

Abstract
A subset of glutamatergic interneurons in the neonatal spinal superficial dorsal horn (SDH) exhibits intrinsic burst-firing (i.e. ‘pacemaker’ activity), which is tightly regulated by persistent, voltage-gated Na+ channels and classic inward-rectifying K+ (Kir2) channels and downregulated over the course of postnatal development. Ascending lamina I projection neurons targeting the parabrachial nucleus (PB) or periaqueductal gray (PAG) can also display pacemaker activity during early life. However, the degree to which the ionic mechanisms driving pacemaker activity are conserved across different cell types in the spinal dorsal horn, as well as whether the intrinsic bursting is restricted to newborn projection neurons, remains to be elucidated. Using in vitro patch clamp recordings from identified lamina I spinoparabrachial neurons in rat spinal cord slices, here we demonstrate that adolescent projection neurons retain their ability to generate pacemaker activity. In contrast to previous findings in lamina I interneurons, pacemaker projection neurons possessed higher membrane capacitance, lower membrane resistance, and a greater Kir-mediated conductance compared to adjacent spinoparabrachial neurons that lacked intrinsic burst-firing. Nonetheless, as previously seen in interneurons, the bath application of riluzole to block persistent Na+ channels significantly dampened pacemaker activity in projection neurons. Collectively, these results suggest that intrinsic burst-firing in the developing dorsal horn can be generated by multiple combinations of ionic conductances, and highlight the need for further investigation into the mechanisms governing pacemaker activity within the major output neurons of the SDH network.
Keywords: dorsal horn, spinal cord, projection neuron, burst-firing, patch clamp
Introduction
Spontaneous activity is critical for the proper establishment and refinement of neuronal circuits in the developing CNS [1–3]. Within the superficial dorsal horn (SDH) of the neonatal rat spinal cord, a subpopulation of glutamatergic interneurons displays intrinsic, oscillatory burst-firing that progressively disappears over the first three weeks of life [4]. This ‘pacemaker’ activity is tightly regulated by the balance between persistent, voltage-gated Na+ channels and leak membrane conductance [4]. Classic inward-rectifying K+ (Kir2) channels make a key contribution to the level of leak membrane conductance, as pacemakers express a lower density of Kir2-mediated conductance compared to adjacent, non-pacemaker interneurons in the SDH, and blocking Kir2 channels with extracellular barium can unmask latent burst-firing [5]. These rhythmically active interneurons project to both flexor and extensor motor pathways in the spinal ventral horn, suggesting the possibility that pacemakers serve as endogenous drivers of the developing spinal networks which ultimately mediate nociceptive withdrawal reflexes [6].
Subsequent work revealed that a small fraction (~10%) of both spinoparabrachial and spino-PAG neurons could exhibit intrinsic bursting during early life [6]. However, it remains unclear if projection neurons lose their ability to generate pacemaker activity during development as previously seen in interneurons. Notably, while the intrinsic membrane properties of SDH interneurons change substantially during the early postnatal period [7, 8], lamina I projection neurons display relatively stable membrane properties throughout the first three weeks of life [9]. It is also unknown if the same intrinsic membrane properties (such as lower membrane capacitance and higher membrane resistance) define pacemaker projection neurons compared to non-pacemaker projection neurons, as previously documented for glutamatergic interneurons within the region. It has been shown that the intrinsic membrane properties of projection neurons, as well as the overall distribution of firing patterns, differ substantially from adjacent propriospinal neurons and interneurons in the SDH [10]. Furthermore, while Kir2 channels are known to be expressed within spinoparabrachial neurons where they strongly regulate intrinsic excitability [11], the ion channels responsible for shaping rhythmic burst-firing in projection neurons are poorly understood. For example, the degree to which persistent, voltage-gated Na+ channels are also essential for pacemaker activity in this neuronal population has yet to be explored.
Therefore the present study sought to determine if pacemaker activity persists into adolescence within lamina I spinoparabrachial neurons, and to identify the degree to which the biophysical signature of pacemaker projection neurons (relative to neighboring projection neurons) might be distinct from that previously seen in SDH interneurons possessing a similar ability to generate intrinsic burst-firing. While the results point to some similarities in the ionic mechanisms governing pacemaker activity in projection neurons, such as a key role for persistent Na+ currents, the intrinsic membrane properties of pacemaker projection neurons are not distinguished from neighboring, non-pacemaker projection neurons in the same manner as previously documented in interneurons, thereby suggesting that the overall complement of ion channels driving pacemaker activity may vary across different cell types in the SDH. This emphasizes the importance of further studies into the ionic mechanisms underlying pacemaker activity in spinal projection neurons, which could have important implications for the level of ascending nociceptive transmission to the brain.
Materials and Methods
All animals in this study were treated in accordance with welfare guidelines established by the University of Cincinnati Institutional Animal Care and Use Committee.
Retrograde labeling of spinoparabrachial neurons
At postnatal days (P) 31–34, Sprague-Dawley rats of either sex (n = 7 males and n = 13 females) were anesthetized via an intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg), positioned in a stereotaxic apparatus, and secured with non-rupture ear bars (World Precision Instruments). An incision was made through the scalp to visualize both lambda and bregma, and a small hole was made in the skull using an OmniDrill35 (World Precision Instruments). A single injection (50–100 nl) of FAST DiI oil (2.5 mg/ml; Invitrogen) was administered into the parabrachial nucleus (PB) using a Hamilton microsyringe (62RN; 2.5 μl volume) equipped with a 28 gauge needle. The following stereotaxic coordinates were used (in mm; relative to bregma): 8.0 caudal, 2.0 lateral, and 6.5 ventral. The skin was closed with Vetbond and the rats returned to the home cage before the beginning of the electrophysiological experiments.
