

Abstract
Cardiovascular disease (CVD) remains the primary cause of death worldwide. Specifically, atherosclerosis is a CVD characterized as a slow progressing chronic inflammatory disease. During atherosclerosis, vascular walls accumulate cholesterol and cause fatty streak formation. The progressive changes in vascular wall stiffness exert alternating mechanical cues on vascular smooth muscle cells (VSMCs). The detachment of VSMCs in the media layer of the vessel and migration toward the intima is a critical step in atherosclerosis. VSMC phenotypic switching is a complicated process that modifies VSMC structure and biomechanical function. These changes affect the expression and function of cell adhesion molecules, thus impacting VSMC migration. Accumulating evidence has shown cholesterol is capable of regulating cellular migration, proliferation, and spreading. However, the interaction and coordinated effects of both cellular cholesterol and the extracellular matrix (ECM) stiffness/composition on VSMC biomechanics remains to be elucidated.
1. Introduction
CVD in the United States had been rising steadily from the 1900s to the 1980s and declined into the 2010s. However, CVD is still responsible for more deaths each year than chronic pulmonary disease and cancer combined (Benjamin et al., 2019). Atherosclerosis is an inflammatory disease characterized the buildup of plaque and subsequent narrowing of the artery. During the progression of atherosclerosis, vascular smooth muscle cells (VSMCs) switch from their normal contractile phenotype to a synthetic one, where they proliferate and migrate toward the innermost layer of the artery (Rudijanto, 2007). This switch is governed by changes in extracellular matrix (ECM) stiffness and composition as well as the accumulation of cholesterol and lipids in the nascent plaque. Together, this causes alterations in VSMC biomechanics including adhesion to the ECM and neighboring cells through integrins and cadherins, respectively, VSMC stiffness, and cytoskeletal architecture. The purpose of this review is to further explore the alterations in VSMC biomechanics during atherosclerosis and possible cellular mechanisms governing these changes.
2. Alterations in VSMC biomechanics in the progression of atherosclerosis
In the United States, coronary heart disease is the leading cause of death (43.2%), trailed by stroke (16.9%), high blood pressure (9.8%), heart failure (9.3%), diseases of the arteries (3.0%), and several minor CVD causes combined (17.7%). The prevalence of CVD increases the older an individual gets. It is projected that there is a 20% risk of CVD in the early 20s, 50% chance at the age of 45, and over 90% of individuals will have some form of CVD over the age of 80. Moreover, the annual direct and indirect cost of CVD in the United States is estimated to be $351.2 billion. Between 2015 and 2035, total direct medical costs of CVD are predicted to surge from $318 billion to $749 billion (Benjamin et al., 2019; Mozaffarian et al., 2015). Thus, the need to understand and reduce CVD occurrence and progression is essential.
Blood vessels are made up of three layers. Moving from the outermost layer toward the inner lumen are the adventitia, the tunica media, and the tunica intima. The adventitia is primarily composed of collagen. The tunica media is composed of VSMCs which control internal vessel diameter and are separated by collagen and elastic fibers. The tunica intima, the innermost layer, contains a single layer of endothelial cells and is responsible for the smooth flow of blood. Depending on location, size, and type of the vessel, the amount of ECM protein composition is different within each layer (Xu & Shi, 2014).
Blood vessels are biological systems that detect and react to mechanical cues and exert forces that control their surroundings’ mechanical properties (Humphrey, Dufresne, & Schwartz, 2014). Mechanobiology, also known as biomechanics, spans a range of biological organization ranging from molecules to cells to organisms that convert mechanical stimuli into biochemical cues. Cells maintain homeostasis during normal physiological function by continuously assessing and reading environmental cues especially their ECM mechanical properties. This process is called mechanosensing and is mediated by cell-ECM adhesion proteins known as integrins. Integrins detect and respond to ECM mechanical cues with cytoskeletal re-organization, force generation, and gene expression thus affecting the cell biomechanics (Humphrey et al., 2014). During the progression of atherosclerosis, vessel walls undergo extensive modification including intimal stiffening, calcification, and lipid streak formation altering the underlying ECM and activating mechanosensitive proteins in VSMCs. This will lead to disruptions in the cytoskeletal architecture and cellular adhesion in addition to changes in the mechanical and elastic properties of the cell, contributing to further disease progression. Ultimately, studying cellular biomechanics will further untangle the mechanisms governing atherosclerosis development.
2.1. Modifications in cell-ECM and cell-cell adhesions of VSMCs in the progression of atherosclerosis
Primarily, VSMCs retain a contractile phenotype as they reside within the media layer of the vascular wall. The “phenotypic switching” of VSMCs from a contractile to a synthetic/non-contractile phenotype (i.e., migratory and proliferative) occurs in a number of CVDs. This phenotype transition is an important step in the progression of atherosclerosis, which entails the detachment, proliferation, and migration of VSMCs toward the intima of the blood vessel, thus participating in plaque formation (Rudijanto, 2007).
During phenotypic switching, VSMCs decrease their expression of contractile protein specific genes and/or enhanced expression of synthetic/proliferative VSMCS or other cell type markers in response to arterial injury or lipid infiltration within the arterial wall (Basatemur, Jorgensen, Clarke, Bennett, & Mallat, 2019). It is essential to recognize that phenotypic switching is not a binary process but rather a spectrum ranging from contractile to synthetic. There are several altered molecular mechanisms and events that lead to phenotypic switching. Transcription factors such as Myocardin (MYOCD) that drives VSMCs contraction-related genes are often altered in expression or activity (Parmacek, 2008). Another transcription factor, Krüppel-like factor 4 (KLF4), blocks the expression of contraction-related gene. VSMC-specific deletion of KLF4 in Apolipoprotein E knockout (Apoe−/−) mice results in reduced numbers of VSMC-derived, macrophage-marker, and mesenchymal stem-cell-marker-positive cells in atherosclerotic plaques (Shankman et al., 2015). In addition to altered transcription factors, phenotypic switching is promoted by external stimuli. Transforming growth factor-β (TGFβ) and platelet-derived growth factor (PDGF) signaling were shown to promote synthetic VSMC phenotype formation (He et al., 2015). Also, growing evidence indicated that VSMC phenotype switching is prone to epigenetic regulation. Studies have suggested that VSCMs retain cellular memory of their contractility state after changing phenotype indicating histone-modifying enzymes and DNA methylation directly affects VSMC behavior (Gomez, Shankman, Nguyen, & Owens, 2013; Liu et al., 2013). Including an access state of inflammation and hypercholesterolemia, VSMCs will undergo a phenotypic shift and migrate toward the intima.
The migration of VSMCs is a complex process that entails the modification of cell adhesion protein molecules (Schwartz & Ginsberg, 2002). The detachment of cell-cell adhesions forms the initial step of VSMC migration toward the blood vessel intima (Johnson, 2014). N-cadherin is the major cell-cell adhesion molecule in VSMCs. The cleavage of N-cadherin-mediated cell-cell adhesion directly affects VSMC behavior including migration, proliferation as well as apoptosis leading to plaque rupture (Brown et al., 2010; Dwivedi, Slater, & George, 2009; Lyon, Johnson, White, Sala-Newby, & George, 2014). A number of studies have shown that cholesterol plays an important role in stabilizing cadherin-mediated cell-cell adhesion, facilitated by the formation of cholesterol-enriched plasma membrane microdomains such as lipid rafts (Baumgartner, Weth, Gutberlet, Harms, & Groschner, 2014; Taulet et al., 2009). Furthermore, both in vitro and ex vivo studies have suggested that substrate stiffness may regulate the stability of cadherin-mediated cell-cell adheren junctions in endothelial cells (Huynh et al., 2011). In vitro studies demonstrated cell-cell adhesion in VSMCs is impacted in a substrate stiffness dependent manner. The alterations in cell density exerts a strong effect on cellular morphology. Thus, it is thought that cell-cell contacts are essential to maintaining vascular function and important in mediating VSMCs response to vascular injury (Sazonova et al., 2011). However, the impact of ECM stiffness on VSMCS cell-cell adhesion is not fully deciphered. During atherosclerosis, vascular walls experience gradual increase in stiffness. Mui et al., demonstrated that N-cadherin is induced in VSMCs in response to vascular injury using an in vivo model of tissue stiffening and proliferation (Mui et al., 2015). In addition, N-cadherin expression is regulated by stiffness-sensitive focal adhesion (FA) kinase (FAK)-p130Cas-Rac signaling pathway. The deletion of N-cadherin in VSMCs in vivo, strongly reduced vascular injury-induced cell cycling, while the inhibition of FAK in VSMCs reduced proliferation after injury and accompanied by reduced induction of N-cadherin. Thus, N-cadherin participates in cell-cycle control within the vasculature and mediates the degree of cell spreading (Mui et al., 2015).
