

Abstract
The alternation of substrate specificity expands the application range of enzymes in industrial, medical, and pharmaceutical fields. l‐Glutamate oxidase (LGOX) from Streptomyces sp. X‐119‐6 catalyzes the oxidative deamination of l‐glutamate to produce 2‐ketoglutarate with ammonia and hydrogen peroxide. LGOX shows strict substrate specificity for l‐glutamate. Previous studies on LGOX revealed that Arg305 in its active site recognizes the side chain of l‐glutamate, and replacement of Arg305 by other amino acids drastically changes the substrate specificity of LGOX. Here we demonstrate that the R305E mutant variant of LGOX exhibits strict specificity for l‐arginine. The oxidative deamination activity of LGOX to l‐arginine is higher than that of l‐arginine oxidase form from Pseudomonas sp. TPU 7192. X‐ray crystal structure analysis revealed that the guanidino group of l‐arginine is recognized not only by Glu305 but also Asp433, Trp564, and Glu617, which interact with Arg305 in wild‐type LGOX. Multiple interactions by these residues provide strict specificity and high activity of LGOX R305E toward l‐arginine. LGOX R305E is a thermostable and pH stable enzyme. The amount of hydrogen peroxide, which is a byproduct of oxidative deamination of l‐arginine by LGOX R305E, is proportional to the concentration of l‐arginine in a range from 0 to 100 μM. The linear relationship is maintained around 1 μM of l‐arginine. Thus, LGOX R305E is suitable for the determination of l‐arginine.
Keywords: crystal structure, l‐arginine oxidase, l‐glutamate oxidase, modification of substrate specificity, substrate recognition
Short abstract
Abbreviations
- AROD
l‐arginine oxidase
- LAAO
l‐amino acid oxidase
- LGOX
l‐glutamate oxidase
1. INTRODUCTION
l‐Arginine is a semi‐essential amino acid for humans and is involved in various metabolic pathways to make products of biological importance, including nitric oxide, agmatine, creatine, citrulline, ornithine, and polyamines. 1 , 2 The cardiovascular system and the nerve system are affected by l‐arginine through the production of nitric oxide. 3 , 4 l‐Arginine can be a medical biomarker because patients with argininemia or cancer show unusual concentrations of l‐arginine in the blood. 5 , 6 Thus, various enzymatic methods have been developed for quick and easy determination of l‐arginine levels. The method using arginase (E.C. 3.5.3.1) and urease (E.C. 3.5.1.5) detects the ammonia produced from l‐arginine by the two coupling reactions: arginase degrades l‐arginine to urea and l‐ornithine, and urease produces ammonia from urea. 7 , 8 The l‐arginine decarboxylase (EC 4.1.1.19) method detects the carbonate ion, which is produced by the decarboxylation of l‐arginine, by ion selective membrane electrode. 9 The method using l‐arginine deiminase (EC 3.5.3.6) and argininosuccinate synthetase (EC 6.3.4.5) measures the pyrophosphate produced from l‐arginine thorough the two‐step reaction. Pyrophosphate is a byproduct of the argininosuccinate synthesis from l‐citrulline by argininosuccinate synthetase, and l‐citrulline is converted from l‐arginine by l‐arginine deiminase. 10 However, these methods need complicated multiple enzymatic reactions or special electrodes, and therefore their application is limited. Recently, Matsi et al. purified l‐arginine oxidase (AROD, EC 1.4.3.25), which is a flavoenzyme that catalyzes the oxidative deamination of l‐arginine to 2‐ketoarginine, from Pseudomonas sp. TPU 7192 and demonstrated that AROD can be used for the determination of l‐arginine. 11 However, the specific activity to l‐arginine of AROD is rather low (0.19 U mg−1). Thus, a new enzymatic method is desired for l‐arginine determination.
l‐Amino acid oxidases (LAAO) are widely distributed flavoenzymes that catalyze the oxidative deamination of an l‐amino acid to produce a 2‐oxo acid with hydrogen peroxide and ammonia as byproducts. 12 LAAOs are involved in various biological and physiological functions in many organisms, including venomous snakes, mammals, insects, fish, mollusks, fungi, and bacteria. 13 While many LAAOs exert broad substrate specificity, several LAAOs, such as l‐glutamate oxidase from Streptomyces sp. X‐119‐6 (LGOX; EC 1.4.3.11), 14 l‐lysine‐α‐oxidase from Trichoderma viride (LysOX) 15 , 16 and l‐phenylalanine oxidase from Pseudomonas sp. P‐501 (PAO), 17 , 18 show strict substrate specificity.
LGOX is a member of the LAAO family and catalyzes the oxidative deamination of the α‐amino group of l‐glutamate to 2‐ketoglutarate via an imino acid intermediate. LGOX is produced as a single polypeptide precursor of 701 residues and is processed into three polypeptide chains to exert the enzymatic activity. The N‐terminal 14 residues, the C‐terminal 18 residues, and 40 residues between the second and the third chains are removed in the mature enzyme. LGOX shows strict substrate specificity for l‐glutamate, and high thermal and pH stability. 14 Therefore, LGOX has been used as a biosensor to quantify l‐glutamate and been applied in quality management of foods and fermentation processes. LGOX can also detect glutamic oxaloacetic transaminase, glutamic pyruvic transaminase and γ‐glutamyl transpeptidase activities, which are diagnostic markers of liver function. 19 , 20 Thus, LGOX is used in biosensors for clinical testing apparatus. We determined the crystal structure of LGOX and found that Arg305 is located in the potential substrate binding site of LGOX. 21 The following mutation study based on the structure and a substrate‐docking simulation revealed that Arg305 plays a key role in the recognition of the side chain of l‐glutamate. 22 Interestingly, point mutation of Arg305 drastically changed the substrate specificity of LGOX. R305L, R305A, and R305K mutant variants of LGOX lost the activity to l‐glutamate, but gained an activity to l‐histidine. LGOX R305D showed an activity to l‐arginine although the activity to l‐glutamate was completely lost. 22 These results suggest that various LAAOs specific to certain l‐amino acids can be developed by mutation of LGOX.
Here we show that the R305E mutant variant of LGOX exhibits strict specificity for l‐arginine. The specific activity of LGOX R305E to l‐arginine is much higher than that of AROD and that of LGOX R305D. The crystal structures of LGOX R305E and its complex with l‐arginine revealed that the molecular mechanism of strict specificity for l‐arginine. These findings indicate that LGOX R305E is a useful enzyme for determination of l‐arginine.
