0 | ||
| rFVIIa and TXA | 25 926 ± 4401 | 29.3 ± 2.6 | 1.4 ± 0.31 | 161.5 ± 18.6 | ||
| WT | 10 939 ± 2424 | 6.3 ± 0.4 | 0.09 ± 0.09 | 0 | ||
| 3A-D | FVIII−/− mice | Vehicle | 4745 ± 545.9 | 23.3 ± 2.8 | 1.06 ± 0.14 | 0 |
| TXA | 4115 ± 439 | 23.3 ± 2.1 | 0.87 ± 0.19 | 104.9 ± 9.57 | ||
| rFVIIa | 3449 ± 599 | 19.9 ± 2.8 | 0.8 ± 0.14 | 0 | ||
| rFVIIa and TXA | 1500 ± 419 | 11.2 ± 2.5 | 0.73 ± 0.28 | 118.9 ± 11.5 | ||
| WT | 366 ± 85 | 3.7 ± 0.4 | 0 ± 0 | 0 | ||
| 3E-H | FVIII−/− mice | Vehicle | 5326 ± 546 | 20.56 ± 1.8 | 1.33 ± 0.43 | 0 |
| TXA | 4355 ± 628 | 20.28 ± 2.7 | 1 ± 0.21 | 150 ± 37.5 | ||
| rFVIII | 3740 ± 724 | 16.7 ± 2.9 | 1.25 ± 0.35 | 0 | ||
| rFVIII and TXA | 2322 ± 600 | 12.8 ± 2.7 | 1.25 ± 0.33 | 139.9 ± 15.2 | ||
| WT | 657 ± 127 | 4.9 ± 0.4 | 0 ± 0 | 0 | ||
| 4 | C57Bl/6 | Vehicle | 6064 ± 967 | 29 ± 1.1 | 0.75 ± 0.22 | 0 |
| TXA | 5356 ± 790 | 25.41 ± 2.6 | 1.25 ± 0.28 | 182.9 ± 33.21 | ||
| 0.4 mg kg−1 rFVIII | 4581 ± 662 | 22.41 ± 1.8 | 2.33 ± 0.74 | 0 | ||
| 0.4 mg kg−1 rFVIII and TXA | 4011 ± 873 | 20.95 ± 3.2 | 1 ± 0.33 | 157.8 ± 29.86 | ||
| 0.7 mg kg−1 rFVIII | 1907 ± 558 | 8.72 ± 1.1 | 2.58 ± 0.7 | 0 | ||
| 0.7 mg kg−1 rFVIIa and TXA | 2761 ± 972 | 10.99 ± 3.3 | 0.5 ± 0.29 | 130.4 ± 15.32 | ||
| WT | 506 ± 107 | 4.87 ± 0.6 | 0 ± 0 | 0 | ||
| 5A-D | t-PA WT mice | Vehicle | 8793 ± 981 | 26.2 ± 2 | 2.13 ± 0.4 | - |
| t-PA−/− mice | 7744 ± 484 | 32.3 ± 1.3 | 0.88 ± 0.023 | - | ||
| t-PA WT mice | 0.7 mg kg−1 rFVIIa | 3975 ± 934 | 17.7 ± 3.2 | 2.5 ± 0.85 | - | |
| t-PA−/− mice | 4509 ± 910 | 20.7 ± 3 | 3 ± 1.36 | - | ||
| t-PA WT mice | 1.5 mg kg−1 rFVIIa | 2627 ± 877 | 13 ± 3.7 | 2 ± 0.78 | - | |
| t-PA−/− mice | 2280 ± 788 | 9.2 ± 1.9 | 4 ± 1.19 | - | ||
| t-PA WT mice | 2.7 mg kg−1 rFVIIa | 710 ± 216 | 6.1 ± 1 | 2 ± 1.09 | - | |
| t-PA−/− mice | 765 ± 208 | 5.8 ± 1.1 | 1 ± 0.86 | - | ||
| t-PA WT mice | No induced HA | 759 ± 123 | 5 ± 0.3 | 0.13 ± 0.13 | - | |
| t-PA−/− mice | 1191 ± 591 | 4.7 ± 0.3 | 0 ± 0 | - | ||
| 5E-H | Plg WT mice | Vehicle | 5477 ± 468 | 25.4 ± 2.4 | 1.38 ± 0.26 | - |
| Plg−/− mice | 5437 ± 606 | 27.2 ± 1.5 | 1.81 ± 0.35 | - | ||
| Plg WT mice | 0.7 mg kg−1 rFVIIa | 4797 ± 1034 | 17.6 ± 2.8 | 3.1 ± 0.72 | - | |
| Plg−/− mice | 4975 ± 1138 | 16 ± 3.8 | 1.57 ± 0.65 | - | ||
| Plg WT mice | 1.5 mg kg−1 rFVIIa | 3087 ± 1305 | 13.1 ± 3.6 | 3.14 ± 1.03 | - | |
| Plg−/− mice | 2262 ± 957 | 10.2 ± 2.7 | 1.13 ± 0.55 | - | ||
| Plg WT mice | 2.7 mg kg−1 rFVIIa | 1735 ± 325 | 9.4 ± 1.8 | 3.13 ± 1.04 | - | |
| Plg−/− mice | 2288 ± 803 | 10.1 ± 2.8 | 2.67 ± 1.07 | - | ||
| Plg WT mice | No induced HA | 760 ± 189 | 5.0 ± 0.4 | 0.14 ± 0.14 | - | |
| Plg−/− mice | 983 ± 344 | 5.3 ± 0.4 | 0 ± 0 | - |
FVIII, coagulation factor VIII; Hgb, hemoglobin; Plg, plasminogen; rFVIIa, activated recombinant factor VII; rFVIII, recombinant factor VIII; t-PA, tissue-type plasminogen activator; WT, wild type. Values are shown as mean ± standard error of the mean.
