

Abstract
Myeloid derived suppressor cells (MDSC) suppress the ability of cytotoxic T cells to attack and clear tumor cells from the body. The active form of vitamin D, 1,25 dihydroxyvitamin D (1,25(OH)2D), regulates myeloid cell biology and previous research showed that in mouse models 1,25(OH)2D reduced the tumor level of CD34+ cells, an MDSC precursor, and reduced metastasis. We tested whether MDSC are vitamin D target cells by examining granulocytic- (G-MDSC) and monocytic (M-MDSC) MDSC from tumors, spleen, and bone marrow. Vitamin D receptor (VDR) mRNA levels are low in MDSC from bone marrow and spleen but are 20-fold higher in tumor MDSC. At all sites, M-MDSC have 4-fold higher VDR mRNA expression than G-MDSC. Bone marrow MDSC were induced to differentiate in vitro into tumor MDSC-like cells by treating with IFN-γ, IL-13, and GM-CSF for 48 h. This treatment significantly elevated Arg1 and Nos2 levels, activated the T cell-suppressive function of MDSC, increased VDR expression 50-fold, and made the MDSC responsive to 1,25(OH)2D treatment. Importantly, 1,25(OH)2D treatment reduced the T cell suppressive capacity of cytokine-induced total MDSC and M-MDSC by ≥70 % and tumor-derived M-MDSC by 30–50 %. Consistent with this finding, VDR deletion (KO) increased T cell suppressive function of in vitro M-MDSC by 30 % and of tumor-derived M-MDSC by 50 % and G-MDSC by 400 %. VDR KO did not alter Nos2 mRNA levels but significantly increased Arg1 mRNA levels in tumor M-MDSC by 100 %. In contrast, 1,25(OH)2D treatment reduced nitric oxide production in both in vitro derived M- and G-MDSC. The major finding of this study is that 1,25(OH)2D signaling through the VDR decreases the immunosuppressive capability of MDSC. Collectively, our data suggest that activation of vitamin D signaling could be used to suppress MDSC function and release a constraint on T-cell mediated clearance of tumor cells.
Keywords: Vitamin D receptor, Myeloid derived suppressor cell, Cancer, Immunology
1. Introduction
Some tumors express antigens that can be recognized by the immune system and this leads to the activation of cytotoxic T cells that kill and clear tumor cells from the body [1]. This knowledge has led to the development of therapies that enhance anti-tumor immunity e.g. immune checkpoint blockade [2]. Unfortunately, not all tumors respond to immunotherapy. This resistance is due in part to the fact that the tumor can combat host immunity by producing immunomodulatory proteins that establish an immunosuppressive environment in the tumor [3]. One of the immunosuppressive cells in the tumor microenvironment is the myeloid derived suppressor cell (MDSC). MDSC have several functions that permit tumor survival including the ability to promote angiogenesis, stimulate the production of regulatory T cells (Treg), and inhibit T cell antitumor activity [4]. There are two MDSC subtypes; monocyte-like MDSC (M-MDSC) have a greater ability to suppress T cell proliferation and function than granulocyte-like MDSC (G-MDSC) [5,6]. As a result of their pro-tumor functions, the presence of MDSC in tumors is associated with cancer progression in mice and humans [7–10] and researchers are actively exploring how to block MDSC function to enhance T cell-driven immunotherapy in humans [11].
Vitamin D is produced in ultraviolet B exposed skin or obtained from the diet. Its metabolite, 1,25 dihydroxyvitamin D (1,25 (OH)2D), is a hormone that regulates gene transcription through the vitamin D receptor (VDR) to control calcium and bone metabolism [12,13]. However, in addition to this classical role, 1,25 (OH)2D can also target immune cells to alter their fate or function [14,15]. In adaptive immunity, vitamin D may create a more tolerogenic T helper cell profile. Consistent with this concept, 1,25 (OH)2D treatment in vitro suppresses NFκB signaling necessary for Th1 cell activation [16] and blocks development of Th17 and Th9 cells implicated in the pathogenesis of different types of autoimmunity and inflammatory diseases [17]. In the innate immune system 1,25(OH)2D stimulates differentiation of monocytes to macrophages [18] and regulates genes crucial for autophagy and anti-microbial actions [19–22]. In addition, 1,25(OH)2D reprograms dendritic cells (DC) to become tolerogenic in an inflammatory setting [23] by altering DC differentiation as well as the function of tolerogenic DC [24]. However, in tumors DC serve as essential antigen presenting cells that assist with effective T cell surveillance [25,26] and there is some evidence that 1,25 (OH)2D promotes DC development in this context [27].
There is growing evidence that vitamin D signaling may be a regulator of MDSC biology. The earliest of these studies examined the impact of vitamin D on CD34+ cells that are precursors for MDSC [28]. 1,25(OH)2D treatment can block the development of immunosuppressive function in cultured CD34+ bone marrow cells [29] and reduce CD34+ cell number while increasing the number of CD8 + T cells in mouse Lewis Lung Carcinoma cell (LLC-LN7) tumors [30]. Consistent with these actions, 1,25(OH)2D treatment in mice with 4-NQO-induced squamous cell carcinoma decreased invasive cancer, reduced tumor MDSC number, blocked IL-6 induced recruitment of MDSC, and reduced the T cell suppressive capacity of MDSC from the tumor. [31]. Several groups have also demonstrated that 1,25(OH)2D may interfere with the production of tumor-derived signals that promote MDSC differentiation, e.g. GM-CSF [29,30], IL-6 [31], and miR155-containing exosomes [32]. However, formal proof that tumor MDSC or their precursors in the periphery are vitamin D target cells is lacking. Here we report evidence for the direct actions of 1,25(OH)2D on MDSC.
2. Materials and methods
2.1. Reagents
Routine laboratory chemicals and 1,25 dihyroxyvitamin D were purchased from Sigma Chemical (St. Louis, MO). 0.25 % Trypsin-EDTA, Hepes, sodium pyruvate, L-Glutamine, Penicillin/Streptomycin (10,000 units/mL), fetal bovine serum (FBS), RPMI 1640 (without glutamine, HEPES, or sodium pyruvate) and high glucose DMEM (4.5 g/L, without glutamine, HEPES, or sodium pyruvate) were purchased from Thermo Fisher Scientific (Waltham, MA). The EdU Click-iT kit, M-MLV reverse transcriptase, 5X first strand buffer, BSA, deoxynucleotide triphosphate, Rnasin, random hexamers, and oligo-dT primers were purchased from Thermo Fisher Scientific (Waltham, MA). Tri-reagent and Direct-zol RNA MiniPrep Plus kits were purchased from Zymo Research (Irvine, CA). Antibodies against CD11b (clone M1/70), Gr-1 (clone RB 6–8 C5), Ly6C (clone HK1.4) Ly6G (clon 1A8), CD8 (clone 53–6.7), TruStain FcX (clone 93), and Zombie Violet were purchased from Biolegend (San Diego, CA) while anti-Rat IgG compensation beads were purchased from BD Biosciences (San Jose, CA). All recombinant murine cytokines were purchased through PeproTech (Rocky Hill, NJ).
Animals: C57BL/6 J (WT) and VDR KO mice (JAX stock number: 006133) on the C57BL/6 J genetic background were maintained in breeding colonies. Male WT mice for routine experiments were fed a standard chow diet (Teklad 2018) and water ad libitum. When males WT mice were compared to VDR KO mice, littermates were maintained on AIN93 G or AIN93G-based rescue diet (20 % lactose, 2 % Ca, 1.25 % P, and 1000 IU/kg VD, Research Diets Inc.), respectively. Prostate OVA-expressing mice-3 (POET-3) mice were generated and maintained as previously described [33]. Rag−/− OT-I mice were generated and maintained as previously described [34]. Mice were used for experiments between 10 and 14 weeks of age. Mice were housed in specific pathogen-free conditions with a 12 h light/dark cycle. The Purdue University Animal Care and Use Committees approved all of the mouse use described in this paper.
2.2. Cell culture
2.2.1. RM-1 cells
a Ras+, Myc-induced mouse prostate cancer cell line from C57BL/6 mice (ATCC CRL-3310), were cultured in DMEM with 10 % fetal bovine serum, 1 mM sodium pyruvate, 10 mM HEPES, and 1 % penicillin-streptomycin at 37 °C in 5 % CO2. RM-1 cells were chosen as the experimental model because they are a model for a solid tumor, because RM-1 tumors have been shown to recruit active MDSC [35], and because they come from the same genetic background as VDR knockout mice.