2. RESULTS
2.1. LGOX R305E shows strict specificity toward l‐arginine
LGOX R305E was prepared by site‐directed mutagenesis. LGOX R305E was expressed in E. coli JM109 and was purified by two‐step column chromatography after maturation using a metalloprotease from Streptomyces griseus 21 (Table 1). We obtained 13 mg of purified enzyme from 7 g of cell pellet. The purified protein showed a single band in Native‐PAGE analysis (Figure 1(a)) and three bands (relative molecular weights of 40, 17, and 10 kDa) corresponding to α, β, and γ‐fragments in SDS‐PAGE analysis (Figure 1(b)), indicating that mature LGOX R305E was successfully purified. The final purified enzyme exhibited specific activity of 7.5 U mg−1 (Table 2).
TABLE 1.
Summary of purification of LGOX R305E
| Total protein (mg) | Total activity (U) | Specific activity (Umg−1) | Yield (%) | Purification (fold) | |
|---|---|---|---|---|---|
| Crude extract | 937 | 59 | 0.063 | 100 | 1 |
| Protease digestion | 125 | 553 | 4.4 | 936 | 70 |
| Ammonium sulfate (30%–60%) | 76 | 184 | 2.4 | 311 | 38 |
| DEAE Toyopearl 650 M | 19 | 110 | 5.7 | 186 | 91 |
| Sephacryl S 300 HR | 13 | 99 | 7.5 | 168 | 118 |
Note: The activity of LGOX R305E was measured at 40°C, pH 8.0.
FIGURE 1.
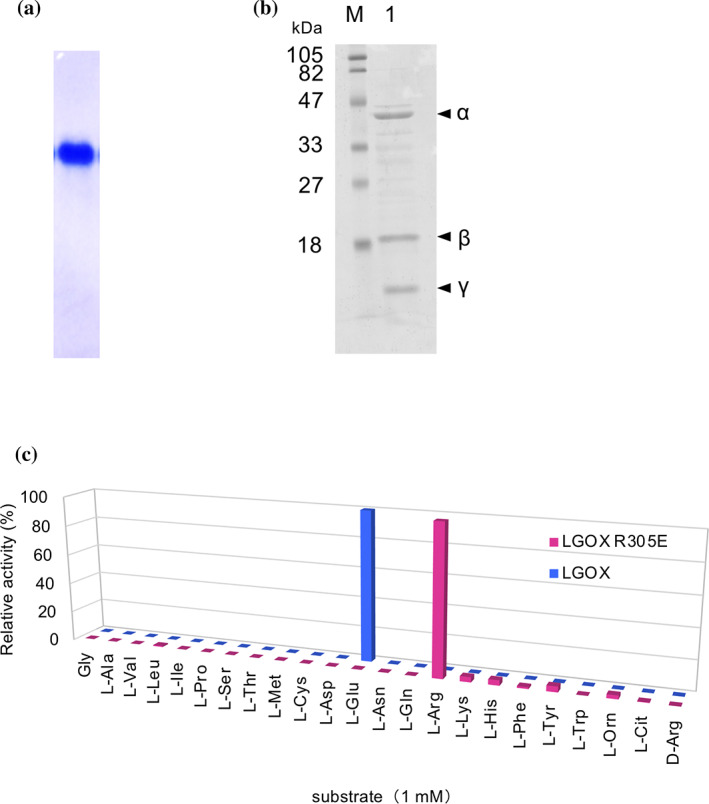
LGOX R305E shows strict specificity for l‐arginine. (a) Blue native‐PAGE analysis of LGOX R305E. In 10 μg of protein was applied. (b) SDS‐PAGE analysis of purified LGOX R305E: lane M, molecular mass marker; lane 1, purified LGOX R305E. (c) Comparison of substrate specificity profiles of LGOX R305E and wild‐type LGOX. Relative oxidative activities of LGOX R305E and wild‐type LGOX are indicated in red, and blue bars, respectively. The substrate that showed the highest activity was set as 100% relative activity. Enzyme activities were measured using 1 mM substrates at 40°C
TABLE 2.
Kinetic parameters of LGOX, LGOX R305E, R305D, and AROD
| Substrate | Specific activity (Umg−1) | K m (mM) | k cat (s−1) | k cat/K m (M−1 s−1) | |
|---|---|---|---|---|---|
| LGOX R305E | l‐Arg | 7.5 | 0.223 ± 0.071 | 3.42 ± 0.61 | 1.53 × 104 |
| LGOX R305D 22 | l‐Arg | 1.5 | 7.20 | 1.65 | 2.29 × 102 |
| LGOX 22 | l‐Glu | 57 | 0.173 | 53.2 | 3.08 × 105 |
| AROD 11 | l‐Arg | 0.19 | 0.149 | ‐ | ‐ |
Note: Kinetic parameters of LGOX R305E was measured at 40°C, pH 8.0. Each data represents the mean ± SD of triplicate measurements.
Oxidative deamination activity of LGOX R305E for various l‐amino acids were assessed using the 4‐aminoantipyrine phenol method. 21 We tested 23 l‐α‐amino acids and found that LGOX R305E exhibits the highest activity toward l‐arginine and low activity toward l‐tyrosine, l‐histidine, l‐lysine, and l‐ornithine with relative activities of 3.7% ± 0.29, 3.5% ± 0.11, 3.4% ± 0.25, and 2.4% ± 0.12 of l‐arginine, respectively. No activity was observed for the other l‐α‐amino acids including the original substrate, l‐glutamate. Therefore, the R305E mutation changes the substrate specificity of LGOX from l‐glutamate to l‐arginine.
2.2. Kinetic properties of LGOX R305E
The kinetic parameters of LGOX R305E for l‐arginine under the optimum condition (pH 8 and 40°C) were determined by the 4‐aminoantipyrine phenol method and are summarized in Table 2. The K m value of LGOX R305E for l‐arginine (0.22 mM) is close to that of LGOX for l‐glutamte, (0.17 mM) and that of AROD for l‐arginine (0.15 mM), indicating that the binding affinity of l‐arginine to LGOX R305E is comparable to that of the original substrate to the wild‐type enzyme. In contrast, the catalytic efficiency (k cat/K m) of LGOX R305E for l‐arginine was 5% of that of LGOX for l‐glutamate.
2.3. The effect of pH and temperature on the enzymatic activity of LGOX R305E
We next characterized the enzyme activity of LGOX R305E toward l‐arginine by monitoring the hydrogen peroxide production using the 4‐aminoantipyrine phenol method. 21 LGOX R305E exhibited the highest specific activity of 7.5 U mg−1 for l‐arginine at pH 8 and 40°C. This value is 40‐fold higher than that of AROX from Pseudomonas sp. TPU 7192 (0.19 U mg−1). 11
The enzyme activity was retained more than 90% of the highest activity from 30°C to 50°C at pH 7.4 and was dropped to 75% at 60°C (Figure 2(a)). The remaining activity was 100% after 1 h incubation at 50°C and 88% at 60°C. The enzyme still showed 60% remaining activity after 1 h incubation at 70°C (Figure 2(b)). The melting temperature (Tm) estimated from circular dichroism (CD) data at 212 nm was 71°C for LGOX R305E and 74°C for the wildtype enzyme (Figure 2(c)). Therefore, LGOX R305E is a thermostable enzyme, although it is slightly thermally unstable compared with wild‐type LGOX (Figure 2(a), (b)).