Preparation of spinal cord slices
At P34–38, rats were deeply anesthetized with sodium pentobarbital (60 mg/kg) and transcardially perfused with ice-cold dissection solution consisting of (in mM): 250 sucrose, 2.5 KCl, 25 NaHCO3, 1.0 NaH2PO4, 6 MgCl2, 0.5 CaCl2, and 25 glucose continuously bubbled with 95% O2 / 5% CO2. The spinal cord was rapidly removed from the vertebral column, the dorsal and ventral roots were cut, and the dura mater was carefully removed. The lumbar enlargement was isolated and immersed in low-melting-point agarose (3% in above solution) at 37°C, and then cooled on ice to solidify. Parasagittal slices (300–350 μm) were cut from the side contralateral to the DiI injection site using a vibrating microtome (7000smz-2; Campden Instruments) and placed in an oxygenated recovery solution containing the following (in mM): 92 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na ascorbate, 2 thiourea, 3 Na pyruvate, 10 MgSO4, and 0.5 CaCl2 for ≤ 15 min and then placed in an oxygenated aCSF solution (composition in mM: 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.0 NaH2PO4, 1.0 MgCl2, 2.0 CaCl2, and 25 glucose) for a further recovery period of ≥ 1 hr at room temperature. Following recovery, slices were transferred to a submersion-type recording chamber (RC-22; Warner Instruments), mounted on the stage of an upright microscope (BX51WI; Olympus) equipped with fluorescence to identify DiI-labeled lamina I projection neurons, and perfused with oxygenated aCSF at a rate of 2 – 3 ml/min.
Patch clamp recordings from lamina I spinoparabrachial neurons
Patch clamp electrodes were constructed using thin-walled, single-filament borosilicate glass (1.5 mm outer diameter, 1.12 mm inner diameter; World Precision Instruments), with a microelectrode puller (P-97; Sutter Instruments). Current clamp and voltage clamp recordings used an internal recording solution composed of (in mM): 130 K-gluconate, 10 KCl, 10 HEPES, 4 MgATP, 10 Na-phosphocreatine, and 0.3 Na2-GTP (pH 7.2–7.4, 295–300 mOsm). Pipette resistances ranged from 4 – 6 MΩ and formed seal resistances ≥ 1 GΩ.
Whole-cell patch clamp recordings were obtained from DiI-labeled projection neurons residing in lamina I of the L4–L6 dorsal horn (i.e. within 50 μm of the edge of the dorsal white matter) which were visualized with infrared-differential interference contrast. Membrane resistance (Rm) was calculated from a hyperpolarization produced in response to a −20 pA current injection from the resting membrane potential (Vrest), while capacitance (Cm) was measured in voltage clamp using the pClamp membrane test (33.3–200 Hz, −10 mV). Spontaneous activity in projection neurons was observed at the resting membrane potential. In neurons displaying irregular action potential discharge or lacking spontaneous firing, current was then slowly injected via the patch electrode (at an approximate rate of 1–2 pA/sec) to evoke membrane depolarization to determine if intrinsic burst-firing could be evoked. Bath perfusion with a cocktail containing 10 μM NBQX, 25 μM AP-5, 10 μM gabazine and 0.5 μM strychnine (to block AMPA, NMDA, GABAA and glycine receptors, respectively) was used to confirm that any observed rhythmic burst-firing reflected intrinsic pacemaker activity.
Inward-rectifying K+ (Kir) currents were isolated as described previously [5, 12]. Briefly, neurons were voltage-clamped at −55 mV in the presence of the above cocktail to block fast synaptic transmission. Negative voltage ramps (from −55 to −155 mV) were applied at a rate of 0.2 mV/ms. BaCl2 (200 μM) was bath-applied to block Kir [13] and the Ba2+-sensitive component of the current was subsequently isolated via electronic subtraction (see Fig. 2A). Conductance (gBa-sensitive) was calculated as: g = I / (Vm − Erev) at two different membrane potentials that were equidistant (25 mV) from the reversal potential. The degree of inward rectification was calculated as a ratio of these conductances (i.e. gBa ratio) as follows: g(E−25) / g(E+25). The potential role of persistent, voltage-gated Na+ currents in the generation of pacemaker activity within projection neurons was examined via the bath application of the anti-convulsant riluzole (10 μM), which shows a greater selectivity for persistent Na+ currents over transient Na+ currents [14]. However, it should be noted that while our prior work has demonstrated that this concentration of riluzole blocks persistent, voltage-gated Na+ currents in unidentified lamina I neurons during early life [4], these currents were not directly isolated in the present study.