2.2.2. MDSC generation and isolation
RM-1 cells were harvested during log phase growth and then 106 cells were suspended in 100 μL sterile PBS for injection into the peritoneal space of mice. 7 day later, MDSC were harvested via peritoneal lavage or isolated from the bone marrow or spleen. Cells were treated with ACK buffer (150 mM ammonium chloride, 10 mM potassium carbonate, 0.1 mM EDTA) to remove contaminating red cells then passed through a 70 u M filter. Cells were treated with TruStain FcX to block Fc receptors (1:50) and stained with Zombie Violet (1:100) in the dark at RT for 10 min. Single cell suspensions were labeled with fluorescence–labeled antibodies against CD11b, Ly6G, and Ly6C for MDSC subtypes or with fluorescence–labeled antibodies against CD11b and Gr-1 for total MDSC. Cells were sorted using fluorescence-activated cell sorting (FACS) at the Purdue Flow Core Facility (BD FACS Aria III). Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
2.3. In vitro MDSC
Bone marrow cells from 3 to 5 naïve, male WT mice were isolated and cultured in RPMI-1640 (10 % FBS, 5 % sodium pyruvate, 5 % Pen/Step, and 10 mM HEPES) with IFN-γ (25 ng/ml), IL-13 (33 ng/ml), and GM-CSF (10 ng/ml) for 48–72 h. During the last 24 h of the cytokine treatment, cells were treated with 0.1 % ethanol (control) or 10 nM 1,25(OH)2D. For all experiments, 105 cells were seeded into each well of a 96 well plate; within each replicate experiment, three wells were used for each treatment and wells were pooled at harvest for analysis. Following the treatment period, media and non-adherent cells were collected. Adherent cells were harvested with 0.25 % Trypsin-EDTA for 10–15 min or until cell layer had dispersed. Adherent and non-adherent cells were pooled and prepared for flow cytometry analysis or qPCR analysis.
2.4. RNA isolation and analysis
Cells were harvested into Tri-reagent, flash frozen in liquid nitrogen, and stored at −80 °C until all biological replicates were collected. RNA was isolated using the Direct-zol RNA MiniPrep Plus kit (Zymo Research, Irvine CA) following the manufacturer’s protocol. cDNA was created as we have previously reported (23) using an 1.5 h incubation at 37 °C. qPCR analysis was conducted as previously described (23) using premade PrimeTime® Primer/probe sets (IDT, Skokie, IL): Nos2 (IDT Assay: Mm.PT.56a.43705194), Arg1 (Mm.PT.58.8651372), VDR (Mm.PT.58.7050931),Cyp24a1 (Mm.PT.58.30780707). r18 s mRNA level (Hs.PT.39a.22214856.g) was used as a house keeping gene. Relative expression levels were determined using the −ΔΔCt method [36].
2.5. T cell proliferation suppression assays
An in vitro test was conducted to determine whether MDSC suppress the proliferation of activated T cells. MDSC were isolated from tumor or in vitro MDSC were prepared as described above. Splenocytes were isolated from Rag1−/− OT-I mice and resuspended to 1 × 106/ml in RPMI-1640 medium with 10 % fetal bovine serum, 5 % sodium pyruvate, 5 % penicillin-streptomycin, and 5 % 1 M HEPES. Live cells were purified on Fico/Lite gradients (Atlanta Biologicals). For short-term assays, tumor MDSC or in vivo generated MDSC were co-cultured with activated OT-I cells for 16 h. Prior to co-culture splenocytes were activated with 1 μg/mL SIINFEKL (ovalbumin peptide 357–264, American Peptide) for 48 h. In one experiment, co-cultured tumor MDSC and activated OT-I splenocytes were treated +/− 10 nM 1,25(OH)2D during the 18 h suppression assay. In this experiment, an additional set of wells with just activated OT-I splenocytes was examined to evaluate the direct impact of 1,25(OH)2D on T cell proliferation; these wells were also used as the reference to calculate suppression in the MDSC + OT1 + 1,25(OH)2D group. For long-term assays, tumor MDSC or in vitro generated MDSC were co-cultured with naive splenocytes for 24 h followed by an additional 24 h in the presence of 1 μg/mL SIINFEKL. A common method was used to assess T cell proliferation in both the short and long-term T cell suppression assays. At 1.5 h prior to cell harvest, 10 μM EdU was added and 1.5 h later cells were fixed and analyzed for EdU levels using the EdU Click-iT staining kit, following manufacturer instructions (Life Science Technologies, Waltham MA). In brief, following fixation cells were resuspended in permeabilization buffer for 10 min then samples were incubated with Click-iT reaction master mix for 30 min at RT. Samples were washed and resuspended in 150 ul permeabilization buffer then analyzed for CD8, CD11b, and EdU on a BD Fortessa cell analyzer. Data were analyzed using Flow Jo v.10 (Tree Star; Ashland, OR), i.e. selection of non-debris events, gating for single cells, biplots for cytotoxic T cells (CD11b- CD8+ cells) and for proliferating cells (SSC-A x EdU+). OT-I splenocytes incubated without EdU were used to create the negative EdU gate.
2.6. Griess Assay for nitrite production
Tumor M- and G-MDSC were isolated from the ip space of RM-1 tumor-bearing mice. Cells were cultured in RPMI + 20 % EL-4 tumor extract supernatant for 24 h in the presence or absence of 10 nM 1,25(OH)2D. Afterwards cell supernatants were harvested and analyzed for nitrite levels (a stable, non-volitile breakdown product of nitric oxide) using a Griess Assay (Promega, Madison, WI).
2.7. Statistics:
Statistical analyses were conducted using SAS Enterprise Guide v 5.1 (SAS Institute Inc. Cary, NC). Evaluation of histograms and the Shapiro-Wilk test of normality (p < 0.05) were used to assess whether the data were normally distributed. When data from flow cytometry were not normally distributed, data were transformed using cube-root, natural log, or (26) transformation. Significance was determined for T-cell suppression tests and for mRNA levels using one-way ANOVA or t-test. Some analyzes were analyzed with a split-plot design to account for the interdependence of responses within each replicate (i.e. due to day-to-day variability in the assay). Results are expressed as Mean +/− SEM of non-transformed data. A p-value < 0.05 was considered statistically significant.
3. Results
We examined the RNA from M- and G-MDSC isolated from bone marrow and spleen of C57BL/6 mice bearing RM-1 tumors as well as from the RM-1 tumors of these mice. In all three tissue compartments, VDR mRNA level was higher in M-MDSC compared to G-MDSC (23-, 8.9-, and 2.9 fold in bone marrow, spleen, and tumor, respectively). In addition, while the VDR mRNA levels of bone marrow and spleen MDSC were comparable, tumor MDSC VDR mRNA levels were significantly higher than in MDSC from the peripheral tissues for both G- (78–115 fold) and M- subtypes (15–26 fold) (Fig. 1). In contrast, mRNA levels for two vitamin D metabolizing enzymes, Cyp27b1 and Cyp24a1, were very low in both MDSC subtypes from all tissues and their levels were not increased in the tumor MDSC population.
Fig. 1.