The attachment of VSMCs to the ECM is mediated by a variety of integrins and integrin β1 is predominantly expressed on VSMCs’ surfaces (Hillis, Mlynski, Simpson, & MacLeod, 1998). α5β1 is an important integrin that binds to fibronectin and promotes FA formation (Sun, Martinez-Lemus, Hill, & Meininger, 2008). In addition to integrin β1, integrins β3 and β5 also conduct key roles in VSMC adhesion and migration (Kappert et al., 2001; Liaw et al., 1995). During phenotypic switching of VSMCs, integrins αVβ3 and αVβ5 are up-regulated, while α5β1 is down-regulated (Kappert et al., 2001). Furthermore, the presence of αVβ3 has been demonstrated in atherosclerotic lesions (Hoshiga, Alpers, Smith, Giachelli, & Schwartz, 1995) and if blocked, reduces plaque growth (Choi et al., 2004) and VSMC migration toward the intima (Kappert et al., 2001). A previous study found that integrin adhesion was rapidly altered after stimulation with contractile or dilatory vasoactive agonists with an increased adhesion during contractile activation and decreased adhesion during relaxation. Parallel changes in cell stiffness was also observed as cell adhesion was altered (Hong et al., 2012). This supports the concept that rapid switching of integrin activity in VSMCs is associated with the cellular cytoskeletal structure. In addition to the effects on integrin-mediated cell-ECM adhesion, cadherin-mediated cell-cell adhesions also play a critical role in the progression of atherosclerosis.
2.2. VSMC cytoskeletal remodeling in the progression of atherosclerosis
VSMCs are dynamic cellular systems that constantly change their shape through coordinated rearrangement of their cellular components and position within the vascular wall to adapt to alterations in their chemical and mechanical environment (Martinez-Lemus, Hill, & Meininger, 2009). During the early stages of atherosclerosis, following the infiltration of monocytes and T lymphocytes into the vascular wall, leukocytes release several cytokines and reactive oxygen species that impact VSMC phenotype allowing their proliferation and migration. These changes subsequently promote the deposition of additional fibrous ECM proteins (e.g., collagen) in the intima (Hillis & Flapan, 1998). During phenotypic switching, not only do VSMCs undergo quantitative changes in contractility and but also significantly remodel their cytoskeletal architecture (Worth, Rolfe, Song, & Campbell, 2001). In the contractile state, α-actin was primarily expressed while non-muscle β-actin became much more prevalent in the synthetic state along with redistribution of these and other contractile proteins (Worth et al., 2001). Earlier studies also showed that depleting cellular cholesterol reduced endothelial cell spreading compared to cholesterol-enriched cells (Hong et al., 2013; Sun et al., 2007). In our recent publication, we found that depleting cellular cholesterol and decreased substrate stiffness significantly reduced the cortical cytoskeletal alignment in VSMCs (Sanyour et al., 2019).
The mechanism governing cytoskeletal organization in response to substrate stiffness and cholesterol is not fully understood. Actin is linked to the ECM through FA complexes containing various proteins including talin, vinculin, and various integrins (Critchley, 2000). At sufficiently high forces, talin unfolds allowing vinculin to bind, stabilizing the FA and increasing force transmission to actin (del Rio et al., 2009; Yao et al., 2014). These mechanosensitive proteins likely have a role in cytoskeleton organization as talin has been shown to mediate the co-alignment of integrins in FAs and F-actin and the growth of FAs parallel to strain. Additionally, vinculin, through its link to actin, has been shown to coordinate cell polarization in response to stretching forces (Carisey et al., 2013). FAs are also known to cluster with lipid rafts, which are membrane domains rich in cholesterol and sphingolipids (Simons & Ikonen, 1997). Lipid rafts themselves may be mechanosensitive with the ganglioside GM1 having been shown to accumulate in lipid rafts in endothelial cells in response to deformation, prior to talin accumulation (Fuentes & Butler, 2012). Clustering of GM1 lipid rafts has also been associated with enhanced β1 integrin adhesion, cortical F-actin polymerization, and shear resistance in T cells (Mitchell, Brown, Woodside, Vanderslice, & McIntyre, 2009). Cholesterol depletion from lipid rafts has been shown to inhibit phosphorylation of Src and FAK and cause subsequent dislocation of FA components (Jeon et al., 2010). Actin disruption following cholesterol depletion has also been association with the reorganization of the phospholipid phosphatidylinositol 4,5-bisphosphate, a key F-actin regulatory molecule (Kwik et al., 2003). Furthermore the cholesterol metabolite 25-hydroxycholesterol has been implicated in activation of integrin-FAK signaling (Pokharel et al., 2019).
2.3. Progressive stiffening of vessel walls
The vessel wall experiences progressive stiffening with aging associated with changes to ECM composition (Dao, Essalihi, Bouvet, & Moreau, 2005; Gaballa et al., 1998). Increased vascular stiffness is associated with hypertension (Benetos, Laurent, Asmar, & Lacolley, 1997; Lemarie, Tharaux, & Lehoux, 2010) and atherosclerosis development (Glasser, 2000; Wang, Keith Jr., Struthers, & Feuerstein, 2008). The increase in vessel wall stiffness alters VSMCs’ microenvironment. In fact, a number of recent studies have shown that VSMCs become intrinsically stiffer with aging and hypertension (Sehgel et al., 2013; Zhu et al., 2012). Both chemical and mechanical factors (Sazonova et al., 2011) lead to changes in VSMC characteristics (Brown et al., 2010; Isenberg, Dimilla, Walker, Kim, & Wong, 2009; McDaniel et al., 2007; Peyton, Kim, Ghajar, Seliktar, & Putnam, 2008; Peyton & Putnam, 2005; Stegemann, Hong, & Nerem, 2005). A possible mechanism governing VSMC stiffness increase is through a mechanotransduction involving the FA complex. Through FAs, cells sense and respond to mechanical changes in their surrounding environment. Our recent study showed that VSMCs cultured on collagen 1 (COL-1) coated gel substrates with variable stiffness are able to sense the substrate stiffness and respond with changes in cell stiffness and adhesion (Fig. 1) (Sanyour et al., 2019).
Fig. 1.
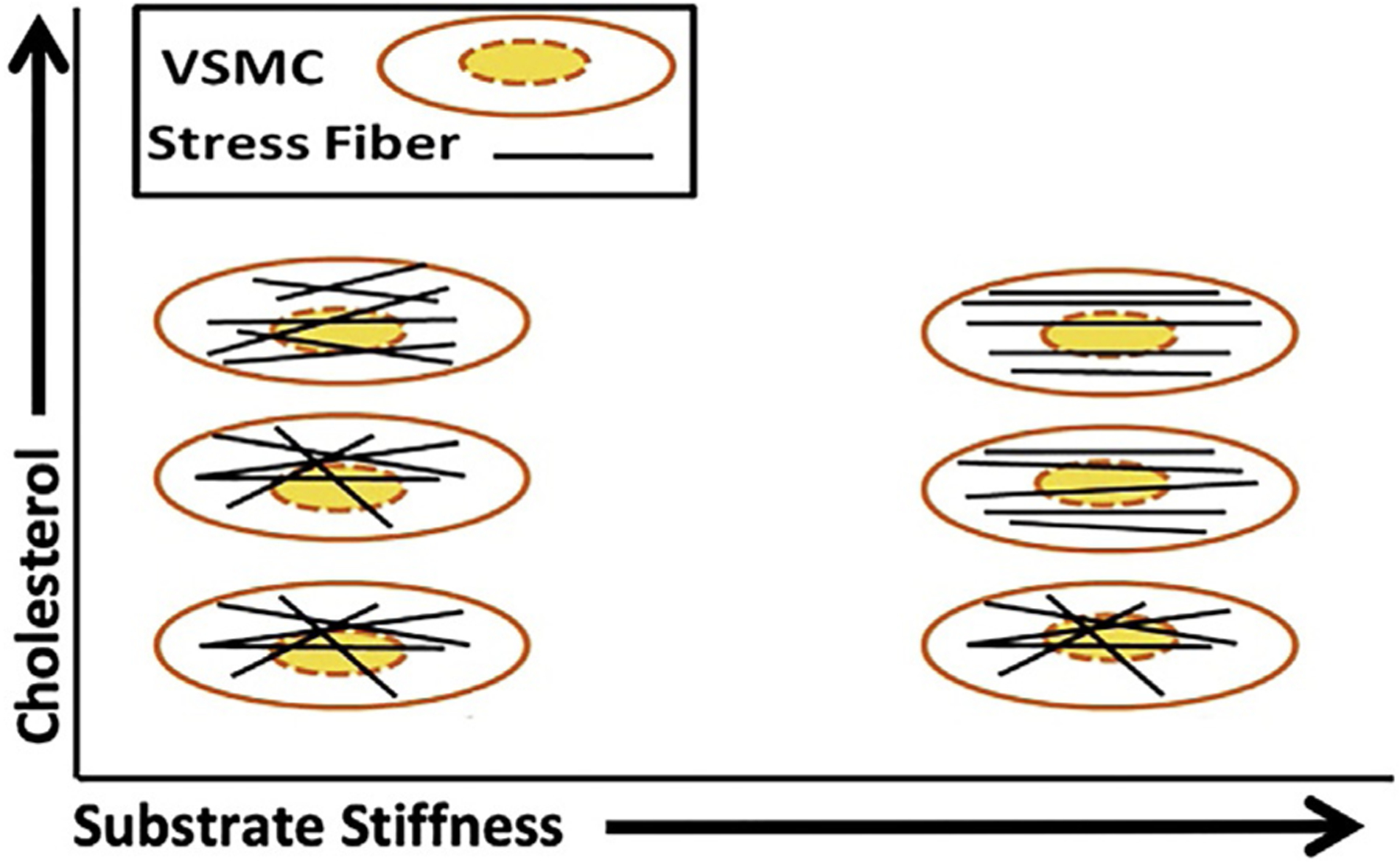
Effect of substrate stiffness and cholesterol on VSMC cytoskeleton. On low stiffness substrates, both normal and cholesterol-depleted VSMCs present a disorganized cytoskeleton orientation, while cholesterol-enriched cells present a more organized cytoskeleton. On stiff substrates, the cytoskeleton becomes more orientated for normal and cholesterol-enriched VSMCs.
