

Abstract
Jalili syndrome is a rare multisystem disorder with the most prominent features consisting of cone-rod dystrophy and amelogenesis imperfecta. Few cases have been reported in the Americas. Here we describe a case series of patients with Jalili syndrome examined at the National Eye Institute’s Ophthalmic Genetics clinic between 2016 and 2018. Three unrelated sporadic cases were systematically evaluated for ocular phenotype and determined to have cone-rod dystrophy with bull’s eye maculopathy, photophobia, and nystagmus. All patients had amelogenesis imperfecta. Two of these patients had Guatemalan ancestry and the same novel homozygous CNNM4 variant (p.Arg236Trp c.706C > T) without evidence of consanguinity. This variant met likely pathogenic criteria by the American College of Medical Genetics guidelines. An additional patient had a homozygous deleterious variant in CNNM4 (c.279delC p.Phe93Leufs*31), which resulted from paternal uniparental isodisomy for chromosome 2p22–2q37. This individual had additional syndromic features including developmental delay and spastic diplegia, likely related to mutations at other loci. Our work highlights the genotypic variability of Jalili syndrome and expands the genotypic spectrum of this condition by describing the first series of patients seen in the United States.
Keywords: amelogenesis, CNNM4, cone-rod dystrophy, Jalili syndrome, retinal degeneration, uniparental isodisomy
1 |. INTRODUCTION
Jalili syndrome is a rare multisystem disorder characterized by a childhood cone-rod retinal degeneration and amelogenesis imperfecta (Jalili, 2010). It was initially described in a large extended family in Gaza, as an autosomal recessive condition in which all affected family members had photophobia, nystagmus, and achromatopsia along with abnormal tooth enamel (Jalili & Smith, 1988). Since that time, most cases have been described in large consanguineous families on multiple continents, including one family in Newfoundland (Doucette et al., 2013) and one family in Guatemala (Parry et al., 2009), with few reported sporadic cases (Daneshmandpour, Darvish, Pashazadeh, & Emamalizadeh, 2019).
Though variants in many genes can lead to amelogenesis imperfecta or cone-rod dystrophy, the constellation of these two findings in the absence of other syndromic features is highly suggestive of one gene, CNNM4, as the underlying cause (Daneshmandpour et al., 2019; Parry et al., 2009; Polok et al., 2009). CNNM4 encodes a transmembrane protein with a critical role in magnesium homeostasis and is expressed in photoreceptors and in the dental enamel (Gomez Garcia, Oyenarte, & Martinez-Cruz, 2011; Parry et al., 2009; Polok et al., 2009). Here, we describe a series of sporadic cases with Jalili syndrome in a cohort of patients originating in North and Central America. These patients carry novel variants in CNNM4 and further highlight the variable phenotypic and genotypic spectrum of the condition.
2 |. METHODS
This study was carried out according to the standards of the Common Rule of the United States Federal Government (46CFR45) under protocols approved by the Institutional Review Board at the National Institutes of Health and informed consent was obtained for all participants. Patients were evaluated in the Ophthalmic Genetics and Visual Function Branch Clinic between 2016 and 2018. All patients underwent ophthalmic clinical evaluation, including best corrected visual acuity (BCVA) using the Early Treatment of Diabetic Retinopathy Study (ETDRS) chart and guidelines, refraction, color vision testing by monocular Farnsworth D15, slit-lamp biomicroscopy, dilated eye exam, Goldmann visual field testing, fundus color, and autofluorescence (FAF) imaging (Topcon, Tokyo, Japan; Optos, Dunfermline, Scotland), optical coherence tomography (Cirrus HD-OCT, Carl Zeiss Meditec, Dublin, CA; Bioptigen Inc., Research Triangle Park, NC), electroretinography (ERG) (LKC, Gaithersburg, MD), and medical evaluation by a clinical geneticist. ERGs were recorded using Burian-Allen electrodes and according to the International Society for Clinical Electrophysiology of Vision standards. Ocular, medical, and dental health records were also reviewed, including panoramic and bite wing X-rays when available. Patients without molecular results underwent clinical genetic testing with an inherited retinal dystrophy panel (Molecular Vision Laboratory [MVL]; Hillsboro, OR; Clinical Laboratory Improvement Amendments identifier, 38D2059762). CNNM4 variants were evaluated for pathogenicity using established American College of Medical Genetics (ACMG) criteria (Richards et al., 2015).