Antifibrinolytic therapy did not improve the response to rFVIIa in vivo in rats with congenital HA
Next, we examined whether TXA could augment the therapeutic effect of a bypassing agent (rFVIIa) in vivo. As expected, rFVIIa treatment decreased the length of the bleeding episodes in some FVIII−/− rats (Fig. 2C), and, as compared with vehicle-treated and TXA-treated FVIII−/− rats, also significantly decreased the overall blood loss, to 18 821 ± 3108 nmol of Hgb (Fig. 2D; Table 3). However, coadministration of TXA did not improve the response to rFVIIa, as the blood loss (25 926 ± 4401 nmol of Hgb) and number of spontaneous bleeds in rFVIIa-treated and TXA-treated FVIII−/− rats were similar to that in rFVIIa-treated FVIII−/− rats (Fig. 2D; Table 3). The mean TXA plasma concentration was determined to be 166.1 μg mL−1 (Table 3), again corresponding to a clinically efficacious plasma level [23].
Antifibrinolytic therapy alone did not improve the in vivo bleeding phenotype of mice with congenital or induced HA
To confirm the absence of observed efficacy of TXA in the non-mucosal TVT bleeding model in rats, FVIII−/− mice with or without TXA treatment were also subjected to the TVT bleeding model in two separate studies. The illustrated visualization of bleeding episodes revealed that vehicle-treated and TXA-treated mice had similar bleeding phenotypes (Fig. 3A,E). This observation was confirmed by further data analyses, as vehicle-treated and TXA-treated FVIII−/− mice had similar levels of blood loss (Fig. 3B,F; Table 3), bleeding times (Fig. 3C,G; Table 3), and numbers of spontaneous bleeds (Fig. 3D,H; Table 3). TXA-treated FVIII−/− mice had a mean TXA plasma concentration of 105 or 150 μg mL−1 (Table 3), corresponding to clinically efficacious plasma levels [23].
Fig. 3.
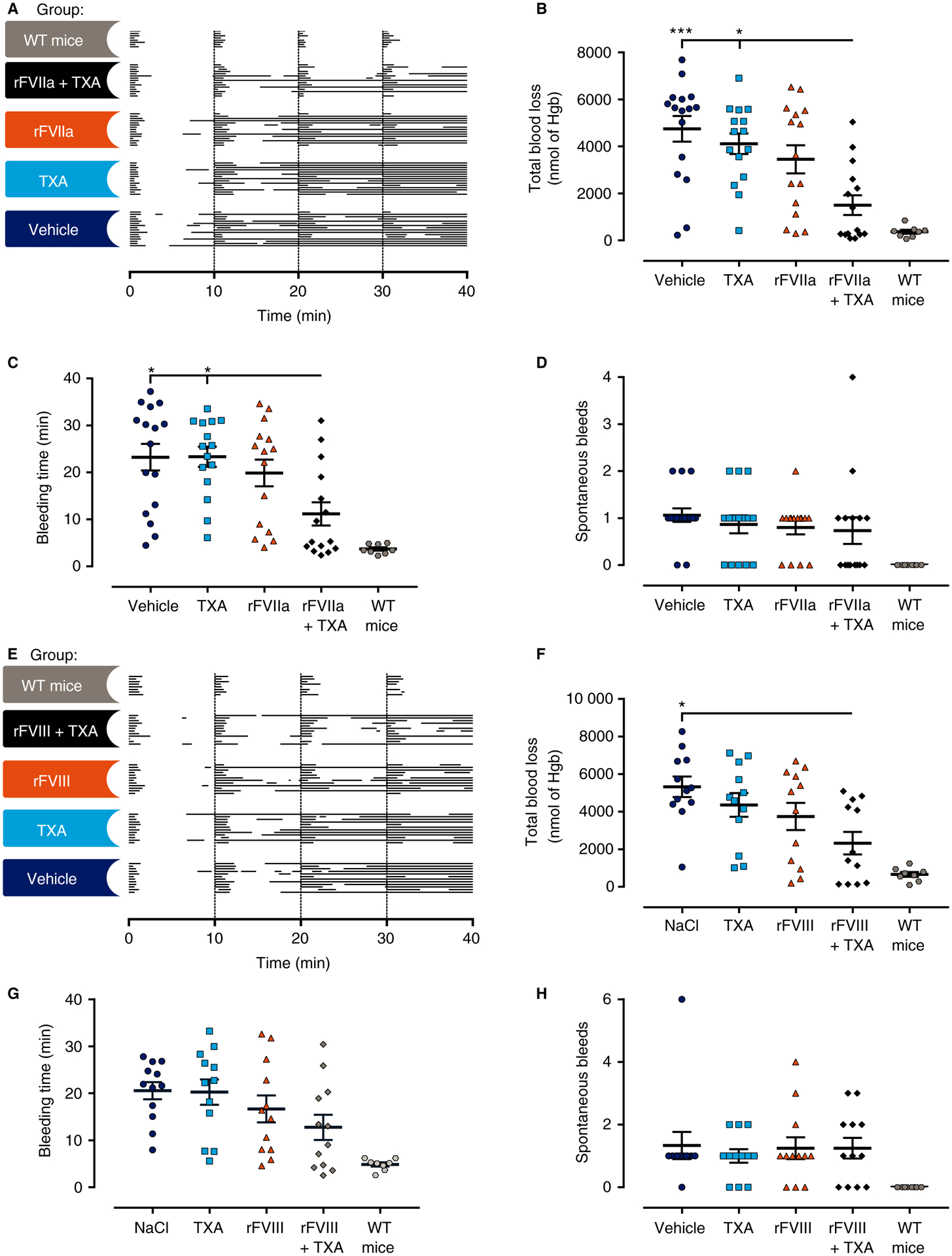
Antifibrinolytic therapy did not significantly improve the bleeding phenotype or the response to recombinant activated factor VII (rFVIIa) or recombinant factor VIII (rFVIII) in mice with congenital hemophilia A. (A, E) Individual bleeding profiles of treated mice: graphical representation of bleeding profiles, in which horizontal lines represent the entire bleeding profile of a single mouse, and each bar in a line represents a single bleeding episode for that mouse. (B–D, F–H) Total blood loss (B, F), bleeding time (C, G) and spontaneous bleeds (D, H) of FVIII−/− mice are shown as individual observations, with horizontal and error bars representing mean ± standard error of the mean. No significant differences for tranexamic acid (TXA) alone versus vehicle or for factor treatment alone versus factor treatment + TXA were detected for blood loss, bleeding time, or the number of spontaneous bleeds. Data were analyzed with the Kruskal–Wallis test, with Dunn’s test to adjust for multiple comparisons between all groups, except for wild-type (WT) mice. *P < 0.05, **P < 0.01, and ***P < 0.001. FVIII−/− mice were used, unless noted otherwise. Hgb, hemoglobin.