2.4. VSMC migration in the progression of atherosclerosis
As atherosclerosis progresses, VSMCs proliferate, migrate, and secrete new ECM proteins that contribute to fibrous plaque formation (Glass & Witztum, 2001; Willis, Pierre-Paul, Sumpio, & Gahtan, 2004). The process of cell migration can be summarized as the protrusion of leading edge lamellipodia, attachment of the lamellipodia to the substrate through integrin interaction with ECM, formation of FAs, stress fiber alignment, and contraction. This is accompanied by the disassembly of FAs and disengagement of integrin-substrate binding at the trailing edge of cells (Horwitz & Parsons, 1999; Stossel, 1993) as cells move forward. We have also shown that VSMC migration behavior is dependent upon both the underlying substrate stiffness and the ECM protein composition. VSMCs were observed to migrate further on stiffer fibronectin-coated polyacrylamide (PA) gels, while the opposite behavior was observed on COL-1 coated PA gels (Rickel, Sanyour, Leyda, & Hong, 2020). This is vital to understanding atherosclerosis progression as both stiffness and protein compositions of atherosclerotic lesions vary with different regions and over time (Doddapattar et al., 2015; Lakatta, 2003; Tracqui et al., 2011; Wang et al., 2006).
3. The role of lipids and cholesterol in driving cardiovascular disease
Generally, cholesterol synthesis is divided into two major components: pre-squalene cholesterol synthesis and post-squalene cholesterol synthesis. Pre-squalene cholesterol synthesis contributes to both sterol and isoprenoid formation, whereas post-squalene cholesterol synthesis commits to vitamin D and sterol synthesis. Changes in cellular cholesterol homeostasis and cholesterol accumulation possess a significant clinical relevance. Not only is cholesterol a major factor involved in fatty deposition in the atherosclerotic lesion, but also plays an important role in regulating cellular mechanics (Byfield, Tikku, Rothblat, Gooch, & Levitan, 2006; Khatibzadeh, Spector, Brownell, & Anvari, 2013; Kowalsky, Byfield, & Levitan, 2008; Kwik et al., 2003; Levitan & Shentu, 2011; Sun et al., 2007), spreading (Hong et al., 2013), and migration (Nagao, Qin, Grosheva, Maxfield, & Pierini, 2007; Ramprasad et al., 2007; Sekine et al., 2010; Yan, Xia, Duan, Li, & Mei, 2011). However, reports about the effect of cholesterol on cell mechanics and migration are varied and controversial and even contradictory in different cell types underscoring the need for further study. In addition, a further complication is the vascular wall mechanical environment that undergoes progressive stiffening with aging and hypertension, therefore, may accelerate the development of atherosclerosis (Dao et al., 2005; Glasser, 2000; Wang et al., 2008). As the major cellular component of blood vessel, VSMCs carry out vital roles at each stage of atherosclerosis development (Willis et al., 2004).
A recent study reported that the amount of excess cholesterol loaded into human atherosclerotic VSMCs was much larger than previously known, with about 40% of the total foam cells in the atherosclerotic plaque being VSMC-derived (Allahverdian, Chehroudi, McManus, Abraham, & Francis, 2014). Similarly, arterial VSMCs isolated from cholesterol-fed atherosclerotic rabbits showed a progressive elevation in the membrane unesterified cholesterol-phospholipid mole ratio (Chen, Preston Mason, & Tulenko, 1995; Tulenko, Chen, Mason, & Mason, 1998). These findings suggest that dietary cholesterol induces changes in the structure and organization of VSMC plasma membrane in vivo. We and others propose that these alterations may cause functional and phenotypic switching of aortic VSMCs during atherogenesis. Another important finding is that excess membrane cholesterol decreases membrane fluidity (Gleason, Medow, & Tulenko, 1991) and alters Ca2+ channels in arterial VSMCs (Bialecki & Tulenko, 1989), changing Ca2+ movements and increasing cytosolic Ca2+ levels. Furthermore, changes in cortical stiffness and adhesion following contractile activation have been shown to occur in sync with changes in cytosolic Ca2+ (Huang, Sun, Hill, & Meininger, 2018). These changes may have important effects on vascular wall properties. In relation to atherosclerosis, Ca2+ is known to participate in signal transduction that promotes VSMC proliferation and migration (Mason & Jacob, 2003). In contrast to VSMCs, most prior studies in relation to the effect of cholesterol on the vascular cell biomechanics have focused on the endothelial cells (Byfield, Aranda-Espinoza, Romanenko, Rothblat, & Levitan, 2004; Hong et al., 2013; Kowalsky et al., 2008; Levitan & Shentu, 2011; Sun et al., 2007) and neglected possible direct effects of cholesterol on VSMCs.
4. Atomic Force Microscope, a powerful tool to study live VSMC biomechanics
Atomic Force Microscope (AFM) is a unique instrument utilized for a plethora of applications. These include contact imaging, tapping imaging, viscoelastic measurements, single force molecule measurement, lithography, and several other modes (Binnig, Quate, & Gerber, 1986; Krieg et al., 2019). AFM is an ideal tool for understanding the scale and size of conducting biomechanical testing of cells. AFM can be used to indent the VSMC surface and conduct nanoscale resolution imaging (Hong et al., 2012). Contact mode imaging is a powerful method that generates high-resolution three-dimensional (3D) topographical images of samples or cells (Hong et al., 2016). This is achieved by applying a constant force across the surface to indent approximately 180–300nm into the VSMCs. The obtained images will show cortical actin and stress fibers that can be analyzed for their orientation and density (Sanyour et al., 2019). Nanoindentation allows for the determination of viscoelastic (stiffness) properties of VSMCs. In addition, AFM can be used to conduct single molecule spectroscopy (Neumann, 2008). Essentially, an AFM stylus tip is coated with a protein that is complementary to cellular surface adhesion receptors, such as integrins or cadherins. This allows for the characterization and quantification of cell-cell and cell-ECM adhesion forces. Recently, we were able to develop an automated approach to filter background noise and extract dynamic biomedical signals/oscillations from large sets of real-time AFM data (Sanyour, Childs, Meininger, & Hong, 2018). Earlier work describes conducting AFM studies to measure the elastic modulus (E-modulus) (Zhu et al., 2012), stress fiber orientation (Gupta et al., 2015), and adhesion of fibronectin to integrins (Helenius, Heisenberg, Gaub, & Muller, 2008) cultured on different substrate stiffnesses. Nevertheless, the combined effect of substrate stiffness and cellular lipid/cholesterol regulations is yet to be assessed. Deciphering the impact of both cholesterol management and substrate stiffness on VSMC biomechanics will better define VSMCs’ experience in the progression of atherosclerosis. Ultimately, this might provide a better understating of the mechanical mechanisms underlying atherosclerosis.
5. Impact of vascular ECM stiffness and cholesterol on VSMC biomechanics
To study the impact of cholesterol on cellular mechanics, cyclodextrins (CD) are widely used to manipulate cell membrane cholesterol by directly removing cholesterol from the cell membrane (Zidovetzki & Levitan, 2007). CDs are made up of α-(1–4)-linked D-glycopyranose units in a cyclic arrangement. Because of their water soluble nature and hydrophobic cavity, they have been used as hydrophobic molecules and drugs carriers (Zidovetzki & Levitan, 2007). Methyl-β-cyclodextrin (MβCD) in particular is widely used to either deplete or enrich cellular membrane cholesterol. However, CD’s are also reported to have toxic effects on cells and induce cell death (Zidovetzki & Levitan, 2007).