FIGURE 2.
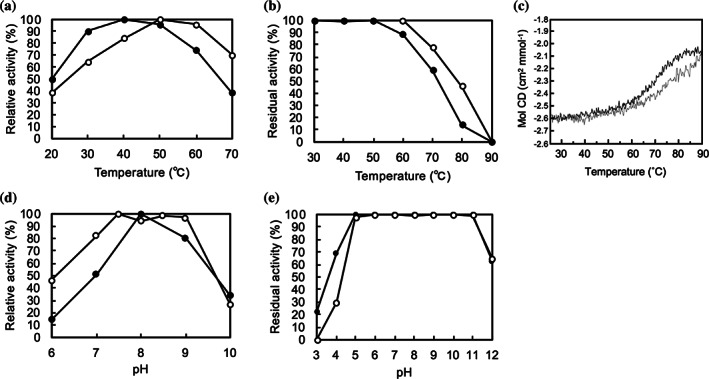
Comparison of enzyme properties of LGOX R305E with wild‐type LGOX. (a) Temperature dependence of the enzyme activities of LGOX R305E (closed circle) and wild‐type LGOX (open circle). Relative activities at pH 7.4 from 20°C to 70°C are plotted. The maximal activity is set as 100% relative activity (40°C for LGOX R305E and 50°C for wild‐type LGOX). (b) The remaining activities after incubation at various temperatures (30–70°C) for 1 h. Relative activities of LGOX R305E (closed circle) and wild‐type LGOX (open circle) are plotted. The activity without heat treatment was set as 100% remaining activity. (c) Far‐UV CD curves of LGOX R305E (black) and wild‐type LGOX (gray) at 210 nm obtained by temperature scan from 25°C to 90°C. (d) Effect of pH on the activity of LGOX R305E (closed circle) and wild‐type LGOX (open circle). The activity at optimum pH was set as 100% relative activity. (d) The remaining activities after incubation at various pH for 1 h on ice on stability. Relative activities of LGOX R305E (closed circle) and wild‐type LGOX (open circle) are plotted. The activity at pH 7.4 was set as 100% remaining activity
The enzyme activity was kept more than 80% of the highest activity in the pH range from 8.0 to 9.0 at 40°C and was reduced to about 15% of the maximum at pH 6 and 35% at pH 10 (Figure 2(c)). The residual activity was over 80% in the pH range of 5.0–11.0 (Figure 2(d)). Thus, LGOX R305E is pH stable, comparable to the wild‐type LGOX.
2.4. Determination of l‐arginine concentration using LGOX R305E
LGOX R305E shows strict substrate specificity for l‐arginine, and its enzyme activity on l‐arginine is much higher than that of AROD. Oxidative deamination of l‐arginine by LGOX R305E produces hydrogen peroxide, which can easily be detected. Thus, LGOX R305E is expected to be useful for determining the concentration of l‐arginine. We prepared solutions with various concentrations of l‐arginine (0–100 μM) and tested LGOX R305E to measure the concentration of l‐arginine using the 4‐aminoantipyrine‐TOOS (N ‐ethyl‐N‐[2‐hydroxy‐3‐sulfopropyl]‐m‐toluidine) method, which quantifies hydrogen peroxide. 23 Each l‐arginine solution was added to the reaction mixture containing 0.25 U ml−1 of LGOX R305E. Then, we monitored absorbance at 555 nm at 28°C and found that the absorbance reached a maximum after around 5 min incubation. The maximum values were recorded and plotted against the l‐arginine concentration. The plots indicated a linear relationship between the 555 nm absorbance and l‐arginine concentration in a range from 0 to 100 μM of l‐arginine concentration with a coefficient of determination of 0.9993 (Figure 3). The plot also showed liner relationship between 0 and 10 μM of l‐arginine, indicating that LGOX R305E is potentially useable to determine l‐arginine concentration.
FIGURE 3.
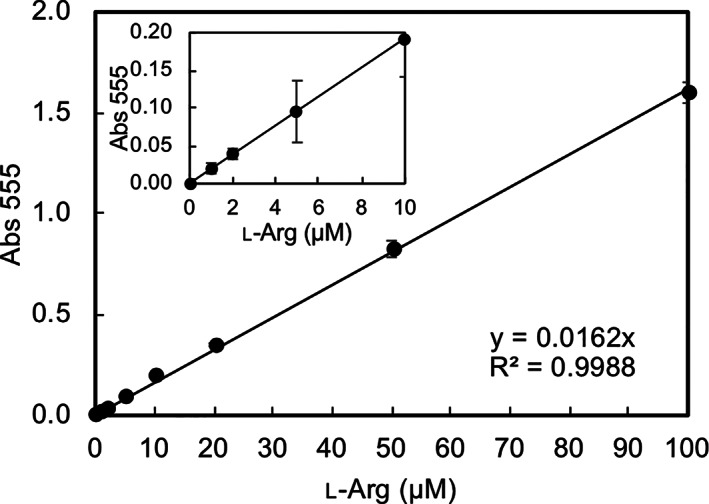
Linear relationship between hydrogen peroxide production by LGOX R305E and l‐arginine concentration. The absorbance at 555 nm is plotted against the l‐arginine concentration from 0 to100 μM. The magnified plot in the range from 0 to 10 μM of l‐arginine is shown as an inset. The linear regression equation with the coefficient of determination is also shown in the graph
2.5. Crystal structure of LGOX R305E
The structure of LGOX R305E was determined at 2.65 Å resolution (Figure 4(a), Table 3). We crystallized LGOX R305E composed of three fragments, α (residue 15–390), γ (391–480), and β (521–683). However, residues 15–16, 363–371, 387–390, 471–480, 521, and 672–683 were not modeled, because these residues did not appear in the electron density map.
FIGURE 4.
