

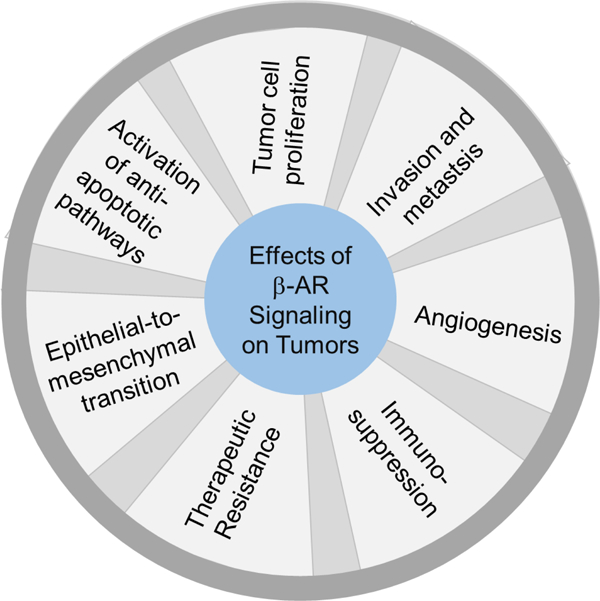
Abstract
Lung cancer results in more patient deaths each year than any other cancer type. Additional treatment strategies are needed to improve clinical responses to approved treatment modalities and prevent the emergence of resistant disease. Catecholamines including norepinephrine and epinephrine are elevated as a result of chronic stress and mediate their physiological effects through activation of adrenergic receptors on target tissues. Lung cancer cells express β-adrenergic receptors (β-ARs), and numerous preclinical studies indicate that β2-AR signaling on lung cancer cells facilities cellular programs including proliferation, motility, apoptosis resistance, epithelial-to-mesenchymal transition, metastasis, and the acquisition of an angiogenic and immunosuppressive phenotype. Here, we review the preclinical and clinical evidence supporting a potential role for beta-blockers in improving the clinical outcome of lung cancer patients.
Keywords: NSCLC, beta-adrenergic receptors, beta-blockers, catecholamines
Graphical Abstract.
Catecholamines including norepinephrine and epinephrine act of β-ARs expressed on NSCLC tumor cells and activate pathways regulating tumor progression.
Non-small cell lung cancer
Lung cancer is the leading cause of cancer death worldwide, resulting in an estimated 1.7 million deaths per year. In the United States alone, there are estimated to be 230,000 new cases of lung cancer and over 140,000 deaths annually (Siegel, Miller, and Jemal 2019). Lung cancer can be largely divided to two categories, small-cell lung cancer (about 15% of the cases) and non-small cell lung cancer (about 85% of the cases). Non-small cell lung cancer (NSCLC) is further classified into histological subtypes including adenocarcinoma, squamous cell carcinoma, large-cell carcinoma and carcinoma not otherwise specified (NOS). In addition to pathological classification, molecule classification for adenocarcinomas of the lung defines subgroups of lung cancer with distinct patient characteristics, disease course and response to targeted therapy. The high mortality rate associated with lung cancer is related to the fact that the majority of the NSCLC patients are diagnosed at metastatic stage. Once metastasis has occurred, NSCLC patients are generally not considered curable. Cytotoxic chemotherapy, immune therapy and targeted therapy treatment strategies have efficacy in NSCLC patients. However, tumors frequently develop therapeutic resistance and patients eventually succumb to their disease. The identification of additional treatment strategies is desperately needed to inhibit the development of resistant disease and to improve the clinical outcome for lung cancer patients.
The potential impact of chronic stress on NSLC
Chronic stress results in increased production of catecholamines including norepinephrine and epinephrine from the adrenal medulla and sympathetic neurons that innervate organs and tissue (Figure 1). Increased levels of stress hormones has been long believed to adversely impact general health (Kemp and Hatmaker 1989) and is an established risk factor for heart disease and infection (McEwen 2002; Sapolsky 1993; Weiner 1992; Robinson and Cinciripini 2006). Cancer patients experience elevated stress levels as part of their cancer diagnosis and treatment. While numerous studies have provided evidence that environmental, genetic, lifestyle and socioeconomic factors impact tumor progression and clinical outcome, the role of psychological distress on malignant disease is not completely understood. Studies have suggested that chronic psychological stress and psychosocial factors correlate with an increased risk of cancer (Penninx et al. 1998). A prospective study of elderly individuals found that individuals with chronic depression had a significantly increased incidence of cancer (Penninx et al. 1998). In a 2009 study by Hamer, et al. psychological distress including ongoing depression and anxiety were associated with increased cancer mortality with the greatest effect seen in lung cancer patients (Hamer, Chida, and Molloy 2009). However, the association between chronic stress and cancer development and progression remains controversial, as some studies have failed to observe links between stress and cancer risk (Duijts, Zeegers, and Borne 2003; Bleiker et al. 2008). The notion that chronic stress hormones promote tumor progression is supported by numerous pre-clinical studies. As outlined in further detail below, in animal models of carcinogen-induced lung cancer, chronic stress potentiated the effect of carcinogens on lung tumor development, and in genetically engineered mouse models of KRAS mutant NSCLC, chronic stress increased tumor burden (Jang et al. 2016). Likewise, in multiple xenograft and syngeneic tumor models of lung cancer, chronic stress has been shown to increase tumor growth rate (Wu et al. 2015; Nilsson et al. 2017).
Figure 1.
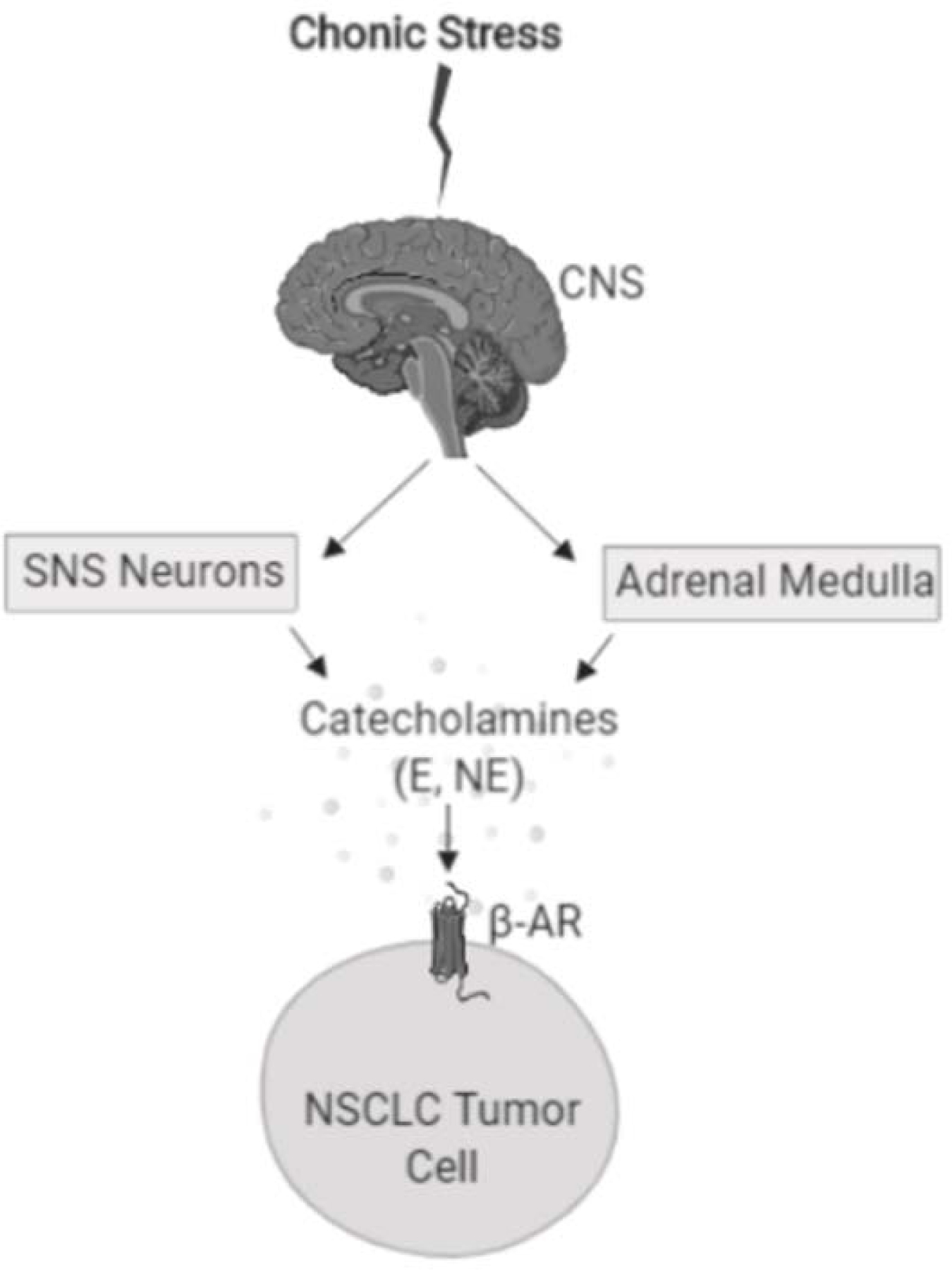
Chronic stress induces the release of catecholamines including epinephrine (E) and norepinephrine (NE) from the sympathetic nervous system (SNS) and adrenal medulla. Epinephrine and norepinephrine can activate β-adrenergic receptors (β-AR) expressed on NSCLC tumor cells.