Figure 2. Intrinsic burst-firing in lamina I projection neurons is associated with greater levels of inward-rectifying K+ conductance.
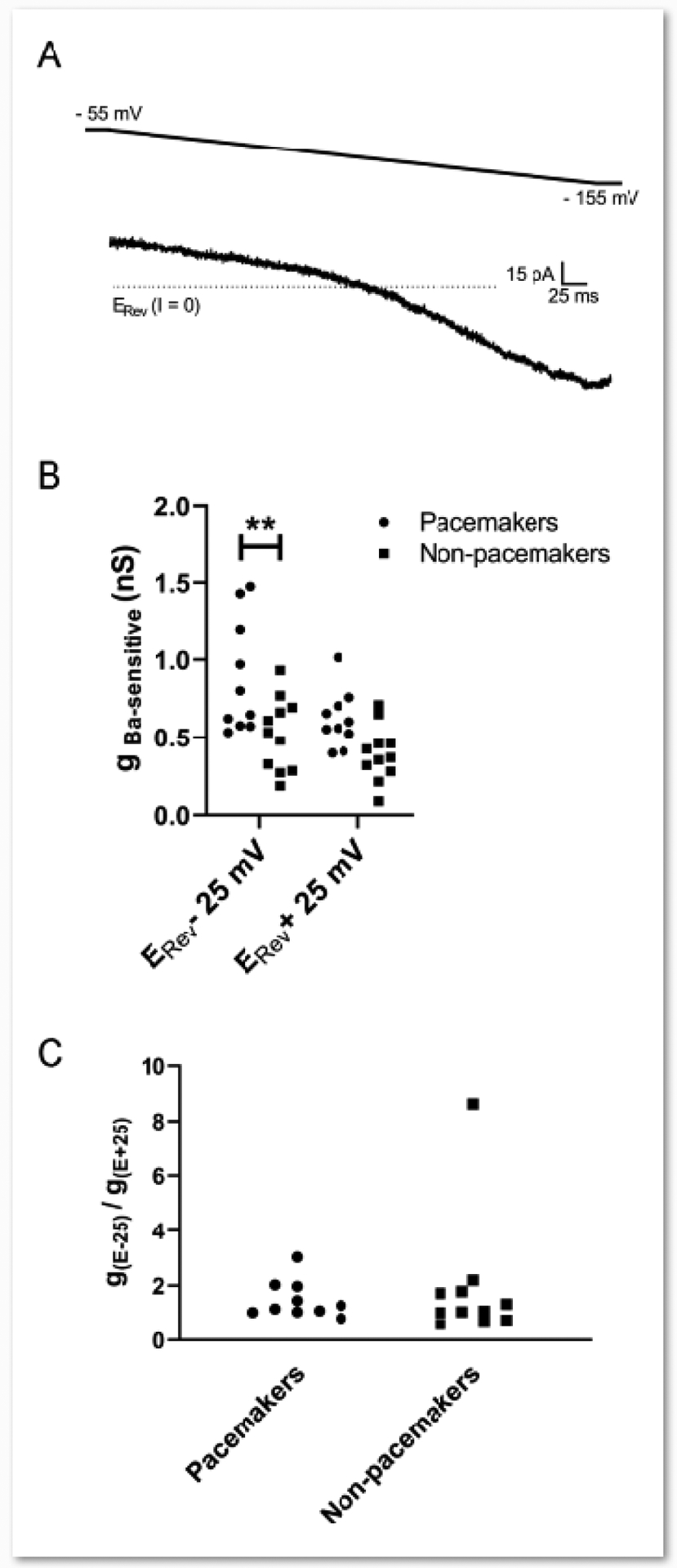
A: Example of the Ba2+-sensitive current (obtained via electronic subtraction) evoked by a voltage ramp from −55 mV to −155 mV which displays marked inward rectification. B: The Ba2+-sensitive conductance was greater in pacemakers compared to non-pacemakers within the population of spinoparabrachial neurons examined (n = 10–11 per group; Cell Type: F(1,19) = 11.10, p = 0.0035, RM two-way ANOVA; **p = 0.0042, Sidak’s multiple comparisons test). C: The degree of inward rectification of the Ba2+ sensitive conductance (as measured by the gBa ratio; see Methods) was similar between bursting and non-bursting spinoparabrachial neurons (U = 46, p = 0.557, Mann Whitney test).
Statistical analysis
The resting membrane potential (Vrest) was compared between silent, burst-firing, and irregular firing spinoparabrachial neurons using the Kruskal-Wallis test. Membrane resistance (Rm) and membrane capacitance were compared between pacemaker and non-pacemaker projection neurons, or between spontaneous vs. induced pacemakers, using the Mann-Whitney test or unpaired t-test depending on the normality of data distribution as evaluated by the D’Agnostino and Pearson test (Prism 8.0, GraphPad Software). The level of Ba2+-sensitive conductance was analyzed using Repeated Measure (RM) two-way analysis of variance (ANOVA) with cell type (i.e. pacemaker vs. non-pacemaker) and holding potential as factors, with the Sidak’s post-test for multiple comparisons. The gBa ratios were compared between pacemakers and non-pacemakers using the Mann-Whitney test. The effects of external Ba2+ on Rm and Vrest were analyzed using RM two-way ANOVA using cell type and time (i.e. before vs. after external Ba2+ application) as factors with Sidak’s post-tests. Finally, the effects of riluzole on rhythmic burst-firing, Vrest, action potential (AP) threshold and AP amplitude were examined using the Wilcoxon Signed Rank test or paired t-test. All data are expressed as mean ± SD.
Results
Higher expression of Ba2+-sensitive leak conductance distinguishes spinal projection neurons exhibiting pacemaker activity
Patch clamp recordings were obtained from 82 lamina I projection neurons retrogradely labeled via DiI injection into the parabrachial nucleus (Fig. 1A, B). The majority of adolescent spinoparabrachial neurons examined were silent at their resting membrane potential (n = 54; 66%), which likely reflects the fact that Vrest was significantly more hyperpolarized within this subpopulation of spinoparabrachial neurons (−70.1 ± 6.9 mV; n = 47) compared to those displaying spontaneous burst-firing (−64.2 ± 7.6 mV; n = 20) or irregular action potential discharge (−63.5 ± 12.1 mV; n = 7; H = 7.22, p = 0.027; Kruskal-Wallis test). The slow depolarization of Vrest via direct current injection evoked rhythmic burst-firing in a subset of previously silent lamina I projection neurons (29 out of 54 sampled; 54%; Fig. 1C). These induced pacemakers possessed more negative resting potentials (−71.4 ± 6.3 mV) compared to spontaneous pacemakers (t(42) = 3.37, p = 0.0016; unpaired t-test). Both spontaneous and induced rhythmic burst-firing occurred despite the block of fast synaptic transmission in the slice, thereby suggesting the presence of intrinsic pacemaker activity in lamina I projection neurons as reported previously [6].
Figure 1. Passive membrane properties of pacemaker vs. non-pacemaker spinoparabrachial neurons during adolescence.