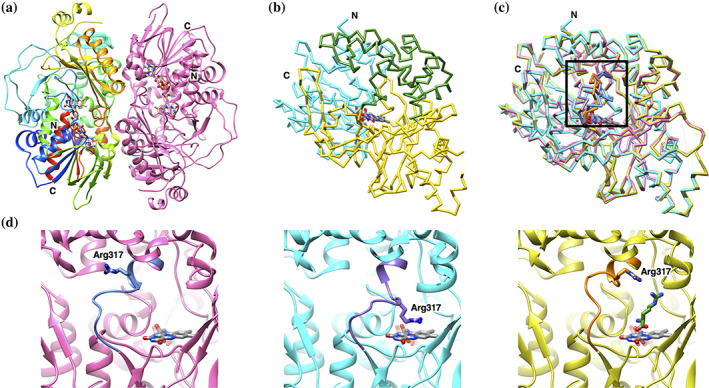
Structure of LGOX R305E. (a) Ribbon representation of the LGOX R305E dimer. One subunit is colored in rainbow from the N‐terminus (blue) to the C‐terminus (red), and the other in pink. (b) Cα trace of a single subunit of LGOX R305E. The domain structure is depicted in different colors: cyan, the FAD‐binding domain; yellow, substrate‐binding domain; green, helical domain. FAD is shown in the stick model colored by element: red, oxygen; blue, nitrogen; gray, carbon. The entrance of the funnel is shown by an arrow. (c) Superposition of Cα trace of wild‐type LGOX (pink), LGOX R305E (cyan), and LGOX R305E in complex with l‐arginine (yellow). The residues S313‐T325 (in the black box) are highlighted in different colors: wild‐type LGOX, pale blue; LGOX R305E, violet; LGOX R305E in complex with l‐arginine, orange. (d) Close‐up view of the loop connecting the helical domain with the substrate binding domain. The residues S313‐T325 are colored different from the rest. Wild‐type LGOX, LGOX R305E, and LGOX R305E in complex with l‐arginine are shown in the left, middle and right panels, respectively. The colors are the same as (c)
TABLE 3.
Data collection and refinement statistics
| LGOX R305E | LGOX R305E‐L‐Arg | |
|---|---|---|
| Data collection | ||
| Space group | P6122 | P6122 |
| Cell dimensions | ||
| a, b, c (Å) | 124.0, 124.0, 168.6 | 124.8, 124.8, 170.2 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 84.3–2.65 (2.78–2.65) | 54.0–2.7 (2.83–2.7) |
| R pim | 0.034 (0.191) | 0.089 (0.317) |
| I / σI | 18.5 (4.6) | 7.7 (2.5) |
| Completeness (%) | 99.9 (100) | 96.4 (99.1) |
| Redundancy | 12.3 (13) | 4.8 (4.8) |
| Refinement | ||
| Resolution (Å) | 66.3–2.65 (2.77–2.65) | 54.0–2.7 (2.82–2.70) |
| No. reflections | 22,853 (2,774) | 21,093 (2,660) |
| R work / R free | 19.1/24.1 (21.6/29.7) | 19.8/25.5 (25.9/35.4) |
| No. atoms | ||
| Protein | 4,723 | 4,800 |
| Ligand/ion | 53 | 65 |
| Water | 87 | 190 |
| B‐factors | ||
| Protein | 45.4 | 30.4 |
| Ligand/ion | 29.4 | 28.1 |
| Water | 40.1 | 30.5 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.006 | 0.002 |
| Bond angles (°) | 0.860 | 0.549 |
| Ramachandran plot (%) | ||
| Preferred regions | 95.2 | 97.0 |
| Allowed regions | 4.5 | 3.0 |
| Outliers | 0.3 | 0 |
Note: Values in parentheses are for highest‐resolution shell.
The LGOX R305E crystal belongs to the space group of P6122, which is the same space group as LGOX, with cell dimensions of a = b = 124.0 and c = 168.6 Å, which are similar to those of the LGOX crystal. 21 A single LGOX R305E molecule exists in an asymmetric unit and forms a dimer with a neighboring molecule related by crystallographic two‐fold symmetry (Figure 4(a)), as seen in the LGOX crystal. The structure of LGOX R305E is almost identical to that of LGOX. LGOX R305E consists of three domains: the FAD‐binding domain (E17–G96, G332–T430, and Y621–A671), the substrate‐binding domain (R97–R204, A324–G331, and H431–V620) and the helical domain (R205–R323) (Figure 4(b)). No significant structural difference was observed between LGOX R305E and LGOX except for the loop region (F314–T325) connecting the helical domain with the substrate binding domain (Figure 4(c), (d)). The loop of LGOX R305E overhangs the substrate binding pocket, and the guanidino group of R317 in the loop lies above the isoalloxazine ring of FAD and forms a cation‐π interaction with FAD (Figure 4(d)). Therefore, a large conformational change of the loop is needed to place a substrate in the active site of LGOX R305E. The loop conformation is further stabilized by the interaction between the carboxy group of E305 and the hydroxyl group of S318. However, it is unclear how the R305E mutation induces the conformational change of the loop from the wild‐type conformation.
2.6. The structural basis of l‐arginine recognition by LGOX R305E
The crystal of LGOX R305E in complex with l‐arginine was prepared by soaking method. The color of the crystals was changed from yellow to colorless after 5–10 min soaking in l‐arginine solution, suggesting that FAD is converted to the reduced form and that the substrate is bound to the enzyme in the crystal. 24 , 25 We collected X‐ray diffraction data from the colorless crystals.
In the substrate‐complex structure, the loop between the helical and the substrate‐binding domains moves away from the substrate binding pocket (Figure 4(d)), and l‐arginine lies on the isoalloxazine ring of FAD. The amino acid backbone of l‐arginine is recognized by LGOX R305E in the same manner as other LAAOs (Figure 5(a)), such as LysOX (Figure 5(b)) 24 and LAAO from Calloselasma rhodostoma venom (CrLAAO). 25 The α‐amino group of l‐arginine forms hydrogen bonds with the carbonyl oxygen of A652 and a cation‐π interaction with its indole ring of W653. The α‐carboxy group of l‐arginine interacts with R124 and Y562. These residues are conserved in other LAAOs. 12 , 24 , 25 The side chain of l‐arginine is recognized by E305, D433, W564, and E617 (Figure 5(a)). The guanidino group stacks on the indole ring of W564 to form a cation‐π interaction and forms hydrogen‐bonds with the side chain carboxy groups of E305, D433, and E617. These multiple interactions contribute to the specific binding of l‐arginine to the active site of LGOX R305E.
FIGURE 5.