3 |. RESULTS
3.1 |. Case reports
3.1.1 |. Patient 1
Patient 1 is a 15-year-old girl born from Guatemalan parents who presented with poor central vision, nystagmus, and light sensitivity. Examination was notable for Snellen BCVA at distance of 20/250 in the right eye and 20/200 in the left eye. Auto-refraction showed a myopic refraction with astigmatism: −2.75 + 2.25 × 103 and −3.00 + 3.00 × 076 in the right and left eye, respectively. She had high frequency, low amplitude horizontal nystagmus and was orthophoric. Anterior segment exam was unremarkable, and fundus exam was notable for bull’s eye maculopathy and granular pigment changes in the periphery. Spectral domain optical coherence tomography (SD-OCT) revealed loss of the ellipsoid zone in the fovea, and FAF demonstrated a bull’s eye pattern of hyper- and hypo-autofluorescence (Figure 1). Farnsworth D15 color vision testing revealed multiple axis errors (achromatic pattern). Loss of the I1e isopter was noted on the Goldmann visual field testing in each eye, with preservation of I4e and V4e isopters. Scotopic electroretinography showed diminished amplitudes (~50%) and mildly delayed implicit times, while photopic bright flash and flicker ERG responses were unrecordable. The dental examination and dental radiographs (bitewings, anterior and posterior periapical radiographs, and panoramic radiograph) revealed an entirely restored dentition with crowns on all molar teeth and composite restorations on the anterior teeth (Figure 2). The pulp chambers appear slightly enlarged on some teeth (20, 23, 26, 29), while others exhibit appearance within normal limits for this age (Figure 2, Figure S1). Clinically, the enamel is thin