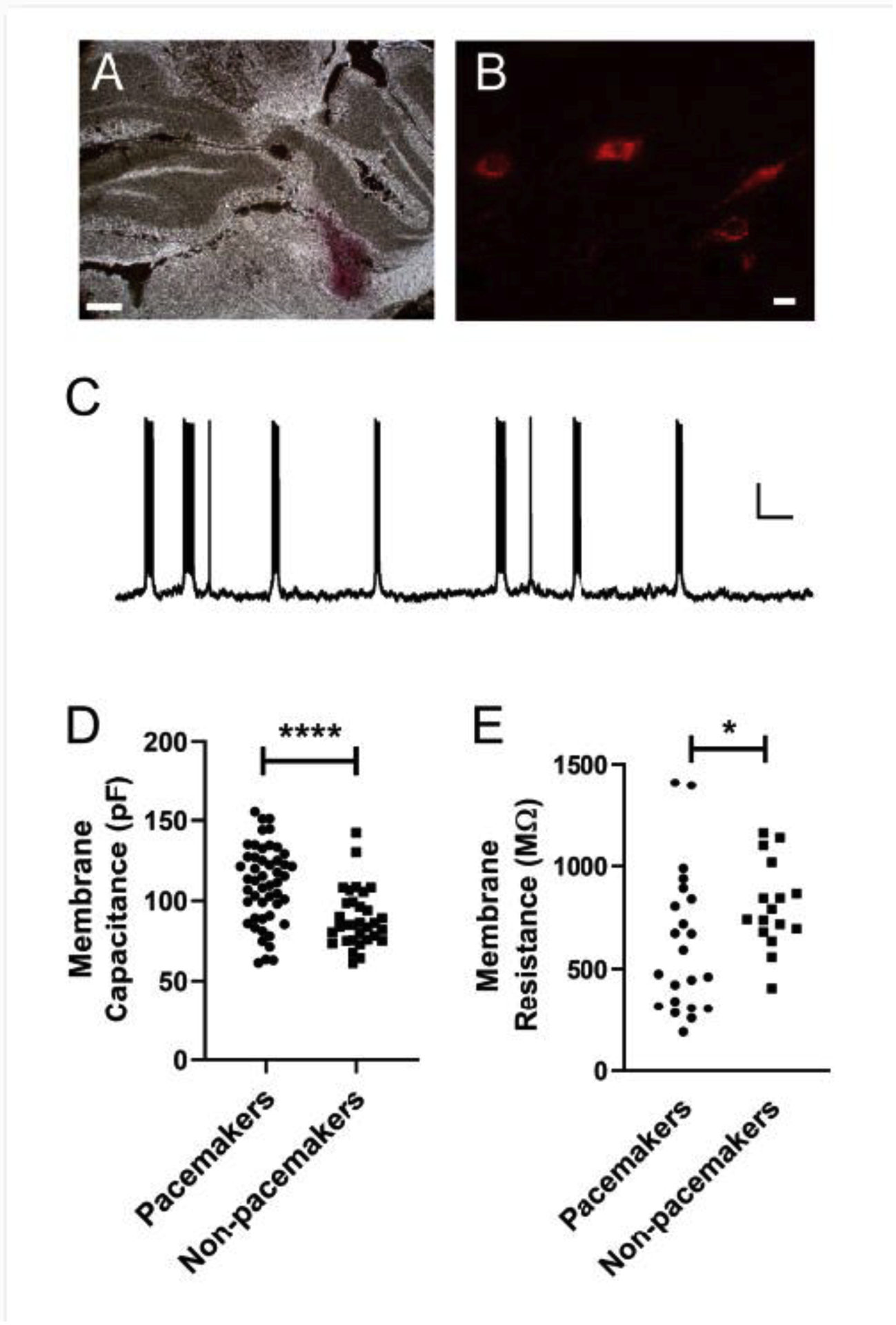
A: Representative example of DiI injection into the rat parabrachial nucleus. Scale bar = 0.5 mm. B: Examples of retrogradely labelled lamina I projection neurons following DiI injection into the parabrachial nucleus. Scale bar = 10 μm. C: Representative current-clamp recording demonstrating the presence of rhythmic burst-firing in a lamina I spinoparabrachial neuron at P35. Scale bar = 15 mV, 5 s. D: Membrane capacitance was significantly higher in projection neurons exhibiting pacemaker activity compared to adjacent projection neurons lacking intrinsic burst-firing (n = 32 – 49 neurons per group; U = 378, p < 0.0001, Mann Whitney test). E: Bursting spinoparabrachial neurons displayed significantly lower membrane resistance compared to adjacent, non-bursting projection neurons (n = 16 – 22 per group; U = 104, p = 0.033, Mann Whitney test).
Interestingly, the population of projection neurons that showed intrinsic bursting (either spontaneous or induced) possessed significantly higher membrane capacitance than adjacent spinoparabrachial neurons that either remained silent or displayed an irregular pattern of spontaneous firing following direct membrane depolarization (Pacemakers: 110.1 ± 24.1 pF, n = 49 neurons; Non-pacemakers: 89.1 ± 18 pF, n = 32, U = 378, p < 0.0001; Mann Whitney test; Fig. 1D), with no significant difference observed between spontaneous and induced pacemakers (t(47) = 1.66, p = 0.103; unpaired t-test). This was accompanied by a significantly lower membrane resistance in the pacemakers compared to non-pacemaker projection neurons (Pacemakers: 624.6 ± 348.1 MΩ, n = 22 neurons; Non-pacemakers: 407 ± 211 MΩ, n = 16, U = 104, p = 0.033; Mann Whitney test; Fig. 1E), with no significant difference observed between spontaneous and induced pacemakers (t(20) = 1.53, p = 0.142; unpaired t-test).
Our prior studies demonstrated that a hallmark feature of pacemaker interneurons in lamina I of the immature SDH was a lower Ba2+-sensitive, inward-rectifying K+ (Kir2) conductance compared to adjacent non-pacemakers in the same neuronal network [5]. To determine if the same differential expression of Ba2+-sensitive Kir channels might be found in pacemakers vs. non-pacemakers within the population of spinoparabrachial neurons, we administered a voltage ramp from −55 mV to −155 mV before and after the bath application of 200 μM BaCl2, and subsequently isolated the Ba2+-sensitive component of the current via electronic subtraction (Fig. 2A) as described previously [5, 12]. As expected when isolating an inward-rectifying K+ current, there was a significant main effect of membrane voltage on the level of Ba2+-sensitive conductance with higher levels seen at more negative membrane potentials (F(1,19) = 9.173, p = 0.0069; RM two-way ANOVA; Fig. 2B). More importantly, there was also a significant main effect of cell type, with pacemakers (n = 10) surprisingly demonstrating a greater Ba2+-sensitive conductance compared to non-pacemakers (n = 11, F(1,19) = 11.10, p = 0.0035; RM two-way ANOVA; Fig. 2B). Furthermore, in contrast to prior observations in burst-firing interneurons of the SDH [5], there was no significant difference in the degree of inward rectification of the Ba2+-sensitive K+ current between pacemakers and non-pacemakers (Pacemakers: 1.49 ± 0.67; Non-pacemakers: 1.89 ± 2.29; U = 46, p = 0.557; Mann Whitney test; Fig. 2C).
Separate current clamp recordings showed that the bath application of Ba2+ led to a significant membrane depolarization in both pacemaker (Fig. 3A) and non-pacemaker spinoparabrachial neurons (n = 21 in each group; Drug: F(1,40) = 33.25, p < 0.0001; Cell type × Drug Interaction: F(1,30) = 0.234, p = 0.631; RM two-way ANOVA; Fig. 3B). Similarly, the addition of external Ba2+ evoked a significant elevation in membrane resistance in lamina I projection neurons regardless of whether or not they displayed intrinsic burst-firing (Pacemakers: n = 19; Non-pacemakers: n = 13; Drug: F(1,30) = 26.78, p < 0.0001; Cell type × Drug Interaction: F(1,30) = 0.437, p = 0.514; RM two-way ANOVA; Fig. 3C).
