

Abstract
Background
Brain tumors are the most common solid tumors of childhood, but little is understood about the factors that influence their development. Pediatric low-grade gliomas in particular display unique temporal and spatial localization associated with different genetic mutations (eg, BRAF genomic alterations, mutations in the neurofibromatosis type 1 [NF1] gene) for reasons that remain unclear. NF1 low-grade gliomas typically arise in the optic pathway of young children as optic pathway gliomas (OPGs), likely from a cell of origin that resides within the third ventricular zone (TVZ). However, the factors that contribute to their distinct temporal patterning and penetrance have not been adequately explored.
Methods
TVZ neuroglial progenitor cells (NPCs) were analyzed over the course of mouse brain development. Progenitors isolated by fluorescence-activated cell sorting (FACS) were assessed for functional and molecular differences. The impact of different germline Nf1 mutations on TVZ NPC properties was analyzed using genetically engineered mice.
Results
We identify 3 individual factors that could each contribute to Nf1 optic glioma temporal patterning and penetrance. First, there are 3 functionally and molecularly distinct populations of mouse TVZ NPCs, one of which (“M” cells) exhibits the highest clonogenic incidence, proliferation, and abundance during embryogenesis. Second, TVZ NPC proliferation dramatically decreases after birth. Third, germline Nf1 mutations differentially increase TVZ NPC proliferation during embryogenesis.
Conclusions
The unique temporal patterning and penetrance of Nf1 optic glioma reflects the combined effects of TVZ NPC population composition, time-dependent changes in progenitor proliferation, and the differential impact of the germline Nf1 mutation on TVZ NPC expansion.
Keywords: germline mutation, gliomagenesis, neurofibromatosis, pediatric brain tumor, third ventricle
Key Points.
1. Three functionally and molecularly distinct mouse TVZ NPC populations exist.
2. “M” TVZ NPCs have the highest proliferation and abundance during embryogenesis.
3. Germline Nf1 mutations differentially affect TVZ NPC proliferation, which decreases after birth.
Importance of the Study.
The factors that contribute to the temporal patterning and penetrance of pediatric gliomas remain unclear. Using Nf1-OPG as a tractable model system, we identified 3 distinct factors—NPC population composition and temporal dynamics, developmental age, and the Nf1 germline mutation—that could each individually influence glioma formation. Future mechanistic studies leveraging this platform will be helpful in understanding how these and other risk factors, including environmental exposures, converge on specific progenitor cells in the developing brain to influence brain tumor formation in children.
Pediatric brain tumors are among the most common tumors of childhood, and represent a significant source of long-term morbidity and mortality for this population.1 In contrast to their adult counterparts, childhood brain tumors display strong temporal, regional, and mutational specificity. As such, brain tumors in children tend to appear at specific ages, in specific locations within the central nervous system, and result from different genetic mutations.2,3 This exquisite spatial and temporal patterning is illustrated by World Health Organization grade I gliomas (pilocytic astrocytomas [PAs]), a benign tumor caused by a limited number of genetic mutations. In the general population, the majority of PAs arise in children younger than 10 years without a familial cancer predisposition and predominate in the cerebellum (36‒42%),4–6 and most commonly harbor a genomic rearrangement involving the BRAF kinase gene.7,8
PAs also arise within the optic pathway (optic nerves, chiasm, tracts, and radiations)9 of young children (mean age, 4.5 y)10 with the tumor predisposition syndrome neurofibromatosis type 1 (NF1), caused by a germline mutation in the NF1 tumor suppressor gene. While somatic NF1 loss is required for NF1 PA formation, it is a stochastic event, reflecting the intrinsic mutation rate at the NF1 locus, similar to retinoblastoma.11 Since this rate would not be expected to vary with brain region or childhood development, other operative factors likely account for the temporal and spatial patterning that characterizes these brain tumors in children.
Using NF1 as an experimental platform, we examined several potential risk factors that could differentially influence pediatric low-grade glioma penetrance, including the cell of origin, the developmental stage, and the specific germline NF1 mutation. Leveraging a series of genetically engineered mouse models, we identified 3 previously undescribed populations of neural progenitor cells (NPCs) within the third ventricular zone (TVZ), the brain region containing the putative cells of origin for murine Nf1-OPG. We demonstrate that these NPC populations are molecularly and phenotypically distinct, and that their abundance and proliferation changes as a function of brain development, declining during postnatal life. Finally, we identified a differential effect of the germline Nf1 mutation on TVZ NPC proliferation, which correlates with murine optic glioma formation. Taken together, these findings establish the foundations for future mechanistic studies aimed at defining the molecular etiologies underlying brain tumor penetrance in children.
Materials and Methods
Mice
All mice were maintained on a C57/BL6 background and housed in barrier facilities with controlled light-dark cycles (12:12 hour) and ad libitum access to food and water in accordance with protocols approved by the Animal Studies Committee at Washington University. Mice used for these studies include GFAP-Cre-IRES-LacZ mice,12BLBP-Cre-IRES-LacZ mice,13 heterozygous Nf1 knockout (Nf1+/-) mice,14 and Nf1 mutant mice heterozygous for NF1 patient-identified gene mutations (Arg681X,15Gly848Arg,15 or Arg1278Pro16). Nestin-CFPnuc mice17 were generously provided by Grigori Enikolopov (Stony Brook University, NY), and were intercrossed with GFAP-Cre, BLBP-Cre, and Nf1-mutant mice.
Human Tissues
Paraffin-embedded human TVZ specimens from third trimester fetuses were obtained for immunohistochemical analysis16 under an approved Washington University institutional review board protocol.
Immunohistochemistry
Brains were harvested from mice at E16.5, E18.5, PN1, PN7, or PN28, fixed with paraformaldehyde, cryopreserved with sucrose and then embedded in optimal cutting temperature compound (OCT; VWR) and stored at −80°C until sectioning at 10–12 microns. Sections were rehydrated, permeabilized, and blocked prior to incubation with primary antibody (Supplementary Table 1) overnight at 4°C. Sections were washed, incubated with secondary antibody for one hour at room temperature, and counterstained with bis-benzimide (Invitrogen) prior to mounting (VectaShield, Vector Laboratories). Images were acquired on a Leica DFC3000G fluorescent microscope using a 20x objective with associated Leica Application Suite X software. Longitudinal TVZ images were photomerged using Adobe Photoshop CS5.1, version 12.1. The percentage of Ki67+ cells were quantified relative to the total number of bis-benzamide+ cells. The average Ki67 index across several sections was determined, with at least 3 mice analyzed per condition. Statistical significance was determined at each age by one-way ANOVA corrected for multiple comparisons with a Tukey post-hoc test.