or absent on the non-restored surfaces of the teeth. Periapical and bitewing radiographs reveal isolated residual enamel on nonrestored and under restored surfaces, respectively (Figure S1, arrows). There were otherwise no systemic abnormalities noted. A 581 gene panel from Molecular Vision Lab (MVL Panel v1) revealed a homozygous CNNM4 c.706C > T (p.Arg236Trp) variant with one copy inherited from each parent.
FIGURE 1.
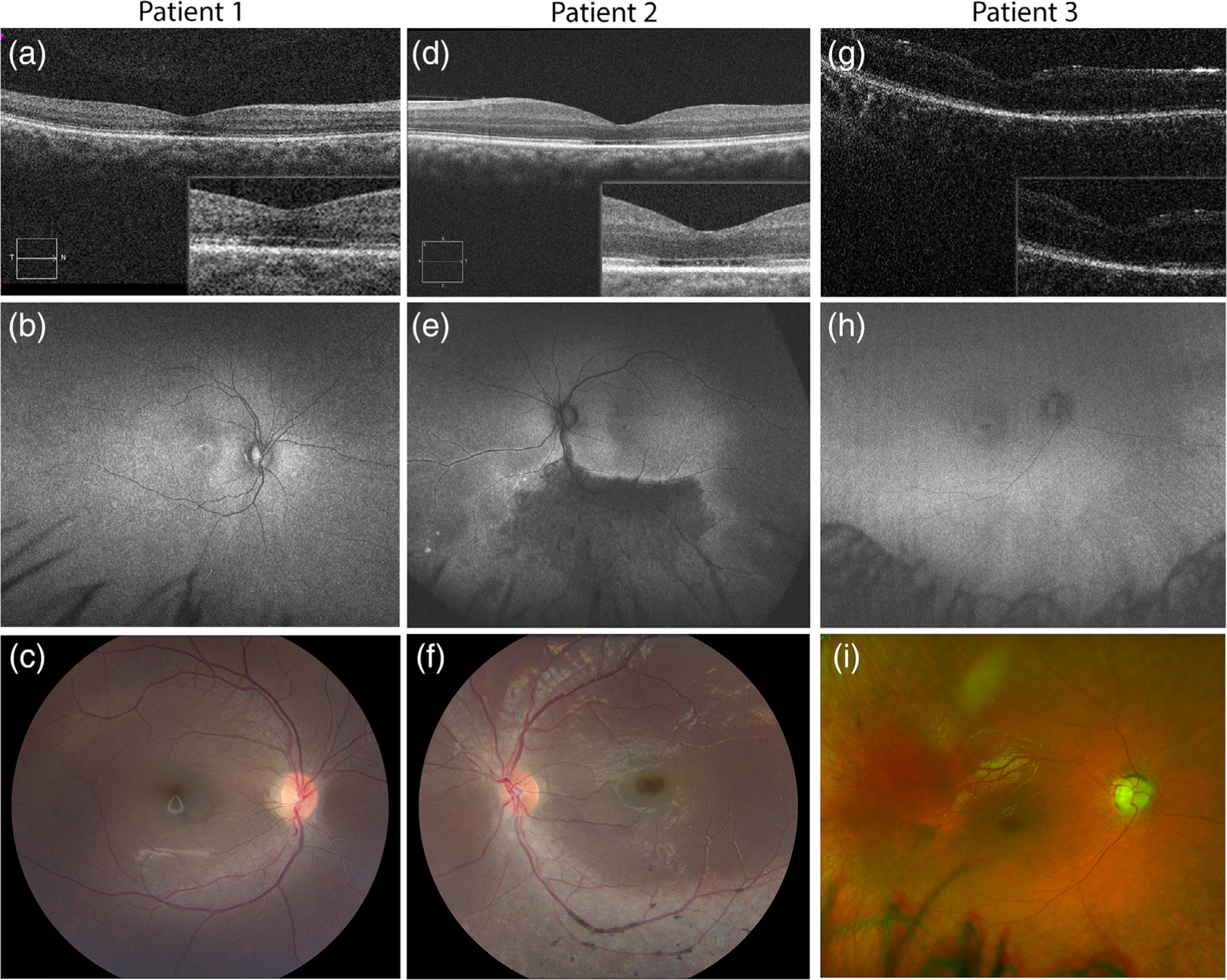
Ocular features of Jalili syndrome cases. (a–c) Multimodal imaging of the right eye of patient 1: SD-OCT (a) demonstrating ellipsoid zone disruption in the fovea; ring of hyper- hypo-autofluorescence on wide-field Optos autofluorescence (b) and bull’s eye maculopathy on 50° color fundus photo (c) are all consistent with the cone-rod dystrophy. (d-f) Multimodal imaging of patient 2’s left eye showing loss of foveal ellipsoid zone on OCT (d), with hypofluorescent sector of inferior retina on wide-field Optos autofluorescence (e), corresponding to the areas of atrophy and retinal pigment migration on color fundus photo (f). (g-i) Multimodal imaging of patient 3’s right eye showing macular changes on Bioptogen OCT (g), with corresponding bull’s eye hyper- hypo-autofluorescence pattern on wide-field Optos autofluorescence (h), and diffuse granular pigment changes and bull’s eye maculopathy on wide-field Optos color photograph (i). All patients share bull’s eye maculopathy, with loss of ellipsoid and interdigitation zone as evidenced by the hyporeflective dark area under the fovea on OCT. However, patient 2 also notably has a large hypoautofluorescent area of atrophy and pigment clumping in the periphery (e,f)
FIGURE 2.