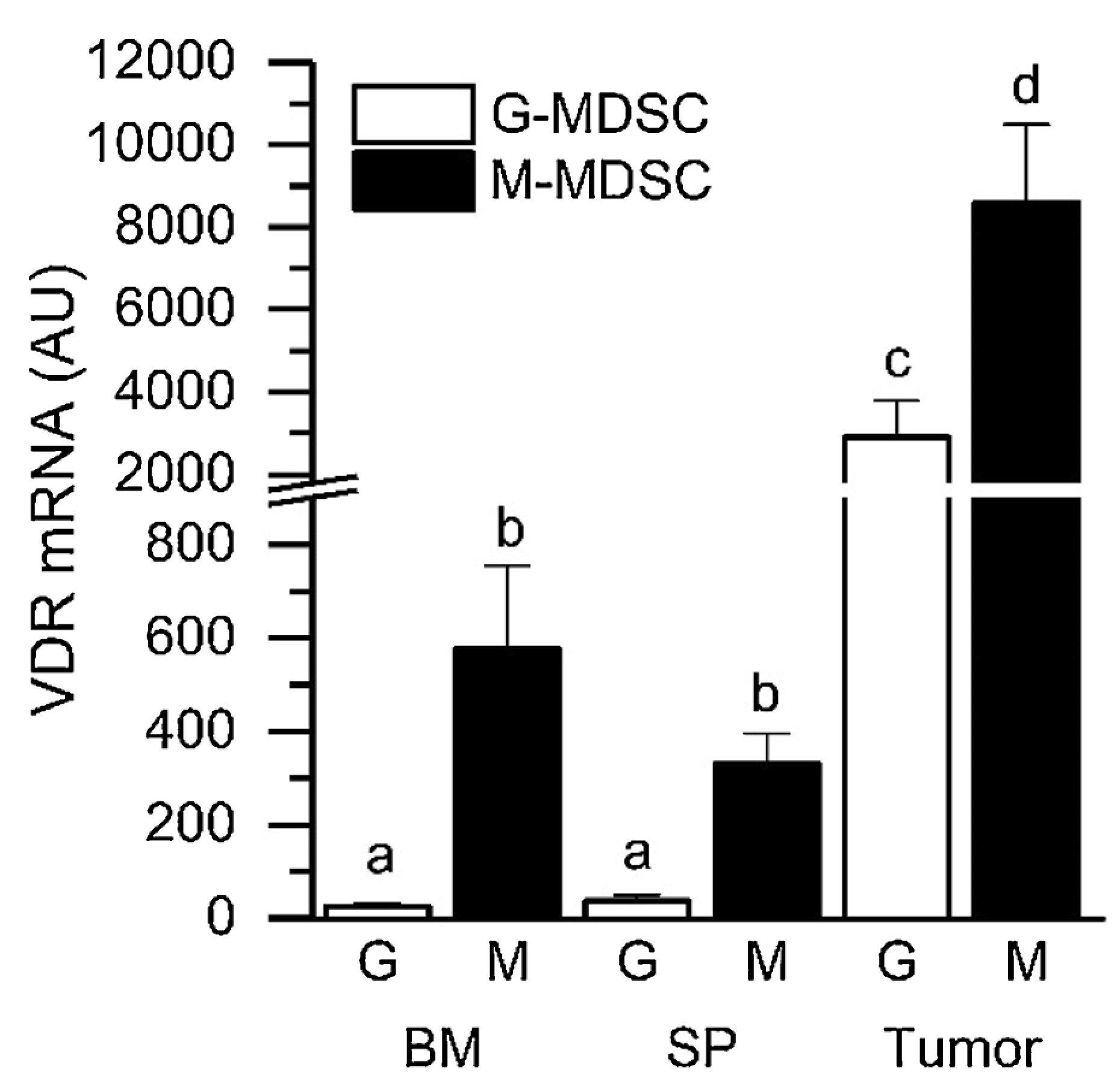
VDR Expression is Elevated in Tumor MDSC Subtypes. RM-1 cells were injected ip into 12–14 wk old male C57BL/6 mice. After 7 d, MDSC subtypes were isolated from the tumor-containing ip space (Tumor), the spleen (SP), and bone marrow (BM) via flow cytometry-based cell sorting. VDR mRNA levels were analyzed by qPCR. n=3 (SP) to 8 (BM, Tumor) biological replicates comprised of pools from 4 to 5 mice each. Bars with different letter superscripts are significantly different (p < 0.05).
To determine whether elevated VDR expression was a feature of MDSC differentiation, we took naïve bone marrow MDSC and induced their differentiation with a cocktail of IFNγ, IL-13, and GM-CSF. As expected, the cytokine treatment induced the expression of two classic markers of mature, tumor MDSC: Nos2 and Arg1. In addition, VDR mRNA levels increased by 108.9 fold after culture compared to pre-culture levels. When 1,25(OH)2D was added for the last 24 h of the 48 h in vitro differentiation protocol, both VDR mRNA and Cyp24 mRNA levels significantly increased (Fig. 2), indicating the treatment made the in vitro MDSC vitamin D target cells.
Fig. 2.
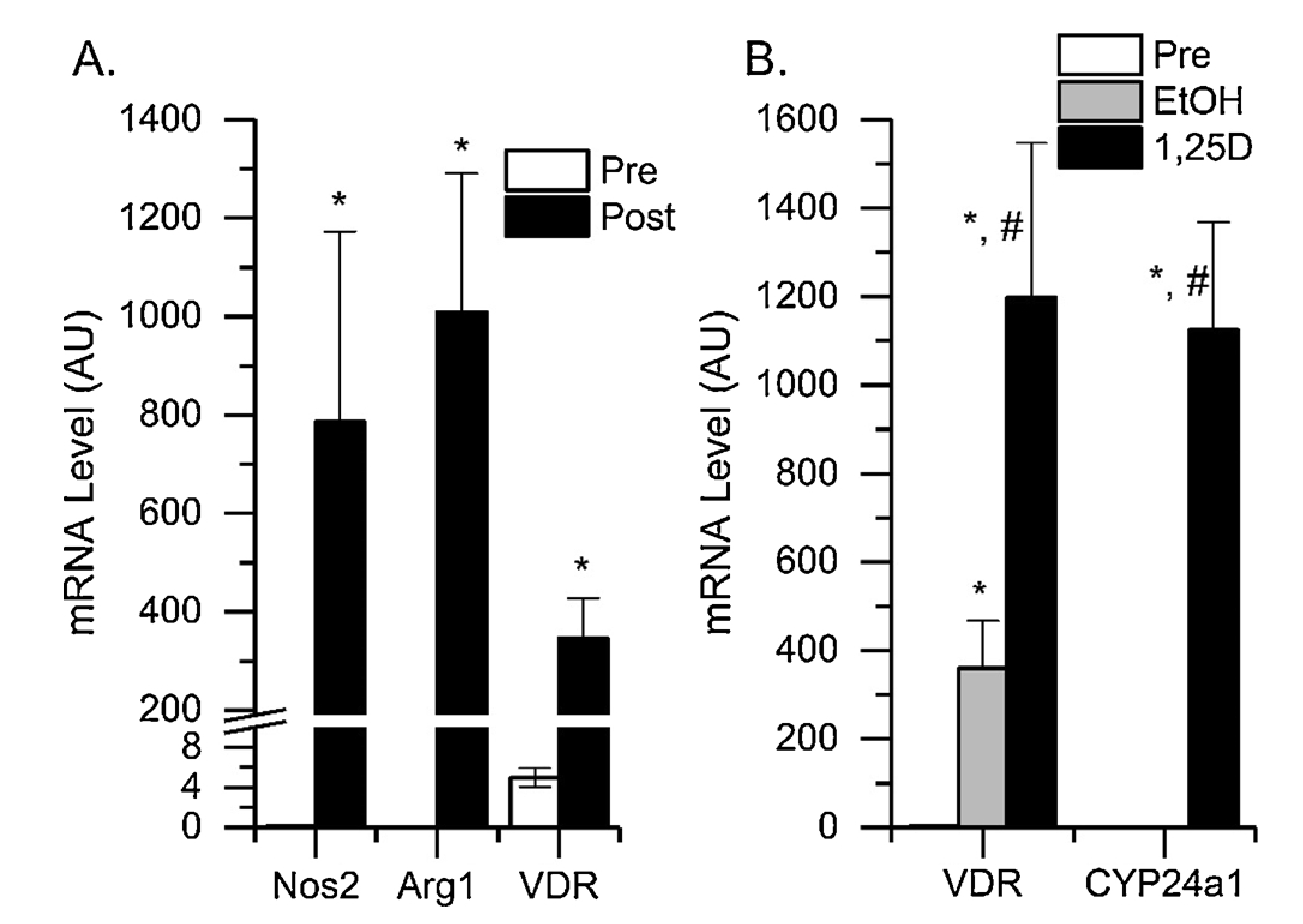
In vitro Induction of MDSC Differentiation Stimulates VDR Expression and Makes MDSC Responsive to 1,25(OH)2D Treatment. Bone marrow MDSC (CD11b+Gr1+) were isolated from naïve male mice by FACS and then treated with a cocktail of 25 ng/ml IFNγ, 33 ng/ml IL-13, and 10 ug/ml GM-CSF for 48 h. (A) RNA was isolated from pre-culture (Pre) and post-culture (Post) cells then analyzed by qPCR for Arg1, Nos2 and VDR mRNA levels. * p < 0.05 vs pre-culture. (B) Bone Marrow MDSC were cultured with the three cytokines for 24 h and then treated for the last 24 h with cytokines combined with 10 nM 1,25(OH)2D (1,25D) or ethanol vehicle (EtOH) prior to analysis of RNA by qPCR for two classical vitamin D target genes, VDR and Cyp24a1. * p < 0.05 vs preculture; #p < 0.05 vs EtOH vehicle.
In vitro generated total MDSC and M-MDSC both develop the ability to suppress the proliferation of activated T cells and this can be detected in a short-term assay (1:1 MDSC:OT-I splenocytes in Fig. 3A and B). In addition, 1,25(OH)2D treatment for the last 24 h of the in vitro differentiation protocol significantly limited the suppressive function of these in vitro activated MDSC. Similarly, when we compared the ability of bone marrow M-MDSC from WT and VDR KO mice to develop T cell suppressive function in a long-term suppression assay, the VDR KO M-MDSC developed greater T cell suppressive function (Fig. 3C). Together, these data suggest that activation of vitamin D signaling through the VDR is an antagonist of the T cell suppressive function of total MDSC and M-MDSC.
Fig. 3.