Fluorescence-Activated Cell Sorting
The TVZ tissue was micro-dissected from PN1 Nestin-CFPnuc mice, dissociated into single cell suspensions by trituration after incubation in Trypsin Digest Medium, washed, resuspended in FACS buffer (Supplementary Methods), and incubated with allophycocyanin-conjugated cluster of differentiation (CD)133/prominin-1 (Prom1; Invitrogen 17-1331-81) antibody for 2 hours on ice. Cells were washed in cold FACS buffer, sorted through a 70-micron tip on a highly modified MoFlo HTS with Cyclone (Beckman Coulter) updated to 4 simultaneous lasers (Propel Labs) following laser-excitation at 488 nm and 640 nm. Gating parameters were empirically set, based on gating controls included with each experiment. Summit v4.3.02 software was used for acquisition.
NSC Neurospheres
FACS-sorted cells were maintained as non-adherent neurospheres in neural stem cell (NSC) medium in low attachment flasks (Corning) in a 37°C tissue culture incubator as previously described.16 The number of neurospheres formed per number of cells seeded was determined, and the results normalized against the Nestin-CFPnuc population clonogenic incidence. Proliferation assays used neurospheres from FACS cells dissociated into single cell suspensions, with 3 replicates per condition plated at a density of 1 × 105 cells/flask. At 13 days post-plating, neurospheres were dissociated and counted manually using a hemocytometer. Results were normalized against the cell count observed in the Nestin-CFPnuc population for each experiment. Each experiment was replicated 3 times with unique biological specimens. Statistical significance was determined at each age by one-way ANOVA corrected for multiple comparisons with a Tukey post-hoc test.
RNA Sequencing and Analysis
RNA was isolated from FACS cells using the Nucleospin RNA Plus XS kit (Takara Bio). A cDNA library was generated using the Clontech-SMARTer kit (Takara Bio), and sequenced on an Illumina HiSeq 2500. Base calls and demultiplexing were performed using Illumina bcl2fastq software and a custom Python demultiplexing program with a maximum of one mismatch in the indexing read. Using Partek Flow software v8.0, RNA-seq reads were aligned to the Ensembl release 95 v2 top-level assembly with STAR v2.5.3a, and gene counts were derived from Ensembl output.
Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. Normalization size factors were calculated for all gene counts by count per million (CPM) to adjust for differences in library size. Ribosomal genes and genes not expressed in the smallest group size minus one samples greater than one CPM were excluded from further analysis. Gene-specific analysis was then performed using the log-normal with shrinkage model (limma-trend method) to analyze for differences between conditions. Results were filtered for only those genes with P-values ≤0.01, log [fold-change] ≥ ± 2 and a false discovery rate [FDR] <5%. Differentially expressed gene contrasts were run for each of the 3 possible contrasts (NCFP vs CD133, NCFP_CD133 vs NCFP, NCFP_CD133 vs CD133) and unique lists for each group were found by filtering the intersections of the contrasts.
The resulting sequencing data are deposited in GEO (GSE149946).
RNAScope In Situ Hybridization
RNA in situ hybridization was performed using the Multiplex Fluorescent V2 Assay (ACDBio) in combination with Opal Dyes (Akoya Biosciences) as per manufacturer’s instructions. Probes are described in the Supplementary Methods. Images were acquired on a Leica DFC3000G fluorescent microscope using a 20x objective with Leica Application Suite X software.
Results