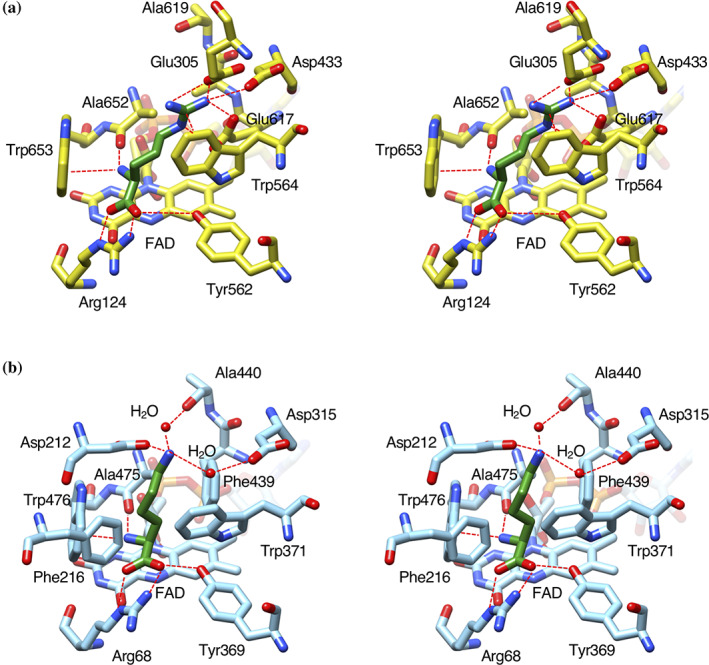
Comparison of the substrate‐binding site structure of LGOX R305E with that of LysOX. Stereo view of the substrate binding structure of LysOX‐Lys (a), and LGOX R305E‐Arg (b). The substrate molecules are colored in green. The protein residues and FAD are shown in yellow for LysOX and cyan for CrLAAO. The nitrogen and oxygen atoms are colored in blue and red, respectively. The bound water molecules are shown as red balls. Possible hydrogen bonds and cation‐π interactions are indicated by red broken lines
The side chain of W371 changes the conformation by substrate binding and blocks the path to the active site. 24 Similar side chain movement by l‐arginine binding was observed in LGOX R305E. However, the path to the active site of LGOX R305E is much wider than LysOX, and therefore, the active site of LGOX R305E is still accessible after l‐arginine binding.
R124, E305, and W564 adopt alternative conformations in the substrate complex structure. This is probably because the substrate does not bind in all molecules in the crystal. In fact, the refined occupancy of the bound l‐arginine is 0.78. Substrate binding may induce conformational change of these residues.
3. DISCUSSION
Previous structural and mutational studies revealed that Arg305 is important for substrate recognition of LGOX and demonstrated that point mutation at Arg305 can drastically change the substrate specificity of LGOX. 21 , 22 Here we found that the R305E mutant variant of LGOX shows strict specificity for l‐arginine. The K m value for l‐arginine is almost the same level as that of LGOX for l‐glutamate. The crystal structures revealed that the molecular mechanism of the substrate specificity change and the structural basis of the high affinity for l‐arginine (Figure 6). The substrate specificity change is not just the result of simple replacement of a basic residue with an acidic residue in the active site. The guanidino group of l‐arginine does not only interact with E305 but also with D433, W564, and E617, which are conserved in wild‐type LGOX (Figure 6(a), (b)). D433 and E617 are trapped by R305 and the negative charge of these residues are neutralized by R305 in the wild‐type LGOX structure (Figure 6(a)). Moreover, R305 sterically blocks W564 to interact with the substrate in LGOX. The R305E mutation releases D433, W564, and E617 and allows them to interact with the substrate bound in the active site of LGOX R305E. These multiple interactions by these residues as well as E305 provide high affinity for l‐arginine. In addition, R305E mutation makes the charge of the active site surface highly negative with the contribution of D433 and E617. This charge distribution attracts the basic amino acids but repels the acidic amino acids from the active site. Due to the charge distribution, LGOX R305E shows low enzymatic activity toward the basic amino acids, such as l‐histidine, l‐lysine, and l‐ornithine, while wild‐type LGOX does not at all.
FIGURE 6.
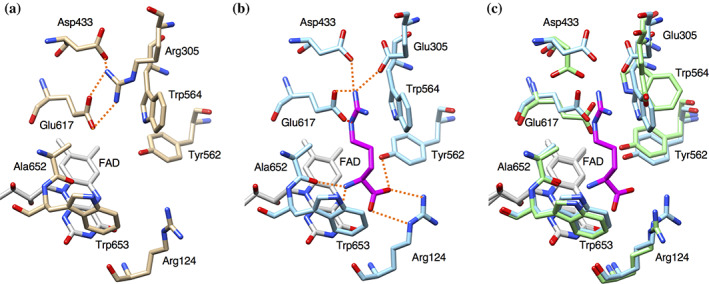
Comparison of the active site structure of l‐arginine complex of LGOX R305E with that of wild‐type LGOX. Active site of ligand‐free wild‐type LGOX (a) and of LGOX R305E with l‐arginine (b). The nitrogen and oxygen atoms are colored in blue and red, respectively. Possible hydrogen bonds are shown as red, dashed lines. (c) Superposition of the active site of LGOX R305E (green) and LGOX R305E in complex with l‐arginine (cyan)
LysOX recognizes the hydrophobic side chain arm of the substrate l‐lysine by hydrophobic hole composed of five hydrophobic residues (F216, W371, F439, A475, and W476) 24 (Figure 5(b)). The hydrophobic hole is also found in other LAAOs that bind hydrophobic amino acids. 25 , 26 , 27 However, LGOX R305E has no such hydrophobic hole (Figure 5(a)). E617 is present in the corresponding position of F439 of LysOX. No residue exists in the corresponding position of F216 of LysOX. Therefore, LGOX R305E does not bind hydrophobic amino acids (Figure 5).
We previously showed that LGOX R305D has activity toward l‐arginine (1.5 U mg−1), 22 but the specific activity is 20% of that of LGOX R305E. The K m value of LGOX R305E for l‐arginine is approximately 3% of that of LGOX R305D, indicating that LGOX R305E has higher substrate‐binding affinity. The catalytic efficiency (k cat/K m) is approximately 65‐fold higher than that of LGOX R305D. The structure of the l‐arginine complex of LGOX R305E suggests that the side chain of D305 is too short to interact with the guanidino group of l‐arginine in the active site and therefore the side chain of the l‐arginine may be recognized only by D433, W564, and E617 in LGOX R305D. Interactions with D433, W564, and E617 may not be enough to bind the l‐arginine in proper orientation for the reaction. Thus, the activity of LGOX R305D toward l‐arginine is weaker than that of LGOX R305E.
In this study, we showed that LGOX R305E has a strict substrate specificity for l‐arginine. The specific activity of LGOX R305E to l‐arginine is much higher than that of native AROD. The thermal and pH stability of LGOX R305E is almost the same level as wild‐type LGOX, which has been widely used for l‐glutamate sensing. These properties are suitable for application to an enzyme‐based sensor. Hydrogen peroxide is produced as a byproduct of oxidative deamination of l‐arginine by LGOX R305E. We demonstrated that the amount of hydrogen peroxide detected by the 4‐aminoantipyrine‐ TOOS method is proportional to the concentration of l‐arginine from 0 to 100 μM. Moreover, the linear relationship is maintained even around 1 μM of l‐arginine. These results indicate that LGOX R305E based method is a powerful tool to determine l‐arginine.