The FVIII−/− mice had a mixed 129SV/C57BL/6 background. Several studies have highlighted interstrain differences in coagulation and fibrinolytic factors in mice [28–30]. To confirm the results obtained in the FVIII−/− mice, the effect of inhibiting fibrinolysis was also evaluated in mice with a pure C57Bl/6 background. A hemophilic condition similar to acquired HA was induced in inbred C57Bl/6 mice, which were then subjected to the TVT bleeding model. The determined TXA plasma concentration of 183 μg mL−1 indicated that these animals were adequately treated with TXA (Table 3). Like the mixed background FVIII−/− mice, vehicle-treated and TXA-treated C57Bl/6 mice had similar bleeding patterns (Fig. 4A), and no significant differences in blood loss (6064 ± 967 nmol of Hgb versus 535 ± 790 nmol of Hgb), bleeding time (29.0 ± 1.1 min versus 25.4 ± 2.6 min), or occurrences of spontaneous bleeds (1.25 ± 0.96 versus 0.75 ± 0.75) were found (Fig. 4B–D; Table 3).
Fig. 4.
Antifibrinolytic therapy did not improve the bleeding phenotype or improve the response to recombinant activated factor VII (rFVIIa) in mice with induced hemophilia A (HA). (A) Individual bleeding profiles of treated mice: graphical representation of bleeding profiles, in which horizontal lines represent the entire bleeding profile of a single mouse, and each bar in a line represents a single bleeding episode for that mouse. (C–D) Total blood loss (B), bleeding time (C) and spontaneous bleeds (D) of C57Bl/6 mice are shown as individual observations, with horizontal and error bars representing mean ± standard error of the mean. No significant differences for tranexamic acid (TXA) alone versus vehicle or rFVIIa alone versus rFVIIa and TXA were detected for blood loss, bleeding time, or the number of spontaneous bleeds. Data points were analyzed with the Kruskal–Wallis test, with Dunn’s test to adjust for multiple comparisons between all groups, except for mice without induced HA. *P < 0.05, **P < 0.01, and ***P < 0.001. Hgb, hemoglobin; WT, wild-type.
Antifibrinolytic therapy did not improve the response to rFVIIa or rFVIII in vivo in mice with congenital or induced HA
The effect of TXA on the response to rFVIIa or rFVIII therapy was also tested in mice with congenital or induced HA. In FVIII−/− mice, neither blood loss nor bleeding time was significantly reduced by the administered dose of rFVIIa, although significance was observed for FVIII−/− mice treated with rFVIIa and TXA (Fig. 3B–C; Table 3). However, no significant difference in blood loss, bleeding time or spontaneous bleeds was observed between rFVIIa-treated mice and mice treated with rFVIIa and TXA (Fig. 3B–D; Table 3). Furthermore, coadministration of TXA did not significantly enhance the therapeutic effect of rFVIII either, as rFVIII-treated FVIII−/− mice and FVIII−/− mice treated with rFVIII and TXA showed comparable bleeding phenotypes (Fig. 3F–H; Table 3). The absence of an effect was not considered to be attributable to an insufficient dose of TXA, as the mean TXA plasma concentrations were 119 μg mL−1 and 140 μg mL−1, corresponding to clinically efficacious plasma levels (Table 3).
In C57Bl/6 mice with induced HA, TXA was not found to improve the response to rFVIIa (Fig. 4B–D). The mean TXA plasma concentrations were determined to be 158 μg mL−1 and 130 μg mL−1, corresponding to clinically efficacious plasma levels (Table 3) [23].
Genetically induced fibrinolytic deficiency did not improve the in vivo bleeding phenotype in mice with induced HA
To confirm the results obtained with pharmacological inhibition of fibrinolysis, mice with complete, genetically imposed deficiency in either t-PA or plasminogen were studied with the TVT bleeding model. Both t-PA-deficient and plasminogen-deficient mice have been shown to have significantly reduced clot lysis [16,31]. Following TVT, no significant difference in bleeding phenotype could be detected between t-PA−/− and t-PA WT mice (Fig. 5A–D; Table 3). Furthermore, in mice with induced HA, the genetic elimination of t-PA did not reduce the total blood loss (8793 ± 981 nmol of Hgb versus 7744 ± 484 nmol of Hgb), bleeding time (26.2 ± 2 min versus 32.3 ± 1.3 min) or the occurrences of spontaneous bleeds (2.13 ± 0.4 versus 0.88 ± 0.023) associated with induced FVIII deficiency (Fig. 5A–D). HA was also induced in plasminogen-deficient mice, which were then subjected to the TVT bleeding model. The complete genetic loss of plasminogen failed to improve the bleeding phenotype associated with loss of FVIII as evaluated by blood loss (5477 ± 468 nmol of Hgb versus 5437 ± 606 nmol of Hgb), bleeding time (25.4 ± 2.4 min versus 27.2 ± 1.5 min), and spontaneous bleeds (1.38 ± 0.26 versus 1.82 ± 0.35) (Fig. 5E–H; Table 3).
Fig. 5.