VSMCs experience a wide variety of environments of varying stiffness and cholesterol concentration within an atherosclerotic plaque (Fig. 2). In a recent study, we reported the combinatorial effects of substrate stiffness and MβCD mediated cholesterol depletion and enrichment on VSMC cytoskeletal remodeling and biomechanics. Rat VSMCs were cultured on COL-1 coated polyacrylamide (PA) gels with a tunable stiffness. AFM was employed to measure cell and PA gel stiffness, α5β1-fibronectin adhesion, and contact mode was used to image cortical cytoskeleton orientation. To further confirm AFM results, confocal microscopy was used to image the entire cell cytoskeleton (Fig. 3E). Upon cholesterol depletion, VSMC stiffness and α5β1-fibronectin adhesion were reduced with the cells displaying a more disorganized cytoskeleton (Fig. 3A–C, green). Cholesterol enrichment with a MβCD-cholesterol saturate solution increased VSMC stiffness, α5β1 adhesion, and cytoskeletal orientation compared to cholesterol-depleted VSMCs (Fig. 3A–C, red). Furthermore, decreasing substrate stiffness resulted in a reduced VSMC stiffness (Fig. 3A), α5β1 adhesion (Fig. 3B), and whole cytoskeletal organization on the softest substrate (Fig. 3D and E) (Sanyour et al., 2019).
Fig. 2.
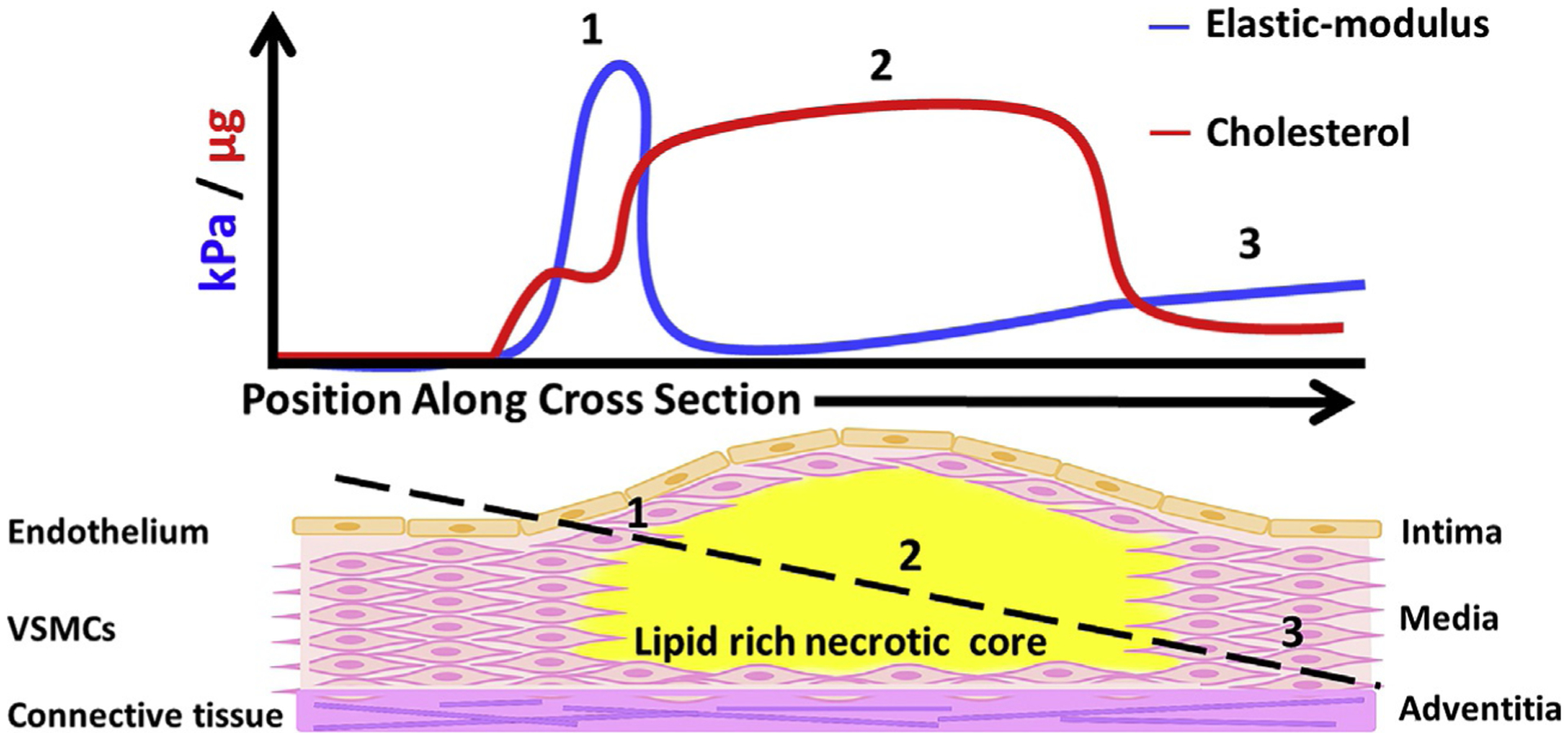
The different microenvironments of an atherosclerotic plaque. The graph in the upper panel follows the black dotted line in the lower panel showing: (1) the fibrous cap is stiff and enriched with cholesterol. (2) The plaque core is rich in cholesterol and very soft. (3) Normal arterial wall structure and cholesterol composition.
Fig. 3.
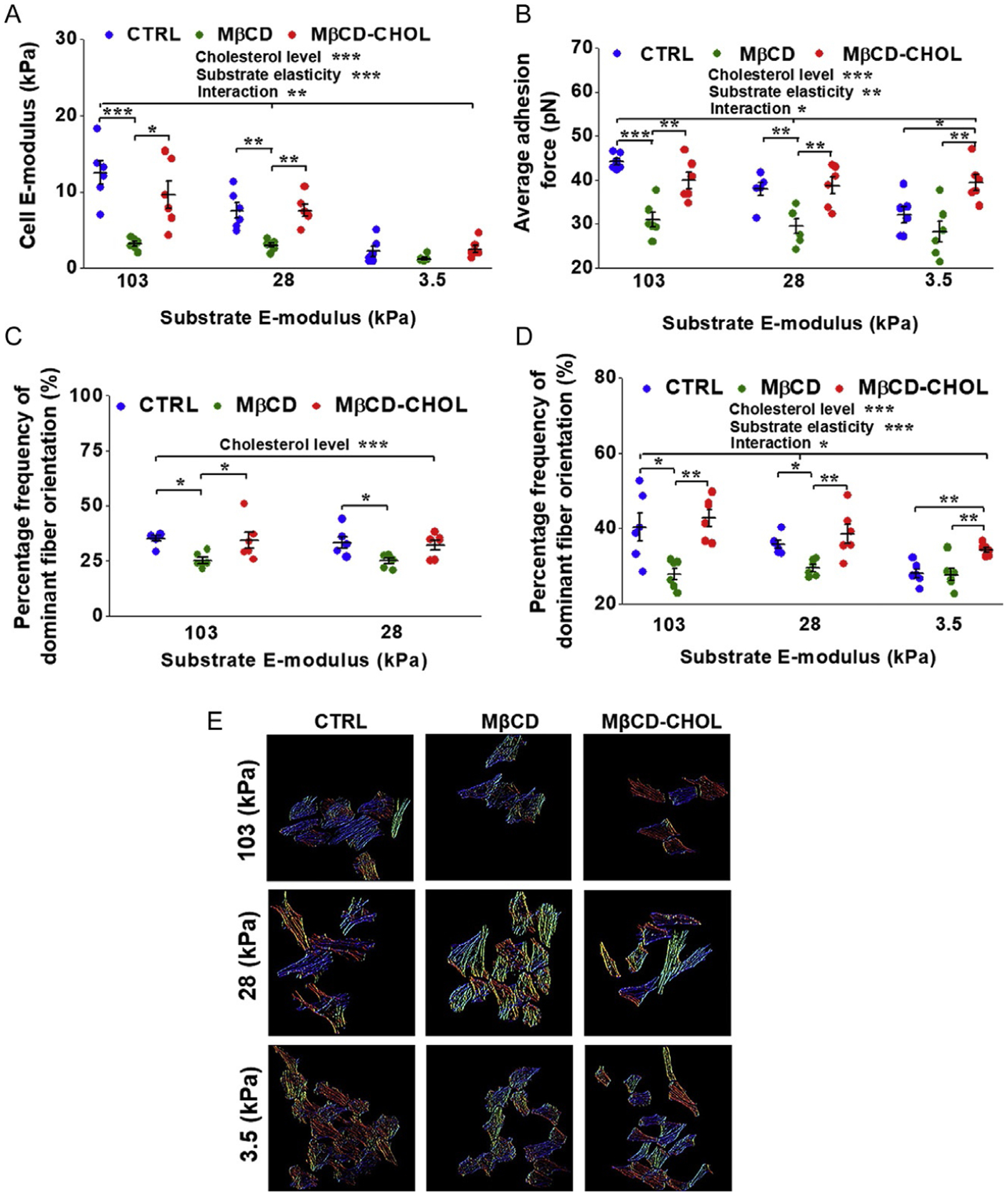
Effects of MβCD mediated cholesterol depletion and enrichment on VSMCs cultured on substrates of varying stiffness. (A) Cholesterol depletion and decreasing substrate stiffness significantly lowered VSMC E-modulus while cholesterol-enriched VSMCs had higher stiffness than cholesterol depleted ones. (B) α5B1-fibronectin adhesion force was significantly reduced upon cholesterol depletion and decreasing substrate stiffness. (C) AFM imaging of submembraneous actin showed cholesterol depletion decreased cytoskeletal organization while enriched VSMCs showed greater organization. (D) Quantification of confocal imaging of the whole cell cytoskeleton revealed VSMCs on the softest substrate had significantly reduced cytoskeletal organization. (E) Representative color map images of actin fiber orientation computed from confocal images. Different colors represent different actin orientation angles. VSMCs on the stiffer substrates showed more uniform color, indicative of a more orientated actin cytoskeleton. Cholesterol-depleted VSMCs showed more variable colors and a more disperse actin alignment, while cholesterol-enriched VSMCs had more uniform color and actin orientation. Figure is adopted from Sanyour H. J., Li N., Rickel A. P., Childs J. D., Kinser C. N. & Hong Z. (2019). Membrane cholesterol and substrate stiffness co-ordinate to induce the remodelling of the cytoskeleton and the alteration in the biomechanics of vascular smooth muscle cells. Cardiovascular Research, 115, 1369–1380.