4. METHODS
4.1. Site‐directed mutagenesis
The expression vector of LGOX (pGOx_mal1) was constructed as described previously. 22 Site‐directed mutagenesis was conducted by inverse PCR using KOD Plus Neo (Takara, Japan), plasmid pGOx_mal1 as a template, and the following oligonucleotide primers: 5′‐CATGACCTCGGAACTCCACCTGC‐3′, and 5′‐CAGGTGGAGTTCCGAGGTCATG‐3′. The template plasmid was digested with Dpn I at 37°C for 1 h. The plasmid carrying LGOX R305E (pGOx_mal1_305E) was transformed into E. coli JM109. The introduction of the mutation was confirmed by DNA sequencing.
4.2. Purification of LGOX R305E
E. coli JM109 harboring pGOx_mal1_305E was cultured at 22°C for 24 h in 1 L of 2 × YT medium containing 50 μg ml−1 ampicillin. Expression of the LGOX R305E gene was induced by the addition of 0.1 mM IPTG for 22 h at 22°C. Cells were harvested by centrifugation and stored at −80°C until use. The pellet was thawed and resuspended in 20 mM potassium phosphate buffer (KPB) at pH 7.4 and sonicated at 150 W for 15 min at 4°C. After centrifugation, the supernatant was stored with metalloprotease from Streptomyses griseus (Sigma‐Aldrich Corp., St. Louis, MO, USA) at 30°C for 18 h for maturation 22 and then was incubated at 60°C for 20 min to inactivate the protease. The solution was centrifuged, and the supernatant was brought to 30% saturation with ammonium sulfate for 30 min on ice. After removal of the precipitate, the supernatant was brought to 60% saturation with ammonium sulfate for 30 min on ice. The precipitate was collected by centrifugation, dissolved in 20 mM KPB (pH 7.4), and dialyzed for 16 h against 20 mM KPB (pH 7.4) with 100 mM NaCl. The solution was loaded on a DEAE Toyopearl 650 M column (Tosoh Corp., Tokyo, Japan) equilibrated with 100 mM NaCl in 20 mM KPB (pH 7.4) and was eluted with 20 mM KPB (pH 7.4) containing 200 mM NaCl. The fractions containing LGOX R305E were concentrated and applied to a Sephacryl S 300 HR column (GE healthcare). The peak fractions were collected and pooled. The protein concentration was determined by the Bradford method using a Bio‐Rad Protein Assay kit (Bio‐Rad Laboratories, Inc., Hercules, CA, USA) with bovine serum albumin as a standard. Wild‐type LGOX was expressed and purified as previously described. 22 The purity of the enzyme was assessed by SDS‐PAGE (15%) and Native‐PAGE (7.5%).
4.3. Enzyme assay
The enzyme activity of LGOX R305E was measured by detecting hydrogen peroxide using the 4‐aminoantipyrine phenol method or by monitoring α‐keto acid using the 3‐methyl‐2‐benzothiazolinone hydrazone (MBTH) method.
The enzyme assay by the 4‐aminoantipyrine phenol method was conducted as described previously with slight modification. 28 0.9 ml of reaction mixture containing 50 mM buffer (borate‐NaOH buffer (pH 8.0) for LGOX R305E and KPB (pH 7.4) for wild‐type LGOX), 2 mM phenol, 2 mM 4‐aminoantipyrine, 10 U ml−1 horseradish peroxidase, and an appropriate amount of enzyme was pre‐incubated at 40°C for 3 min. The reaction was initiated by the addition of 0.1 ml of 100 mM substrate solution, and the formation of red quinoimine dye was monitored at a wavelength of 505 nm by a Shimadzu UVmini‐1,240 UV–VIS spectrophotometer (Shimadzu GLC Ltd., Tokyo, Japan). One unit of activity was defined as the amount of enzyme that catalyzes the formation of 1 μmol of hydrogen peroxide per minute.
The enzyme assay by the MBTH method was performed as previously described with slight modification. 29 0.9 ml of reaction mixture containing 70 mM buffer (borate‐NaOH buffer (pH 8.0) for LGOX R305E and KPB (pH 7.4) for wild‐type LGOX), 300 U ml−1 catalase, and an appropriate amount of enzyme was pre‐incubated at 40°C for 3 min. The reaction was started by the addition of 0.1 ml of 100 mM l‐arginine for LGOX R305E and 10 mM l‐glutamate for wild‐type LGOX followed by incubation at 40°C for 5 min. The reaction was stopped by adding 0.1 ml of 25% trichloroacetic acid to the reaction mixture. Then 1.9 ml of 1 M acetate buffer (pH 5.0) and 0.8 ml of 1 mg ml−1 MBTH aqueous solution were added to the reaction mixture, and the mixture was incubated at 50°C for 30 min. After cooling the mixture at room temperature for 20 min, the formation of an MBTH‐azine α‐keto acid derivative was measured at a wavelength of 316 nm by Shimadzu UVmini‐1,240 UV–VIS spectrophotometer. One unit of activity was taken as the amount of enzyme that liberates 1 μmol of α‐keto acid per minute.
4.4. Determination of substrate specificity
The substrate specificity of LGOX R305E and wild‐type LGOX was determined by measuring enzyme activities for 23 l‐α‐amino acids using the 4‐aminoantipyrine phenol method.
4.5. Determination of optimum pH and temperature
Optimum pH was determined by measuring enzyme activity (LGOX R305E for l‐arginine at 40°C; wild‐type LGOX for l‐glutamate at 50°C) using the 4‐aminoantipyrine method. The pH was adjusted using a universal buffer (50 mM boric acid, 50 mM citric acid, 50 mM acetic acid, 50 mM NaH2PO4, 50 mM CAPS, and 25 mM HEPES) by adding 5 mol L−1 NaOH solution. The optimum temperature was determined by measuring enzyme activity (LGOX R305E for l‐arginine; wild‐type LGOX for l‐glutamate) in 70 mM KPB (pH 7.4) using the MBTH method.
4.6. Measurement of residual activity
To determine the thermostability, enzyme activity (LGOX R305E for l‐arginine; wild‐type LGOX for l‐glutamate) was measured by the MBTH method at 40°C after incubation of the enzyme in KPB (pH 7.4) at various temperatures (30–90°C) for 1 h. To evaluate the pH stability of the enzyme, enzyme activity (LGOX R305E for l‐arginine; wild‐type LGOX for l‐glutamate) was measured by the 4‐aminoantipyrine phenol method at 40°C for LGOX R305E and at 50°C for wild‐type LGOX after incubation of the enzyme in universal buffer at various pH (pH 3.0–12.0) for 1 h on ice. The pH value of the enzyme solution was adjusted to pH 7.4 by adding 100 mM KPB before the enzyme activity assay.