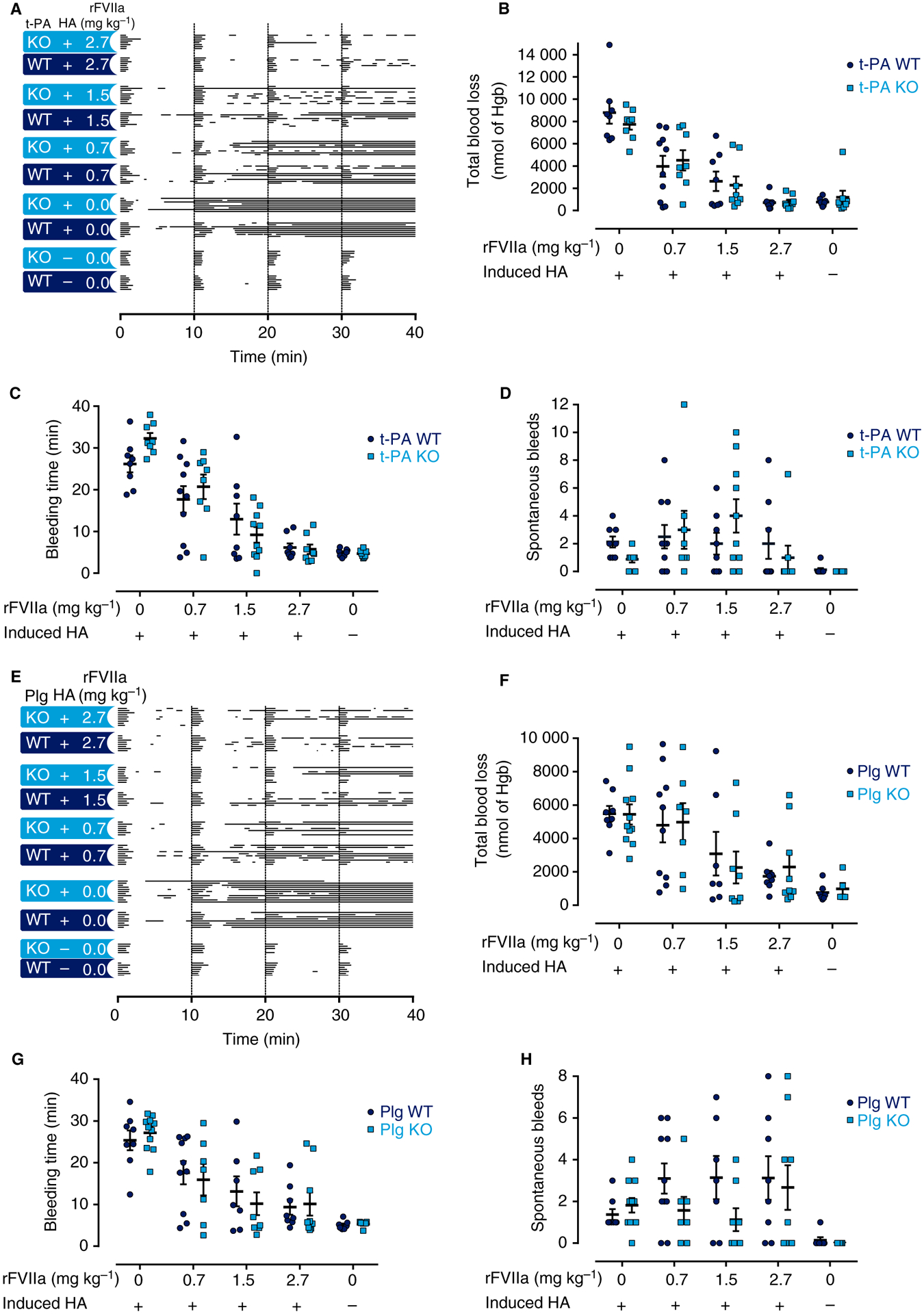
Genetically induced fibrinolytic deficiency did not improve the bleeding phenotype or improve the response to recombinant activated factor VII (rFVIIa) in mice with induced hemophilia A (HA). (A, E) Individual bleeding profiles of treated mice: graphical representation of bleeding profiles, in which horizontal lines represent the entire bleeding profile of a single mouse, and each bar in a line represents a single bleeding episode for that mouse. (B–D, F–H) Total blood loss (B, F), bleeding time (C, G) and spontaneous bleeds (D, H) of mice are shown as individual observations, with horizontal and error bars representing mean ± standard error of the mean. (A–D) Graphs from studies with tissue-type plasminogen (Plg) activator (t-PA) mice. (E–H) Graphs from studies with Plg mice. In no case was a significant difference between vehicle-treated t-PA wild-type (WT) and t-PA−/− mice, or between Plg WT and Plg−/− mice, detected. Similarly, no difference between rFVIIa-treated t-PA WT and t-PA−/− mice, or between Plg WT and Plg−/− mice, was detected. Data were analyzed with a two-way ANOVA with Bonferroni’s correction for multiple comparisons. *P < 0.05. Hgb, hemoglobin; KO, knockout.
Genetically induced fibrinolytic deficiency did not improve the response to pharmacologically administered rFVIIa in vivo in mice with induced HA
HA was induced in WT mice and t-PA−/− mice, which were then treated with rFVIIa. As expected, the mice showed a dose-dependent reduction in lengths of bleeding episodes, blood loss and bleeding times in response to rFVIIa therapy (Fig. 5A–D; Table 3). However, no difference in the hemostatic effect of rFVIIa therapy could be detected between t-PA WT and t-PA−/− mice with induced HA (Fig. 5A–D). Similar observations were made in mice with plasminogen deficiency, as the loss of plasminogen failed to enhance the therapeutic effect of rFVIIa (Fig. 5E–H; Table 3).
Discussion
These studies show that TXA has the capacity to preserve thrombi generated in whole blood sampled from rats with HA. However, the striking findings in the ex vivo assay did not translate to an in vivo decrease in TVT bleeding in hemophilic rats. Furthermore, neither pharmacologically nor genetically induced diminution of fibrinolysis improved TVT bleeding or the response to rFVIIa or rFVIII in mice with congenital or induced HA.