β-adrenergic receptor signaling in physiology and disease
The biological effects of stress hormones are mediated by adrenergic receptors. Adrenergic receptors are G protein-coupled receptors (GPCRs) that are divided into α and β subtypes based on structure and the signaling pathways they activate. The β-adrenergic receptor (β-AR) pathway regulates the sympathetic nervous system-driven fight-or-flight stress responses (Weiner 1992; Sapolsky 1993). β-ARs include 3 subtypes, β1, β2, and β3, which are expressed on numerous tissues including brain, lung, liver, breast, ovary, and lymphoid tissue. Binding of epinephrine or norepinephrine to β-ARs results in activation of Gαs guanine nucleotide-binding protein, which stimulates adenylyl cyclase synthesis of cyclic AMP (cAMP). The subsequent rise in cAMP activates protein kinase A (PKA). In addition, following β-AR activation adenylyl cyclase can activate the guanine nucleotide exchange protein EPAC (de Rooij et al. 1998), which triggers activation of additional signal transduction pathways including the MAPK/ERK pathway (Figure 2). Collectively, these pathways regulate cell survival, motility, and proliferation and can result in activation of transcription factors including NF-kB and the cAMP-responsive element binding protein (CREB) family. These transcriptional regulators modulate the expression of numerous genes including interleukin-6 (IL-6), vascular endothelial growth factor (VEGF), interleukin-8 (IL-8), and matrix metalloproteinases, which promote angiogenesis, cellular invasion, and inflammation (Montminy 1997; Thaker et al. 2006; Nilsson et al. 2007; Cole and Sood 2012).
Figure 2.
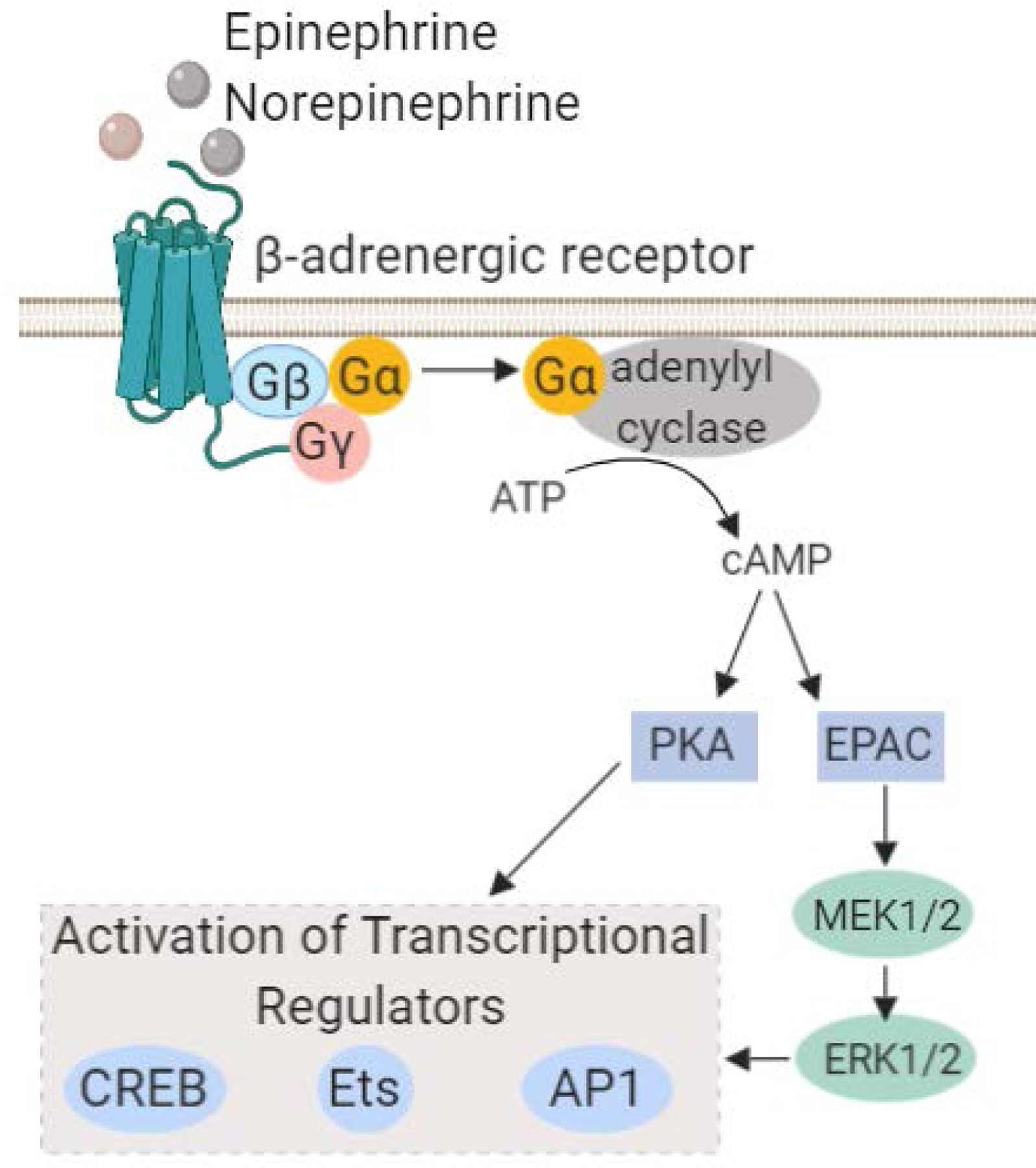
The β-adrenergic signaling pathway. Catacolamines, epinephrine and norepinephrine, act on b-adrenergic receptors expressed on normal tissue or tumor cells, which mediates activation of adenlylyl cyclase. This results in a transient flux in intracellular cAMP resulting in activation of the PKA and MAPK pathways. As a result, transcriptional regulators including CREB, AP-1 and Ets family transcription factors become active which in turn can modulate the expression of numerous genes involved in inflammation and angiogenesis.
In addition to regulating physiological processes, β-ARs are thought to contribute to malignant disease as β1, β2, and β3-ARs are present at sites of tumor growth and metastasis including the brain, lung, liver, breast, and vasculature (Daly and McGrath 2011). Moreover, β-ARs are detected directly on cancer cells. We and others have observed that β-ARs are indeed expressed on NSCLC cell lines in vitro as well in clinical specimens of NSCLC (Nilsson et al. 2017; Kondratenko et al. 1993). These studies indicate that tumors may usurp the physiological signaling pathways of adrenergic receptors to facilitate tumor progression and metastasis. β-ARs have a recognized role in cardiovascular function, and beta-blockers are widely used for the treatment of cardiovascular disease. Given that beta-blockers are generally well-tolerated and that β-ARs activate multiple cellular programs involved in tumor progression, these agents in combination with other treatment modalities could be an inexpensive approach to potentially improve clinical outcomes.