4.7. Circular dichroism spectroscopy
Far‐UV CD data were collected on Jasco‐720 W spectropolarimeter with a Peltier type cell holder for temperature control (JASCO International Co., Tokyo, Japan). A quartz cell with 1 mm path‐length were filled with 0.33 mg ml−1 of protein solutions containing 20 mM sodium phosphate (pH 7.4) and 150 mM NaCl. CD melting curves were measured at 210 nm over the temperature rage 25°C‐90°C at a heating rate of 1°C min−1 with data acquisition interval of 0.2°C, response time 8 s and bandwidth 2 nm.
4.8. Determination of the kinetic parameters
Enzyme activity for l‐arginine with different concentrations was measured at 40°C using the 4‐aminoantipyrine phenol method. Kinetic parameters were determined by Lineweaver–Burk plot.
4.9. L‐arginine determination using LGOX R305E
The amount of hydrogen peroxide produced by oxidative deamination of l‐arginine by LGOX R305E was measured using 4‐aminoantipyrine‐TOOS (N ‐ethyl‐N‐[2‐hydroxy‐3‐sulfopropyl]‐m‐toluidine) method. 23 0.9 ml of reaction mixture containing 20 mM Tris–HCl buffer (pH 9.0), 0.4 mM toos ( N ‐ethyl‐N‐[2‐hydroxy‐3‐sulfopropyl]‐m‐toluidine), 0.4 mM 4‐aminoantipyrine, 10 U ml−1 horseradish peroxidase, and 0.25 U ml−1 was pre‐incubated at 28°C for 3 min. The reaction was started by adding 0.1 ml of l‐arginine solution (0–1 mM) at 28°C, and the formation of quinone diamide was monitored at a wavelength of 555 nm for 10 min by Eppendorf Biospectrometer kinetic (Eppendorf Co., Ltd., Tokyo, Japan). The maximum values were recorded and plotted against the l‐arginine concentration.
4.10. Preparation of crystals
Initial crystallization screening was performed at 277 K and 293 K by the sitting‐drop vapor diffusion method using the following screening kit: Wizard I and II, Cryo I and II (Emerald Biostructures) and Crystal Screen I and II (Hampton Research). Each drop was prepared by mixing 1 μL protein solution (8.7 mg·ml−1) with an equal volume of reservoir solution. Clusters of small crystals were grown from the condition containing 100 mM Tris–HCl (pH 7.0), PEG8000, and MgCl2. The crystallization conditions were optimized by screening additives and the concentration of the precipitants and additives. The best crystals were grown at 277 K from drops prepared by mixing protein solution (8.7 mg·ml−1) containing 20 mM KPB (pH 7.4) with an equivalent volume of reservoir solution containing 100 mM Tris–HCl (pH 7.0), 12% (v/v) PEG8000, and 75 mM MgCl2. The substrate complex crystals were prepared by soaking technique. The crystals were soaked in a reservoir solution containing 10% (v/v) MPD and 35 mM l‐arginine and were stored at 277 K until the yellow color disappeared (typically 5–10 min).
4.11. Data collection and structure determination
The X‐ray data were corrected at SPring‐8 beamlines, BL41XU and BL26B1 (Harima, Japan), with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2016B2719, 2017A2585, and 2017B2585). The crystals were transferred into liquid nitrogen for freezing. The R305E variant crystals were soaked in a cryo‐protectant solution containing 10% (v/v) MPD and 90% (v/v) of the reservoir solution for several seconds before freezing. The diffraction data were collected under nitrogen gas flow at 100 K. The diffraction data were indexed and integrated with MOSFLM, 30 and were scaled with AIMLESS. 31 The diffraction data statistics are summarized in Table 3. The initial phase was obtained by the molecular replacement method with Phaser 32 using the structure of wild type LGOX (PDB: 2E1M) as a search model. The atomic models were built with Coot 33 and refined with PHENIX. 34 The structural refinement statistics are shown in Table 3.
AUTHOR CONTRIBUTIONS
Yoshika Yano: Investigation; writing‐original draft. Shinsaku Matsuo: Investigation. Nanako Ito: Investigation; visualization. Takashi Tamura: Investigation. Hitoshi Kusakabe: Resources. Kenji Inagaki: Conceptualization; project administration; resources; supervision; writing‐original draft; writing‐review & editing. Katsumi Imada: Conceptualization; investigation; project administration; supervision; visualization; writing‐original draft; writing‐review & editing.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We thank SPring‐8 beamline staffs for technical help in use of beamline BL26B1 and BL41XU, Ayaka Yamaguchi and Analytical Instrument Faculty of Graduate School of Science, Osaka University for supporting CD measurements. This work was supported by JSPS KAKENHI Grant Numbers 24560962 (to Kenji Inagaki).
Yano Y, Matsuo S, Ito N, et al. A new l‐arginine oxidase engineered from l‐glutamate oxidase. Protein Science. 2021;30:1044–1055. 10.1002/pro.4070
Yoshika Yano, Shinsaku Matsuo, and Nanako Ito contributed equally to this work.
Funding information Japan Science and Technology Corporation, Grant/Award Number: 24560962
Contributor Information
Kenji Inagaki, Email: kinagaki@okayama-u.ac.jp.
Katsumi Imada, Email: kimada@chem.sci.osaka-u.ac.jp.