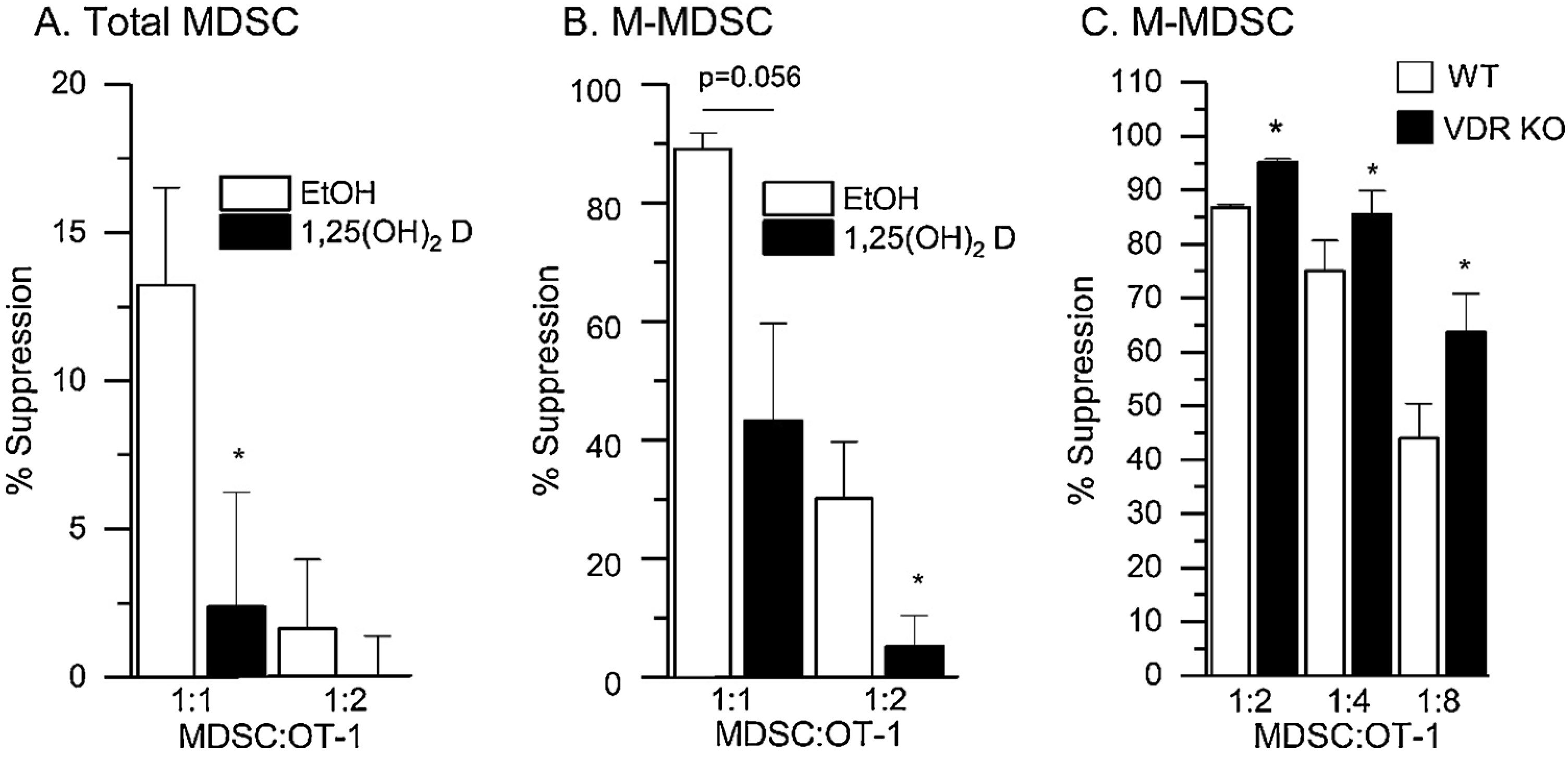
Vitamin D Signaling Inhibits the Suppression of T cell Proliferation by in vitro Induced MDSC. Bone marrow from naïve mice was used to isolate total MDSC (CD11b+Gr1+) or M-MDSC (CD11b+Ly6ChiLy6Glo) and these cells were then treated with a cocktail of 25 ng/ml IFNγ, 33 ng/ml IL-13, and 10 ug/ml GM-CSF for 48 h. After 24 h of cytokine treatment, the cells were treated with 10 nM 1,25(OH)2D or ethanol vehicle (EtOH) in the presence of cytokines for an additional 24 h. Afterwards, total MDSC (A) or M-MDSC (B) were isolated then co-cultured with activated OT-I splenocytes for 18 h. EdU was added for the last 1.5 h to assess T cell proliferation. Two different MDSC:OT-I ratios were used to assess the ability of MDSC to suppress T cell proliferation. (C) M-MDSC were isolated from bone marrow of naïve WT and VDR knockout (KO) Mice. Cells were co-cultured with OT-I splenocytes for 48 h. 24 h before harvest, ovalbumin peptide was added to activate splenocytes (SIINFKL to 1 ug/ml) and EdU was added for the last 1.5 h to assess T-cell proliferation. Differences between groups were determined on ArcSIN-transformed data by t-test (p < 0.05), n = 4 biological replicates of cells pooled from n = 3–5 mice. * p < 0.05 vs EtOH control at the same MDSC:OT-1 ratio.
To test whether vitamin D signaling modifies the T cell suppressive function of MDSC in the tumor microenvironment, we isolated cells from WT and VDR KO mice and examined their T cell suppressive function in a short-term assay. Total, M-, and G-MDSC from tumors were all highly suppressive (Fig. 4), with M-MDSC being more suppressive than G-MDSC (e.g. at 1:2 MDSC:OT-I ratio, M-MDSC were twice as suppressive as G-MDSC) (Fig. 4B, C). In addition, the T cell suppressive function of total, M-, and G-MDSC from tumors of VDR KO mice was significantly higher than for WT mice, e.g. at the 1:4 MDSC:OT-I ratio, VDR KO cells were 2–4 fold more suppressive than WT cells) (Fig. 4).
Fig. 4.
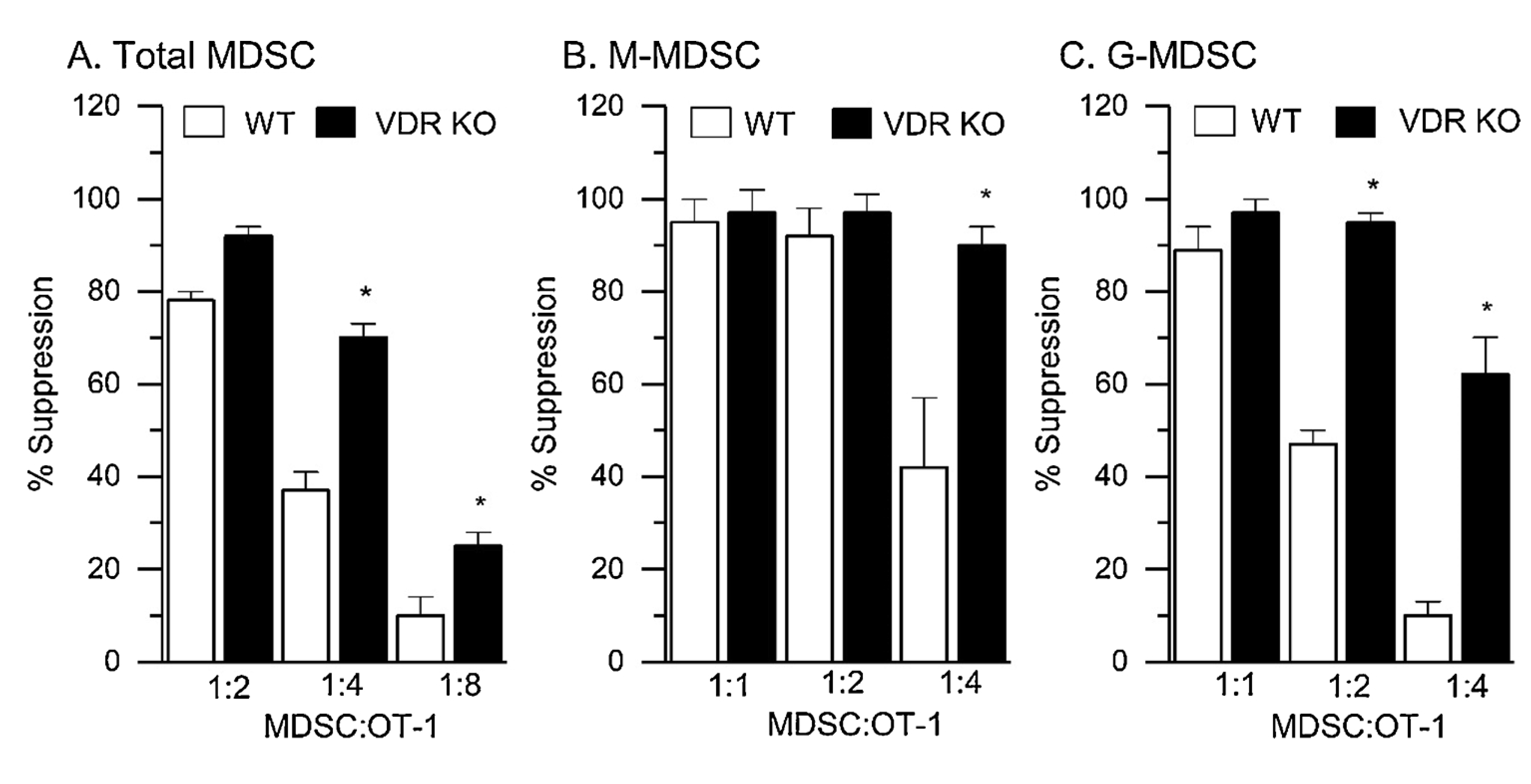
VDR Deletion (VDR KO) Enhances the Ability of Freshly Isolated tumor MDSC to Suppress T Cell Proliferation. RM-1 tumor cells were injected ip into wild-type (WT) and VDR KO mice and after 7 d, tumor MDSC were isolated from the ip space for use in an 18 h T cell proliferation suppression test using pre-activated OT-1 splenocytes. EdU was added for the last 1.5 h to assess T cell proliferation. (A) total tumor MDSC, (B) tumor M-MDSC, and (C) tumor G-MDSC were studied at three MDSC:OT-I cell ratios to capture the maximal suppressive effect on T cell proliferation (n = 3 independent biological replicates from pools of 3–5 mice) * p < 0.05 vs WT at a specific ratio.