β-AR expression as a prognostic marker in NSCLC
β-ARs are detected directly on tumor cells in numerous malignancies including cancers of the breast, prostate, and skin and are associated with tumor recurrence, metastasis and poor clinical outcome (Chen et al. 2012; Ramberg et al. 2008; Shang, Liu, and Liang 2009; Shi et al. 2011; Yang et al. 2009). In a bioinformatics analysis of lung adenocarcinoma datasets, ADRB2, the gene that encodes β2-AR, was found to be overexpressed in lung cancers (Wu et al. 2012). A study by Yazawa et al. investigated the prognostic significance of β-AR expression in lung cancer and evaluated β2-AR expression on 328 primary NSCLC tumors. Membranous and cytoplasmic β2-AR expression was detected on 27% of all tumors analyzed, with 29% of adenocarcinomas and 24% of non-adenocarcinomas showing positive expression of β2-AR (Yazawa et al. 2016). β2-AR expression was significantly correlated with tumor vascularization, lymphatic permeation, and cell proliferation as determined by Ki-67 positivity. Moreover, β2-AR expression was associated with metastasis and a significantly worse overall survival, and in a multivariate analysis, β2-AR expression was an independent prognostic marker for worse progression free survival (PFS) in lung adenocarcinoma patients with stage I disease. However, the relationship between β2-AR expression and progression and metastasis was not observed in non-adenocarcinoma patients in this study.
A separate study similarly evaluated β2-AR expression in pulmonary pleomorphic carcicinoma (PPC) (Kaira et al. 2019), a rare and aggressive form of lung cancer. Here, the investigators detected positive β2-AR expression on 63% of tumors and observed a statistically significant correlation between β2-AR expression and tumor cell proliferation. High levels of β2-AR expression was further associated with lymph node metastasis in patients with early stage disease, and β2-AR was a significant independent predictive marker of worse prognosis for patients with PPC (Kaira et al. 2019).
Interactions between the effects of tobacco carcinogens and chronic stress in NCLC
Smoking is the leading cause of lung cancer and primarily exerts its deleterious effects through induction of DNA damage. However, tobacco carcinogens can also directly impact adrenergic receptor signaling, as nicotine and nicotine derivatives have a high affinity for β-ARs. In an acidic environment, nicotine forms 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). NNK is structurally similar to the catecholamines, norephinephrine and epinephrine, and has been shown to act as an agonist for β-ARs with an affinity 600 fold higher than norephinephrine and 2,200 fold higher than epinephrine (Schuller et al. 1999). Moreover, smoking likely has indirect effects on β-AR signaling as well. Studies using small airway epithelial cells have shown that stimulation with nicotine or NNK induces the synthesis and release of norephinephrine and epinephrine, and this in turn can drive tumor cell proliferation and migration, which was inhibited with the addition of the beta-blocker propranolol (Al-Wadei et al. 2010). Similar findings were reported using other cell types including pancreatic cancer (Al-Wadei, Al-Wadei, and Schuller 2012b), colon cancer (Wong et al. 2007), and gastric cancer cell lines (Shin et al. 2007).
Pre-clinical evidence supports a role for β-AR signaling in NSCLC progression
Numerous pre-clinical studies have demonstrated a role for β-AR signaling in driving lung cancer progression. β-ARs are detected on NSCLC cell lines and clinical specimens (Nilsson et al. 2017; Kondratenko et al. 1993). In a study investigating lung adenocarcinomas induced by NNK, researchers observed expression of β2-ARs but not β1-ARs on tumor cells (Schuller and Cekanova 2005). As detailed below, studies indicate that activation of β-ARs contributes to cancer development, tumor cell proliferation, the acquisition of an invasive phenotype and enhanced tumor growth and therapeutic resistance.
Treatment of normal bronchial epithelial cells with norepinephrine resulted in increased activation of receptor tyrosine kinases including IGF-1R and its downstream signaling cascade and resulted in cellular transformation as indicated by anchorage-independent colony formation (Jang et al. 2016), and this effect was blocked by the addition of the beta-blockers propranolol or atenolol. In addition to activating mitogenic signal transduction pathways, β-ARs may promote tumor development by inducing DNA damage. This is described in an elegant study by Hara et al. where β2-ARs acting through PKA and β-arrestin triggered DNA damage and suppressed expression of the tumor suppressor p53 (Hara et al. 2011). Other investigators have shown that in vitro stimulation of lung cancer cells with epinephrine enhanced cellular proliferation (Park et al. 1995). Lung tumor cell proliferation was enhance by the β-AR agonist, isoproterenol. The pan-beta-blocker, propranolol, or inhibition of adenylyl cyclase or cAMP accumulation blocked the pro-mitotic effects of epinephrine or isoproterenol, whereas the α-adrenergic receptor inhibitor, tolazoline, did not (Park et al. 1995).
Additional studies indicate that stress hormones can also induce an epithelial-to-mesenchymal transition (EMT) in NSCLC tumor cells. EMT is a process in which epithelial tumor cells shift to a mesenchymal phenotype characterized by loss of E-cadherin and increased expression of N-cadherin and vimentin. EMT is an important step in tumor invasiveness and metastasis (Thiery et al. 2009). Treatment of NSCLC cell lines HT-29 and A549 with norepinephrine resulted in morphologic and proteomic changes consistent with having undergone EMT (Zhang et al. 2016). Norepinephrine stimulation increased in vitro cellular migration, indicating a more invasive phenotype, and this effect was diminished by the addition of propanolol (Zhang et al. 2016). Moreover, norepinephrine treatment increased tumor cell expression of transforming growth factor-β(TGF-β), a key driver of EMT, and this too was inhibited by beta blockade. In our own studies using murine NSCLC cell lines, treatment of 393P cells with norepinephrine significantly increased tumor cell migration in a wound healing assay, and the effect of norepinephrine stimulation was blocked by propranolol (data not shown). Consistent with these findings, norepinephrine has been shown to promote an EMT in models of ovarian cancer, prostate cancer, and gastric adenocarcinoma (Choi et al. 2015; Barbieri et al. 2015; Shan et al. 2014).
Another pre-clinical study evaluated the interaction between adrenergic signaling and lung cancer stem cell properties (Banerjee, Papu John, and Schuller 2015). Although cancer stem cells are only a fraction of the cells within tumors and cell lines, these cells have the ability to self-renew and differentiate. Moreover, cancer stem cells play an important role in tumor progression and therapeutic resistance (Eramo et al. 2008; Hassan et al. 2013; Shao et al. 2014; Zhang et al. 2012; Singh and Chellappan 2014). In vitro, epinephrine treatment significantly increased the fraction of cancer stem cells from NSCLC cell lines. In mice, stress reduction was associated with diminished expression of cancer stem cell markers and decreased tumor growth (Banerjee, Papu John, and Schuller 2015).
Stress hormones have also been shown to impact the in vivo growth of lung cancer. In carcinogen-induced models and KRAS mutant genetically engineered mouse models of lung cancer, chronic stress enhanced tumor development and increased tumor burden (Jang et al. 2016). Similar findings demonstrating that chronic stress accelerates the growth of lung tumor models have been generated using the lewis lung cancer (LLC) syngeneic tumor model and EGFR mutant NSCLC xenografts (Nilsson et al. 2017; Wu et al. 2015). In a study by Al-Wadei et al., NSCLC tumor-bearing animals were exposed to control conditions or social stress, which was induced by changing the cage-mates of the animals twice per week (Al-Wadei et al. 2012). Stress-exposed mice had significantly higher serum levels of epinephrine, norepinephrine, and cortisol. Catecholamine levels were elevated within the tumors of stress animals as well. Consistent with this, stressed animals exhibited higher intracellular cAMP levels and increased phosphorylation of MAPK/ERK and CREB indicating that the increased catecholamines observed in stressed animals were inducing adrenergic receptor signaling in tumor cells. Using the H322 NSCLC xenograft model, the investigators demonstrated that social stress significantly accelerated tumor growth. Treatment of mice with γ-aminobutyric acid (GABA), a neurotransmitter that can block this pathway, inhibited the effect of chronic stress on tumors (Al-Wadei et al. 2012).