REFERENCES
- 1. Grillo MA, Colombatto S. Arginine revisited. Amino Acids. 2004;26:345–351. [DOI] [PubMed] [Google Scholar]
- 2. Morris SM Jr. Enzymes of arginine metabolism. J Nutr. 2004;134:2743S–2747S. [DOI] [PubMed] [Google Scholar]
- 3. Wu G, Meininger CJ. Arginine nutrition and cardiovascular function. J Nutr. 2000;130:2626–2629. [DOI] [PubMed] [Google Scholar]
- 4. Böger RH, Bode‐Böger SM. The clinical pharmacology of l‐arginine. Annu Rev Pharmacol Toxicol. 2001;41:79–99. [DOI] [PubMed] [Google Scholar]
- 5. Marescau B, Dedeyn PP, Lowenthal A, et al. Guanidino compound analysis as a complementary diagnostic parameter for hyperargininemia: Follow‐up of guanidino compound levels during therapy. Pediatric Res. 1990;27:297–303. [DOI] [PubMed] [Google Scholar]
- 6. Vissers YL, Dejong CH, Luiking YC, Fearon KC, von Meyenfeldt MF, Deutz NE. Plasma arginine concentrations are reduced in cancer patients: Evidence for arginine deficiency? Am J Clin Nutr. 2005;81:1142–1146. [DOI] [PubMed] [Google Scholar]
- 7. Nikolelis DP, Hadjiioannou TP. Construction of an arginine enzyme electrode and determination of arginine in biological materials. Anal Chim Acta. 1983;147:33–39. [Google Scholar]
- 8. Liu D, Yin A, Ge K, Chen K, Nie L, Yao S. Enzymatic analysis of arginine with the SAW/conductance sensor system. Enzyme Microb Technol. 1995;17:856–863. [Google Scholar]
- 9. Tong SL, Rechnitz GA. Enzymatic l‐arginine and l‐lysine determination using a carbonate ion selective membrane electrode. Anal Lett. 1976;9:1–11. [Google Scholar]
- 10. Kameya M, Asano Y. Rapid enzymatic assays for l‐citrulline and l‐argininebased on the platform of pyrophosphate detection. Enzyme Microb Technol. 2014;57:36–41. [DOI] [PubMed] [Google Scholar]
- 11. Matsui D, Terai A, Asano Y. l‐arginine oxidase from Pseudomonas sp. TPU 7192: Characterization, gene cloning, heterologous expression, and application to l‐arginine determination. Enzyme Microb Technol. 2016;82:151–157. [DOI] [PubMed] [Google Scholar]
- 12. Pollegioni L, Motta P, Molla G. l‐amino acid oxidase as biocatalyst: A dream too far? Appl Microbiol Biotechnol. 2013;97:9323–9341. [DOI] [PubMed] [Google Scholar]
- 13. Yu Z, Qiao H. Advances in non‐snake venom l‐amino acid oxidase. Appl Biochem Biotechnol. 2012;167:1–13. [DOI] [PubMed] [Google Scholar]
- 14. Kusakabe H, Midorikawa Y, Fujishima T, Kuninaka A, Yoshino H. Purification and properties of a new enzyme, l‐glutamate oxidase, from Streptomyces sp. X‐119‐6 grown on wheat bran. Agr Biol Chem. 1983;47:1323–1328. [Google Scholar]
- 15. Kusakabe H, Kodama K, Kuninaka A, Yoshino H, Misono H, Soda K. A new antitumor enzyme, l‐lysine oxidase from Trichoderma viride. Purification and enzymological properties. J Biol Chem. 1980;255:976–981. [PubMed] [Google Scholar]
- 16. Amano M, Mizuguchi H, Sano T, et al. Recombinant expression, molecular characterization and crystal structure of antitumor enzyme, l‐lysine α‐oxidase from Trichoderma viride . J Biochem. 2015;157:549–559. [DOI] [PubMed] [Google Scholar]
- 17. Koyama H. Purification and characterization of a novel l‐phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P‐501. J Biochem. 1982;92:1235–1240. [DOI] [PubMed] [Google Scholar]
- 18. Koyama H. Further characterization of a novel l‐phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P‐501. J Biochem. 1983;93:1313–1319. [DOI] [PubMed] [Google Scholar]
- 19. Upadhyay S, Ohgami N, Kusakabe H, et al. Performance characterization of recombinant l‐glutamate oxidase in a micro GOT/GPT sensing system. Sens Actuators B Chem. 2006;119:570–576. [Google Scholar]
- 20. Upadhyay S, Ohgami N, Kusakabe H, Suzuki H. Electrochemical determination of gamma‐glutamyl transpeptidase activity and its application to a miniaturized analysis system. Biosens Bioelectron. 2006;21:1230–1236. [DOI] [PubMed] [Google Scholar]
- 21. Arima J, Sasaki C, Sakaguchi C, et al. Structural characterization of l‐glutamate oxidase from Streptomyces sp. X‐119‐6. FEBS J. 2009;276:3894–3903. [DOI] [PubMed] [Google Scholar]
- 22. Utsumi T, Arima J, Sakaguchi C, et al. Arg305 of Streptomyces l‐glutamate oxidase plays a crucial role for substrate recognition. Biochem Biophys Res Commun. 2012;417:951–955. [DOI] [PubMed] [Google Scholar]
- 23. Tamaoku K, Ueno K, Akiura K, Ohkura Y. New water‐soluble hydrogen donors for the enzymatic photometric determination of hydrogen peroxide. II. n‐ethyl‐n‐(2‐hydroxy‐3‐sulfopropyl) aniline derivatives. Chem Pharm Bull. 1982;30:2492–2497. [Google Scholar]
- 24. Kondo H, Kitagawa M, Matsumoto Y, et al. Structural basis of strict substrate recognition of l‐lysine α‐oxidase from Trichoderma viride . Protein Sci. 2020;29:2213–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moustafa IM, Foster S, Lyubimov AY, Vrielink A. Crystal structure of LAAO from Calloselasma rhodostoma with an l‐phenylalanine substrate: Insights into structure and mechanism. J Mol Biol. 2006;364:991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang H, Teng M, Niu L, et al. Purification, partial characterization, crystallization and structural determination of AHP‐LAAO, a novel l‐amino‐acid oxidase with cell apoptosis‐inducing activity from Agkistrodon halys pallas venom. Acta Cryst D. 2004;60:974–977. [DOI] [PubMed] [Google Scholar]
- 27. Georgieva D, Murakami M, Perband M, Arni R, Betzel C. The structure of a native l‐amino acid oxidase, the major component of the Vipera ammodytes ammodytes venomic, reveals dynamic active site and quaternary structure stabilization by divalent ions. Mol Biosyst. 2011;7:379–384. [DOI] [PubMed] [Google Scholar]
- 28. Arima J, Tamura T, Kusakabe H, et al. Recombinant expression, biochemical characterization and stabilization through proteolysis of an l‐glutamate oxidase from Streptomyces sp. X‐119‐6. J Biochem. 2003;134:805–812. [DOI] [PubMed] [Google Scholar]
- 29. Soda K. Microdetermination of d‐amino acids and d‐amino acid oxidase activity with 3‐methyl‐2‐benzothiazolone hydrazone hydrochloride. Anal Biochem. 1968;25:228–235. [DOI] [PubMed] [Google Scholar]
- 30. Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: A new graphical interface for diffraction image processing with MOSFLM. Acta Cryst D. 2011;67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Evans P. Scaling and assessment of data quality. Acta Cryst D. 2006;62:72–82. [DOI] [PubMed] [Google Scholar]
- 32. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: A comprehensive python‐based system for macromolecular structure solution. Acta Cryst D. 2010;58:1948–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]