3-Hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase inhibitors, commonly known as statins, block cholesterol synthesis by inhibiting HMG-CoA reductase, the rate-limiting enzyme in the mevalonate pathway. While statin therapy is known to decrease the incidence of clinical cardiovascular disease through the lowering of serum cholesterol levels (Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995), evidence suggests that statins exert additional benefits including decreased VSMC proliferation (Bellosta et al., 2004; Corsini, Pazzucconi, Pfister, Paoletti, & Sirtori, 1996). Apart from lowering cholesterol biosynthesis, statins also halt the synthesis of various metabolic intermediates associated with the mevalonate pathway, such as isoprenoid synthesis (Ward, Watts, & Eckel, 2019). Cholesterol-independent effects of statins potentially mediate pleiotropic effects on cardiovascular physiology by modifying plaque stability, signaling pathways, platelet aggregation, and VSMC function (Oesterle, Laufs, & Liao, 2017). In vascular disease, inflammation plays a key role spanning initial disease events such as endothelial damage and macrophage recruitment to further distal events such as plaque rupture and stability (Libby, Ridker, & Maseri, 2002). Hence, statins were explored for their properties beyond their cholesterol lowering properties as anti-inflammatories. Statins have been extensively reviewed for over two decades and demonstrated to have clinical lipid-independent anti-inflammatory effects. Several different clinical anti-inflammatory mechanisms have been described (Jain & Ridker, 2005). Under normal conditions, VSMCs control vascular tone and blood pressure. Due to inflammatory vascular injury and VSMC phenotypic switching, VSMCs increase their proliferation, migration, and ECM protein secretions. Statins were shown to induce cell-cycle arrest in the G1/S phase though factors such as cyclin dependent kinase inhibitor 1B (p27Kip1) and caspases to inhibit their proliferation (Laufs, Marra, Node, & Liao, 1999). In addition, Sakamoto et al. demonstrated that fluvastatin prevented VSMC hyperplasia, proliferation, and phenotype modulation by ERK1/2 and p38MAPK inactivation (Sakamoto, Murata, Chuma, Hori, & Ozaki, 2005). We previously demonstrated that total VSMCs and aortic tissue cholesterol was reduced and their biomechanical properties altered following fluvastatin treatment (Sanyour et al., 2020). AFM analysis showed fluvastatin lowered VSMC E-modulus and caused them to display a more disorganized submembraneous actin structure (Fig. 4B and D–F). Our results also revealed fluvastatin-mediated cholesterol depletion had differential effects on VSMC responses to COL-1 and fibronectin. VSMC migration distance decreased upon cholesterol depletion on a fibronectin-coated surface while adhesion to fibronectin increased (Fig. 4A and C). No changes were observed for migration distance on COL-1 or VSMC adhesion to COL-1 (Fig. 4A and C) (Sanyour et al., 2020). Yet, our preliminary ELISA (enzyme-linked immunosorbent assay) experiments (not shown) suggested a none-significant effect on proinflammatory tumor necrosis factor (TNF) and interleukin 1 (IL-1) secretion in VSMCs and aortic tissue-culture media after 3 days of statin treatment. Therefore, deciphering the exact cholesterol dependent/independent effects of statins in modifying VSMC biomechanics, is essential.
Fig. 4.
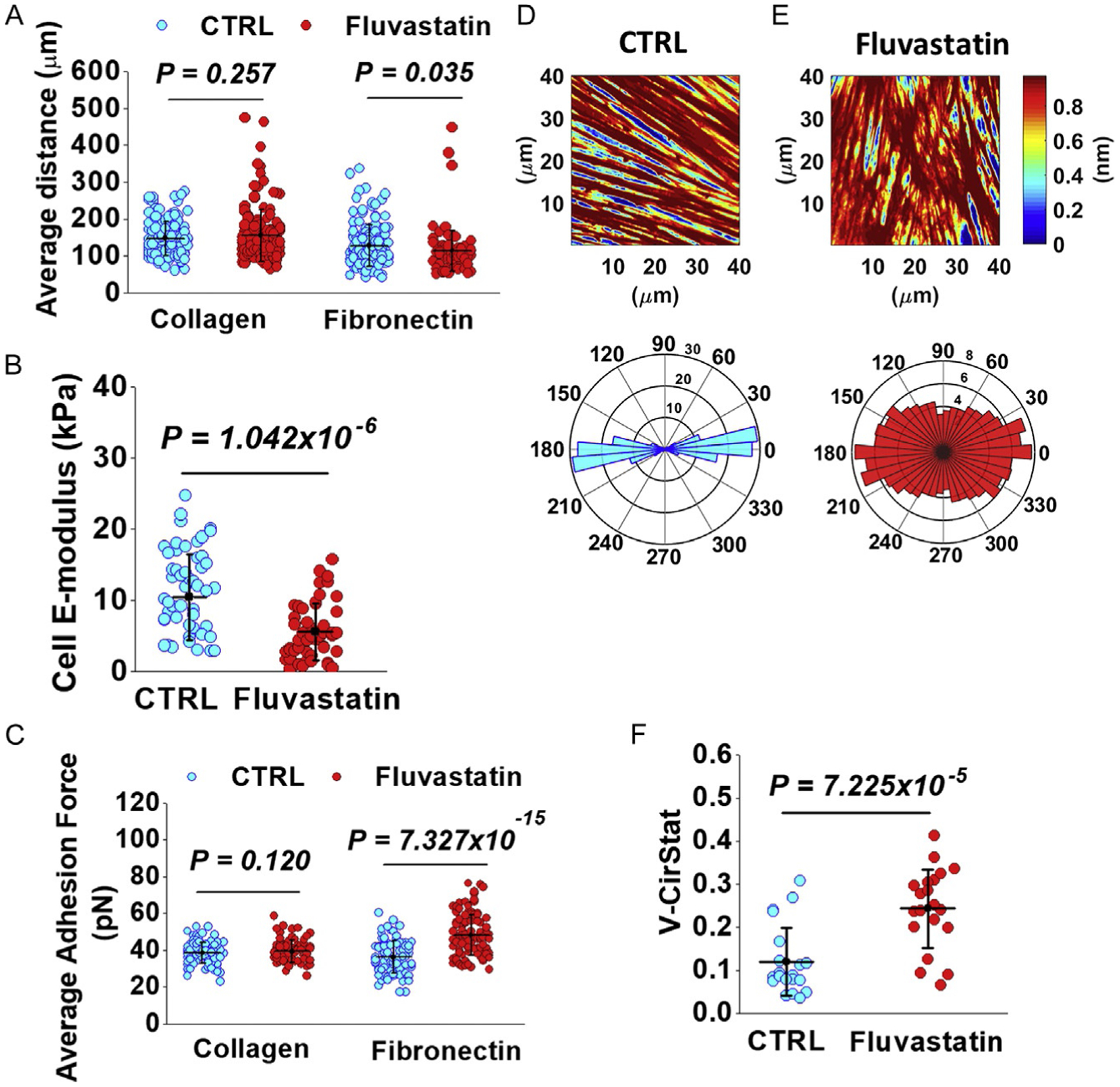
Effects of fluvastatin-mediated cholesterol depletion. (A) Fluvastatin-treated VSMCs migrating on fibronectin had significantly reduced migration distance while those on COL-1 had no difference in migration distance. (B) Fluvastatin significantly reduced VSMC E-modulus. (C) Adhesion to fibronectin significantly increased with fluvastatin treated while adhesion to COL-1 did not change. (D and E) (Top) Representative AFM height images of submembraneous actin for control and fluvastatin treatments, respectively. (Bottom) Corresponding circular distribution plots of actin orientation angles normalized to the dominant orientation angle. (F) Fluvastatin treatment significantly reduced submembraneous actin organization. Figure is adopted from Sanyour H. J., Li N., Rickel A. P., Torres H. M., Anderson R. H., Miles M. R., Childs J. D., Francis K. R., Tao J. & Hong Z. (2020). Statin mediated cholesterol depletion exerts coordinated effects on the alterations in rat vascular smooth muscle cell biomechanics and migration. The Journal of Physiology, 598, 1505–1522.