Antifibrinolytic drugs have been used in the management of hemophilia since the 1960s [10], as a widely held clinical view is that enhanced fibrinolytic activity exacerbates bleeding symptoms in patients with HA [4]. Similarly to our findings, modified TF/t-PA ROTEM ex vivo/in vitro assays have indicated that antifibrinolytic drugs have a clot-stabilizing effect. Thus, the working theory has been that these agents can enhance the efficacy of factor replacement treatment [11–13,32,33]. However, these modified assays rely on artificial activation of the fibrinolytic system, and lack the contribution of the vasculature. Our studies are the first to combine the TVT bleeding model with a ROTEM TF/t-PA assay in the same set of FVIII−/− rats to directly evaluate the predictive potential of the TF/t-PA ROTEM assay. As hypothesized, TXA, both alone and in combination with rFVIIa, induced resistance to t-PA-mediated fibrinolysis in the TF/t-PA assay. However, despite the clear clot-stabilizing effect observed ex vivo, TXA treatment did not diminish bleeding or increase the response to rFVIIa therapy in an in vivo challenge in FVIII−/− rats. Our observation corresponds with the lack of compelling evidence for a beneficial effect of TXA in non-mucosal bleeds in clinical medicine. Although thromboelastography assays can assess the effect of rFVIII treatments and the clinical phenotypes of HA patients [34,35], the comparison of our ex vivo and in vivo results indicates that the modified TF/t-PA assay may not accurately describe the complete clinical situation [11–13]. Consequently, care should be taken when results from the TF/t-PA assay are interpreted and translated in terms of patient outcomes.
Although antifibrinolytic drugs have been proven to be effective as adjunctive HA therapy for mucosal bleeds [5,36–38], in non-mucosal tissues they are mainly used empirically. Only limited evidence supports the use of antifibrinolytic drugs in cases of non-mucosal bleeding and in surgical operations [9,10,39–45]. Moreover, no study has definitively shown the usefulness of systemic TXA therapy in patients with HA. To shed more light on the potential benefits of inhibiting fibrinolysis in non-mucosal HA bleeds, we performed tail vein bleeding studies in hemophilic rodent models with impaired fibrinolysis. Neither TXA treatment, t-PA deficiency nor plasminogen deficiency was observed to significantly reduce bleeding. Furthermore, in neither of the tested animal models was the effect of factor treatment enhanced by genetically or pharmacologically induced inhibition of fibrinolysis, despite the use of doses of TXA sufficient to obtain clinically relevant plasma levels of TXA [22,23], which are known to fully inhibit fibrinolysis in vitro [24]. Consequently, we were unable to demonstrate a significant in vivo value of inhibiting fibrinolysis in non-mucosal hemophilic TVT bleeds.
To confirm that our findings were valid in both congenital HA and an induced condition similar to acquired HA, the role of fibrinolysis in non-mucosal TVT bleeding was examined with complementary approaches and in different species; the inhibition of fibrinolysis and the HA setting were obtained both with pharmacological intervention and with genetically induced deficiencies. The TVT bleeding model employed here has been shown to be sensitive to clinically relevant doses and valuable in assessing the pharmacodynamic effects of rFVIII and rFVIIa [18,46,47]. Importantly, FVIII−/− mice subjected to the TVT bleeding model also develop spontaneous recurrent bleeding, which decreases in a dose-dependent manner with rFVIII replacement [18]. The occurrence of recurrent bleeding should allow for assessment of any potential rebleeds resulting from fibrinolysis, as plasminogen is activated in an early phase of hemostasis [48]. This supports the suitability of the bleeding model for testing our hypothesis, as it recapitulates the bleeding phenotype seen in patients with HA. This model has the further strength of incorporating the contribution of blood flow and platelet function during primary hemostasis [49].
The observation that diminished fibrinolysis failed to reduce bleeding indicates that, in non-mucosal large-vessel injuries, the effect of fibrinolysis may be relatively limited. Insufficient TAFI activation by thrombin has been presumed to increase fibrinolysis [4,50,51], but TAFI is also activated by other proteins. Indeed, plasmin activates TAFI in vitro and in vivo [52,53], and could potentially help to prevent premature clot degradation. Furthermore, TAFI−/− mice have a comparable tail-bleeding phenotype to WT mice, even when bleeding is exacerbated with an antithrombotic agent [49,54], and non-mucosal bleeding models in dogs, rats and rabbits were not affected by a TAFI inhibitor [55]. Results regarding the influence of TAFI on HA pathology are also conflicting [51,56], and it remains to be determined whether activated TAFI actually affects the bleeding symptoms of HA patients.
It is recognized that there are a number of limitations in the current study. For example, the lack of suitable mucosal rodent bleeding models prevented us from examining whether inhibition of fibrinolysis would be effective in preventing mucosal bleeding in hemophilic rodents. Moreover, the model used here examined bleeding following a transection injury to a large vessel. It is possible that antifibrinolytics effectively limit bleeding from some vessel types (i.e. arterial versus venous) or vascular beds (i.e. small vessels or capillaries versus large vessels) in the setting of HA, but not from others. Indeed, the model used here is more akin to a setting of bleeding following a surgical incision, whereas common spontaneous clinical pathologies of HA include synovial microvessel, muscular and parenchymal bleeds. Accordingly, validation of our findings with additional non-mucosal bleeding models, e.g. a joint-bleeding model, would provide further insights. Furthermore, it could be argued that the pathological traits of plasminogen deficiency could constitute a confounding variable, as could the mixed background of the FVIII−/− mice, owing to the increased potential for genetic drift. The FVIII−/− mice with a mixed 129SV/C57BL/6 background are, however, very well characterized, and are widely accepted by the scientific community. Finally, the degree of fibrinolytic inhibition with TXA was not directly measured, but was assumed to be sufficient on the basis of the TXA exposure, the TF/t-PA ROTEM assay, and previous reports [20,21]. The inclusion of additional models could therefore also be useful to confirm the efficacy of TXA dosing in the rodent species employed.