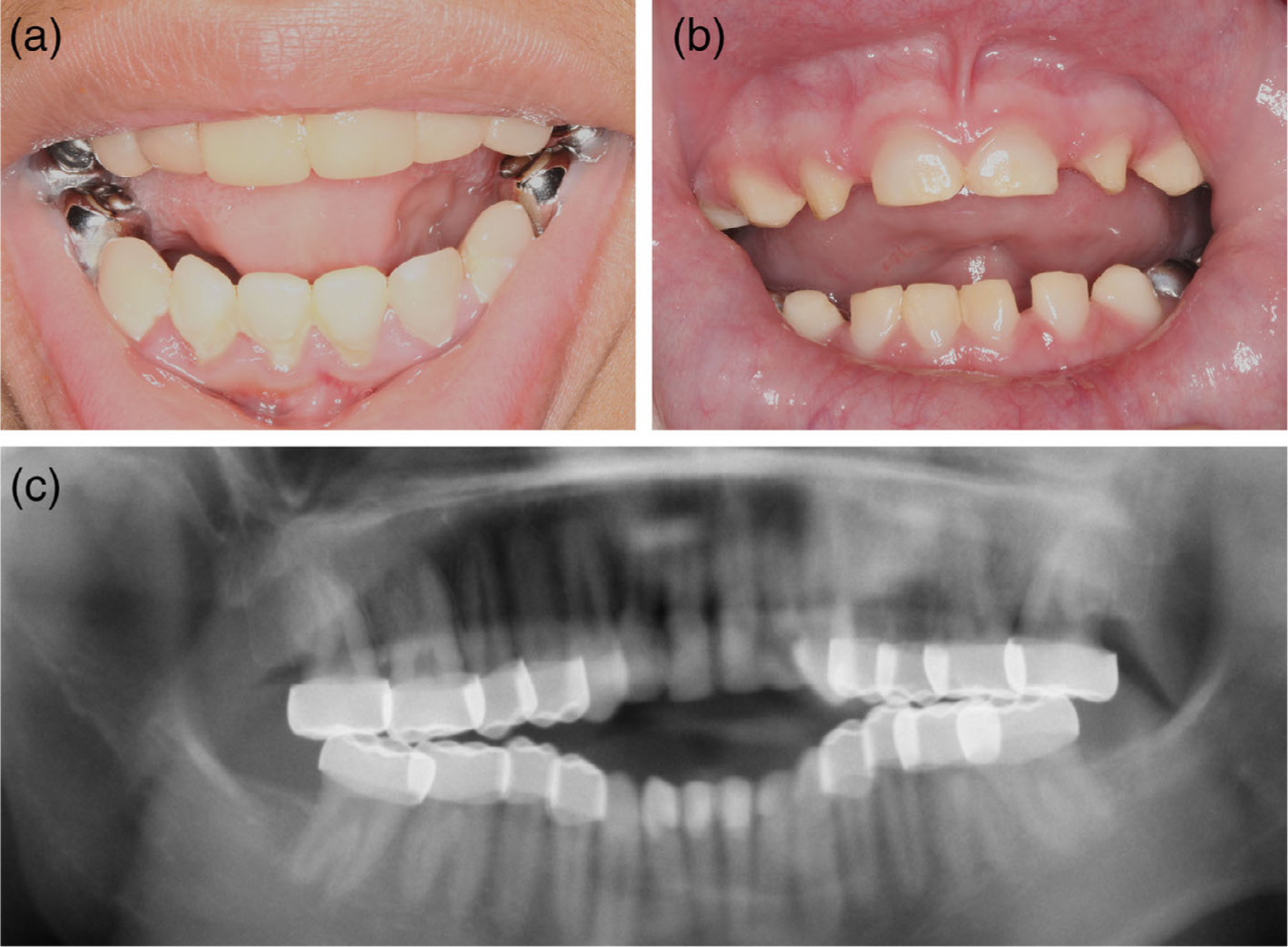
Dental features of Jalili syndrome cases. (a,b) External photos of mouth of patient 1 (a) and patient 3 (b) showing tooth decay and crowns on multiple teeth. (c) Panoramic X-ray of patient 1 showing crowns on all adult molar teeth
3.1.2 |. Patient 2
Patient 2 is a 16-year-old boy from Guatemala, who presented with poor central vision and light sensitivity. Examination was notable for Snellen BCVA at distance of 20/200 in the right eye and 20/160 in the left eye, with a manifest refraction of +1.00 + 4.50×109 and – 0.25 + 4.25×069, respectively. He had mild end-gaze nystagmus and was orthophoric. Anterior segment exam was unremarkable, and fundus exam was notable for bull’s eye maculopathy, perivascular, and segmental pigment deposition in the inferior retina, and mild vascular attenuation (Figure 1ahyphen;d). OCT revealed loss of the ellipsoid zone in the fovea, and FAF imaging revealed a hypofluorescent sector corresponding to the areas of retinal atrophy and pigment deposition. Color vision testing by Farnsworth D15 revealed multiple axis errors. Goldmann visual fields demonstrated superior constriction, and loss of the I1e isopter centrally, consistent with the retinal changes inferiorly (Figure 1). Scotopic ERG showed mildly diminished amplitudes and delayed implicit times, while photopic bright flash and flicker ERG were extinguished. Dental examination revealed crowns on multiple teeth and the absence of enamel. There were otherwise no systemic abnormalities. Molecular Vision Lab NGS Retinal Dystrophy SmartPanel v11 (281 genes) revealed a homozygous CNNM4 c.706C > T (p.Arg236Trp) variant with one copy inherited from each parent.