β-AR signaling likely accelerates tumor growth through both direct tumor cell effects (i.e. as a tumor cell mitogen) and indirect effects on stromal cells including microvascular endothelial cells within the tumor microenvironment. Catecholamines have been shown to upregulate tumor cell expression of angiogenic factors (Lutgendorf et al. 2003; Yang et al. 2006; Yang et al. 2008). In ovarian cancer models, chronic stress significantly increased expression of angiogenic factors including VEGF and enhanced tumor vascularization through β2-AR signaling, and this effect was blocked with the addition of the beta-blocker, propranolol (Thaker et al. 2006). In prostate cancer xenograft models, β-AR signaling similarly enhanced tumor angiogenesis (Hulsurkar et al. 2017). In animal models of lung cancer, chronic stress increased tumor cell expression of angiogenic factors including VEGF, MMP-2, and MMP-9 and enhanced intratumoral vascular density (Wu et al. 2015). Detailed mechanistic studies of endothelial cells have revealed that stimulation of β2-ARs on endothelial cells increases VEGF-induced sprouting and enhanced VEGFR-2 expression, priming the endothelium for angiogenesis (Garg et al. 2017). Additional reports indicate that catecholamines can act on adrenergic receptors on tumor-associated immune cells such as macrophages triggering VEGF production and tumor angiogenesis. β-AR blockade with propranolol inhibited the catecholamine-induced signaling between macrophages and endothelial cells (Xia et al. 2019).
In addition to the paracrine effects of catecholamines on lung cancer cells, some studies have indicated that lung tumor cells have the capacity to produce neurotransmitters for autocrine signaling. A subset of NSCLC tumors and cell lines were found to be positive for expression of dopa-decarboxylase, the enzyme which mediates the conversion of L-Dopa to dopamine which subsequently is used to generate norepinephrine (Gazdar et al. 1988). Moreover, treatment of NSCLC cell lines with nicotine has been shown to induce tumor cell production of norephinephrine (Al-Wadei, Al-Wadei, and Schuller 2012a).
β-AR signaling as a driver of therapeutic resistance in NSCLC
Concurrent chemoradiation is a standard treatment for NSCLC patients with locally advanced disease. Preclinical studies addressing whether adrenergic activators impact resistance to chemotherapy or radiotherapy have been conducted. Using NSCLC cell lines, the addition of the beta-blocker propranolol was found to potentiate the effects of radiation and cisplatin in an in vitro clonogenic assay (Chaudhary et al. 2019). Tumor cells treated with propranolol in combination with radiation had elevated levels of phosphorylated γH2AX, a marker of double-stranded DNA breaks, compared to cells treated with radiation alone, suggesting that β-AR blockade may impair DNA double-stranded break repair (Chaudhary et al. 2019). Two retrospective analyses of NSCLC patients support these pre-clinical findings and provide a clinical association between beta-blocker use and improved clinical outcome (Chaudhary et al. 2019; Wang et al. 2013).
In addition to potentially promoting chemotherapeutic resistance, β-AR signaling can also drive resistance to targeted agents. About 15% of Caucasian patients and up to 50% of Asian patients with lung adenocarcinoma harbor mutations in the epidermal growth factor receptor (EGFR) gene. EGFR-mutant NSCLC represents a molecularly-defined subset of lung cancer. Because the tumor cells are dependent on the EGFR pathway, tumors harboring EGFR activating mutations are initially sensitive to small molecule tyrosine kinase inhibitors, such as erlotinib and osimertinib. However, tumor cells eventually escape the inhibition by developing resistance (Engelman and Janne 2008). Resistance to EGFR TKIs can be mediated by secondary EGFR mutations (i.e. T790M) (Kobayashi et al. 2005; Pao et al. 2005) or through EGFR-independent mechanisms. Interleukin-6 (IL-6) is one driver of EGFR-independent resistance (Yao et al. 2010). We have demonstrated that activation of β2-ARs but not β1-ARs on the surface of EGFR mutant NSCLC cells results in dramatic increases in IL-6 transcription and secretion and this effect can be blocked by the beta-blocker propranolol (Nilsson et al. 2017). Specifically, β2-AR signaling on EGFR mutant tumor cells activates adenylyl cyclase resulting in increased levels of cAMP and activation of protein kinase C (PKC). This subsequently enhances activity of the transcription factor CREB, which elaborates IL-6 expression. Moreover, stimulation of EGFR mutant NSCLC cell lines with β2-AR activators induces resistance to EGFR TKIs both in vitro and in vivo, and this can be blocked with the addition of beta-blockers (propranolol) or IL-6 blocking-antibodies (siltuximab) (Figure 3). Clinical evidence supports these preclinical findings, as incidental use of beta-blockers in NSCLC patients was associated with significantly reduced circulating levels of IL-6. Analysis of incidental beta-blocker use in the randomized phase III LUX-Lung3 study comparing afatinib versus chemotherapy in EGFR mutant NSCLC patients (Sequist et al. 2013) revealed that in patient taking beta-blockers, afatinib was associated with a greater relative PFS benefit for afatinib. In the beta-blocker use group the median PFS was 13.6 and 2.5 months for afatinib and chemotherapy, respectively. Among patients not receiving beta-blockers the median PFS time was 11.1 and 6.9 months for afatinib and chemotherapy, respectively. Although this analysis is limited by the modest number of patients receiving beta-blockers, it agrees with the preclinical findings that β-AR blockade could delay therapeutic resistance to EGFR TKIs and supports the need for future clinical testing of beta-blockers in combination with EGFR TKIs in EGFR-mutant NSCLC patients.
Figure 3.
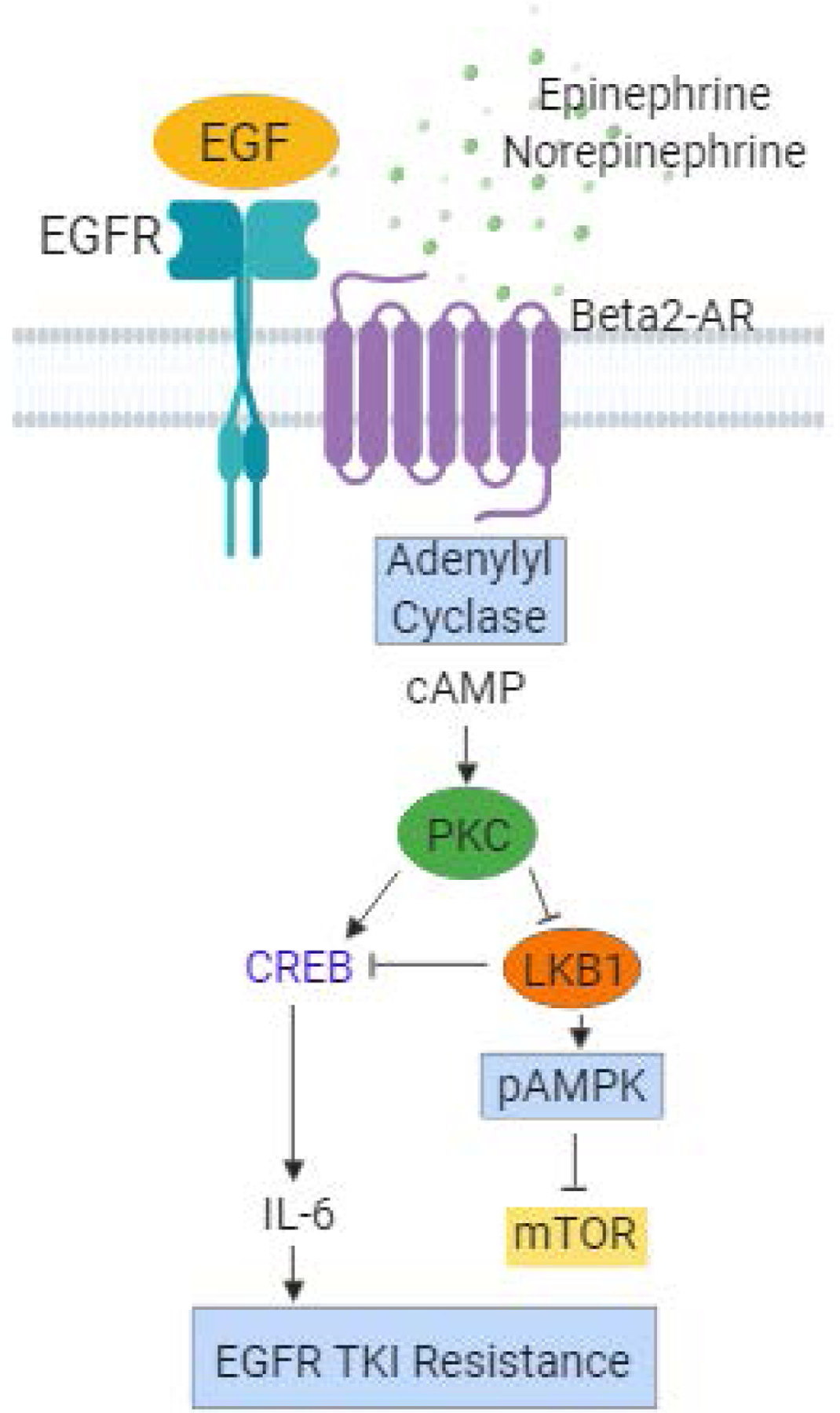
In NSCLC cells with EGFR activating mutations, activation of b2-ARs promotes resistance to EGFR tyrosine kinase inhibitors (TKIs) though an IL-6-dependent mechanism. Chronic stress hormones activate B2-ARs on lung cancer cells triggering activation of adenylyl cyclase and a rise in intracellular cAMP. This in turn activates protein kinase C (PKC), which phosphorylates CREB leading to increased transcription of CREB target genes including IL-6. PKC also phosphorylates the tumor suppressor LKB1 at the S428 inhibitory site, which results in increased mTOR signaling.