Figure 3. Extracellular barium application increases membrane resistance and evokes membrane depolarization in intrinsically bursting and non-bursting spinoparabrachial neurons.
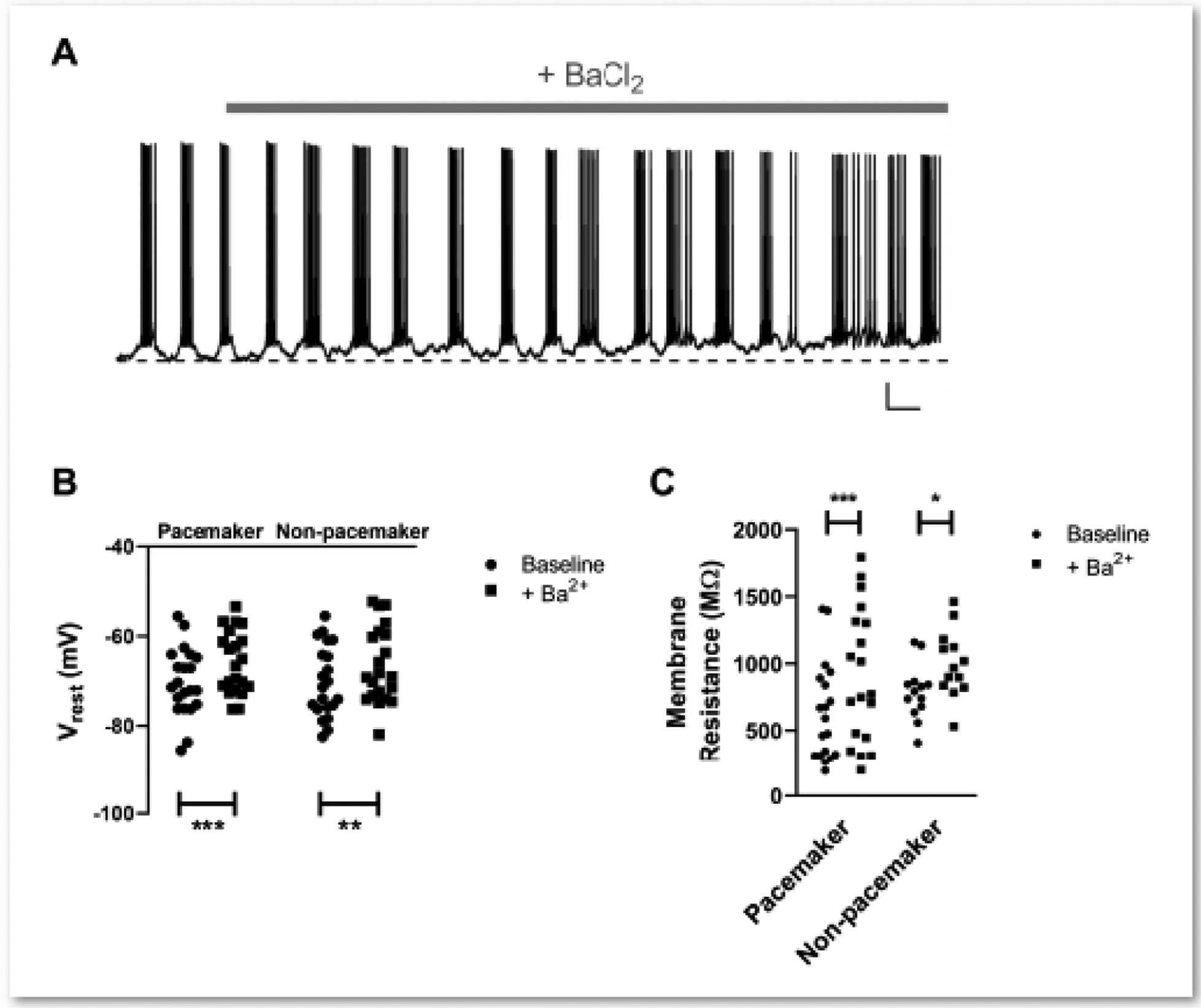
A: Representative trace illustrating the depolarization of resting membrane potential following the bath application of BaCl2 (200 μM) in a pacemaker spinal projection neuron. Dotted line indicates the initial resting membrane potential. Scale bar = 10 mV, 5 s. B: The degree of membrane depolarization evoked by external Ba2+ was similar between the types of projection neuron (n = 21 in each group; Drug: F(1,40) = 33.25, p < 0.0001; Cell type × Drug Interaction: F(1,30) = 0.234, p = 0.631; RM two-way ANOVA; **p = 0.001, ***p = 0.0001, Sidak’s multiple comparisons test). C: Membrane resistance was increased by the presence of extracellular Ba2+ in both pacemakers and non-pacemakers (n = 13–19 per group; Drug: F(1,30) = 26.78, p < 0.0001; Cell type × Drug Interaction: F(1,30) = 0.437, p = 0.514; RM two-way ANOVA; *p = 0.013, ***p = 0.0002, Sidak’s multiple comparisons test).