Three Spatially and Molecularly Distinct NPC Populations Reside Within the Murine TVZ
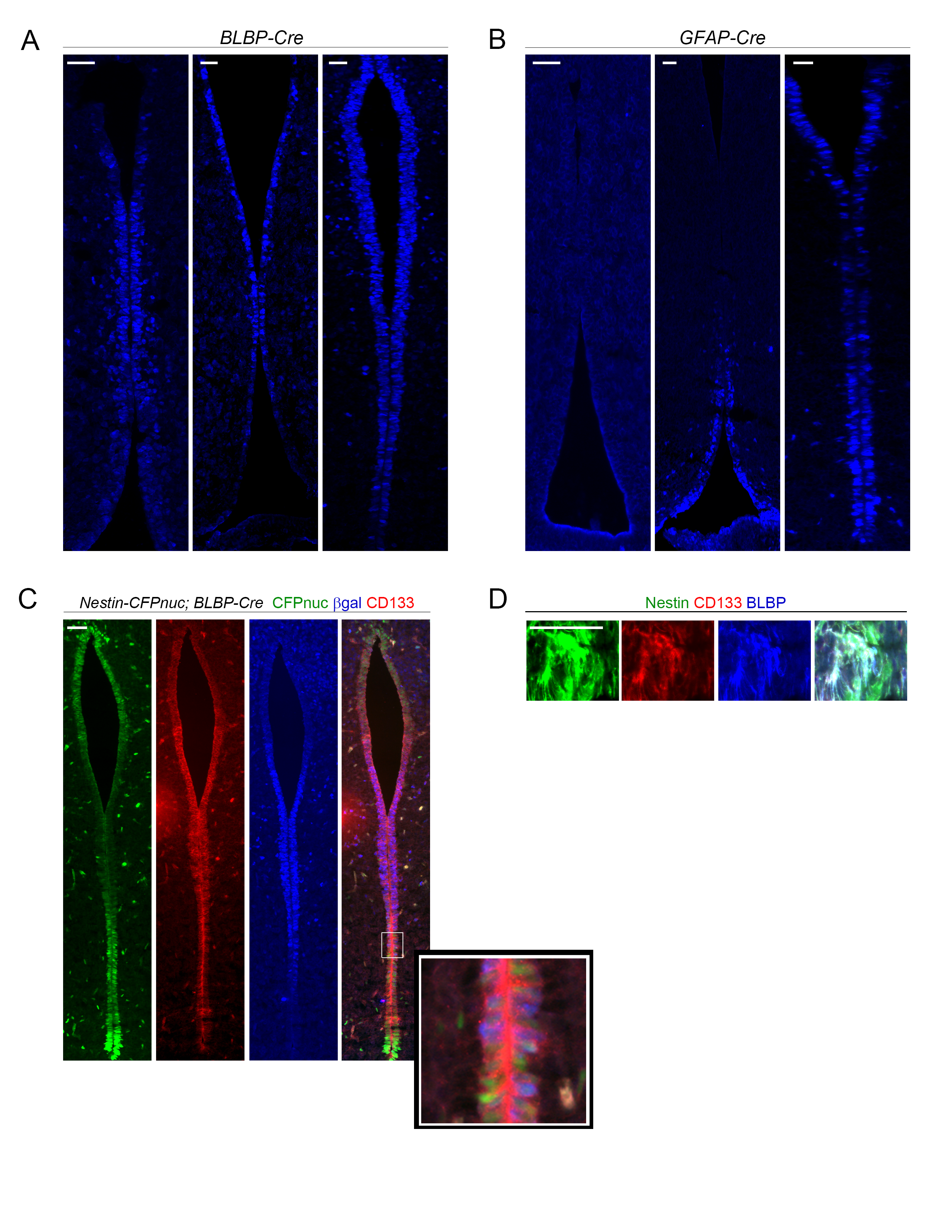
Previous mouse optic glioma modeling studies using the Cre-LoxP system have demonstrated that not all cell types are capable of giving rise to tumors following Nf1 loss.12,13,18,19 While these prior studies identified the TVZ as the region of the brain where the cell(s) capable of transformation upon somatic Nf1 loss reside,20,21 numerous reports have highlighted the striking cellular heterogeneity within this ventricular zone.22,23 To determine which TVZ cells were capable of giving rise to Nf1-OPG, we evaluated β-galactosidase expression as a marker of transgene expression in BLBP-Cre-IRES-LacZ and GFAP-Cre-IRES-LacZ mice, previously shown to generate optic gliomas when used to drive somatic Nf1 loss in Nf1-mutant (Nf1+/−) TVZ neuroglial progenitors (NPCs).12,13
Serial coronal sections were evaluated (schematic with low magnification images, Figure 1A). At postnatal day 1 (PN1) in GFAP-Cre-IRES-LacZ mice, β-galactosidase expression was present predominantly in neuroepithelial cells of the central and posterior TVZ (Figure 1B), whereas a broader distribution was identified in BLBP-Cre-IRES-LacZ mice (Figure 1C). Importantly, there was significant overlap between the cell populations in both Cre strains, predominantly within the central and posterior TVZ (Figure 1B, C, dotted white boxes). All the neuroepithelial cells lining the TVZ were immunopositive for the stem cell marker sex determining region Y–box 2 (Sox2) (Figure 1B, C), consistent with their designation as NPCs. Together with previous observations that BLBP-Cre; Nf1flox/-, and GFAP-Cre; Nf1flox/- mice form tumors with similar characteristics (microglial infiltration, proliferation, tumor volumes) and latency (~3 mo of age), these data suggest a common cell of origin for murine Nf1-OPG NPCs residing in the central-to-posterior TVZ.
Fig. 1.

Cre transgene expression identifies progenitors in the central-to-posterior TVZ as the putative cells of origin for murine Nf1-OPG. (A) Schematic of the TVZ, denoting the approximate anterior-posterior location of serial coronal sections (labeled 1, 2, 3, and 4) within the TVZ (left). Low magnification sagittal (top right) and coronal (bottom right) sections; TVZ, white dotted boxes. (B) β-gal and Sox2 immunostaining of serial coronal sections from PN1 GFAP-Cre-IRES-LacZ mice, demonstrating Cre transgene expression in Sox2+ progenitors in the central-to-posterior TVZ. (C) β-gal and Sox2 immunostaining from PN1 BLBP-Cre-IRES-LacZ mice, showing a broader distribution of Cre transgene expression in Sox2+ progenitors. BLBP- and GFAP-Cre transgene expression overlaps in the central-to-posterior TVZ (B, C, white boxes). At least 3 independent experiments were performed using unique biological replicates. Scale bars, 40 µm.
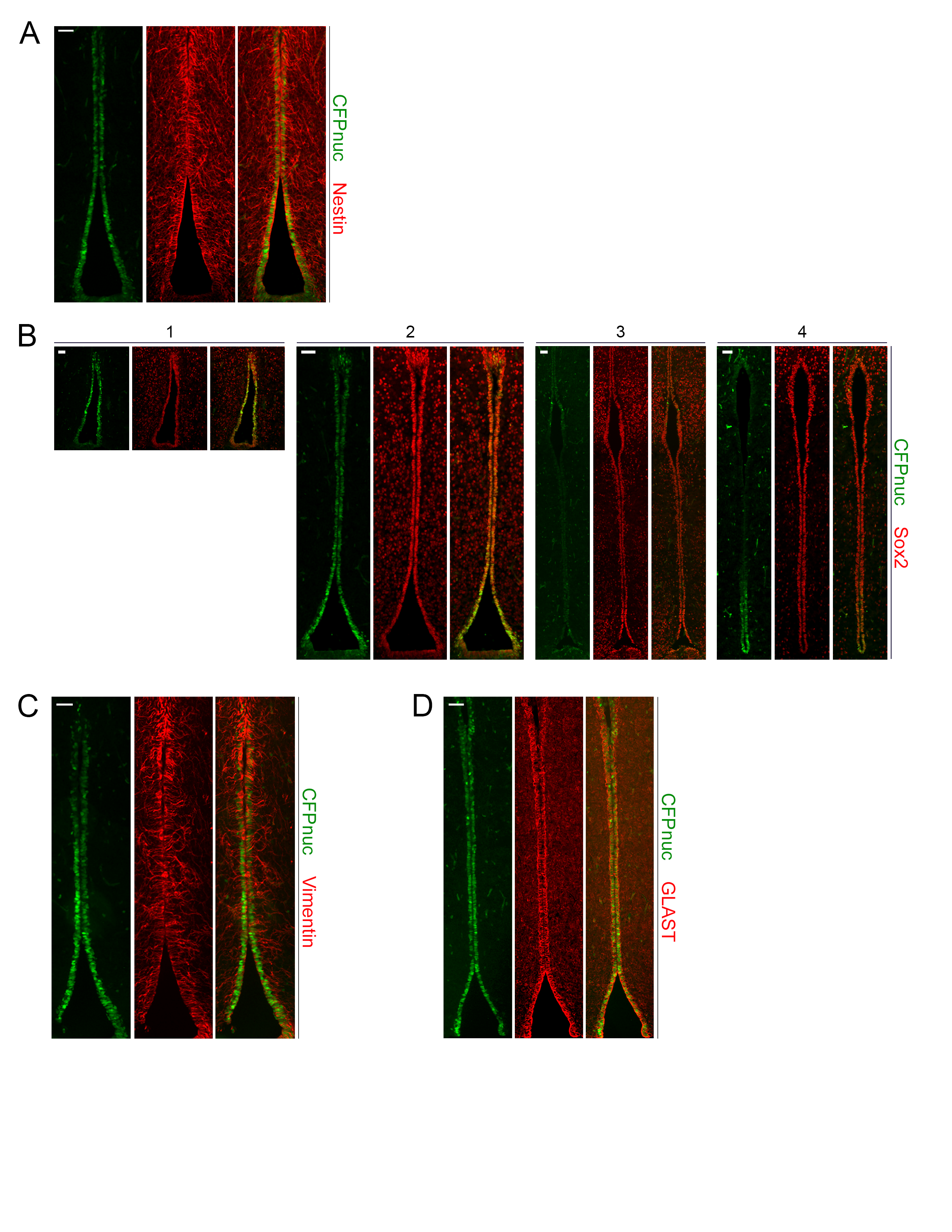
Second, to identify the NPC population(s) residing in the TVZ, we leveraged the Nestin-CFPnuc17 mouse, which expresses a nuclear-localized cyan fluorescent protein under the control of the Nestin promoter. After verifying that Nestin+ cells and CFPnuc+ cells co-localized in NPCs within the murine TVZ (Supplementary Figure 1A), we evaluated the expression of CFPnuc in PN1 mice. As seen in BLBP-Cre-IRES-LacZ and GFAP-Cre-IRES-LacZ transgenic mice, all TVZ neuroepithelial cells were Sox2-immunopositive, but only a subset were positive for CFPnuc (Supplementary Figure 1B). Serial coronal sections were then examined to determine the extent and localization of CFPnuc+ cells within the TVZ (schematic; Figure 2A). CFPnuc expression was