In conclusion, antifibrinolytic drugs are used to stabilize hemostatic plugs in the oral cavities of HA patients. Moreover, they are often applied off-label as systemic agents for the treatment of HA. However, in the present rodent study, the beneficial effect of systemic TXA treatment observed ex vivo could not be confirmed in vivo. Furthermore, completely inhibiting fibrinolysis either genetically or pharmacologically did not improve the bleeding phenotype in HA rodents, as assessed with a sensitive non-mucosal TVT bleeding model that predicts the effect of rFVIIa and rFVIII therapies well. Instead, our observations indicate that, in HA, fibrinolysis may not be a major driver of hemorrhage in non-mucosal bleeds in rodents. Thus, systemic antifibrinolytics may have limited value as stand-alone or adjunctive therapy in hemophilic large-vessel injuries. Further investigation is therefore warranted.
Supplementary Material
Data S1. Validation of the experimentally induced hemophilia A bleeding phenotype.
Data S2. Determination of the median effective dose (ED50) of rFVIII and rFVIIa in the TVT bleeding model.
Fig. S1. The anti-FVIII antibody 4F30 induces a hemophilic state in C57Bl/6 mice in vivo and in vitro.
Fig. S2. Dose response of rFVIIa 5 min after intravenous administration to FVIII−/− rats.
Fig. S3. Dose response of rFVIIa 5 min after intravenous administration to FVIII−/− mice with a mixed 129SV/C57BL/6 background or C57Bl/6 background.
Essentials.
The efficacy of systemic antifibrinolytics for hemophilic non-mucosal bleeding is undetermined.
The effect of systemically inhibiting fibrinolysis in hemophilic mice and rats was explored.
Neither bleeding nor the response to factor treatment was improved after inhibiting fibrinolysis.
The non-mucosal bleeding phenotype in hemophilia A appears largely unaffected by fibrinolysis.
Acknowledgements
The study was sponsored by Novo Nordisk A/S, the University of Copenhagen, and Innovation Fund Denmark. We gratefully acknowledge and thank S. Andersen for helping with the statistical analyses, H. F. Kierkegaard, K. Larsen and D. F. Danielsen for their excellent help with the animal experiments, C. Cruz, C. Rewerts, M. Shaw, M. D. Frederik and M. Smith for their outstanding technical assistance, and E. S. Mullins for his extremely helpful advice and manuscript corrections. The authors would also like to thank Shimadzu Europa GmbH and Shim-pol A. M. Borzymowski Company for supplying the LCMS-8060 Triple Quadrupole LC/MS/MS equipment, as well as MilliporeSigma/Supelco for providing HS F5 columns.
Disclosure of Conflict of Interests
R. Stagaard and C. D. Ley were employees of Novo Nordisk A/S, and report receiving grants from Innovation Fund Denmark during the conduct of the study. T. Knudsen is a previous Novo Nordisk A/S employee and current shareholder. L. H. Olsen was affiliated with the in vivo pharmacology research center LIFEPHARM, which was supported by Novo Nordisk A/S. M. J. Flick reports receiving grants from Novo Nordisk A/S during the conduct of the study, but in a research area separate and distinct from the HA studies reported in the present article. The other authors state that they have no conflict of interest.
References
- 1.Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol 2006; 26: 41–8. [DOI] [PubMed] [Google Scholar]
- 2.He S, Blombäck M, Jacobsson Ekman G, Hedner U. The role of recombinant factor VIIa (FVIIa) in fibrin structure in the absence of FVIII/FIX. J Thromb Haemost 2003; 1: 1215–19. [DOI] [PubMed] [Google Scholar]
- 3.Brummel-Ziedins KE, Branda RF, Butenas S, Mann KG. Discordant fibrin formation in hemophilia. J Thromb Haemost 2009; 7: 825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broze GJ Jr, Higuchi DA. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood 1996; 88: 3815–23. [PubMed] [Google Scholar]
- 5.Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, Ludlam CA, Mahlangu JN, Mulder K, Poon MC, Street A; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia 2013; 19: e1–47. [DOI] [PubMed] [Google Scholar]
- 6.Forbes C, Barr R, Reid G, Thomson C, Prentice C, Mc Nicol G, Douglas A. Tranexamic acid in control of haemorrhage after dental extraction in haemophilia and Christmas disease. Br Med J 1972; 2: 311–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh PN, Rizza C, Matthews J, Eipe J, Kernoff P, Coles M, Bloom A, Kaufman B, Beck P, Hanan C. Epsilon-aminocaproic acid therapy for dental extractions in haemophilia and christmas disease: a double blind controlled trial. Br J Haematol 1971; 20: 463–75. [DOI] [PubMed] [Google Scholar]
- 8.Coetzee MJ. The use of topical crushed tranexamic acid tablets to control bleeding after dental surgery and from skin ulcers in haemophilia. Haemophilia 2007; 13: 443–4. [DOI] [PubMed] [Google Scholar]
- 9.Rainsford SG, Jouhar AJ, Hall A. Tranexamic acid in the control of spontaneous bleeding in severe haemophilia. Thromb Diath Haemorrh 1973; 30: 272–9. [PubMed] [Google Scholar]
- 10.Gordon AM, McNicol GP, Dubber AHC, McDonald GA, Douglas AS. Clinical trial of epsilon-aminocaproic acid in severe haemophilia. Br Med J 1965; 1: 1632–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rea CJ, Foley JH, Bevan DH, Sorensen B. An in-vitro assessment of tranexamic acid as an adjunct to rFVIII or rFVIIa treatment in haemophilia A. Ann Hematol 2014; 93: 683–92. [DOI] [PubMed] [Google Scholar]