3.1.3 |. Patient 3
Patient 3 is a 3-year-old boy born from a Puerto Rican father and Caucasian mother who presented to ophthalmology clinic with light sensitivity and multiple congenital anomalies. On examination, binocular visual acuity was 2.4cy/cm by Teller acuity cards (approximately 20/360 Snellen equivalent). Cycloplegic refraction showed myopia with astigmatism in both eyes: −3.25 + 2.25 × 090 and − 3.25 + 2.50 × 095, in the right and left eye, respectively. Anterior segment exam was unremarkable. Fundus exam was notable for diffuse granular pigment changes and bull’s eye maculopathy, with typical corresponding bull’s eye pattern on FAF (Figure 1). Bioptogen OCT revealed loss of the ellipsoid zone at the fovea (Figure 1). Dental examination revealed crowns on multiple molar teeth and yellow/opaque appearance of the anterior teeth with thinning and complete absence of enamel on some teeth (Figure 2). Systemic evaluation was notable for spastic paraplegia, developmental delay, and fatty liver. The patient was dependent on tracheostomy for respiration and gastrostomy tube for feeding. Clinical whole exome sequencing revealed homozygosity for a frameshift variant in CNNM4: c.279delC (p.Phe93Leufs*31). Segregation analysis revealed that this variant was present in only the patient’s father and not mother. Subsequent analysis examining other single nucleotide polymorphisms (SNPs) revealed paternal uniparental isodisomy (UPD) on chromosome 2p22–2q37. With the exception of homozygosity for the CNNM4 variant, no additional variants within the UPD region were directly linked to the phenotypic features of the patient.
3.2 |. Genetic analysis
We evaluated pathogenicity of CNNM4 variants in each patient using established ACMG criteria. The CNNM4 c.279delC (p.Phe93Leufs*31) creates a frameshift early in the protein, which would be predicted to result in nonsense mediated decay of RNA or generation of a non-functional protein. The CNNM4 c.706C > T (p.Arg236Trp) variant creates a nonconservative substitution in the protein in a highly conserved residue (GERP score 4.99) located in the DUF21 domain of the protein. The missense variant p.Arg236Trp was predicted to be damaging by 14 different in-silico prediction tools (SIFT, PolyPhen2, PROVEAN, DANN, DEOGEN2, EIGEN, FATHMM-MKL, M-CAP, MVP, MutationAssess, MutationTaster, REVEL, MetaSVM, MetaLR) whereas it was predicted benign by only one in-silico tool (PrimateAI) (Kopanos et al., 2019). Also, the missense variant p.Arg236Trp had a high CADD score (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019) of 32, while the gnomAD missense Z score (2.37) indicates that CNNM4 gene is moderately constrained against missense changes. The p.Arg236Trp variant was found to be absent in population databases comprised healthy (gnomAD, https://gnomad.broadinstitute.org) as well as diseased individuals (HGMD, http://www.hgmd.cf.ac.uk; ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/). Notably, limited haplotype analysis on genetic panel data did not reveal additional homozygous variants in CNNM4 or surrounding genes ALMS1 and MERTK on chromosome 2, suggesting this is a founder mutation in the Guatemalan population rather than a result of consanguinity or previously unknown relatedness between families of patients 1 and 2. A previous variant in this same amino acid, CNNM4 c.707G > A (p.Arg236Gln), segregated with disease in a large family with Jalili syndrome, with a 0.517 probability of cosegregation by chance (Polok et al., 2009). This variant was also absent from the gnomAD database. Based on 2015 ACMG/AMP variant interpretation criteria, CNNM4 p.Phe93Leufs*31 is considered pathogenic and p.Arg236Gln is considered likely pathogenic (Richards et al., 2015).
4 |. DISCUSSION
The phenotypic spectrum of Jalili syndrome typically comprises cone-rod dystrophy and amelogenesis imperfecta, but there is significant variability in the severity of the retinal degeneration, even within families with the same mutation (Daneshmandpour et al., 2019; Gerth-Kahlert et al., 2015; Jalili, 2010). Consistent with this observation, we see a difference in structural and functional measures among two patients of similar age and demographic background carrying the same CNNM4 p. Arg236Trp variant. While both have significant cone dysfunction and a bull’s eye maculopathy, Patient 2 has a significant peripheral sector of retinal atrophy and pigment deposition with a corresponding visual field defect, while Patient 1 has disease mostly limited to the posterior pole. This suggests that factors beyond the genotype influence the disease severity. Both probands were of Guatemalan descent, denied consanguinity, and yet carried the same homozygous variant. This suggests a founder effect, though the ancestor may be more distant as neither patient carries homozygous variants in neighboring retinopathy genes ALMS1 nor MERTK, which would constitute a ~39 Mb haplotype block. Likewise, the previously reported Guatemalan family carried two different mutations (Parry et al., 2009), suggesting that there are at least 3 alleles present in this population.