Pacemaker activity is driven by persistent, voltage-gated Na+ currents in lamina I spinoparabrachial neurons
Persistent, voltage-gated Na+ channels have been implicated in the generation of intrinsic burst-firing in multiple neuronal subtypes across the CNS, including spinal interneurons [4] and neurons within the respiratory brainstem [15]. To explore a potential role of these channels in the generation of pacemaker activity within ascending spinal projection neurons, we examined the effects of bath applying riluzole on intrinsic bursting in adolescent spinoparabrachial neurons (Fig. 4A). As reported previously in glutamatergic interneurons of the newborn SDH [4], riluzole (RLZ) significantly decreased burst frequency in projection neurons showing pacemaker activity (Baseline: 9.30 ± 5.10 per min; RLZ: 4.55 ± 4.99 per min; n = 10, W = −43.0, p = 0.027; Wilcoxon Signed Rank test; Fig. 4B), which included the abolition of burst-firing in 3 neurons which showed tonic firing (n = 1) or were silent (n = 2) in the presence of riluzole. Amongst the neurons displaying residual bursting following riluzole administration, the mean frequency of action potential (AP) discharge within an individual burst (i.e. intraburst frequency) was also significantly reduced by riluzole (Baseline: 6.01 ± 2.91 Hz; RLZ: 3.38 ± 2.66 Hz; n = 7, W = −24.0, p = 0.047; Wilcoxon Signed Rank test; Fig. 4C). The ability of riluzole to dampen pacemaker activity is unlikely to reflect a general disruption of AP firing, as AP threshold was not significantly affected by the block of persistent Na+ channels (n = 7, Baseline: −49.7 ± 6.1 mV; Riluzole: −47.9 ± 4.8 mV; W = 14.0, p = 0.297; Wilcoxon Signed Rank test; data not shown). Riluzole also failed to significantly alter Vrest (Baseline: −68.3 ± 4.2 mV; RLZ: −67.0 ± 4.8 mV; t(9) = 1.72, p = 0.119; paired t-test; data not shown) or AP amplitude (Baseline: 77.9 ± 7.8 mV; RLZ: 75.5 ± 8.5 mV; t(7) = 1.31, p = 0.233; paired t-test; data not shown).
Figure 4. Persistent, voltage-gated Na+ channels contribute to the intrinsic burst-firing of adolescent lamina I projection neurons.
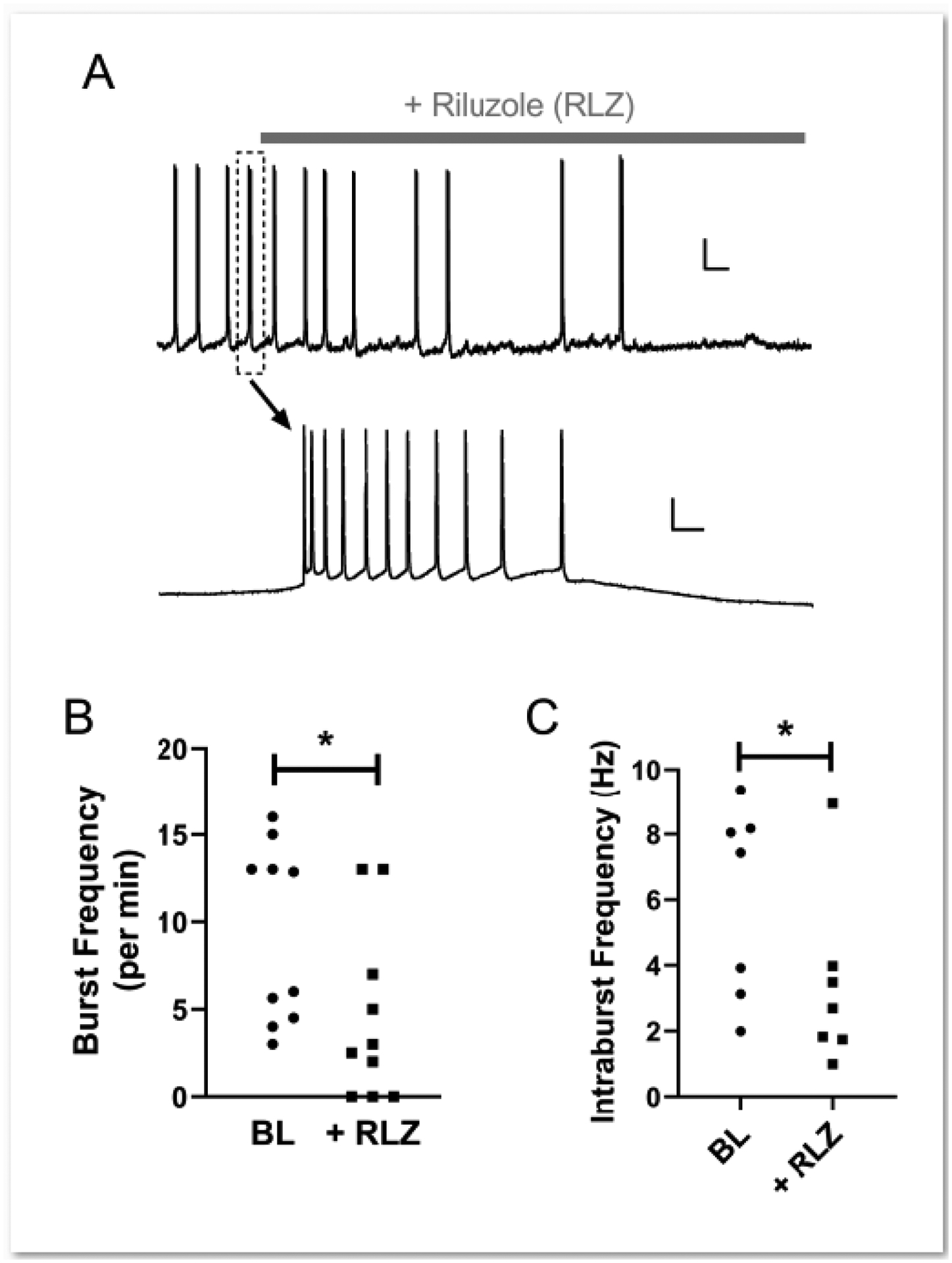
A: Example of pacemaker activity in a lamina I spinoparabrachial neuron before and after the bath application of riluzole (gray bar; top). The dotted rectangle highlights a burst which is displayed at a different time scale below (bottom). Scale bars = 15 mV, 10 s (top); 15 mV, 100 ms (bottom). B, C: Riluzole (RLZ; 10 μM) significantly decreased the burst frequency (n = 10; W = −43.0, p = 0.027, Wilcoxon Signed Rank test; B) as well as the mean frequency of action potential discharge within a burst (n = 7; W = −24.0, p = 0.047; Wilcoxon Signed Rank test; C).