6. Conclusions and future directions
VSMCs undergo a myriad of changes as CVD and especially atherosclerosis progress. Recent discoveries have expanded the role of cholesterol in the progression of atherosclerosis and indicates the need for further exploration. Since VSMCs experience a wide variety of microenvironments in vivo, novel in vitro models need to be developed in order to study the mechanical contribution of VSMCs in CVD development. Using different inhibitors of cholesterol biosynthesis precursors to modulate cholesterol homeostasis might elucidate specific sterols that regulate VSMC functions. Interrogating the changes in VSMCs from atherosclerosis disease models is a logical and essential future direction. Our recent findings that both cholesterol and ECM stiffness coordinate and contribute to the changes in VSMC biomechanics sheds light on the complicated dynamics involved in atherosclerosis development (Fig. 5). Understanding and deciphering the molecular crosstalk mechanisms between the ECM and cholesterol will enhance the discovery of new targets that potentially hinder atherosclerosis development.
Fig. 5.
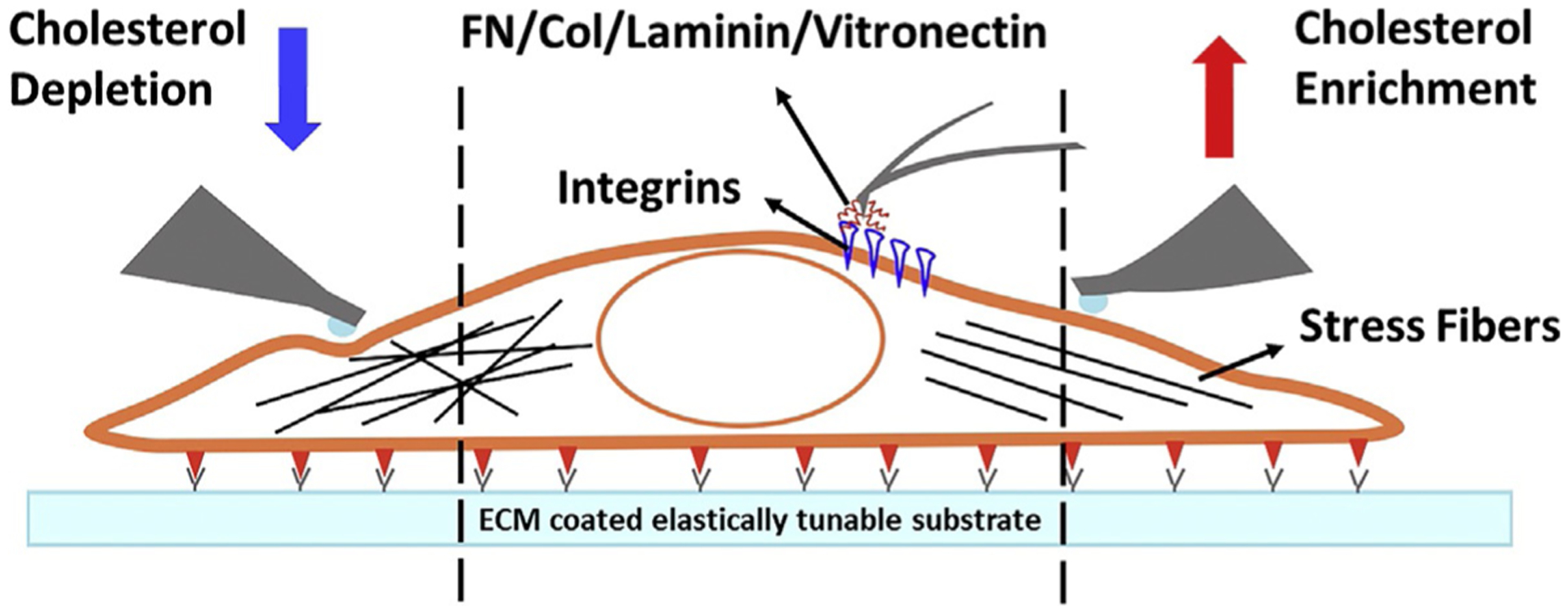
VSMC biomechanics obtained from AFM. (Left) VSMC stiffness and cytoskeletal orientation decreases following cholesterol depletion. (Center) AFM stylus tip is coated with protein to measure adhesion forces. (Right) VSMC stiffness and cytoskeletal orientation increases following cholesterol enrichment.
Acknowledgment
This work was supported, in part, by American Heart Association 15SDG25420001 (to Z. H.), South Dakota Board of Regents UP1600205 (to Z. H.), and NIH R15HL147214 (Z. H.).
References
- Allahverdian S, Chehroudi AC, McManus BM, Abraham T, & Francis GA (2014). Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation, 129, 1551–1559. [DOI] [PubMed] [Google Scholar]
- Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, & Mallat Z (2019). Vascular smooth muscle cells in atherosclerosis. Nature Reviews Cardiology, 16, 727–744. [DOI] [PubMed] [Google Scholar]
- Baumgartner W, Weth A, Gutberlet J, Harms G, & Groschner K (2014). Localization of VE-cadherin in plasmalemmal cholesterol rich microdomains and the effects of cholesterol depletion on VE-cadherin mediated cell-cell adhesion. Biochimica et Biophysica Acta (BBA)—Molecular and Cell Biology of Lipids, 1841, 1725–1732. [DOI] [PubMed] [Google Scholar]
- Bellosta S, Arnaboldi L, Gerosa L, Canavesi M, Parente R, Baetta R, et al. (2004). Statins effect on smooth muscle cell proliferation. Seminars in Vascular Medicine, 4, 347–356. [DOI] [PubMed] [Google Scholar]
- Benetos A, Laurent S, Asmar RG, & Lacolley P (1997). Large artery stiffness in hypertension. Journal of Hypertension Supplement, 15, S89–S97. [DOI] [PubMed] [Google Scholar]
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. (2019). Heart disease and stroke statistics-2019 update: A report from the American Heart Association. Circulation, 139, e56–e528. [DOI] [PubMed] [Google Scholar]
- Bialecki RA, & Tulenko TN (1989). Excess membrane cholesterol alters calcium channels in arterial smooth muscle. American Journal of Physiology-Cell Physiology, 257, C306–C314. [DOI] [PubMed] [Google Scholar]
- Binnig G, Quate CF, & Gerber C (1986). Atomic force microscope. Physical Review Letters, 56, 930. [DOI] [PubMed] [Google Scholar]
- Brown XQ, Bartolak-Suki E, Williams C, Walker ML, Weaver VM, & Wong JY (2010). Effect of substrate stiffness and PDGF on the behavior of vascular smooth muscle cells: Implications for atherosclerosis. Journal of Cellular Physiology, 225, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byfield FJ, Aranda-Espinoza H, Romanenko VG, Rothblat GH, & Levitan I (2004). Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophysical Journal, 87, 3336–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byfield FJ, Tikku S, Rothblat GH, Gooch KJ, & Levitan I (2006). OxLDL increases endothelial stiffness, force generation, and network formation. Journal of Lipid Research, 47, 715–723. [DOI] [PubMed] [Google Scholar]
- Carisey A, Tsang R, Greiner AM, Nijenhuis N, Heath N, Nazgiewicz A, et al. (2013). Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Current Biology, 23, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Preston Mason R, & Tulenko TN (1995). Atherosclerosis alters the composition, structure and function of arterial smooth muscle cell plasma membranes. Biochimica et Biophysica Acta, Molecular Basis of Disease, 1272, 101–112. [DOI] [PubMed] [Google Scholar]
- Choi ET, Khan MF, Leidenfrost JE, Collins ET, Boc KP, Villa BR, et al. (2004). β3-Integrin mediates smooth muscle cell accumulation in neointima after carotid ligation in mice. Circulation, 109, 1564. [DOI] [PubMed] [Google Scholar]
- Corsini A, Pazzucconi F, Pfister P, Paoletti R, & Sirtori CR (1996). Inhibitor of proliferation of arterial smooth-muscle cells by fluvastatin. The Lancet, 348, 1584. [DOI] [PubMed] [Google Scholar]
- Critchley DR (2000). Focal adhesions—The cytoskeletal connection. Current Opinion in Cell Biology, 12, 133–139. [DOI] [PubMed] [Google Scholar]
- Dao HH, Essalihi R, Bouvet C, & Moreau P (2005). Evolution and modulation of age-related medial elastocalcinosis: Impact on large artery stiffness and isolated systolic hypertension. Cardiovascular Research, 66, 307–317. [DOI] [PubMed] [Google Scholar]
- del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, & Sheetz MP (2009). Stretching single talin rod molecules activates vinculin binding. Science, 323, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME, et al. (2015). Fibronectin splicing variants containing extra domain a promote atherosclerosis in mice through toll-like receptor 4. Arteriosclerosis, Thrombosis, and Vascular Biology, 35, 2391–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi A, Slater SC, & George SJ (2009). MMP-9 and −12 cause N-cadherin shedding and thereby beta-catenin signalling and vascular smooth muscle cell proliferation. Cardiovascular Research, 81, 178–186. [DOI] [PubMed] [Google Scholar]
- Fuentes DE, & Butler PJ (2012). Coordinated mechanosensitivity of membrane rafts and focal adhesions. Cellular and Molecular Bioengineering, 5, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaballa MA, Jacob CT, Raya TE, Liu J, Simon B, & Goldman S (1998). Large artery remodeling during aging: Biaxial passive and active stiffness. Hypertension, 32, 437–443. [DOI] [PubMed] [Google Scholar]
- Glass CK, & Witztum JL (2001). Atherosclerosis. The road ahead. Cell, 104, 503–516. [DOI] [PubMed] [Google Scholar]
- Glasser SP (2000). On arterial physiology, pathophysiology of vascular compliance, and cardiovascular disease. Heart Disease, 2, 375–379. [PubMed] [Google Scholar]
- Gleason MM, Medow MS, & Tulenko TN (1991). Excess membrane cholesterol alters calcium movements, cytosolic calcium levels, and membrane fluidity in arterial smooth muscle cells. Circulation Research, 69, 216–227. [DOI] [PubMed] [Google Scholar]
- Gomez D, Shankman LS, Nguyen AT, & Owens GK (2013). Detection of histone modifications at specific gene loci in single cells in histological sections. Nature Methods, 10, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Sarangi BR, Deschamps J, Nematbakhsh Y, Callan-Jones A, Margadant F, et al. (2015). Adaptive rheology and ordering of cell cytoskeleton govern matrix rigidity sensing. Nature Communications, 6, 7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Medley SC, Hu T, Hinsdale ME, Lupu F, Virmani R, et al. (2015). PDGFRβ signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nature Communications, 6, 7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius J, Heisenberg C-P, Gaub HE, & Muller DJ (2008). Single-cell force spectroscopy. Journal of Cell Science, 121, 1785–1791. [DOI] [PubMed] [Google Scholar]