identified predominantly in the anterior and posterior TVZ, with more robust expression observed ventrally (Figure 2B). In contrast to CFPnuc expression, the neural stem cell marker brain lipid binding protein (BLBP) was expressed from the mid-anterior to the posterior TVZ, with stronger expression dorsally (Figure 2C). Similar to BLBP, CD133 was expressed from the mid-anterior through the posterior TVZ, with stronger dorsal expression (Figure 2D). BLBP and CD133 exhibited significant co-localization in the TVZ (Figure 2E), and showed only a small area of overlap with CFPnuc+ cells in the mid-anterior and mid-posterior TVZ. Based on these findings, we identified 3 distinct cell populations: (1) a Nestin-CFPnuc+ population located predominantly in the anterior and posterior ventral TVZ (“V” cells; green), (2) a Nestin-CFPnuc+ BLBP+ CD133+ population (“M” cells; orange) predominantly localized in the transition zones in the anterior and posterior TVZ, and (3) a CD133+ BLBP+ population (“D” cells; red) located throughout the majority of the central TVZ, as well as in the dorsal tips of the anterior and posterior TVZ (Figure 2F). All 3 cell populations localized to the region identified to harbor the putative cell of origin for murine Nf1-OPG (dotted white boxes, Figure 1B, C).
Fig. 2.

Three populations of neuroglial progenitors are present in the TVZ of early postnatal mice. (A) TVZ schematic, denoting the approximate anterior-posterior location of serial coronal sections (labeled 1, 2, 3, and 4). (B) CFPnuc expression is predominantly detected within the anterior and posterior TVZ, with stronger expression in the anterior and ventral regions. (C) BLBP is expressed in progenitors within the mid-anterior to posterior TVZ. (D) Similar to BLBP, CD133 is expressed in progenitors from the mid-anterior to the posterior TVZ, with stronger expression dorsally. (E) BLBP and CD133 co-localize in progenitors within the TVZ. (F) Schematic representation of the 3 progenitor populations: “V” cells (green; CFP+ with predominantly ventral localization), “M” cells (orange; CFP+, BLBP+ and CD133+; intermediate localization), and “D” cells (red; BLBP+ and CD133+, predominantly dorsal localization). All 3 populations are present in the regions of interest identified in Figure 1 (dotted boxes). At least 3 independent experiments were performed using unique biological replicates. Scale bars, 40 µm.
Third, to determine whether these 3 populations are defined by previously described NPC markers, we first assessed for expression of vimentin and glutamate/aspartate transporter (GLAST) (radial glial cells). Similar to Sox2, vimentin and GLAST were evenly distributed throughout the TVZ (Supplementary Figure 1C, D). Next, we evaluated the expression of Vglut3 and Vcan, which identify subsets of adult tanycytes in the TVZ.23 Vglut3 and Vcan exhibited a spatial expression gradient in the adult TVZ, but were not expressed in the PN1 mouse brain (Supplementary Figure 2A, B). Lastly, we leveraged the Allen Brain Atlas to discover additional genes to define these PN1 TVZ NPC populations. Of these, Ddr1 was detected only in rare cells within the TVZ (both “M” and “D” cells, Supplementary Figure 2C), while Slit2 was detected only in the optic chiasm (Supplementary Figure 2D). Other genes diffusely expressed in the adult mouse TVZ were either not expressed (CD9 and Clic1; Supplementary Figure 2E) or expressed in all cells of neuroepithelial origin (Dyrk1b, Hdac6; Supplementary 2F), while those with a more spatially restricted expression profile (Cntfr, Gpr50; Supplementary Figure 2G) were not detected in the PN1 TVZ.