Elevated Nos2 and Arg1 mRNA levels are thought to be essential for the T cell suppressive function of MDSC [37]. We tested whether VDR deletion would increase the expression of these essential genes. Nos2 and Arg1 mRNA levels were significantly higher in tumor MDSC compared to bone marrow MDSC, regardless of the subtype (Fig. 5). Although Nos2 mRNA levels were lower in bone marrow MDSC from VDR KO mice, they were not altered in tumor MDSC from VDR KO mice. In contrast, Arg1 mRNA levels were not altered by VDR KO in bone marrow MDSC or tumor G-MDSC but they were 46 % higher in VDR KO tumor M-MDSC than WT tumor M-MDSC.
Fig. 5.
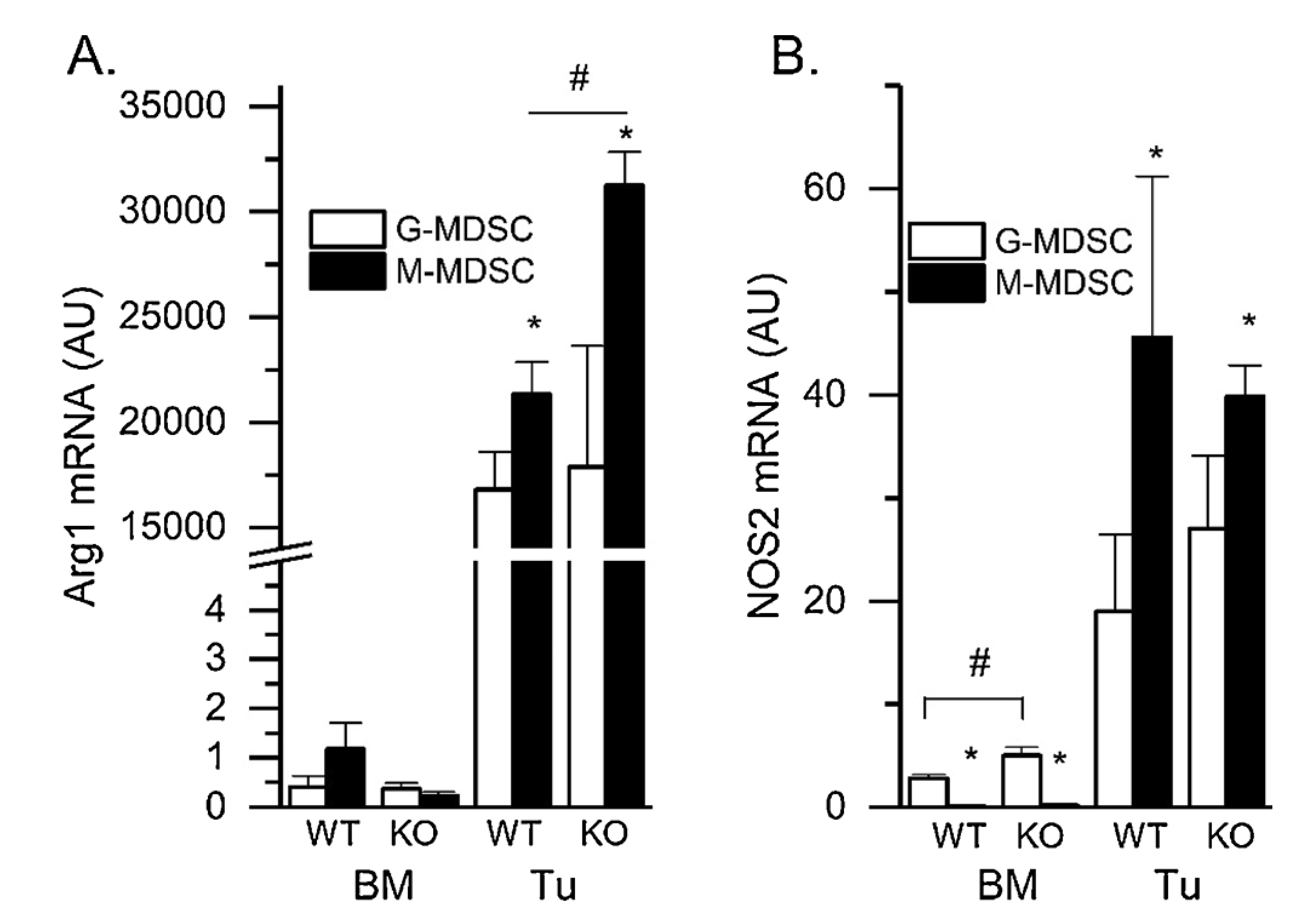
Effects of VDR Deletion (KO) on Arg1 and Nos2 mRNA levels from Freshly Isolated Tumor MDSC. RM-1 tumor cells were injected ip into wild type (WT) and VDR KO mice. After 7 d, tumor MDSC subtypes were isolated from the ip space for RNA isolation and PCR analysis. (A) Arg1 mRNA and (B) Nos2 mRNA levels were assessed by qPCR (n = 4–6 biological replicates from pools of 3–5 mice). * p < 0.05 G vs M; # p < 0.05 WT vs VDR KO for a subtype within a tissue.
In a final test of vitamin D action on tumor MDSC, we treated cocultures of M-MDSC and activated OT-I splenocytes with 10 nM 1,25(OH)2D or vehicle during an 18 h T cell proliferation suppression test. 1,25(OH)2D treatment did not alter T cell proliferation in the control cultures (i.e. OT-I splenocytes without MDSC co-culture; EtOH = 65.0±1.6 %; 125(OH)2D = 64.9±2.4 %). However, 1,25(OH)2D treatment significantly reduced the suppression of T-cell proliferation caused by co-culture with tumor M-MDSC (Fig. 6a; by −37.5 % at 1:2 MDSC:OT-I ratio) (Fig. 6). When MDSC were cultured for 24 h and treated with 1,25(OH)2D, nitrite production was reduced in both M-MDSC (−22.3 %, p = 0.052) and G-MDSC (−48.7 %, p = 0.058); the reduction in G-MDSC was significantly greater than that seen in M-MDSC (p < 0.05).
Fig. 6.