Discussion
The present results clearly indicate that lamina I spinoparabrachial neurons retain their ability to generate pacemaker activity through adolescence. Although the extent to which pacemaker activity occurs in vivo remains to be determined, this raises the interesting possibility that the rhythmic bursting of spinoparabrachial neurons can provide an endogenous excitatory drive to the immature brain, thereby avoiding the need to endure noxious stimulation during development in order to promote the activity-dependent refinement of supraspinal nociceptive circuits. Overall, the maturation of pain networks in the parabrachial nucleus (PB) remains poorly understood, although it has been shown that noxious mechanical stimulation can evoke cfos expression in both NK1R-expressing lamina I projection neurons and PB neurons by P3 [16]. Age-dependent changes in the efficacy or frequency of ascending nociceptive inputs to the PB could also have important implications for the development of descending inhibitory pathways originating in the brainstem [17], given the presence of direct connections between the PB and rostroventromedial medulla [18, 19]. Interestingly, these descending inhibitory pathways are known to mature slowly during early postnatal development [20], as descending facilitation dominates throughout the first three weeks of life in the rat [21].
The precise role that pacemaker activity within projection neurons might play in nociceptive processing within the CNS remains to be identified. The lateral PB is a highly complex structure which is involved in the modulation of salt appetite, thirst and the regulation of blood pressure in addition to nociception [22]. The presence of burst-firing in ascending spinoparabrachial neurons may help to entrain subsets of PB neurons to respond in concert to peripheral noxious stimulation. This could occur by facilitating higher action potential frequency, as seen in intrinsically bursting neurons within the anterior cingulate cortex (ACC) following noxious mechanical stimulation [23], and/or enhancing the reliability of excitatory synaptic signaling [24] which likely results from increased glutamate release. For example, spontaneous burst-firing in inner hair cells of the developing cochlea [25, 26] leads to the synchronized activation of spiral ganglion neurons that ultimately encode similar sound frequencies [27].
Nonetheless, oscillatory burst-firing of spinal projection neurons could also disrupt high-fidelity transmission of ascending nociceptive input to the brain, as prior studies show that high-threshold primary afferent input to SDH interneurons can be heavily filtered by the presence of this rhythmic activity [4]. Notably, in vivo recordings from pyramidal neurons in the electrosensory lateral line lobe of weakly electric fish have demonstrated that intrinsic burst-firing interferes with the ability of neurons to precisely encode the temporal dynamics of sensory input [28]. Therefore, lamina I projection neurons may be able to switch between two distinct states of neuronal activity and information transfer, similar to that previously proposed for thalamic neurons in the lateral geniculate nucleus [29]: (1) a ‘pacemaker mode’, which is characterized by intrinsic, rhythmic bursting and a decreased sensitivity to the peripheral sensory receptive fields; and (2) a ‘transfer mode’ which maintains neuronal sensitivity to excitatory synaptic inputs and faithfully transmits incoming sensory information to higher order nociceptive circuits. Given that many pacemaker projection neurons required slow membrane depolarization from Vrest in order to trigger the intrinsic burst-firing, it will ultimately be important to better understand how metabotropic signaling onto projection neurons might regulate the transition between these two modes of information transfer, as described for burst-firing neurons of the deep dorsal horn [12]. It would also be interesting to elucidate if persistent aberrations in sensory input alters the generation of pacemaker activity in spinoparabrachial neurons, as seen in the rodent somatosensory cortex where sensory deprivation elevates the prevalence of burst-firing in layer 5 pyramidal neurons [30].
The ability of riluzole to dampen rhythmic burst-firing strongly suggests a key role for persistent, voltage-gated Na+ channels in the generation of pacemaker activity in lamina I projection neurons, as reported for glutamatergic interneurons in the newborn SDH [4], although we cannot exclude the possibility that riluzole modulates rhythmic burst-firing of projection neurons via actions at other types of voltage-dependent or voltage-independent ion channels. Persistent Na+ currents have also been previously implicated in the intrinsic bursting of spinal neurons involved in central pattern generation, including commissural and Hb9 interneurons [31]. This burst-firing can be triggered by changes in the extracellular concentrations of K+ or Ca2+ [32], the latter of which reflects a shift in the voltage dependence of activation of persistent, voltage-gated Na+ channels [33]. However, the observation that membrane resistance was significantly lower in pacemaker projection neurons compared to non-pacemaker projection neurons, while the inward-rectifying K+ conductance was greater, suggests that a low level of leak conductance is not a hallmark feature of pacemaker neurons within the population of spinoparabrachial neurons. In contrast, neonatal SDH interneurons exhibiting pacemaker activity were distinguished by both a high ratio of persistent Na+ to leak membrane conductance (gNa,P / gleak) and a low expression of classic Kir2-mediated currents. Nonetheless, it should be noted that persistent voltage-gated Na+ currents were not isolated, and gNa,P / gleak ratios were not calculated, in spinal projection neurons in the present experiments, thereby making direct comparisons across studies more difficult. A multitude of other ionic conductances have been shown to underlie the generation of rhythmic bursting in other types of projection neurons across the CNS, ranging from R-type voltage-gated Ca2+ currents and large conductance Ca2+-activated K+ (i.e. BK) currents in the accessory olfactory bulb [34] to heteromeric Kv1 channels in the output neurons of the deep cerebellar nuclei [35]. Further investigation is clearly needed to delineate the full complement of ion channels which shape intrinsic burst-firing in ascending spinal projection neurons.
In conclusion, the current results demonstrate that adolescent spinoparabrachial neurons can generate intrinsic burst-firing that is driven in part by persistent, voltage-gated Na+ channels, despite the presence of lower membrane resistance compared to adjacent lamina I projection neurons lacking pacemaker activity. These findings suggest that the developing spinal cord may persistently retain the ability to drive neuronal activation in supraspinal nociceptive circuits independently of peripheral sensory input.
Highlights:
Spinoparabrachial neurons exhibit pacemaker activity through adolescence.
Pacemakers exhibit different intrinsic membrane properties than non-pacemakers.
Persistent Na+ currents contribute to pacemaker activity in projection neurons.