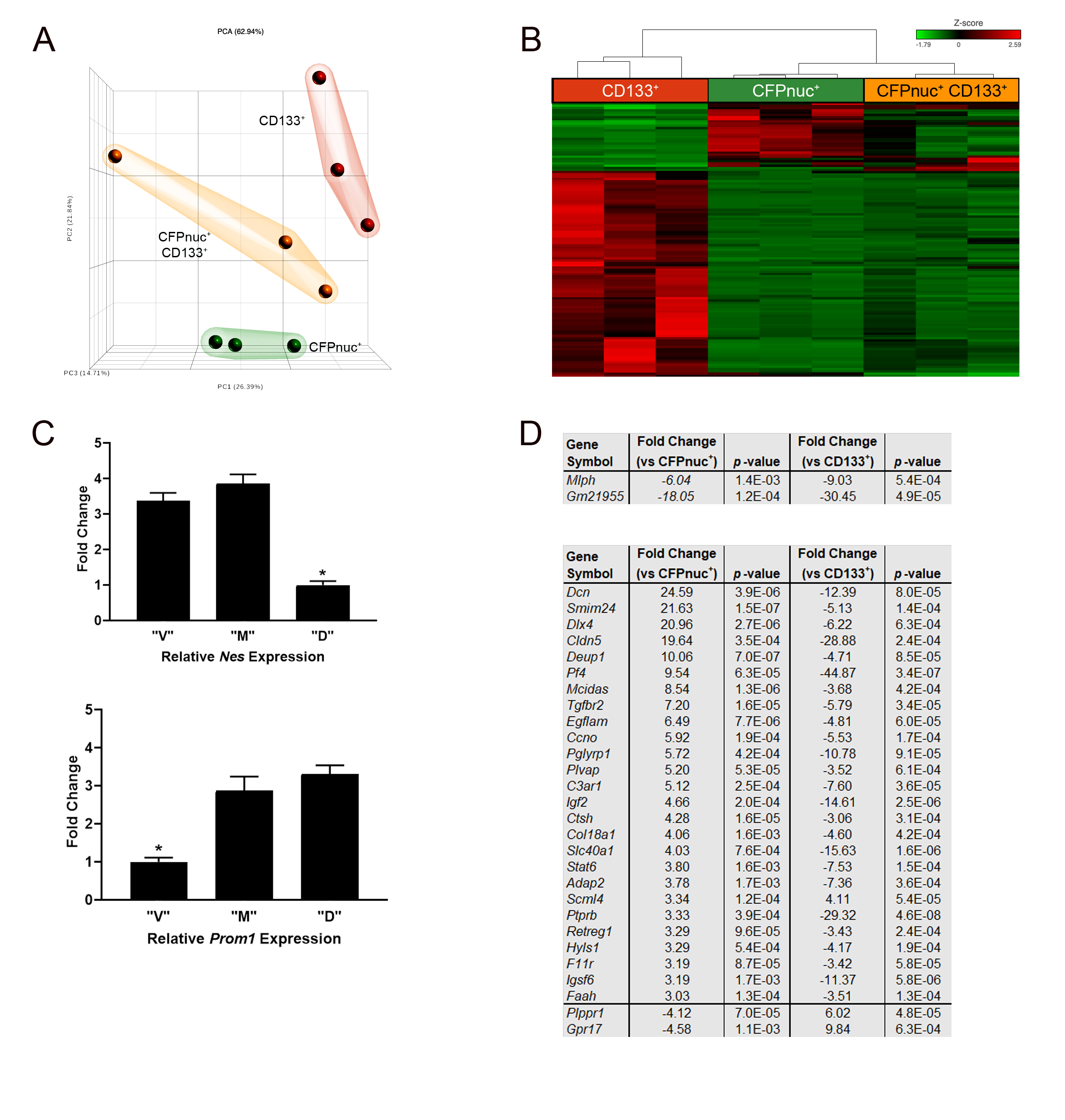
Finally, to identify potential biologic differences between these TVZ NPC populations, we employed FACS to isolate each population based on the expression of the Cyan Fluorescent Protein or the cell surface marker CD133 (Figure 3A). Using clonogenic incidence and proliferation assays, “M” type cells were functionally distinct, displaying both a higher rate of clonogenic incidence (Figure 3B) and greater proliferation (Figure 3C) than either the “V” or “D” cells. Principal component analysis (Supplementary Figure 3A) and hierarchical clustering of genes differentially expressed in these 3 NPC populations by RNA sequencing (Supplementary Figure 3B) revealed that “M” type cells were most similar to “V” type cells. Nes showed >3-fold enrichment in “V” and “M” cells compared with “D” cells (Supplementary Figure 3C, top panel), while Prom1 (CD133) showed >2-fold enrichment in “M” and “D” cells compared with “V” cells (Supplementary Figure 3C, bottom panel), confirming the sorting efficiency. At a higher cutoff, a total of 37 genes were identified as differentially expressed (≥3-fold change, P < 0.01, FDR <5%) in “M” type cells relative to the other 2 NPC populations, of which 28 appeared to be “transition” states (eg, increased compared with “V” cells and decreased compared with “D” cells or vice versa; Supplementary Figure 3D, bottom table), 2 were downregulated (Supplementary Figure 3C, top table), and 7 were upregulated (Figure 3D). Validation of these targets was performed by RNAScope in situ hybridization. Of the 3 differentially expressed genes for which suitable RNA probes could be generated, Msx1 and Npy were enriched in “M” type cells (Figure 3E, F, top panels; higher magnification images on the right), but not in “V” type cells (Figure 3E, F, bottom panels), whereas Wnt8b expression fell below the threshold of detection (data not shown).
Fig. 3.

The 3 populations of TVZ neuroglial progenitors are functionally and molecularly distinct. (A) FACS separation of the 3 TVZ NPC populations (left), with the percentage of each population relative to the entire population (right). (B) “M” (CFPnuc+ CD133+) cells have a higher clonogenic incidence (*p < 0.05, one-way ANOVA with Tukey post-hoc test) following normalization to the CFPnuc+ population. (C) “M” (CFPnuc+ CD133+) cells demonstrate higher proliferation (*P < 0.05, one-way ANOVA with Tukey post-hoc test) following normalization to the CFPnuc+ population. (D) Genes upregulated in the “M” (CFPnuc+ CD133+) population (fold-change ≥ ±3, false discovery rate <5%, P value <0.01). Using RNAScope, (E) Msx1 and (F) Npy are detected in progenitor cells coexpressing CFPnuc and CD133 (“M” cells, top panels), but not in cells expressing CFPnuc only (“V” cells, bottom panels). Higher magnification overlay images are included to the right. Scale bars, 40 µm.