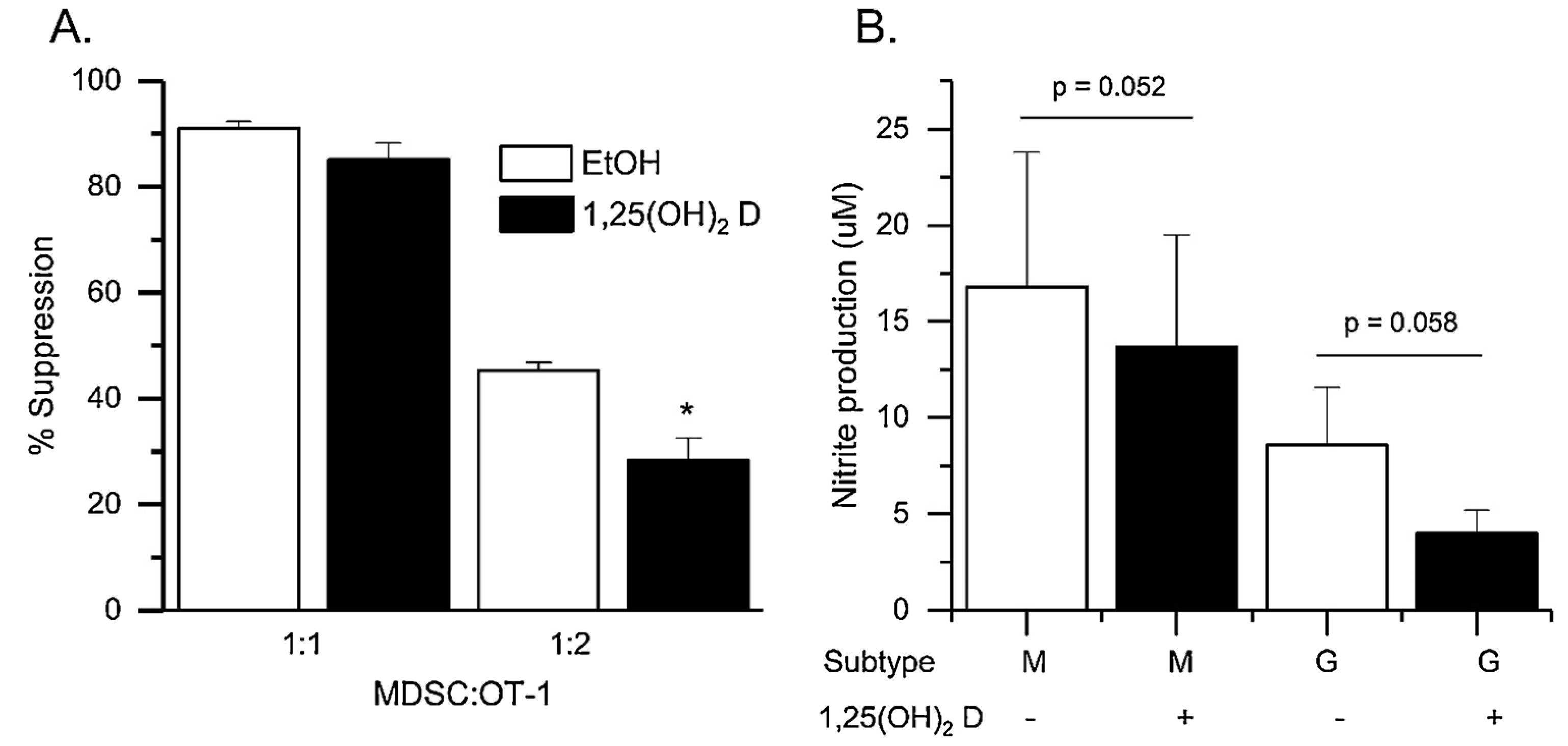
1,25(OH)2D Treatment Reduces the Ability of Freshly Isolated Tumor M-MDSC to Suppress T Cell Proliferation. M-MDSC were isolated from the ip space of mice with RM-1 ip tumors by FACS. Cells were incubated with activated OT-I splenocytes for 18 h in a short-term T cell suppression test. EdU was added for the last 1.5 h to assess T cell proliferation. Different MDSC:OT-I ratios were used to capture the maximal suppressive effect on T cell proliferation. During this time, cells were co-treated with 10 nM 1,25(OH)2D or ethanol (EtOH) vehicle. (A) Percent suppression of T cell proliferation (n = 4 biological replicates of pooled samples from 3 to 5 mice per replicate). * p < 0.05 vs EtOH group; (B) A separate set of tumor G- or M-MDSC were cultured in the presence of absence of 1,25(OH)2D for 24 h at which point nitrite production was assessed with the Griess assay. (n = 3 biological replicates from pools of 3–5 mice). Paired t-tests were conducted on log10 transformed data. P values provided for comparisons.
4. Discussion
In the context of cancer, vitamin D signaling has traditionally been studied for its impact on tumor cells. These studies reveal that 1,25(OH)2D treatment has anti-proliferative, pro-apoptotic, and pro-differentiating effects on cancer cells [38]. However, the tumor is a complex microenvironment that includes immune cells in addition to cancer cells. The tumor immune cell population represents a complex dynamic among T cells seeking to kill tumor cells and immunosuppressive cells that battle T cells to permit tumor growth [1]. MDSC are central to the immunosuppressive environment of tumors; they expand and are recruited in response to tumor-derived signals [39]. While others have shown that 1,25(OH)2D is immunomodulatory [14,15], our data are the first to show that vitamin D signaling is directly acting on MDSC by modifying their biology. Here we show that both subtypes of MDSC express the VDR and that increased VDR expression is a feature of MDSC differentiation. In addition, we used a variety of approaches to demonstrate that MDSC respond to 1,25(OH)2 D by reducing their T cell suppressive function.
Previous research suggests that activating vitamin D signaling influences the tumor microenvironment at multiple levels. First, vitamin D can suppress production of tumor cell-derived signals that promote MDSC function. Initially, Young et al. [40,41] found that 1,25(OH)2D suppressed production of GM-CSF, a driver of MDSC differentiation [42], by cultured LLC-LN7 cells and by LLC-LN7 tumor fragments. Later, Chen et al. [31] found that 1,25(OH)2D treatment reduced tumor cell production of IL-6, a cytokine that stimulates MDSC generation from their precursors [43] by reducing p38 signaling and phospho-STAT3 levels. More recently, Bruns et al. [32] reported that tumor-derived, miR155-containing exosomes can promote production of M-MDSC from monocytes and that 1,25(OH)2D suppresses NFkB-mediated production of miR155-containing exosomes by chronic lymphocytic leukemia cells. A second way that 1,25(OH)2D could influence tumors is through a direct effect on tumor immune cells. For example, Weirs et al. [44] showed that 1,25(OH)2D increased cell proliferation and IFNγ production in T cells isolated from LLC-LN7 tumor-bearing mice and activated in vitro with anti-CD3 antibody and IL-2. In addition, 1,25(OH)2D treatment may shift the differentiation of CD34+ myeloid precursor cells. CD34+ from the bone marrow of tumor-bearing mice do not differentiate into dendritic cells when cultured with GM-CSF, SCF, and TNFα (22 % vs 48 % conversion in bone marrow CD34+ cells from naïve mice) [27]. However, treatment with 1,25(OH)2D restored dendritic cell development (to 46 % conversion) and increased antigen presentation to normal levels. Our data extend the role that 1,25(OH)2 D plays in the modulation of the tumor immune microenvironment by demonstrating that the hormone directly regulates MDSC.
Our data are consistent with earlier data that suggested, but did not provide direct supportive evidence, that vitamin D signaling inhibits MDSC function. Several groups have previously demonstrated that high dose 1,25(OH)2D treatment in mice can reduce the growth of established lished LLC-LN7 lung tumors [40] or 4-NQO-induced squamous cell esophageal tumors [31], and inhibit lung metastases and tumor recurrence after resection of a primary tumor [41]. 1,25(OH)2D treatment is also associated with reduced CD34+ cell numbers in mouse LLC-LN7 tumors [30] and human head and neck squamous cell carcinoma (HNSCC) patients [45] as well as lower natural suppressor cell activity in bone marrow from LLC-LN7 tumor-bearing mice [46]. Finally, these changes were also associated with increased intratumor CD8+ cells in mice [30,47] and humans [48]. However, while these data suggest that activation of vitamin D signaling suppresses tumor growth and expansion by reducing MDSC-mediated immunosuppression and enhancing T cell-mediated immune surveillance, there is one study that contradicts this narrative. Cao et al. [49] reported that in mice with 4T1 tumors, vitamin D treatment increased tumor volume, reduced numbers of both CD4+ and CD8+ cells, and increased MDSC numbers.
Our data demonstrate that vitamin D signaling directly targets MDSC to inhibit their development or function but the mechanism for its suppressive function is not clear. Several studies show that increased activity of arginase 1 (ARG1) and inducible nitric oxide synthase 2 (NOS2) in tumor MDSC can deplete L-arginine from the tumor microenvironment as well as produce nitric oxide (NO), reactive oxygen species (ROS) and reactive nitrogen species (RNS), all of which can suppress T-cell function [50]. Our data suggest that at least part of the reason that vitamin D modulates MDSC function is due to its effects on this system (i.e. Arg1 mRNA levels are elevated in VDR KO M-MDSC with greater T cell suppressive function; 1,25(OH)2D treatment reduces the production of NO in both G- and M-MDSC). However, while it is not clear that these outcomes are direct effects, potential mechanisms of vitamin D signaling on MDSC can be inferred from the reported effects of vitamin D in other cell types. MDSC expansion and activation is regulated by a number of different transcriptional pathways including C/EBPα; STAT1, 3, and 6; NF-kB; and HIF1α [51]. Interestingly, there is evidence that vitamin D signaling can interfere with several of these regulatory pathways. Vitamin D signaling inhibits STAT1 and STAT3 phosphorylation and activation in a variety of cell types including T cells [52] and cancer cells [31] and blocks HIF1α signaling in various human cancer cell lines [53]. In addition, vitamin D signaling interferes with NFkB signaling in two ways. First, VDR interacts with the p50 subunit of NFkB to block LPS-induced macrophage proliferation [54] as well as TNFα-mediated NFkB activation and IkB degradation in human PBMC [55]. In addition, 1,25(OH)2D treatment increases IkB gene expression to inhibit TNFα-induced NFkB activity in MCF-7 breast cancer cells [56]. These potential mechanisms should be explored in future studies. In addition, many vitamin D regulated genes have been identified in human THP-1 monocytes [57] and these genes may also provide clues to the mechanism of vitamin D action in developing and mature MDSC.