- 12.Tran HT, Sorensen B, Rea CJ, Bjornsen S, Ueland T, Pripp AH, Tjonnfjord GE, Holme PA. Tranexamic acid as adjunct therapy to bypassing agents in haemophilia A patients with inhibitors. Haemophilia 2014; 20: 369–75. [DOI] [PubMed] [Google Scholar]
- 13.Hvas AM, Sorensen HT, Norengaard L, Christiansen K, Ingerslev J, Sorensen B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J Thromb Haemost 2007; 5: 2408–14. [DOI] [PubMed] [Google Scholar]
- 14.Bi L, Lawler A, Antonarakis S, High K, Gearhart JA, Kazazian HH. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet 1995; 10: 119–21. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen LN, Wiinberg B, Hager M, Holmberg HL, Hansen JJ, Roepstorff K, Tranholm M. A novel F8−/− rat as a translational model of human hemophilia A. J Thromb Haemost 2014; 12: 1274–82. [DOI] [PubMed] [Google Scholar]
- 16.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994; 368: 419–24. [DOI] [PubMed] [Google Scholar]
- 17.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 1995; 9: 794–807. [DOI] [PubMed] [Google Scholar]
- 18.Johansen PB, Tranholm M, Haaning J, Knudsen T. Development of a tail vein transection bleeding model in fully anaesthetized haemophilia A mice – characterization of two novel FVIII molecules. Haemophilia 2016; 22: 625–31. [DOI] [PubMed] [Google Scholar]
- 19.Tsuji K, Aoki T, Tejima E, Arai K, Lee SR, Atochin DN, Huang PL, Wang X, Montaner J, Lo EH. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke 2005; 36: 1954–9. [DOI] [PubMed] [Google Scholar]
- 20.Illanes S, Zhou W, Schwarting S, Heiland S, Veltkamp R. Comparative effectiveness of hemostatic therapy in experimental warfarin-associated intracerebral hemorrhage. Stroke 2011; 42: 191–5. [DOI] [PubMed] [Google Scholar]
- 21.Sperzel M, Huetter J. Evaluation of aprotinin and tranexamic acid in different in vitro and in vivo models of fibrinolysis, coagulation and thrombus formation. J Thromb Haemost 2007; 5: 2113–18. [DOI] [PubMed] [Google Scholar]
- 22.Dowd NP, Karski JM, Cheng DC, Carroll JA, Lin Y, James RL, Butterworth J. Pharmacokinetics of tranexamic acid during cardiopulmonary bypass. Anesthesiology 2002; 97: 390–9. [DOI] [PubMed] [Google Scholar]
- 23.Grassin-Delyle S, Tremey B, Abe E, Fischler M, Alvarez JC, Devillier P, Urien S. Population pharmacokinetics of tranexamic acid in adults undergoing cardiac surgery with cardiopulmonary bypass. Br J Anaesth 2013; 111: 916–24. [DOI] [PubMed] [Google Scholar]
- 24.Andersson L, Nilsoon IM, Colleen S, Granstrand J, Melander B. Role of urokinase and tissue activator in sustaining bleeding and the management thereof with EACA and AMCA. Ann N Y Acad Sci 1968; 146: 642–56. [DOI] [PubMed] [Google Scholar]
- 25.Elm T, Karpf DM, Ovlisen K, Pelzer H, Ezban M, Kjalke M, Tranholm M. Pharmacokinetics and pharmacodynamics of a new recombinant FVIII (N8) in haemophilia A mice. Haemophilia 2012; 18: 139–45. [DOI] [PubMed] [Google Scholar]
- 26.Whiting D, DiNardo JA. TEG and ROTEM: technology and clinical applications. Am J Hematol 2014; 89: 228–32. [DOI] [PubMed] [Google Scholar]
- 27.Gorynski K, Bojko B, Kluger M, Jerath A, Wasowicz M, Pawliszyn J. Development of SPME method for concomitant sample preparation of rocuronium bromide and tranexamic acid in plasma. J Pharm Biomed Anal 2014; 92: 183–92. [DOI] [PubMed] [Google Scholar]
- 28.White TA, Pan S, Witt TA, Simari RD. Murine strain differences in hemostasis and thrombosis and tissue factor pathway inhibitor. Thromb Res 2010; 125: 84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrios M, Rodriguez-Acosta A, Gil A, Salazar AM, Taylor P, Sanchez EE, Arocha-Pinango CL, Guerrero B. Comparative hemostatic parameters in BALB/c, C57BL/6 and C3H/He mice. Thromb Res 2009; 124: 338–43. [DOI] [PubMed] [Google Scholar]
- 30.Hoover-Plow J, Shchurin A, Hart E, Sha J, Hill AE, Singer JB, Nadeau JH. Genetic background determines response to hemostasis and thrombosis. BMC Hematology 2006; 6.1: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation 1995; 92: 2585–93. [DOI] [PubMed] [Google Scholar]
- 32.Parcq J, Petersen KU, Borel-Derlon A, Gautier P, Ebel M, Vivien D, Repesse Y. F376A/M388A-solulin, a new promising antifibrinolytic for severe haemophilia A. Haemophilia 2017; 23: 319–25. [DOI] [PubMed] [Google Scholar]
- 33.Foley JH, Petersen KU, Rea CJ, Harpell L, Powell S, Lillicrap D, Nesheim ME, Sorensen B. Solulin increases clot stability in whole blood from humans and dogs with hemophilia. Blood 2012; 119: 3622–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chitlur M, Warrier I, Rajpurkar M, Hollon W, Llanto L, Wise-man C, Lusher JM. Thromboelastography in children with coagulation factor deficiencies. Br J Haematol 2008; 142: 250–6. [DOI] [PubMed] [Google Scholar]
- 35.Ingerslev J, Poulsen LH, Sorensen B. Potential role of the dynamic properties of whole blood coagulation in assessment of dosage requirements in haemophilia. Haemophilia 2003; 9: 348–52. [DOI] [PubMed] [Google Scholar]
- 36.Mahlangu JN, Gilham A. Guideline for the treatment of haemophilia in South Africa. S Afr Med J 2008; 98: 126–40. [PubMed] [Google Scholar]