Dynamic Changes in NPC Populations Occur During Mouse Brain Development
Previous studies have shown that the timing of somatic Nf1 loss during mouse brain development is critical for Nf1-OPG formation.19,20 Based on this temporal patterning, we evaluated serial coronal sections of the embryonic TVZ (schematic, Figure 4A) in Nestin-CFPnuc mice for the 3 populations identified at PN1. At E16.5, a well-defined TVZ could be appreciated with a thicker progenitor cell layer than observed in older mice. In contrast to PN1 mice, CFPnuc was expressed more broadly throughout the anterior, central and a significant portion of the posterior TVZ (Figure 4B), while BLBP expression showed a similar pattern to PN1 mice (mid-anterior to posterior, increased dorsal expression; Figure 4C). CD133 protein expression was predominantly detected in the posterior TVZ at E16.5 (data not shown), increasing over time to resemble the PN1 TVZ by E18.5 (Figure 4D), at which time CFPnuc remained broadly distributed. While all 3 populations were identified during late embryogenesis (Figure 4E), the “M” population was more abundant and declined in relative abundance by PN1. This decline continued over postnatal development due to an age-dependent decrease in BLBP and CD133 expression, with low levels of expression detected in the central-to-posterior region by PN7 (Figure 5A, B), and virtually no expression by PN28 (Figure 5C, D). In contrast, CFPnuc expression begins to rebound by PN7 and is diffusely positive in TVZ tanycytes at PN28 (Figure 5C, D), consistent with prior reports.24–26
Fig. 4.

Increased “M” cells are observed during embryogenesis. (A) Schematic of the TVZ during embryogenesis, denoting the approximate anterior-posterior location of serial coronal sections (labeled 1, 2, 3, and 4) within the TVZ. (B) CFPnuc is broadly expressed throughout the embryonic TVZ, whereas (C) BLBP expression is localized to progenitors from the mid-anterior to posterior TVZ. (D) CD133 is expressed in progenitors from the mid-anterior to posterior TVZ. (E) Schematic representation of the 3 progenitor populations: “V” cells (green; CFP+), “M” cells (orange; CFP+, BLBP+ and CD133+), and “D” cells (red; BLBP+ and CD133+) – with magnified images showing staining patterns. At least 3 independent experiments were performed using unique biological replicates. Scale bars, 40 µm.
Fig. 5.

“M” cells (CFPnuc+ BLBP+ CD133+) decrease during postnatal life. Relative to embryonic stages, PN mice demonstrate time-dependent reductions in BLBP (A, C) and CD133 (B, D) expression in TVZ NPCs (A, B, PN1; C, D, PN28). At least 3 independent experiments were performed using unique biological replicates. (E) Ki67 indices decline over time in the wild-type TVZ, showing the highest proliferation rate during embryogenesis when “M” cells are more abundant (*P < 0.05, one-way ANOVA with Tukey post-hoc test). Inset panel shows representative staining for the TVZ at each time point. Scale bars, 40 µm.
Since NPCs with higher rates of growth would be preferentially expanded in the setting of Nf1 loss, we assessed the proliferation of TVZ progenitors over time. As previously reported in the mouse and human TVZ,20 proliferation, as determined by Ki67 immunolabeling, is greater in the embryonic TVZ and declines by PN1. Taken together, these data demonstrated that “M” type cells are most abundant at the time when proliferation in the TVZ is high (Figure 5E).
Consistent with embryonic “M” cells potentially serving as the cells of origin for murine Nf1-OPG, Cre transgene expression (β-galactosidase+, marking the cells with biallelic Nf1 loss in OPG mice12,13) was detected in overlapping distributions in the central and posterior TVZ of embryonic BLBP-Cre-IRES-LacZ and GFAP-Cre-IRES-LacZ mice Supplementary Figure 4A,B including in a subset of “M” type cells (white box and inset, Supplementary Figure 4C). “M” cells were also observed in the late embryonic optic nerve (Supplementary Figure 4D), consistent with the migration of these pre-neoplastic precursors to the site of later tumor formation. Finally, “M” cells could also be detected in TVZ tissue obtained from third trimester human fetuses (Supplementary Figure 5A, B), further supporting their role in human NF1-OPG formation.
Nf1 Germline Mutations Differentially Increase TVZ NPC Proliferation
Emerging evidence from population-based studies,27–29 human induced pluripotent stem cells,30 and genetically engineered mice15 has revealed that germline NF1 mutations differentially affect tumor development and cell function. In this regard, we have previously shown that Nf1-mutant mice harboring different germline Nf1 gene mutations exhibit differences in optic glioma formation and proliferation.15 To determine whether the germline Nf1 gene mutation might differentially affect NPCs relevant to the risk of tumor formation, we crossed Nestin-CFPnuc mice with mice heterozygous for several different germline Nf1 mutations (Figure 6A), including an artificial null allele (neomycin insertion cassette; Nf1+/−) or one of 3 different mutations identified in NF1 patients (Gly848Arg, Arg681X, Arg1278Pro). 3 of these mutations (neo, Arg681X, Arg1278Pro) cause Nf1-OPG formation in GFAP-Cre; Nf1flox/mut mice,15,16,31 while GFAP-Cre; Nf1flox/Gly848Arg mice do not develop optic gliomas.15
Fig. 6.

Germline Nf1 mutations differentially increase TVZ progenitor proliferation in a time-dependent manner. (A) The mice heterozygous for different Nf1 germline gene mutations used, segregated by OPG formation in GFAP-Cre; Nf1flox/mut mice. (B) All germline Nf1 gene mutations increased BLBP expression in progenitors in the anterior ventral TVZ by immunostaining (top panel), resulting in decreased “V” cell expansion (bottom panel). (C) Increased Ki67 indices in mice heterozygous for tumor-causing Nf1 mutations (Arg681X, n = 3; Arg1278Pro, n = 4; neo, n = 3) at E16.5 (top panel), but not in mice heterozygous for a non-tumor-causing mutation (Gly848Arg, n = 3). This pattern persisted at PN1 (middle panel). At least 3 independent experiments were performed using unique biological replicates. Significance was determined by one-way ANOVA with correction for multiple comparisons using a Tukey post-hoc test, *P < 0.05. (D) Schematic model of the 3 factors that partially dictate Nf1-OPG penetrance. Scale bars, 40 µm.