Although additional research is needed on the impact of vitamin D on MDSC biology, our data, coupled with previous reports in the literature, suggest that this work has translational potential. Several groups have previously reported that improved vitamin D nutritional status suppresses expansion of MDSC or MDSC precursors. In one case-study of an infant with severe rickets, vitamin D therapy reduced CD34+ cell number in blood and spleen by 75 percent [58]. Similarly, two small human pilot studies suggest that 25OHD treatment (40, 60 ug/d; 6–8 wks) may reduce CD34+ cells in blood of HNSCC patients [59,60]. In addition, Bruns et al. [32] found blood M-MDSC levels were lower in people with serum 25OHD levels above 50 nM compared to those lower than 50 nM. Consistent with a loss of T cell inhibitory MDSC with high vitamin D status, Alves et al. [61] reported that serum 25OHD levels were positively correlated to total CD8+ cell number and negatively correlated to senescent, effector memory T cells re-expressing CD45RA. Finally, two animal studies suggest that 1,25(OH)2D treatment may even improve the efficacy of immunotherapy. When Wiers et al. [30,46] compared how adoptive transfer of tumor-reactive lymph node cells plus IL-2 could influence metastasis of LLC-LN7 tumors to the lung of mice, they found that the adoptive transfer therapy was not effective unless combined with 1,25(OH)2D treatment (5 ug/kg/every other day for 22 d). Future studies should explore both how low vitamin D status might limit the effectiveness of immunotherapies and how activation of vitamin D signaling with 1,25(OH)2D or vitamin D analogs might enhance the efficacy of immunotherapies.
5. Conclusions
Our data show clearly that MDSC are direct vitamin D target cells. Both G- and M-MDSC express the VDR, which we show is also a marker of MDSC differentiation. In addition, by using both VDR deletion and 1,25(OH)2D treatment, we show that vitamin D signaling is an important inhibitor of MDSC function. This work expands our understanding of how vitamin D can modify the tumor immune microenvironment. In addition, this work sets the foundation for additional research to test whether vitamin D status and activating the VDR with 1,25(OH)2D or vitamin D analog treatment can be used to augment immunotherapies designed to enhance T cell function.
Acknowledgements
We thank Dr. Jill Hutchcroft and the staff of the Purdue Flow Cytometry Core facility for their assistance with the experiments.
Funding
This work was supported by grants from the American Institute for Cancer Research (AICR, ID# 359587, JCF) and the National institute of Diabetes and Digestive and Kidney Diseases (DK084454, TLR). FACS analysis and sorting was conducted at the Flow Cytometry and Cell Separation Facility, a core service of the PUCCR (P30 CA023168). GNB was supported by an NIH Training Grant (5T32OD011122).
Abbreviations:
- 1,25(OH)2D
1,25 dihydroxyvitamin D
- 25OHD
25 hydroxyvitamin D
- HNSCC
head and neck squamous cell carcinoma
- MDSC
myeloid derived suppressor cell
- PBMC
peripheral blood mononuclear cell
- VDR
vitamin D receptor
References
- [1].Schreiber RD, Old LJ, Smyth MJ, Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion, Science 331 (2011) 1565–1570. [DOI] [PubMed] [Google Scholar]
- [2].Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade, Science 359 (2018) 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ, The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells, Immunology 138 (2013) 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, Utikal J, Umansky V, Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors, Front. Immunol 9 (2018) 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De BP, Van Ginderachter JA, Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity, Blood 111 (2008) 4233–4244. [DOI] [PubMed] [Google Scholar]
- [6].Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E, Denstman F, Xu X, Altieri DC, Du H, Yan C, Gabrilovich DI, CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation, Immunity 44 (2016) 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Condamine T, Ramachandran I, Youn JI, Gabrilovich DI, Regulation of tumor metastasis by myeloid-derived suppressor cells, Annu. Rev. Med 66 (2015) 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF, Understanding the tumor immune microenvironment (TIME) for effective therapy, Nat. Med 24 (2018) 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ostrand-Rosenberg S, Fenselau C, Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment, J. Immunol 200 (2018) 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Marvel D, Gabrilovich DI, Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected, J. Clin. Invest 125 (2015) 3356–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gabrilovich DI, Myeloid-derived suppressor cells, Cancer Immunol. Res 5 (2017) 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fleet JC, Molecular regulation of calcium metabolism, in: Weaver CM, Heaney RP (Eds.), Calcium in Human Health, Humana Press, Totowa, NJ, 2006, pp. 163–190. [Google Scholar]
- [13].Fleet JC, Schoch RD, Molecular mechanisms for regulation of intestinal calcium absorption by vitamin D and other factors, Crit. Rev. Clin. Lab. Sci 47 (2010) 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cantorna MT, Zhu Y, Froicu M, Wittke A, Vitamin D status, 1,25-dihydroxyvitamin D3, and the immune system, Am. J. Clin. Nutr 80 (2004) 1717S–1720S. [DOI] [PubMed] [Google Scholar]
- [15].Wright AF, Carothers AD, Pirastu M, Population choic in mapping genes for complex diseases, Nat. Genet 23 (1999) 397–404. [DOI] [PubMed] [Google Scholar]
- [16].Griffin MD, Dong X, Kumar R, Vitamin D receptor-mediated suppression of RelB in antigen presenting cells: a paradigm for ligand-augmented negative transcriptional regulation, Arch. Biochem. Biophys 460 (2007) 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Palmer MT, Lee YK, Maynard CL, Oliver JR, Bikle DD, Jetten AM, Weaver CT, Lineage-specific effects of 1,25-dihydroxyvitamin D(3) on the development of effector CD4 T cells, J. Biol. Chem 286 (2011) 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hewison M, Vitamin D and the intracrinology of innate immunity, Mol. Cell. Endocrinol 321 (2010) 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH, Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression, J. Immunol 173 (2004) 2909–2912. [DOI] [PubMed] [Google Scholar]
- [20].Gombart AF, Borregaard N, Koeffler HP, Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly upregulated in myeloid cells by 1,25-dihydroxyvitamin D3, FASEB J 19 (2005) 1067–1077. [DOI] [PubMed] [Google Scholar]
- [21].Wang TT, Dabbas B, Laperriere D, Bitton AJ, Soualhine H, Tavera-Mendoza LE, Dionne S, Servant MJ, Bitton A, Seidman EG, Mader S, Behr MA, White JH, Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease, J. Biol. Chem 285 (2010) 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lagishetty V, Misharin AV, Liu NQ, Lisse TS, Chun RF, Ouyang Y, McLachlan SM, Adams JS, Hewison M, Vitamin D deficiency in mice impairs colonic antibacterial activity and predisposes to colitis, Endocrinology 151 (2010) 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Szeles L, Keresztes G, Torocsik D, Balajthy Z, Krenacs L, Poliska S, Steinmeyer A, Zuegel U, Pruenster M, Rot A, Nagy L, 1,25-Dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype, J. Immunol 182 (2009) 2074–2083. [DOI] [PubMed] [Google Scholar]
- [24].Adams JS, Liu PT, Chun R, Modlin RL, Hewison M, Vitamin D in defense of the human immune response, Ann. N. Y. Acad. Sci 1117 (2007) 94–105. [DOI] [PubMed] [Google Scholar]
- [25].Ma Y, Aymeric L, Locher C, Kroemer G, Zitvogel L, The dendritic cell-tumor cross-talk in cancer, Curr. Opin. Immunol 23 (2011) 146–152. [DOI] [PubMed] [Google Scholar]
- [26].Petersen TR, Dickgreber N, Hermans IF, Tumor antigen presentation by dendritic cells, Crit. Rev. Immunol 30 (2010) 345–386. [DOI] [PubMed] [Google Scholar]