ACKNOWLEDGEMENTS:
This work was supported by the National Institutes of Health (NS072202 to MLB). The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Blanquie O, et al. , Homeostatic interplay between electrical activity and neuronal apoptosis in the developing neocortex. Neuroscience, 2017. 358: p. 190–200. [DOI] [PubMed] [Google Scholar]
- [2].Di Guilmi MN, et al. , Strengthening of the Efferent Olivocochlear System Leads to Synaptic Dysfunction and Tonotopy Disruption of a Central Auditory Nucleus. J Neurosci, 2019. 39(36): p. 7037–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jamann N, Jordan M, and Engelhardt M, Activity-dependent axonal plasticity in sensory systems. Neuroscience, 2018. 368: p. 268–282. [DOI] [PubMed] [Google Scholar]
- [4].Li J and Baccei ML, Pacemaker neurons within newborn spinal pain circuits. J Neurosci, 2011. 31(24): p. 9010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li J, Blankenship ML, and Baccei ML, Inward-rectifying potassium (Kir) channels regulate pacemaker activity in spinal nociceptive circuits during early life. J Neurosci, 2013. 33(8): p. 3352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li J, et al. , Connectivity of pacemaker neurons in the neonatal rat superficial dorsal horn. J Comp Neurol, 2015. 523(7): p. 1038–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Safronov BV, Wolff M, and Vogel W, Axonal expression of sodium channels in rat spinal neurones during postnatal development. J Physiol, 1999. 514 (Pt 3)(Pt 3): p. 729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Walsh MA, et al. , Evidence for a critical period in the development of excitability and potassium currents in mouse lumbar superficial dorsal horn neurons. J Neurophysiol, 2009. 101(4): p. 1800–12. [DOI] [PubMed] [Google Scholar]
- [9].Li J and Baccei ML, Developmental regulation of membrane excitability in rat spinal lamina I projection neurons. J Neurophysiol, 2012. 107(10): p. 2604–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ruscheweyh R, et al. , Distinctive membrane and discharge properties of rat spinal lamina I projection neurones in vitro. J Physiol, 2004. 555(Pt 2): p. 527–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ford NC and Baccei ML, Inward-rectifying K(+) (K(ir)2) leak conductance dampens the excitability of lamina I projection neurons in the neonatal rat. Neuroscience, 2016. 339: p. 502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Derjean D, et al. , Dynamic balance of metabotropic inputs causes dorsal horn neurons to switch functional states. Nat Neurosci, 2003. 6(3): p. 274–81. [DOI] [PubMed] [Google Scholar]
- [13].Coetzee WA, et al. , Molecular diversity of K+ channels. Ann N Y Acad Sci, 1999. 868: p. 233–85. [DOI] [PubMed] [Google Scholar]
- [14].Urbani A and Belluzzi O, Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci, 2000. 12(10): p. 3567–74. [DOI] [PubMed] [Google Scholar]
- [15].Del Negro CA, et al. , Persistent sodium current, membrane properties and bursting behavior of pre-bötzinger complex inspiratory neurons in vitro. J Neurophysiol, 2002. 88(5): p. 2242–50. [DOI] [PubMed] [Google Scholar]
- [16].Man SH, Géranton SM, and Hunt SP, Lamina I NK1 expressing projection neurones are functional in early postnatal rats and contribute to the setting up of adult mechanical sensory thresholds. Mol Pain, 2012. 8: p. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Millan MJ, Descending control of pain. Prog Neurobiol, 2002. 66(6): p. 355–474. [DOI] [PubMed] [Google Scholar]
- [18].Chen Q, et al. , Optogenetic Evidence for a Direct Circuit Linking Nociceptive Transmission through the Parabrachial Complex with Pain-Modulating Neurons of the Rostral Ventromedial Medulla (RVM). eNeuro, 2017. 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roeder Z, et al. , Parabrachial complex links pain transmission to descending pain modulation. Pain, 2016. 157(12): p. 2697–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fitzgerald M and Koltzenburg M, The functional development of descending inhibitory pathways in the dorsolateral funiculus of the newborn rat spinal cord. Brain Res, 1986. 389(1–2): p. 261–70. [DOI] [PubMed] [Google Scholar]
- [21].Hathway GJ, et al. , The changing balance of brainstem-spinal cord modulation of pain processing over the first weeks of rat postnatal life. J Physiol, 2009. 587(Pt 12): p. 2927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Davern PJ, A role for the lateral parabrachial nucleus in cardiovascular function and fluid homeostasis. Front Physiol, 2014. 5: p. 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Koga K, et al. , In vivo whole-cell patch-clamp recording of sensory synaptic responses of cingulate pyramidal neurons to noxious mechanical stimuli in adult mice. Mol Pain, 2010. 6: p. 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Izhikevich EM, et al. , Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci, 2003. 26(3): p. 161–7. [DOI] [PubMed] [Google Scholar]
- [25].Johnson SL, et al. , Position-dependent patterning of spontaneous action potentials in immature cochlear inner hair cells. Nat Neurosci, 2011. 14(6): p. 711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang HC and Bergles DE, Spontaneous activity in the developing auditory system. Cell Tissue Res, 2015. 361(1): p. 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Babola TA, et al. , Homeostatic Control of Spontaneous Activity in the Developing Auditory System. Neuron, 2018. 99(3): p. 511–524.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Toporikova N and Chacron MJ, SK channels gate information processing in vivo by regulating an intrinsic bursting mechanism seen in vitro. J Neurophysiol, 2009. 102(4): p. 2273–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McCormick DA and Feeser HR, Functional implications of burst firing and single spike activity in lateral geniculate relay neurons. Neuroscience, 1990. 39(1): p. 103–13. [DOI] [PubMed] [Google Scholar]
- [30].Breton JD and Stuart GJ, Loss of sensory input increases the intrinsic excitability of layer 5 pyramidal neurons in rat barrel cortex. J Physiol, 2009. 587(Pt 21): p. 5107–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tazerart S, Vinay L, and Brocard F, The persistent sodium current generates pacemaker activities in the central pattern generator for locomotion and regulates the locomotor rhythm. J Neurosci, 2008. 28(34): p. 8577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Brocard F, et al. , Activity-dependent changes in extracellular Ca2+ and K+ reveal pacemakers in the spinal locomotor-related network. Neuron, 2013. 77(6): p. 1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Su H, et al. , Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J Neurosci, 2001. 21(12): p. 4173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gorin M, et al. , Interdependent Conductances Drive Infraslow Intrinsic Rhythmogenesis in a Subset of Accessory Olfactory Bulb Projection Neurons. J Neurosci, 2016. 36(11): p. 3127–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ovsepian SV, et al. , A defined heteromeric KV1 channel stabilizes the intrinsic pacemaking and regulates the output of deep cerebellar nuclear neurons to thalamic targets. J Physiol, 2013. 591(7): p. 1771–91. [DOI] [PMC free article] [PubMed] [Google Scholar]