Increased BLBP expression was observed at PN1 in all Nf1-mutant strains relative to WT controls, extending into the ventral-anterior region of the TVZ that is normally populated by “V” cells (CFPnuc+ only; Figure 6B). CFPnuc and CD133 expression were unchanged (data not shown), indicating that Nf1 mutation induces aberrant BLBP expression but does not overtly increase “M” cell abundance. In contrast, mice heterozygous for the neo, Arg681X, and Arg1278Pro mutations (OPG-forming mice) exhibited increased proliferation (% Ki67+ cells at E16.5 and PN1; Figure 6C, top and middle panels), whereas the proliferation of TVZ progenitors from mice heterozygous for the Gly848Arg mutation (non-OPG-forming mice) was indistinguishable from wild-type controls.
Discussion
Prior epidemiologic and clinical investigations have identified numerous potential factors that could influence the risk of developing a brain tumor during childhood. However, defining the individual contributions of each of these elements is challenging in human studies, where the data may be insufficiently granular or powered to tease apart the relative contributions of multiple variables. In this study, we leveraged genetically engineered mouse strains to create a model in which murine Nf1-OPG penetrance is partly determined by a confluence of individual factors, including the abundance of “M” cells within the TVZ, the level of TVZ NPC proliferation as a function of age, and the differential effects of the germline Nf1 mutation on these TVZ NPCs (Figure 6D). These findings raise several important points relevant to brain tumor pathogenesis in children.
First, the cell of origin is a critical determinant of brain tumor penetrance. In this report, we identify 3 functionally and molecularly distinct populations of progenitors within the TVZ. Consistent with prior Nf1 optic glioma modeling experiments using BLBP-Cre13 and prominin-1 (CD133)-Cre19 lines, our data suggest that one of these TVZ NPC populations, the Nestin+/BLBP+/CD133+ “M” cells, is the most likely cell of origin for murine Nf1-OPG. These findings add to the growing recognition that not all cells are capable of generating brain tumors, with more differentiated cells showing decreasing susceptibility to malignant transformation,32 reflecting a transformational hierarchy where stem cells sit at the apex. In situations where multiple cell types can give rise to brain tumors, transforming events often yield different tumor latencies or subtypes. For example, while both Olig2+ progenitors and Nestin+/BLBP+/CD133+ “M” cells can generate Nf1 optic gliomas, somatic Nf1 loss in Olig2+ progenitors delays tumor latency from 3 to 6 months of age.19 Similarly, in mouse models of adult high-grade gliomas, identical driver mutations produce phenotypically and molecularly distinct glioblastoma subtypes when expressed in different pools of adult CNS progenitors.33 Distinct tumor types may also be driven by identical driver mutations in different cells of origin, such that EGFRvIII and TP53 mutations induce benign infiltrative astrocytomas or malignant astrocytomas/ oligodendrogliomas, depending on the cell type being transformed (glial restricted progenitors, gastrin-releasing peptides (GRPs), astrocytes derived from mutant GRPs, or OPCs, respectively).34
Second, the developmental stage at which somatic mutation occurs is also critical for brain tumor formation in children. Consistent with prior studies from our laboratory using an inducible neural stem cell Cre transgenic mouse (prominin-1 (CD133)-CreER) to demonstrate that optic glioma formation requires somatic Nf1 loss during embryonic development,19 we now show that “M” cells are most abundant during embryogenesis and disappear during early postnatal life. Moreover, this pattern of “M” cell loss mirrors the temporal course of proliferation within the TVZ, which sharply declines by PN1. The finding that the cells most likely to give rise to Nf1-OPGs are both more plentiful and continuing to proliferate during late embryogenesis suggests that the young age of NF1-OPG development (mean, 4.5 y9) in children might reflect the presence of a greater number of dividing progenitor cells capable of generating these tumors during late fetal life. While human and murine TVZ development are not identical (ie, early presence of glial fibrillary acidic protein–positive (GFAP+) radial glia-like cells22), the age-dependent decline in proliferation is similar22 and “M” cells were detected in human fetal TVZ tissue.
Third, mutational specificity is another under-recognized risk factor for cancer formation. In addition to population-based genotype-phenotype correlations in NF1 demonstrating that germline mutations in particular regions of the NF1 gene might predispose to optic gliomas,29 the impact of mutational specificity is best illustrated by the Arg1809 missense mutation,35 where individuals with this germline mutations do not develop neurofibromas or OPGs. These clinical observations, coupled with the findings reported herein, support the conclusion that NF1 germline mutations may not be equivalent in their ability to promote tumor development. Understanding how these mutations differentially expand the progenitors for somatic Nf1 inactivation may provide important insights relevant to future predictive risk assessment for children with NF1.
Taken together, these results suggest that pediatric glioma (Nf1 optic glioma) development requires a “perfect storm,” where multiple conditions need to coexist in order for genetic mutation to result in tumor formation. Some of these factors, such as the cell of origin and age, are inextricably linked, given the evolution of cellular identity in the developing brain, whereas others (germline Nf1 mutation) could operate independently. The fact that each of these factors converge on the cells of origin raises the intriguing possibility that other risk factors, including maternal exposures (eg, infection, drug exposure, dietary changes) could similarly impact on gliomagenesis in children by acting on susceptible progenitor cell populations. In this regard, maternal infection has been associated with reduced NPC proliferation,36 whereas maternal high-fat diet during gestation increases hypothalamic NPC proliferation and differentiation.37 Future mechanistic work will be required to determine how each of these risk factors modulate NPC function relevant to the patterning and biology of pediatric low-grade glioma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank the Alvin J. Siteman Cancer Center (SCC) at Washington University School of Medicine and Barnes-Jewish Hospital for the use of the Siteman Flow Cytometry Core facility, and the SCC and the Washington University Institute of Clinical and Translational Sciences (ICTS) for the use of the Genome Technology Access Center. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant (#P30 CA091842), while the ICTS is funded by the National Institutes of Health CATS Clinical and Translational Science Award program (#UL1 TR002345). A portion of this work was presented at the 2019 Society for Neuro-Oncology meeting (abstract #694309).