The finding that β-AR signaling promotes resistance to EGFR targeted therapies implies that stress hormones may similarly drive resistance to other targeted agents. This notion is supported by studies in other disease settings. Specifically, β2-AR signaling has been shown to drive resistance to the HER2 targeting antibody, trastuzumab, in breast cancer models through re-activation of the PI3K/Akt/mTOR signaling pathways (Liu et al. 2016). High expression of β2-AR was negatively associated with trastuzumab response in patients with HER2 overexpressing breast cancer (Liu et al. 2016).
β-AR signaling and the tumor immune microenvironment
In addition to being expressed on normal tissue and tumor cells, adrenergic receptors are also present on immune cells. Therefore, in addition to indirect effects of catecholamines on immune cells through the release of tumor-associated cytokines, stress hormones directly impact immune cell populations. Although acute stress prompted by an injury for example activates the immune response, chronic stress is typically immunosuppressive (Dhabhar 2014). Numerous studies have documented the expression of both α- and β-ARs on innate immune cells including neutrophils, monocytes, NK cells, macrophages, and mature dendritic cells (Qiao et al. 2018), β2-ARs are the most highly expressed adrenergic receptor subtype on immune cells. Moreover, T and B cells express β2-ARs but not other adrenergic receptors (Cremaschi, Fisher, and Boege 1991; Kohm and Sanders 1999; Van Tits et al. 1990). Thus, the effects of stress hormones on immune function are believed to be primarily mediated by β2-ARs. Moreover, catecholamines also act on hematopoietic stem cells or progenitor cells in the bone marrow, thus increasing myelopoiesis and the production of inflammatory cells (Maestroni 2019). These expanded populations of inflammatory cells could, in turn, modulate the tumor microenvironment and promote tumor growth (Granot 2019).
Catecholamines act on innate immune cells resulting in pro-tumor effects. Adrenergic signaling in macrophages promotes differentiation toward an M2 phenotype, potentially driving an angiogenic and metastatic tumor phenotype (Qin et al. 2015; Sloan et al. 2010). Catecholamines reduce antigen presentation of other innate cells including dendritic cells, and this effect is blocked by β-AR blockade (Seiffert et al. 2002; Herve et al. 2013). Whole animal studies using rats demonstrated that β-AR activation suppresses natural killer (NK) cell activity for prolonged periods (Shakhar and Ben-Eliyahu 1998), compromising resistance to metastasis, an effect that was reversed with the addition of a beta-blocker (Shakhar and Ben-Eliyahu 1998; Ben-Eliyahu et al. 2000). Another pre-clinical study evaluated lung micrometastasis and NK cell activity in a breast cancer model using surgical stress. Surgery increased lung tumor cell retention and decreased NK cell cytotoxicity, and the beta-blocker propranolol attenuated the effect (Benish et al. 2008). Additional in vivo studies using the LLC model of lung cancer have similarly show that surgical stress reduced NK cell function and propranolol counteracted the changes (Glasner et al. 2010).
Adrenergic signaling also affects the adaptive immune system and can directly inhibit T-cell activation, differentiation and function. In animal models, chronic adrenergic signaling suppressed effector CD8+ T cells within the tumor-microenvironment. Moreover, beta-blocker treatment converted tumors to an immunologically active phenotype (Bucsek et al. 2017). This is consistent with other preclinical studies showing increased intra-tumoral cytotoxic lymphocytes and a shift toward anti-tumor immunity following beta-blocker treatment (Jean Wrobel et al. 2016). Given the effects of catecholamines on antigen presentation and T-cell function, it is feasible the stress hormones contribute to tumor cell immune escape, and beta-blockers could potentially enhance the efficacy of immunotherapy regimens in NSCLC patients. Additional studies investigating the effects of β-AR signaling on anti-tumoral immunity in NSCLC are warranted.
Retrospective analyses of beta-blocker use in NSCLC patients and clinical outcome
A population-based cohort study of 6,771individuals by Lin et al. demonstrated that long-term use of the pan beta-blocker carvedilol was associated with a reduced risk of lung cancer and upper gastrointestinal tract cancer risk, suggesting a potential role for β-AR blockade in cancer prevention (Lin et al. 2015). Likewise, incidental use of propranolol has been shown to be associated with reduced risk of multiple cancer types in a separate population-based cohort study, which included 24,238 individuals (Chang et al. 2015), and beta-blocker use has been shown to be associated with a reduced cancer recurrence and cancer-related mortality in patients with breast cancer (Powe et al. 2010; Melhem-Bertrandt et al. 2011; Ganz et al. 2011). As with other malignancies, the results from retrospective studies assessing the impact of beta-blocker use on clinical outcome of NSCLC patients have been mixed, and conflicting findings have been reported. Multiple population-based cohort studies have observed no association between beta-blocker use and reduced mortality among lung cancer patients (Weberpals et al. 2017; Shah et al. 2011; Yang et al. 2017; Musselman et al. 2018). One such retrospective cohort study of 435 lung cancer patients indicated no correlation between beta-blocker use and overall survival in patients undergoing lung cancer resection (Cata et al. 2014).
In contrast, other retrospective studies have indicated that beta-blocker use is indeed associated with an improved clinical outcome in lung cancer patients (Table 1). For example, beta-blocker use during chemotherapy treatment may improve overall survival in patients with metastatic NSCLC (Aydiner et al. 2013). In this retrospective univariate analysis of 107 NSCLC patients with metastatic disease, the investigators found a significantly improved overall survival (OS) in patients receiving beta-blockers compared to patients not receiving beta-blockers (Aydiner et al. 2013), and beta-blocker use provided a six-month survival benefit (Aydiner et al. 2013). Wang et al. conducted a separate retrospective analysis of 722 NSCLC patients treated with definitive radiotherapy with or without chemotherapy (Wang et al. 2013). Of the 722 patients included in the analysis, 155 had received beta-blockers during radiotherapy. In both univariate analysis and multivariate analyses, which adjusted for stage, histology, performance status and treatment regimen, incidental beta-blocker use was associated with a significantly improved distant metastasis-free survival, disease-free survival, and overall survival. However, beta-blocker use did not affect locoregional progression, suggesting that beta-blocker use may have a greater impact on metastatic programs than primary tumor growth.
Table 1.