- 37.Stubbs M, Lloyd J. A protocol for the dental management of von Willebrand’s disease, haemophilia A and haemophilia B. Aust Dent J 2001; 46: 37–40. [DOI] [PubMed] [Google Scholar]
- 38.Ghosh K Management of haemophilia and its complications in developing countries. Int J Lab Hematol 2004; 26: 243–51. [DOI] [PubMed] [Google Scholar]
- 39.Schulman S Continuous infusion. Haemophilia 2003; 9: 368–75. [DOI] [PubMed] [Google Scholar]
- 40.Schulman S, d’Oiron R, Martinowitz U, Pasi J, Briquel M, Mauser-Bunschoten E, Morfini M, Ritchie B, Goudemand J, Lloyd J. Experiences with continuous infusion of recombinant activated factor VII. Blood Coagul Fibrinolysis 1998; 9: S97–101. [PubMed] [Google Scholar]
- 41.Ghosh K, Shetty S, Jijina F, Mohanty D. Role of epsilon amino caproic acid in the management of haemophilic patients with inhibitors. Haemophilia 2004; 10: 58–62. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Merchan EC, Romero-Garrido JA, Gomez-Cardero P. Multimodal blood loss prevention approach including intra-articular tranexamic acid in primary total knee arthroplasty for patients with severe haemophilia A. Haemophilia 2016; 22: e318–20. [DOI] [PubMed] [Google Scholar]
- 43.Abe T, Sato A, Kazama M, Matsumura T. Effect of Σ-aminocaproic acid in haemophilia. Lancet 1962; 280: 405. [Google Scholar]
- 44.Mainwaring D, Keidan S. Fibrinolysis in haemophilia: the effect of ε-amino-caproic acid. Br J Haematol 1965; 11: 682–8. [DOI] [PubMed] [Google Scholar]
- 45.Reid W, Holburn R, DeSipin M, Tocantins L. The role of fibrinolysin and profibrinolysin activator in hemophilia. Am J Med Sci 1965; 249: 518–33. [DOI] [PubMed] [Google Scholar]
- 46.Elm T, Behrens C, Dainiak M, Akesson M, Hansen JJ, Jensen MS, Hilden I, Ostergaard H, Hermit MB. Dose-response and duration of effect of a half-life extended recombinant factor VIIa in the tail vein transection bleeding model in haemophilia rats. Res Pract Thromb Haemost 2017; 1: 772. [Google Scholar]
- 47.Johansen PB, Tranholm M, Ley CD, Elm T, Haaning J, Ezban M, Knudsen T. In vivo effect of recombinant FVIIa (NovoSeven®) and rFIX in a refined tail vein transection bleeding model in anesthetized mice with hemophilia A or B. J Thromb Haemost 2015; 13: 315. [Google Scholar]
- 48.Brzoska T, Tanaka-Murakami A, Suzuki Y, Sano H, Kanayama N, Urano T. Endogenously generated plasmin at the vascular wall injury site amplifies lysine binding site-dependent plasminogen accumulation in microthrombi. PLoS One 2015; 10: e0122196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Broze GJ Jr, Yin ZF, Lasky N. A tail vein bleeding time model and delayed bleeding in hemophiliac mice. Thromb Haemost 2001; 85: 747–8. [PubMed] [Google Scholar]
- 50.Lisman T, Mosnier LO, Lambert T, Mauser-Bunschoten EP, Meijers JC, Nieuwenhuis HK, de Groot PG. Inhibition of fibrinolysis by recombinant factor VIIa in plasma from patients with severe hemophilia A. Blood 2002; 99: 175–9. [DOI] [PubMed] [Google Scholar]
- 51.Mosnier LO, Lisman T, van den Berg HM, Nieuwenhuis HK, Meijers JC, Bouma BN. The defective down regulation of fibrinolysis in haemophilia A can be restored by increasing the TAFI plasma concentration. Thromb Haemost 2001; 86: 1035–9. [PubMed] [Google Scholar]
- 52.Vercauteren E, Emmerechts J, Peeters M, Hoylaerts MF, Declerck PJ, Gils A. Evaluation of the profibrinolytic properties of an anti-TAFI monoclonal antibody in a mouse thromboembolism model. Blood 2011; 117: 4615–22. [DOI] [PubMed] [Google Scholar]
- 53.Vercauteren E, Mutch NJ, Declerck PJ, Gils A. Plasmin and the thrombin–thrombomodulin complex both contribute to thrombin-activatable fibrinolysis inhibitor activation in whole blood model thrombi. J Thromb Haemost 2013; 11: 190–2. [DOI] [PubMed] [Google Scholar]
- 54.Nagashima M, Yin Z-F, Zhao L, White K, Zhu Y, Lasky N, Halks-Miller M, Broze GJ, Fay WP, Morser J. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficiency is compatible with murine life. J Clin Invest 2002; 109: 101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y-X, da Cunha V, Vincelette J, Zhao L, Nagashima M, Kawai K, Yuan S, Emayan K, Islam I, Hosoya J, Sullivan ME, Dole WP, Morser J, Buckman BO, Vergona R. A novel inhibitor of activated thrombin activatable fibrinolysis inhibitor (TAFIa) – part II: enhancement of both exogenous and endogenous fibrinolysis in animal models of thrombosis. Thromb Haemost 2007; 97: 54–61. [PubMed] [Google Scholar]
- 56.Foley JH, Nesheim ME, Rivard GE, Brummel-Ziedins KE. Thrombin activatable fibrinolysis inhibitor activation and bleeding in haemophilia A. Haemophilia 2012; 18: e316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Validation of the experimentally induced hemophilia A bleeding phenotype.
Data S2. Determination of the median effective dose (ED50) of rFVIII and rFVIIa in the TVT bleeding model.
Fig. S1. The anti-FVIII antibody 4F30 induces a hemophilic state in C57Bl/6 mice in vivo and in vitro.
Fig. S2. Dose response of rFVIIa 5 min after intravenous administration to FVIII−/− rats.
Fig. S3. Dose response of rFVIIa 5 min after intravenous administration to FVIII−/− mice with a mixed 129SV/C57BL/6 background or C57Bl/6 background.