- Hillis G, & Flapan A (1998). Cell adhesion molecules in cardiovascular disease: A clinical perspective. Heart, 79, 429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis GS, Mlynski RA, Simpson JG, & MacLeod AM (1998). The expression of β1 integrins in human coronary artery. Basic Research in Cardiology, 93, 295–302. [DOI] [PubMed] [Google Scholar]
- Hong Z, Ersoy I, Sun M, Bunyak F, Hampel P, Hong Z, et al. (2013). Influence of membrane cholesterol and substrate elasticity on endothelial cell spreading behavior. Journal of Biomedical Materials Research. Part A, 101, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Sun Z, Li Z, Mesquitta W-T, Trzeciakowski JP, & Meininger GA (2012). Coordination of fibronectin adhesion with contraction and relaxation in microvascular smooth muscle. Cardiovascular Research, 96, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Zhao G, Hong Z, Sun Z, Yang Y, Clifford PS, et al. (2016). Mechanical activation of angiotensin II type 1 receptors causes actin remodelling and myogenic responsiveness in skeletal muscle arterioles. The Journal of Physiology, 594, 7027–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz AR, & Parsons JT (1999). Cell migration—Movin’ on. Science, 286, 1102–1103. [DOI] [PubMed] [Google Scholar]
- Hoshiga M, Alpers CE, Smith LL, Giachelli CM, & Schwartz SM (1995). Integrin expression in normal and atherosclerotic artery. Circulation Research, 77, 1129. [DOI] [PubMed] [Google Scholar]
- Huang H, Sun Z, Hill MA, & Meininger GA (2018). A calcium mediated mechanism coordinating vascular smooth muscle cell adhesion during KCl activation. Frontiers in Physiology, 9, 1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey JD, Dufresne ER, & Schwartz MA (2014). Mechanotransduction and extracellular matrix homeostasis. Nature Reviews. Molecular Cell Biology, 15, 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, et al. (2011). Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Science Translational Medicine, 3, 112ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg BC, Dimilla PA, Walker M, Kim S, & Wong JY (2009). Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength. Biophysical Journal, 97, 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain MK, & Ridker PM (2005). Anti-inflammatory effects of statins: Clinical evidence and basic mechanisms. Nature Reviews. Drug Discovery, 4, 977–987. [DOI] [PubMed] [Google Scholar]
- Jeon JH, Kim SK, Kim HJ, Chang J, Ahn CM, & Chang YS (2010). Lipid raft modulation inhibits NSCLC cell migration through delocalization of the focal adhesion complex. Lung Cancer, 69, 165–171. [DOI] [PubMed] [Google Scholar]
- Johnson JL (2014). Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovascular Research, 103, 452–460. [DOI] [PubMed] [Google Scholar]
- Kappert K, Blaschke F, Meehan WP, Kawano H, Grill M, Fleck E, et al. (2001). Integrins αvβ3 and αvβ5 mediate VSMC migration and are elevated during neointima formation in the rat aorta. Basic Research in Cardiology, 96, 42–49. [DOI] [PubMed] [Google Scholar]
- Khatibzadeh N, Spector AA, Brownell WE, & Anvari B (2013). Effects of plasma membrane cholesterol level and cytoskeleton F-actin on cell protrusion mechanics. PLoS One, 8, e57147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalsky GB, Byfield FJ, & Levitan I (2008). oxLDL facilitates flow-induced realignment of aortic endothelial cells. American Journal of Physiology. Cell Physiology, 295, C332–C340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg M, Fläschner G, Alsteens D, Gaub BM, Roos WH, Wuite GJL, et al. (2019). Atomic force microscopy-based mechanobiology. Nature Reviews. Physics, 1, 41–57. [Google Scholar]
- Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, & Edidin M (2003). Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proceedings of the National Academy of Sciences of the United States of America, 100, 13964–13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG (2003). Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part III: Cellular and molecular clues to heart and arterial aging. Circulation, 107, 490–497. [DOI] [PubMed] [Google Scholar]
- Laufs U, Marra D, Node K, & Liao JK (1999). 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1). The Journal of Biological Chemistry, 274, 21926–21931. [DOI] [PubMed] [Google Scholar]
- Lemarie CA, Tharaux PL, & Lehoux S (2010). Extracellular matrix alterations in hypertensive vascular remodeling. Journal of Molecular and Cellular Cardiology, 48, 433–439. [DOI] [PubMed] [Google Scholar]
- Levitan I, & Shentu T-P (2011). Impact of oxLDL on cholesterol-rich membrane rafts. Journal of Lipids, 2011, 730209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw L, Skinner MP, Raines EW, Ross R, Cheresh DA, Schwartz SM, et al. (1995). The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins. Role of alpha v beta 3 in smooth muscle cell migration to osteopontin in vitro. The Journal of Clinical Investigation, 95, 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, & Maseri A (2002). Inflammation and atherosclerosis. Circulation, 105, 1135–1143. [DOI] [PubMed] [Google Scholar]
- Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. (2013). Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation, 128, 2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon CA, Johnson JL, White S, Sala-Newby GB, & George SJ (2014). EC4, a truncation of soluble N-cadherin, reduces vascular smooth muscle cell apoptosis and markers of atherosclerotic plaque instability. Molecular Therapy—Methods & Clinical Development, 1, 14004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Hill MA, & Meininger GA (2009). The plastic nature of the vascular wall: A continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology (Bethesda), 24, 45–57. [DOI] [PubMed] [Google Scholar]
- Mason RP, & Jacob RF (2003). Membrane microdomains and vascular biology: Emerging role in atherogenesis. Circulation, 107, 2270–2273. [DOI] [PubMed] [Google Scholar]
- McDaniel DP, Shaw GA, Elliott JT, Bhadriraju K, Meuse C, Chung KH, et al. (2007). The stiffness of collagen fibrils influences vascular smooth muscle cell phenotype. Biophysical Journal, 92, 1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JS, Brown WS, Woodside DG, Vanderslice P, & McIntyre BW (2009). Clustering T-cell GM1 lipid rafts increases cellular resistance to shear on fibronectin through changes in integrin affinity and cytoskeletal dynamics. Immunology and Cell Biology, 87, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. (2015). Heart disease and stroke statistics—2015 Update: A report from the American Heart Association. Circulation, 131, e29–322. [DOI] [PubMed] [Google Scholar]
- Mui KL, Bae YH, Gao L, Liu S-L, Xu T, Radice GL, et al. (2015). N-cadherin induction by ECM stiffness and FAK overrides the spreading requirement for proliferation of vascular smooth muscle cells. Cell Reports, 10, 1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Qin C, Grosheva I, Maxfield FR, & Pierini LM (2007). Elevated cholesterol levels in the plasma membranes of macrophages inhibit migration by disrupting RhoA regulation. Arteriosclerosis, Thrombosis, and Vascular Biology, 27, 1596–1602. [DOI] [PubMed] [Google Scholar]
- Neumann T (2008). Determining the elastic modulus of biological samples using atomic force microscopy. In JPK instruments application report (pp. 1–9). [Google Scholar]
- Oesterle A, Laufs U, & Liao JK (2017). Pleiotropic effects of statins on the cardiovascular system. Circulation Research, 120, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmacek MS (2008). Myocardin: Dominant driver of the smooth muscle cell contractile phenotype. Arteriosclerosis, Thrombosis, and Vascular Biology, 28, 1416–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton SR, Kim PD, Ghajar CM, Seliktar D, & Putnam AJ (2008). The effects of matrix stiffness and RhoA on the phenotypic plasticity of smooth muscle cells in a 3-D biosynthetic hydrogel system. Biomaterials, 29, 2597–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyton SR, & Putnam AJ (2005). Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. Journal of Cellular Physiology, 204, 198–209. [DOI] [PubMed] [Google Scholar]