Population-based and retrospective studies indicating that beta-blocker use is associated with improved clinical outcome in lung cancer patients.
| Reference | Number of Patients | Cancer Type | Therapeutic Effect |
|---|---|---|---|
| Lin et al. 2015 | 6,771 | Lung and other cancers | Long-term treatment with the pan-beta-blocker carvedilol is associated with reduced lung cancer risk |
| Wang et al. 2013 | 722 | NSCLC | In NSLC patients receiving definitive radiotherapy, incidental beta-blocker use was associated with improved DMFS, DFS, and OS. |
| Aydiner et al. 2013 | 107 | NSCLC | In NSCLC patients receiving chemotherapy, beta-blocker use was associated with an improved OS in a univariate analysis |
| Chaudhary et al. 2019 | 77 | NSCLC | Beta blocker use was associated with decreased distant metastasis in patients with Stage IIIA NSCLC |
DMFS = distant metastasis free survival; DFS = disease free survival; OS = overall survival
A separate retrospective analysis of NSCLC patients receiving neoadjuvant chemoradiation and surgery was also conducted by Chaudhary et al. Although this study was limited in patient size (n=77; 16 patients receiving beta-blockers; 61 patients not receiving beta-blockers), beta-blocker use was associated with decreased distant metastasis (Chaudhary et al. 2019). Moreover, the investigators observed trends associated with beta-blocker use and improved response by CT imaging, improved overall survival at 1 year.
Conclusions
A growing body of preclinical evidence strongly supports a role for β-AR signaling on lung cancer cells in facilitating multiple aspects of tumor progression including tumor cell proliferation, the acquisition of a mesenchymal and invasive phenotype, angiogenesis, immune evasion, and resistance to chemotherapy, radiation and targeted agents. These findings have important clinical implications as beta-blockers are well-tolerated, inexpensive, and clinically available. While some retrospective clinical analyses have indeed shown an association between incidental beta-blocker use and improved clinical outcome in lung cancer patients, results from other studies have failed to observe these associations. Several factors may contribute to these discrepancies. Preclinical studies indicate that the direct effects of stress hormones on tumor cells are predominantly mediated by β2-ARs. Thus, the use of cardio-selective versus nonselective beta-blockers likely have differential effects on tumor progression, with beta-blockers having activity against β2-ARs predicted to be the most effective. Moreover, patients receiving beta-blockers may have confounding health conditions that may influence clinical outcomes. Given the potential benefits of beta-blockers in cancer patients as well as the favorable safety profile of these agents, the current evidence supports the future direct clinical testing of beta-blockers in combination with other therapeutic agents.
Funding:
This work was funded by Lung SPORE grant 5 P50 CA070907, Lung Cancer Moon Shot Program, NIH CCSG (CA016672), and the Hanlon Fund.
Footnotes
Conflicts of Interest/Disclosures: MN and JVH have filed a patent for the use of poziotinib for treating EGFR and HER2 mutant cancers, and licensed the technology to Spectrum Pharmaceuticals. JVH also has/had the following disclosures: grant or research support from AstraZeneca, Bayer, and GlaxoSmithKline and has served on advisory committees for AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Exelixis, Hengrui Therapeutics, Genentech, GSK, Guardant Health, Lilly, Novartis, Spectrum, Takeda, and Synta. XL is a consultant for Eli Lilly and AztraZeneca.
Ethical approval: No animals were utilized in this manuscript.
Figures: Created with BioRender.com
References
- Al-Wadei HA, Al-Wadei MH, Masi T, and Schuller HM. 2010. ‘Chronic exposure to estrogen and the tobacco carcinogen NNK cooperatively modulates nicotinic receptors in small airway epithelial cells’, Lung Cancer, 69: 33–9. [DOI] [PubMed] [Google Scholar]
- Al-Wadei HA, Al-Wadei MH, and Schuller HM. 2012a. ‘Cooperative regulation of non-small cell lung carcinoma by nicotinic and beta-adrenergic receptors: a novel target for intervention’, PLoS One, 7: e29915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wadei HA, Plummer HK 3rd, Ullah MF, Unger B, Brody JR, and Schuller HM. 2012. ‘Social stress promotes and gamma-aminobutyric acid inhibits tumor growth in mouse models of non-small cell lung cancer’, Cancer Prev Res (Phila), 5: 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wadei MH, Al-Wadei HA, and Schuller HM. 2012b. ‘Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors alpha3, alpha5, and alpha7’, Mol Cancer Res, 10: 239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydiner A, Ciftci R, Karabulut S, and Kilic L. 2013. ‘Does beta-blocker therapy improve the survival of patients with metastatic non-small cell lung cancer?’, Asian Pac J Cancer Prev, 14: 6109–14. [DOI] [PubMed] [Google Scholar]
- Banerjee J, Papu John AM, and Schuller HM. 2015. ‘Regulation of nonsmall-cell lung cancer stem cell like cells by neurotransmitters and opioid peptides’, Int J Cancer, 137: 2815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri A, Bimonte S, Palma G, Luciano A, Rea D, Giudice A, Scognamiglio G, La Mantia E, Franco R, Perdona S, De Cobelli O, Ferro M, Zappavigna S, Stiuso P, Caraglia M, and Arra C. 2015. ‘The stress hormone norepinephrine increases migration of prostate cancer cells in vitro and in vivo’, Int J Oncol, 47: 527–34. [DOI] [PubMed] [Google Scholar]
- Ben-Eliyahu S, Shakhar G, Page GG, Stefanski V, and Shakhar K. 2000. ‘Suppression of NK cell activity and of resistance to metastasis by stress: a role for adrenal catecholamines and beta-adrenoceptors’, Neuroimmunomodulation, 8: 154–64. [DOI] [PubMed] [Google Scholar]
- Benish M, Bartal I, Goldfarb Y, Levi B, Avraham R, Raz A, and Ben-Eliyahu S. 2008. ‘Perioperative use of beta-blockers and COX-2 inhibitors may improve immune competence and reduce the risk of tumor metastasis’, Ann Surg Oncol, 15: 2042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleiker EM, Hendriks JH, Otten JD, Verbeek AL, and van der Ploeg HM. 2008. ‘Personality factors and breast cancer risk: a 13-year follow-up’, J Natl Cancer Inst, 100: 213–8. [DOI] [PubMed] [Google Scholar]
- Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, Liu H, Kokolus KM, Eng JW, Messmer MN, Attwood K, Abrams SI, Hylander BL, and Repasky EA. 2017. ‘beta-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8(+) T Cells and Undermines Checkpoint Inhibitor Therapy’, Cancer Res, 77: 5639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cata JP, Villarreal J, Keerty D, Thakar DR, Liu DD, Sood AK, and Gottumukkala V. 2014. ‘Perioperative beta-blocker use and survival in lung cancer patients’, J Clin Anesth, 26: 106–17. [DOI] [PubMed] [Google Scholar]
- Chang PY, Huang WY, Lin CL, Huang TC, Wu YY, Chen JH, and Kao CH. 2015. ‘Propranolol Reduces Cancer Risk: A Population-Based Cohort Study’, Medicine (Baltimore), 94: e1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary KR, Yan SX, Heilbroner SP, Sonett JR, Stoopler MB, Shu C, Halmos B, Wang TJC, Hei TK, and Cheng SK. 2019. ‘Effects of beta-Adrenergic Antagonists on Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer’, J Clin Med, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xing W, Hong J, Wang M, Huang Y, Zhu C, Yuan Y, and Zeng W. 2012. ‘The beta2-adrenergic receptor is a potential prognostic biomarker for human hepatocellular carcinoma after curative resection’, Ann Surg Oncol, 19: 3556–65. [DOI] [PubMed] [Google Scholar]
- Choi MJ, Cho KH, Lee S, Bae YJ, Jeong KJ, Rha SY, Choi EJ, Park JH, Kim JM, Lee JS, Mills GB, and Lee HY. 2015. ‘hTERT mediates norepinephrine-induced Slug expression and ovarian cancer aggressiveness’, Oncogene, 34: 3402–12. [DOI] [PubMed] [Google Scholar]