Funding
This work is funded by grants from Alex’s Lemonade Stand Foundation (18-12558 to N.M.B.), Hyundai Hope on Wheels (DR-2019–672 to N.M.B.) and the National Institute of Neurological Disorders and Stroke (1-R35-NS07211-01 to D.H.G).
Conflict of interest statement. D.H.G. has a licensing agreement with the Tuberous Sclerosis Alliance (GFAP-Cre mice).
Authorship statement. Conceptualization: N.M.B., D.H.G. Methodology: N.M.B., S.T. Writing: N.M.B., D.H.G. Investigation, N.M.B. performed most of the experiments; S.T. performed the quantitative analyses; O.C. performed the RNA sequencing analysis; S.D. procured the human specimens. Resources: D.H.G. Supervision: D.H.G. Funding: N.M.B., D.H.G.
References
- 1. Armstrong GT. Long-term survivors of childhood central nervous system malignancies: the experience of the Childhood Cancer Survivor Study. Eur J Paediatr Neurol. 2010;14(4):298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17(9):503–511. [DOI] [PubMed] [Google Scholar]
- 3. Makino K, Nakamura H, Yano S, Kuratsu J; Kumamoto Brain Tumor Group . Population-based epidemiological study of primary intracranial tumors in childhood. Childs Nerv Syst. 2010;26(8):1029–1034. [DOI] [PubMed] [Google Scholar]
- 4. Georgakis MK, Karalexi MA, Kalogirou EI, et al. Incidence, time trends and survival patterns of childhood pilocytic astrocytomas in Southern-Eastern Europe and SEER, US. J Neurooncol. 2017;131(1):163–175. [DOI] [PubMed] [Google Scholar]
- 5. Ostrom QT, de Blank PM, Kruchko C, et al. Alex’s Lemonade Stand Foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2015;16(Suppl 10):x1–x36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burkhard C, Di Patre PL, Schüler D, et al. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J Neurosurg. 2003;98(6):1170–1174. [DOI] [PubMed] [Google Scholar]
- 7. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang J, Wu G, Miller CP, et al. ; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project . Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125(1):63–66. [DOI] [PubMed] [Google Scholar]
- 10. Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61(3):189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63(24):8573–8577. [PubMed] [Google Scholar]
- 13. Hegedus B, Dasgupta B, Shin JE, et al. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell. 2007;1(4):443–457. [DOI] [PubMed] [Google Scholar]
- 14. Brannan CI, Perkins AS, Vogel KS, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8(9):1019–1029. [DOI] [PubMed] [Google Scholar]
- 15. Toonen JA, Anastasaki C, Smithson LJ, et al. NF1 germline mutation differentially dictates optic glioma formation and growth in neurofibromatosis-1. Hum Mol Genet. 2016;25(9):1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo X, Pan Y, Gutmann DH. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol. 2019;21(10):1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci U S A. 2006;103(21):8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solga AC, Gianino SM, Gutmann DH. NG2-cells are not the cell of origin for murine neurofibromatosis-1 (Nf1) optic glioma. Oncogene. 2014;33(3):289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Solga AC, Toonen JA, Pan Y, et al. The cell of origin dictates the temporal course of neurofibromatosis-1 (Nf1) low-grade glioma formation. Oncotarget. 2017;8(29):47206–47215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee DY, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22(1):131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen YH, Gianino SM, Gutmann DH. Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev. 2015;29(16):1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dahiya S, Lee DY, Gutmann DH. Comparative characterization of the human and mouse third ventricle germinal zones. J Neuropathol Exp Neurol. 2011;70(7):622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen R, Wu X, Jiang L, Zhang Y. Single-cell RNA-seq reveals hypothalamic cell diversity. Cell Rep. 2017;18(13):3227–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furube E, Ishii H, Nambu Y, et al. Neural stem cell phenotype of tanycyte-like ependymal cells in the circumventricular organs and central canal of adult mouse brain. Sci Rep. 2020;10(1):2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robins SC, Stewart I, McNay DE, et al. α-Tanycytes of the adult hypothalamic third ventricle include distinct populations of FGF-responsive neural progenitors. Nat Commun. 2013;4:2049. [DOI] [PubMed] [Google Scholar]
- 26. Mirzadeh Z, Kusne Y, Duran-Moreno M, et al. Bi- and uniciliated ependymal cells define continuous floor-plate-derived tanycytic territories. Nat Commun. 2017;8:13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharif S, Upadhyaya M, Ferner R, et al. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J Med Genet. 2011;48(4):256–260. [DOI] [PubMed] [Google Scholar]
- 28. Bolcekova A, Nemethova M, Zatkova A, et al. Clustering of mutations in the 5’ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma. 2013;60(6):655–665. [DOI] [PubMed] [Google Scholar]
- 29. Anastasaki C, Morris SM, Gao F, Gutmann DH. Children with 5′-end NF1 gene mutations are more likely to have glioma. Neurol Genet. 2017;3(5):e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anastasaki C, Wegscheid ML, Hartigan K, et al. Human iPSC-derived neurons and cerebral organoids establish differential effects of germline NF1 gene mutations. Stem Cell Reports. 2020;14(4):541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22(14):5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alcantara Llaguno S, Sun D, Pedraza AM, et al. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat Neurosci. 2019;22(4):545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alcantara Llaguno SR, Wang Z, Sun D, et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell. 2015;28(4):429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J, Bushman J, Wang X, Mayer-Proschel M, Johnson M, Noble M. Oligodendrocyte/type-2 astrocyte progenitor cells and glial-restricted precursor cells generate different tumor phenotypes in response to the identical oncogenes. J Neurosci. 2013;33(42):16805–16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rojnueangnit K, Xie J, Gomes A, et al. High incidence of noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat. 2015;36(11):1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stolp HB, Turnquist C, Dziegielewska KM, Saunders NR, Anthony DC, Molnár Z. Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain. 2011;134(Pt 11):3236–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci. 2008;28(46):12107–12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.