- Pokharel SM, Shil NK, Gc JB, Colburn ZT, Tsai SY, Segovia JA, et al. (2019). Integrin activation by the lipid molecule 25-hydroxycholesterol induces a proinflammatory response. Nature Communications, 10, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramprasad OG, Srinivas G, Rao KS, Joshi P, Thiery JP, Dufour S, et al. (2007). Changes in cholesterol levels in the plasma membrane modulate cell signaling and regulate cell adhesion and migration on fibronectin. Cell Motility and the Cytoskeleton, 64, 199–216. [DOI] [PubMed] [Google Scholar]
- Rickel AP, Sanyour HJ, Leyda NA, & Hong Z (2020). Extracellular matrix proteins and substrate stiffness synergistically regulate vascular smooth muscle cell migration and cortical cytoskeleton organization. ACS Applied Bio Materials, 3, 2360–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudijanto A (2007). The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Medica Indonesiana, 39, 86–93. [PubMed] [Google Scholar]
- Sakamoto K, Murata T, Chuma H, Hori M, & Ozaki H (2005). Fluvastatin prevents vascular hyperplasia by inhibiting phenotype modulation and proliferation through extracellular signal-regulated kinase 1 and 2 and p38 mitogen-activated protein kinase inactivation in organ-cultured artery. Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 327–333. [DOI] [PubMed] [Google Scholar]
- Sanyour H, Childs J, Meininger GA, & Hong Z (2018). Spontaneous oscillation in cell adhesion and stiffness measured using atomic force microscopy. Scientific Reports, 8, 2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyour HJ, Li N, Rickel AP, Childs JD, Kinser CN, & Hong Z (2019). Membrane cholesterol and substrate stiffness co-ordinate to induce the remodelling of the cytoskeleton and the alteration in the biomechanics of vascular smooth muscle cells. Cardiovascular Research, 115, 1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyour HJ, Li N, Rickel AP, Torres HM, Anderson RH, Miles MR, et al. (2020). Statin mediated cholesterol depletion exerts coordinated effects on the alterations in rat vascular smooth muscle cell biomechanics and migration. The Journal of Physiology, 598, 1505–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazonova OV, Lee KL, Isenberg BC, Rich CB, Nugent MA, & Wong JY (2011). Cell-cell interactions mediate the response of vascular smooth muscle cells to substrate stiffness. Biophysical Journal, 101, 622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scandinavian Simvastatin Survival Study Group. (1994). Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). The Lancet, 344, 1383–1389. [PubMed] [Google Scholar]
- Schwartz MA, & Ginsberg MH (2002). Networks and crosstalk: Integrin signalling spreads. Nature Cell Biology, 4, E65–E68. [DOI] [PubMed] [Google Scholar]
- Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP, Hong Z, Hunter WC, et al. (2013). Increased vascular smooth muscle cell stiffness: A novel mechanism for aortic stiffness in hypertension. American Journal of Physiology. Heart and Circulatory Physiology, 305, H1281–H1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Demosky SJ, Stonik JA, Furuya Y, Koike H, Suzuki K, et al. (2010). High-density lipoprotein induces proliferation and migration of human prostate androgen-independent cancer cells by an ABCA1-dependent mechanism. Molecular Cancer Research, 8, 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. (2015). KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature Medicine, 21, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. (1995). Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. The New England Journal of Medicine, 333, 1301–1307. [DOI] [PubMed] [Google Scholar]
- Simons K, & Ikonen E (1997). Functional rafts in cell membranes. Nature, 387, 569–572. [DOI] [PubMed] [Google Scholar]
- Stegemann JP, Hong H, & Nerem RM (2005). Mechanical, biochemical, and extracellular matrix effects on vascular smooth muscle cell phenotype. Journal of Applied Physiology, 98, 2321–2327. [DOI] [PubMed] [Google Scholar]
- Stossel TP (1993). On the crawling of animal cells. Science, 260, 1086–1094. [DOI] [PubMed] [Google Scholar]
- Sun Z, Martinez-Lemus LA, Hill MA, & Meininger GA (2008). Extracellular matrix-specific focal adhesions in vascular smooth muscle produce mechanically active adhesion sites. American Journal of Physiology. Cell Physiology, 295, C268–C278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Northup N, Marga F, Huber T, Byfield FJ, Levitan I, et al. (2007). The effect of cellular cholesterol on membrane-cytoskeleton adhesion. Journal of Cell Science, 120, 2223–2231. [DOI] [PubMed] [Google Scholar]
- Taulet N, Comunale F, Favard C, Charrasse S, Bodin S, & Gauthier-Rouviere C (2009). N-cadherin/p120 catenin association at cell-cell contacts occurs in cholesterol-rich membrane domains and is required for RhoA activation and myogenesis. The Journal of Biological Chemistry, 284, 23137–23145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracqui P, Broisat A, Toczek J, Mesnier N, Ohayon J, & Riou L (2011). Mapping elasticity moduli of atherosclerotic plaque in situ via atomic force microscopy. Journal of Structural Biology, 174, 115–123. [DOI] [PubMed] [Google Scholar]
- Tulenko TN, Chen M, Mason PE, & Mason RP (1998). Physical effects of cholesterol on arterial smooth muscle membranes: Evidence of immiscible cholesterol domains and alterations in bilayer width during atherogenesis. Journal of Lipid Research, 39, 947–956. [PubMed] [Google Scholar]
- Wang X, Keith JC Jr., Struthers AD, & Feuerstein GZ (2008). Assessment of arterial stiffness, a translational medicine biomarker system for evaluation of vascular risk. Cardiovascular Therapeutics, 26, 214–223. [DOI] [PubMed] [Google Scholar]
- Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, et al. (2006). Matrix metalloproteinase 2 activation of transforming growth factor-beta1 (TGF-beta1) and TGF-beta1-type II receptor signaling within the aged arterial wall. Arteriosclerosis, Thrombosis, and Vascular Biology, 26, 1503–1509. [DOI] [PubMed] [Google Scholar]
- Ward NC, Watts GF, & Eckel RH (2019). Statin toxicity. Circulation Research, 124, 328–350. [DOI] [PubMed] [Google Scholar]
- Willis AI, Pierre-Paul D, Sumpio BE, & Gahtan V (2004). Vascular smooth muscle cell migration: Current research and clinical implications. Vascular and Endovascular Surgery, 38, 11–23. [DOI] [PubMed] [Google Scholar]
- Worth NF, Rolfe BE, Song J, & Campbell GR (2001). Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motility and the Cytoskeleton, 49, 130–145. [DOI] [PubMed] [Google Scholar]
- Xu J, & Shi G-P (2014). Vascular wall extracellular matrix proteins and vascular diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1842, 2106–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, Xia C, Duan C, Li S, & Mei Z (2011). Biological characteristics of foam cell formation in smooth muscle cells derived from bone marrow stem cells. International Journal of Biological Sciences, 7, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Goult BT, Chen H, Cong P, Sheetz MP, & Yan J (2014). Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Scientific Reports, 4, 4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Qiu H, Trzeciakowski JP, Sun Z, Li Z, Hong Z, et al. (2012). Temporal analysis of vascular smooth muscle cell elasticity and adhesion reveals oscillation waveforms that differ with aging. Aging Cell, 11, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, & Levitan I (2007). Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1768, 1311–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]