- Cole SW, and Sood AK. 2012. ‘Molecular pathways: beta-adrenergic signaling in cancer’, Clin Cancer Res, 18: 1201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremaschi GA, Fisher P, and Boege F. 1991. ‘Beta-adrenoceptor distribution in murine lymphoid cell lines’, Immunopharmacology, 22: 195–206. [DOI] [PubMed] [Google Scholar]
- Daly CJ, and McGrath JC. 2011. ‘Previously unsuspected widespread cellular and tissue distribution of beta-adrenoceptors and its relevance to drug action’, Trends Pharmacol Sci, 32: 219–26. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, and Bos JL. 1998. ‘Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP’, Nature, 396: 474–7. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS 2014. ‘Effects of stress on immune function: the good, the bad, and the beautiful’, Immunol Res, 58: 193–210. [DOI] [PubMed] [Google Scholar]
- Duijts SF, Zeegers MP, and Borne BV. 2003. ‘The association between stressful life events and breast cancer risk: a meta-analysis’, Int J Cancer, 107: 1023–9. [DOI] [PubMed] [Google Scholar]
- Engelman JA, and Janne PA. 2008. ‘Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer’, Clin Cancer Res, 14: 2895–9. [DOI] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, and De Maria R. 2008. ‘Identification and expansion of the tumorigenic lung cancer stem cell population’, Cell Death Differ, 15: 504–14. [DOI] [PubMed] [Google Scholar]
- Ganz PA, Habel LA, Weltzien EK, Caan BJ, and Cole SW. 2011. ‘Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: results from the LACE cohort’, Breast Cancer Res Treat, 129: 549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg J, Feng YX, Jansen SR, Friedrich J, Lezoualc’h F, Schmidt M, and Wieland T. 2017. ‘Catecholamines facilitate VEGF-dependent angiogenesis via beta2-adrenoceptor-induced Epac1 and PKA activation’, Oncotarget, 8: 44732–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Helman LJ, Israel MA, Russell EK, Linnoila RI, Mulshine JL, Schuller HM, and Park JG. 1988. ‘Expression of neuroendocrine cell markers L-dopa decarboxylase, chromogranin A, and dense core granules in human tumors of endocrine and nonendocrine origin’, Cancer Res, 48: 4078–82. [PubMed] [Google Scholar]
- Glasner A, Avraham R, Rosenne E, Benish M, Zmora O, Shemer S, Meiboom H, and Ben-Eliyahu S. 2010. ‘Improving survival rates in two models of spontaneous postoperative metastasis in mice by combined administration of a beta-adrenergic antagonist and a cyclooxygenase-2 inhibitor’, J Immunol, 184: 2449–57. [DOI] [PubMed] [Google Scholar]
- Granot Z 2019. ‘Neutrophils as a Therapeutic Target in Cancer’, Front Immunol, 10: 1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer M, Chida Y, and Molloy GJ. 2009. ‘Psychological distress and cancer mortality’, J Psychosom Res, 66: 255–8. [DOI] [PubMed] [Google Scholar]
- Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, Shenoy SK, Gregory SG, Ahn S, Duckett DR, and Lefkowitz RJ. 2011. ‘A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1’, Nature, 477: 349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, and Wicha MS. 2013. ‘Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma’, Clin Cancer Res, 19: 1972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herve J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, Rozec B, Gauthier C, Bach JM, and Blancou P. 2013. ‘beta2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells’, J Immunol, 190: 3163–71. [DOI] [PubMed] [Google Scholar]
- Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D, and Li W. 2017. ‘Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1’, Oncogene, 36: 1525–36. [DOI] [PubMed] [Google Scholar]
- Jang HJ, Boo HJ, Lee HJ, Min HY, and Lee HY. 2016. ‘Chronic Stress Facilitates Lung Tumorigenesis by Promoting Exocytosis of IGF2 in Lung Epithelial Cells’, Cancer Res, 76: 6607–19. [DOI] [PubMed] [Google Scholar]
- Jean Wrobel L, Bod L, Lengagne R, Kato M, Prevost-Blondel A, and Le Gal FA. 2016. ‘Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma’, Oncotarget, 7: 77825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaira K, Kamiyoshihara M, Kawashima O, Endoh H, Imaizumi K, Sugano M, Tanaka S, Fujita A, Kogure Y, Shimizu A, Oyama T, Asao T, Shimizu K, and Mogi A. 2019. ‘Prognostic Impact of beta2 Adrenergic Receptor Expression in Surgically Resected Pulmonary Pleomorphic Carcinoma’, Anticancer Res, 39: 395–403. [DOI] [PubMed] [Google Scholar]
- Kemp VH, and Hatmaker DD. 1989. ‘Stress and social support in high-risk pregnancy’, Res Nurs Health, 12: 331–6. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, and Halmos B. 2005. ‘EGFR mutation and resistance of non-small-cell lung cancer to gefitinib’, N Engl J Med, 352: 786–92. [DOI] [PubMed] [Google Scholar]
- Kohm AP, and Sanders VM. 1999. ‘Suppression of antigen-specific Th2 cell-dependent IgM and IgG1 production following norepinephrine depletion in vivo’, J Immunol, 162: 5299–308. [PubMed] [Google Scholar]
- Kondratenko TY, Zacharova IV, Kuzina NV, Katukov VYu, Severin ES, Kornilova ZCh, and Perelman MI. 1993. ‘Alterations in human lung adrenergic receptors in cancer’, Biochem Mol Biol Int, 29: 123–30. [PubMed] [Google Scholar]
- Lin CS, Lin WS, Lin CL, and Kao CH. 2015. ‘Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study’, Int J Cardiol, 184: 9–13. [DOI] [PubMed] [Google Scholar]
- Liu D, Yang Z, Wang T, Yang Z, Chen H, Hu Y, Hu C, Guo L, Deng Q, Liu Y, Yu M, Shi M, Du N, and Guo N. 2016. ‘beta2-AR signaling controls trastuzumab resistance-dependent pathway’, Oncogene, 35: 47–58. [DOI] [PubMed] [Google Scholar]
- Lutgendorf SK, Cole S, Costanzo E, Bradley S, Coffin J, Jabbari S, Rainwater K, Ritchie JM, Yang M, and Sood AK. 2003. ‘Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines’, Clin Cancer Res, 9: 4514–21. [PubMed] [Google Scholar]
- Maestroni GJM 2019. ‘Adrenergic Modulation of Hematopoiesis’, J Neuroimmune Pharmacol. [DOI] [PubMed]
- McEwen BS 2002. ‘Sex, stress and the hippocampus: allostasis, allostatic load and the aging process’, Neurobiol Aging, 23: 921–39. [DOI] [PubMed] [Google Scholar]
- Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, and Gonzalez-Angulo AM. 2011. ‘Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer’, J Clin Oncol, 29: 2645–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy M 1997. ‘Transcriptional regulation by cyclic AMP’, Annu Rev Biochem, 66: 807–22. [DOI] [PubMed] [Google Scholar]
- Musselman RP, Bennett S, Li W, Mamdani M, Gomes T, van Walraven C, Boushey R, Al-Obeed O, Al-Omran M, and Auer RC. 2018. ‘Association between perioperative beta blocker use and cancer survival following surgical resection’, Eur J Surg Oncol, 44: 1164–69. [DOI] [PubMed] [Google Scholar]
- Nilsson MB, Armaiz-Pena G, Takahashi R, Lin YG, Trevino J, Li Y, Jennings N, Arevalo J, Lutgendorf SK, Gallick GE, Sanguino AM, Lopez-Berestein G, Cole SW, and Sood AK. 2007. ‘Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism’, J Biol Chem, 282: 29919–26. [DOI] [PubMed] [Google Scholar]
- Nilsson MB, Sun H, Diao L, Tong P, Liu D, Li L, Fan Y, Poteete A, Lim SO, Howells K, Haddad V, Gomez D, Tran H, Pena GA, Sequist LV, Yang JC, Wang J, Kim ES, Herbst R, Lee JJ, Hong WK, Wistuba I, Hung MC, Sood AK, and Heymach JV. 2017. ‘Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers’, Sci Transl Med, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, and Varmus H. 2005. ‘Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain’, PLoS Med, 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PG, Merryman J, Orloff M, and Schuller HM. 1995. ‘Beta-adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: implications for individuals with preexisting chronic lung disease’, Cancer Res, 55: 3504–8. [PubMed] [Google Scholar]
- Penninx BW, Guralnik JM, Pahor M, Ferrucci L, Cerhan JR, Wallace RB, and Havlik RJ. 1998. ‘Chronically depressed mood and cancer risk in older persons’, J Natl Cancer Inst, 90: 1888–93. [DOI] [PubMed] [Google Scholar]
- Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, and Entschladen F. 2010. ‘Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival’, Oncotarget, 1: 628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao G, Chen M, Bucsek MJ, Repasky EA, and Hylander BL. 2018. ‘Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response’, Front Immunol, 9: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JF, Jin FJ, Li N, Guan HT, Lan L, Ni H, and Wang Y. 2015. ‘Adrenergic receptor beta2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment’, BMB Rep, 48: 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramberg H, Eide T, Krobert KA, Levy FO, Dizeyi N, Bjartell AS, Abrahamsson PA, and Tasken KA. 2008. ‘Hormonal regulation of beta2-adrenergic receptor level in prostate cancer’, Prostate, 68: 1133–42. [DOI] [PubMed] [Google Scholar]
- Robinson JD, and Cinciripini PM. 2006. ‘The effects of stress and smoking on catecholaminergic and cardiovascular response’, Behav Med, 32: 13–8. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM 1993. Why zebras don’t get ulcers: A guide to stress, stress‐related disease and coping (W. H. Freeman & Co: New York: ). [Google Scholar]
- Schuller HM, and Cekanova M. 2005. ‘NNK-induced hamster lung adenocarcinomas over-express beta2-adrenergic and EGFR signaling pathways’, Lung Cancer, 49: 35–45. [DOI] [PubMed] [Google Scholar]
- Schuller HM, Tithof PK, Williams M, and Plummer H 3rd. 1999. ‘The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid’, Cancer Res, 59: 4510–5. [PubMed] [Google Scholar]
- Seiffert K, Hosoi J, Torii H, Ozawa H, Ding W, Campton K, Wagner JA, and Granstein RD. 2002. ‘Catecholamines inhibit the antigen-presenting capability of epidermal Langerhans cells’, J Immunol, 168: 6128–35. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, Massey D, Zazulina V, Shahidi M, and Schuler M. 2013. ‘Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations’, J Clin Oncol, 31: 3327–34. [DOI] [PubMed] [Google Scholar]
- Shah SM, Carey IM, Owen CG, Harris T, Dewilde S, and Cook DG. 2011. ‘Does beta-adrenoceptor blocker therapy improve cancer survival? Findings from a population-based retrospective cohort study’, Br J Clin Pharmacol, 72: 157–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhar G, and Ben-Eliyahu S. 1998. ‘In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats’, J Immunol, 160: 3251–8. [PubMed] [Google Scholar]
- Shan T, Cui X, Li W, Lin W, Li Y, Chen X, and Wu T. 2014. ‘Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines’, Cancer Sci, 105: 847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang ZJ, Liu K, and Liang DF. 2009. ‘Expression of beta2-adrenergic receptor in oral squamous cell carcinoma’, J Oral Pathol Med, 38: 371–6. [DOI] [PubMed] [Google Scholar]
- Shao C, Sullivan JP, Girard L, Augustyn A, Yenerall P, Rodriguez-Canales J, Liu H, Behrens C, Shay JW, Wistuba II, and Minna JD. 2014. ‘Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway’, Clin Cancer Res, 20: 4154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Liu D, Duan H, Qian L, Wang L, Niu L, Zhang H, Yong Z, Gong Z, Song L, Yu M, Hu M, Xia Q,Shen B, and Guo N. 2011. ‘The beta2-adrenergic receptor and Her2 comprise a positive feedback loop in human breast cancer cells’, Breast Cancer Res Treat, 125: 351–62. [DOI] [PubMed] [Google Scholar]
- Shin VY, Wu WK, Chu KM, Koo MW, Wong HP, Lam EK, Tai EK, and Cho CH. 2007. ‘Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells’, Toxicol Sci, 96: 21–9. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A. 2019. ‘Cancer statistics, 2019’, CA Cancer J Clin, 69: 7–34. [DOI] [PubMed] [Google Scholar]
- Singh S, and Chellappan S. 2014. ‘Lung cancer stem cells: Molecular features and therapeutic targets’, Mol Aspects Med, 39: 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, Arevalo JM, Morizono K, Karanikolas BD, Wu L, Sood AK, and Cole SW. 2010. ‘The sympathetic nervous system induces a metastatic switch in primary breast cancer’, Cancer Res, 70: 7042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN, Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM, Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW, and Sood AK. 2006. ‘Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma’, Nat Med, 12: 939–44. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, and Nieto MA. 2009. ‘Epithelial-mesenchymal transitions in development and disease’, Cell, 139: 871–90. [DOI] [PubMed] [Google Scholar]
- Van Tits LJ, Michel MC, Grosse-Wilde H, Happel M, Eigler FW, Soliman A, and Brodde OE. 1990. ‘Catecholamines increase lymphocyte beta 2-adrenergic receptors via a beta 2-adrenergic, spleen-dependent process’, Am J Physiol, 258: E191–202. [DOI] [PubMed] [Google Scholar]
- Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C, and Gomez DR. 2013. ‘Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy’, Ann Oncol. [DOI] [PMC free article] [PubMed]
- Weberpals J, Jansen L, Haefeli WE, Hoffmeister M, Wolkewitz M, Herk-Sukel Mppv, Vissers PAJ, and Brenner H. 2017. ‘Pre- and post-diagnostic beta-blocker use and lung cancer survival: A population-based cohort study’, Sci Rep, 7: 2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner H . 1992. Perturbing the organism: The biology of stressful experience (University of Chicago Press.: Chicago, IL, US: ). [Google Scholar]
- Wong HP, Yu L, Lam EK, Tai EK, Wu WK, and Cho CH. 2007. ‘Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells’, Toxicol Appl Pharmacol, 221: 261–7. [DOI] [PubMed] [Google Scholar]
- Wu X, Liu BJ, Ji S, Wu JF, Xu CQ, Du YJ, You XF, Li B, Le JJ, Xu HL, Duan XH, and Dong JC. 2015. ‘Social defeat stress promotes tumor growth and angiogenesis by upregulating vascular endothelial growth factor/extracellular signal-regulated kinase/matrix metalloproteinase signaling in a mouse model of lung carcinoma’, Mol Med Rep, 12: 1405–12. [DOI] [PubMed] [Google Scholar]
- Wu X, Zang W, Cui S, and Wang M. 2012. ‘Bioinformatics analysis of two microarray gene-expression data sets to select lung adenocarcinoma marker genes’, Eur Rev Med Pharmacol Sci, 16: 1582–7. [PubMed] [Google Scholar]
- Xia Y, Wei Y, Li ZY, Cai XY, Zhang LL, Dong XR, Zhang S, Zhang RG, Meng R, Zhu F, and Wu G. 2019. ‘Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages’, Brain Behav Immun. [DOI] [PubMed]
- Yang EV, Donovan EL, Benson DM, and Glaser R. 2008. ‘VEGF is differentially regulated in multiple myeloma-derived cell lines by norepinephrine’, Brain Behav Immun, 22: 318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang EV, Kim SJ, Donovan EL, Chen M, Gross AC, Webster Marketon JI, Barsky SH, and Glaser R. 2009. ‘Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression’, Brain Behav Immun, 23: 267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, Lemeshow S, and Glaser R. 2006. ‘Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells’, Cancer Res, 66: 10357–64. [DOI] [PubMed] [Google Scholar]
- Yang P, Deng W, Han Y, Shi Y, Xu T, Shi J, Elhalawani H, Zhao Y, Xie X, Lou F, Zhang R, and Jin H. 2017. ‘Analysis of the correlation among hypertension, the intake of beta-blockers, and overall survival outcome in patients undergoing chemoradiotherapy with inoperable stage III non-small cell lung cancer’, Am J Cancer Res, 7: 946–54. [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L, and Sordella R. 2010. ‘TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer’, Proc Natl Acad Sci U S A, 107: 15535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa T, Kaira K, Shimizu K, Shimizu A, Mori K, Nagashima T, Ohtaki Y, Oyama T, Mogi A, and Kuwano H. 2016. ‘Prognostic significance of beta2-adrenergic receptor expression in non-small cell lung cancer’, Am J Transl Res, 8: 5059–70. [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Deng YT, Liu J, Wang YQ, Yi TW, Huang BY, He SS, Zheng B, and Jiang Y. 2016. ‘Norepinephrine induced epithelial-mesenchymal transition in HT-29 and A549 cells in vitro’, J Cancer Res Clin Oncol, 142: 423–35. [DOI] [PubMed] [Google Scholar]
- Zhang S, Wang Y, Mao JH, Hsieh D, Kim IJ, Hu LM, Xu Z, Long H, Jablons DM, and You L. 2012. ‘Inhibition of CK2alpha down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells’, PLoS One, 7: e38996. [DOI] [PMC free article] [PubMed] [Google Scholar]