- [27].Young MR, Wright MA, Vellody K, Lathers DM, Skewed differentiation of bone marrow CD34+ cells of tumor bearers from dendritic toward monocytic cells, and the redirection of differentiation toward dendritic cells by 1alpha,25-dihydroxyvitamin D3, Int. J. Immunopharmacol 21 (1999) 675–688. [DOI] [PubMed] [Google Scholar]
- [28].Park MY, Lim BG, Kim SY, Sohn HJ, Kim S, Kim TG, GM-CSF promotes the expansion and differentiation of cord blood myeloid-derived suppressor cells, which attenuate xenogeneic graft-vs.-host disease, Front. Immunol 10 (2019) 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wright MA, Wiers K, Vellody K, Djordjevic D, Young MR, Stimulation of immune suppressive CD34+ cells from normal bone marrow by Lewis lung carcinoma tumors, Cancer Immunol. Immunother 46 (1998) 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wiers KM, Lathers DM, Wright MA, Young MR, Vitamin D3 treatment to diminish the levels of immune suppressive CD34+ cells increases the effectiveness of adoptive immunotherapy, J. Immunother 23 (2000) 115–124. [DOI] [PubMed] [Google Scholar]
- [31].Chen PT, Hsieh CC, Wu CT, Yen TC, Lin PY, Chen WC, Chen MF, 1alpha,25-dihydroxyvitamin D3 inhibits esophageal squamous cell carcinoma progression by reducing IL6 signaling, Mol. Cancer Ther 14 (2015) 1365–1375. [DOI] [PubMed] [Google Scholar]
- [32].Bruns H, Bottcher M, Qorraj M, Fabri M, Jitschin S, Dindorf J, Busch L, Jitschin R, Mackensen A, Mougiakakos D, CLL-cell-mediated MDSC induction by exosomal miR-155 transfer is disrupted by vitamin D, Leukemia 31 (2017) 985–988. [DOI] [PubMed] [Google Scholar]
- [33].Lees JR, Charbonneau B, Hayball JD, Diener K, Brown M, Matusik R, Cohen MB, Ratliff TL, T-cell recognition of a prostate specific antigen is not sufficient to induce prostate tissue destruction, Prostate 66 (2006) 578–590. [DOI] [PubMed] [Google Scholar]
- [34].Burcham GN, Cresswell GM, Snyder PW, Chen L, Liu X, Crist SA, Henry MD, Ratliff TL, Impact of prostate inflammation on lesion development in the POET3(+)Pten(+/−) mouse model of prostate carcinogenesis, Am. J. Pathol 184 (2014) 3176–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Haverkamp JM, Crist SA, Elzey BD, Cimen C, Ratliff TL, In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site, Eur. J. Immunol 41 (2011) 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) method, Methods 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [37].Cai W, Qin A, Guo P, Yan D, Hu F, Yang Q, Xu M, Fu Y, Zhou J, Tang X, Clinical significance and functional studies of myeloid-derived suppressor cells in chronic hepatitis C patients, J. Clin. Immunol 33 (2013) 798–808. [DOI] [PubMed] [Google Scholar]
- [38].Fleet JC, DeSmet M, Johnson R, Li Y, Vitamin D and cancer: a review of molecular mechanisms, Biochem. J 441 (2012) 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tcyganov E, Mastio J, Chen E, Gabrilovich DI, Plasticity of myeloid-derived suppressor cells in cancer, Curr. Opin. Immunol 51 (2018) 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Young MR, Halpin J, Hussain R, Lozano Y, Djordjevic A, Devata S, Matthews JP, Wright MA, Inhibition of tumor production of granulocyte-macrophage colony-stimulating factor by 1 alpha, 25-dihydroxyvitamin D3 reduces tumor motility and metastasis, Invasion Metastasis 13 (1993) 169–177. [PubMed] [Google Scholar]
- [41].Young MR, Ihm J, Lozano Y, Wright MA, Prechel MM, Treating tumor-bearing mice with vitamin D3 diminishes tumor-induced myelopoiesis and associated immunosuppression, and reduces tumor metastasis and recurrence, Cancer Immunol. Immunother 41 (1995) 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH, GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells, Breast Cancer Res. Treat 123 (2010) 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V, Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor, Immunity 32 (2010) 790–802. [DOI] [PubMed] [Google Scholar]
- [44].Wiers KM, Lozano Y, Messingham KA, Metz RJ, Young MR, 1alpha,25-Dihydroxyvitamin D3 activates T cells of tumor bearers through protein phosphatase 2A, Cancer Immunol. Immunother 44 (1997) 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kulbersh JS, Day TA, Gillespie MB, Young MR, 1alpha,25-Dihydroxyvitamin D (3) to skew intratumoral levels of immune inhibitory CD34(+) progenitor cells into dendritic cells, Otolaryngol. Head. Neck Surg 140 (2009) 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wiers K, Wright MA, Vellody K, Young MR, Failure of tumor-reactive lymph node cells to kill tumor in the presence of immune-suppressive CD34+ cells can be overcome with vitamin D3 treatment to diminish CD34+ cell levels, Clin. Exp. Metastasis 16 (1998) 275–282. [DOI] [PubMed] [Google Scholar]
- [47].Young MR, Lozano Y, Ihm J, Wright MA, Prechel MM, Vitamin D3 treatment of tumor bearers can stimulate immune competence and reduce tumor growth when treatment coincides with a heightened presence of natural suppressor cells, Cancer Lett 104 (1996) 153–161. [DOI] [PubMed] [Google Scholar]
- [48].Walsh JE, Clark AM, Day TA, Gillespie MB, Young MR, Use of alpha,25-dihydroxyvitamin D3 treatment to stimulate immune infiltration into head and neck squamous cell carcinoma, Hum. Immunol 71 (2010) 659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cao Y, Du Y, Liu F, Feng Y, Cheng S, Guan S, Wang Y, Li X, Li B, Jin F, Lu S, Wei M, Vitamin D aggravates breast cancer by inducing immunosuppression in the tumor bearing mouse, Immunotherapy 10 (2018) 555–566. [DOI] [PubMed] [Google Scholar]
- [50].Bronte V, Zanovello P, Regulation of immune responses by L-arginine metabolism, Nat. Rev. Immunol 5 (2005) 641–654. [DOI] [PubMed] [Google Scholar]
- [51].Condamine T, Mastio J, Gabrilovich DI, Transcriptional regulation of myeloid-derived suppressor cells, J. Leukoc. Biol 98 (2015) 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Olson KC, Kulling PM, Olson TL, Tan SF, Rainbow RJ, Feith DJ, Loughran TP Jr., Vitamin D decreases STAT phosphorylation and inflammatory cytokine output in T-LGL leukemia, Cancer Biol. Ther 18 (2017) 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ben-Shoshan M, Amir S, Dang DT, Dang LH, Weisman Y, Mabjeesh NJ, 1alpha,25-Dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells, Mol. Cancer Ther 6 (2007) 1433–1439. [DOI] [PubMed] [Google Scholar]
- [54].Ma D, Zhang RN, Wen Y, Yin WN, Bai D, Zheng GY, Li JS, Zheng B, Wen JK, 1, 25(OH)2D3-induced interaction of vitamin D receptor with p50 subunit of NF-kappaB suppresses the interaction between KLF5 and p50, contributing to inhibition of LPS-induced macrophage proliferation, Biochem. Biophys. Res. Commun 482 (2017) 366–374. [DOI] [PubMed] [Google Scholar]
- [55].Stio M, Martinesi M, Bruni S, Treves C, Mathieu C, Verstuyf A, d’Albasio G, Bagnoli S, Bonanomi AG, The Vitamin D analogue TX 527 blocks NF-kappaB activation in peripheral blood mononuclear cells of patients with Crohn’s disease, J. Steroid Biochem. Mol. Biol 103 (2007) 51–60. [DOI] [PubMed] [Google Scholar]
- [56].Lundqvist J, Yde CW, Lykkesfeldt AE, 1alpha,25-Dihydroxyvitamin D3 inhibits cell growth and NFkappaB signaling in tamoxifen-resistant breast cancer cells, Steroids 85 (2014) 30–35. [DOI] [PubMed] [Google Scholar]
- [57].Nurminen V, Seuter S, Carlberg C, Primary vitamin d target genes of human monocytes, Front. Physiol 10 (2019) 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yetgin S, Yalcin SS, The effect of vitamin D3 on CD34 progenitor cells in vitamin Ddeficiency rickets, Turk. J. Pediatr 46 (2004) 164–166. [PubMed] [Google Scholar]
- [59].Lathers DM, Clark JI, Achille NJ, Young MR, Phase IB study of 25-hydroxyvitamin D(3) treatment to diminish suppressor cells in head and neck cancer patients, Hum. Immunol 62 (2001) 1282–1293. [DOI] [PubMed] [Google Scholar]
- [60].Lathers DM, Clark JI, Achille NJ, Young MR, Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3, Cancer Immunol. Immunother 53 (2004) 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Alves AS, Ishimura ME, Duarte YAO, Bueno V, Parameters of the immune system and vitamin D levels in old individuals, Front. Immunol 9 (2018) 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]