We report the first case of uniparental isodisomy associated with Jalili syndrome. This patient had additional features including spastic paraplegia, fatty liver, and developmental delay, while both his father and mother were phenotypically normal. Though the patient carried a large stretch of UPD on chromosome 2p22–2q37, complete UPD of chromosome 2 can be tolerated (Ou et al., 2013; Zhang et al., 2019). Uniparental isodisomy causes disease by two predominant mechanisms, loss of heterozygosity uncovering rare deleterious variants and through disruption of imprinting. In our case, a frameshift variant in CNNM4, which was paternally inherited, was uncovered and led to the Jalili syndrome phenotype. Exome failed to reveal any rare coding variants that were thought to cause the nonretinal and nondental phenotypes. Likewise, no imprinting disorders with overlapping features have been described for these regions on chromosome 2. It remains possible that homozygosity for noncoding variants explain the other neurological phenotypes.
Our work describes the detailed phenotypic features of this condition in an underreported patient population, identifies a novel missense variant shared by two of the patients, and highlights an unusual case of UPD leading to the disorder. Future efforts will be necessary to identify the full spectrum of UPD-associated disorders on chromosome 2 and identify the precise cause of the additional features in this patient. Additionally, mechanistic studies exploring the structure and function of the DUF21 domain of CNNM4 will be necessary to identify the function of this critical portion of the protein.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the individuals and families for participating in the study; to Denise Cunningham, Mike Arango and the ophthalmic photographers and technicians at the National Institutes of Health Ophthalmic Genetics Clinic. Dr. Lev Prasov is supported in part by NEI K12-EY022299.
Funding information
NEI Intramural Research Grant, Grant/Award Number: NEI K12-EY022299
Footnotes
WEB RESOURCES
gnomAD Database: http://gnomad.broadinstitute.org/
NCBI Human Reference Genome Build 37.1: http://www.ncbi.nlm.nih.gov/genome/assembly/2928/.
Clinvar: https://www.ncbi.nlm.nih.gov/clinvar/HGMD: http://www.hgmd.cf.ac.uk
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Daneshmandpour Y, Darvish H, Pashazadeh F, & Emamalizadeh B (2019). Features, genetics and their correlation in Jalili syndrome: A systematic review. Journal of Medical Genetics, 56(6), 358–369. [DOI] [PubMed] [Google Scholar]
- Doucette L, Green J, Black C, Schwartzentruber J, Johnson GJ, Galutira D, & Young TL (2013). Molecular genetics of achromatopsia in Newfoundland reveal genetic heterogeneity, founder effects and the first cases of Jalili syndrome in North America. Ophthalmic Genetics, 34(3), 119–129. [DOI] [PubMed] [Google Scholar]
- Gerth-Kahlert C, Seebauer B, Dold S, Hanson JV, Wildberger H, Sporri A, … Berger W (2015). Intra-familial phenotype variability in patients with Jalili syndrome. Eye, 29(5), 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Garcia I, Oyenarte I, & Martinez-Cruz LA (2011). Purification, crystallization and preliminary crystallographic analysis of the CBS pair of the human metal transporter CNNM4. Acta Crystallographica Section F, Structural Biology and Crystallization Communications, 67(3), 349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili IK (2010). Cone-rod dystrophy and amelogenesis imperfecta (Jalili syndrome): Phenotypes and environs. Eye, 24(11), 1659–1668. [DOI] [PubMed] [Google Scholar]
- Jalili IK, & Smith NJ (1988). A progressive cone-rod dystrophy and amelogenesis imperfecta: A new syndrome. Journal of Medical Genetics, 25(11), 738–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, & Massouras A (2019). VarSome: The human genomic variant search engine. Bioinformatics, 35(11), 1978–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X, Liu C, Chen S, Yu J, Zhang Y, Liu S, & Sun H (2013). Complete paternal uniparental isodisomy for chromosome 2 revealed in a parentage testing case. Transfusion, 53(6), 1266–1269. [DOI] [PubMed] [Google Scholar]
- Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, Dollfus H, … Inglehearn CF (2009). Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. American Journal of Human Genetics, 84(2), 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polok B, Escher P, Ambresin A, Chouery E, Bolay S, Meunier I, … Schorderet DF (2009). Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. American Journal of Human Genetics, 84(2), 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee ALQA (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ding Z, He R, Qi J, Zhang Z, & Cui B (2019). Complete paternal Uniparental Disomy of chromosome 2 in an Asian female identified by short tandem repeats and whole genome sequencing. Cytogenetic and Genome Research, 157(4), 197–202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.