

Abstract
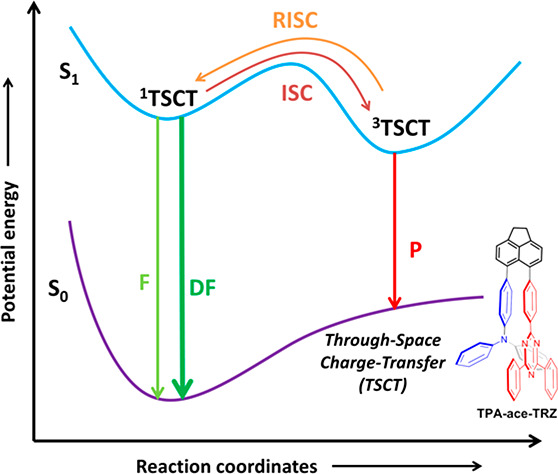
Commonly, thermally activated delayed fluorescence (TADF) emitters present a twisted donor–acceptor structure. Here, electronic communication is mediated through-bond via π-conjugation between donor and acceptor groups. A second class of TADF emitters are those where electronic communication between donor and acceptor moieties is mediated through-space. In these through-space charge-transfer (TSCT) architectures, the donor and acceptor groups are disposed in a pseudocofacial orientation and linked via a bridging group that is typically an arene (or heteroarene). In most of these systems, there is no direct evidence that the TSCT is the dominant contributor to the communication between the donor and acceptor. Herein we investigate the interplay between through-bond localized excited (LE) and charge-transfer (CT) states and the TSCT in a rationally designed emitter, TPA-ace-TRZ, and a family of model compounds. From our photophysical studies, TSCT TADF in TPA-ace-TRZ is unambiguously confirmed and supported by theoretical modeling.
The use of metal-free TADF emitters in organic light-emitting diodes (OLEDs) has attracted significant attention within the OLED community following the first reports from Goushi et al.1 and Uoyama et al.,2 which demonstrated the potential of E-type delayed emission exciplexes and donor–acceptor emitters to produce high-efficiency devices. The reason TADF emitters have become so attractive is due to their capacity to harvest up to 100% of triplet excitons.3 This is made possible by the small energy gaps (ΔEST), generally less than 100 meV, among the lowest CT singlet, triplet, and local triplet excited states of these materials, enabling efficient upconversion of the electrically generated CT triplet states through a vibrationally coupled spin–orbit coupling mechanism where the local triplet excited state (T1) mediates the otherwise forbidden 3CT triplet to 1CT singlet transition via reverse intersystem crossing (RISC).4,5 TADF-OLEDs have achieved maximum external quantum efficiencies (EQEmax) comparable with those of OLEDs based on phosphorescent emitters.6 The requisite small 1CT–3CT ΔEST is achieved within the emitter by spatially separating the HOMO and LUMO on donor (D) and acceptor (A) moieties, respectively, of D–A molecules, thereby minimizing the electron exchange integral of the frontier molecular orbitals.7 However, this strategy can lead to a compromise with the photoluminescence quantum yield (ΦPL) of the emitter due to a reduction of the oscillator strength (f) for this transition, which corresponds to a slower radiative decay rate. Therefore, a balance in modulating ΔEST, f, and slow nonradiative decay is required to produce highly efficient TADF emitters8 and OLEDs.
While a typical intramolecular D–A TADF emitter is monomolecular, with its emissive singlet excited state having CT character, CT excited states can also be realized in intermolecular bimolecular exciplex systems, which likewise show TADF.9,10 A wide variety of intermolecular exciplex TADF emitters have been developed with appropriate combination of D and A molecules;11−13 however, several technical issues remain in converting exciplex emitters to high-efficiency OLEDs. These include low ΦPL and very broad emission envelopes due to the inhomogeneity of the donor–acceptor distances in the exciplex.14
Designing TADF emitters based on TSCT is an alternative approach to overcoming many of the issues associated with exciplex-OLEDs.15 Compared to a traditional twisted intramolecular charge-transfer TADF emitter design, where D and A moieties are directly attached to each other and the CT transition takes place through-bond, in a TSCT-TADF emitter the D and A moieties are separated by a electronically benign spacer but remain in close proximity to each other such that electronic communication and electron transfer are mediated through-space in an analogous manner to exciplexes.16 This approach not only results in emitters with small ΔEST values but also overcomes the issues of inhomogeneous D–A distances in the exciplex emitters in thin films.
Herein we report a study of TSCT and model compounds using acenaphthene as a scaffold and triphenylamine (TPA) as the donor. TSCT TADF in TPA-ace-TRZ is unambiguously confirmed by comparison to the family of materials investigated. The structural, electrochemical, and photophysical properties of TPA-ace-TRZ were investigated and compared with those of model compounds TPA-ace-CN, TPA-ace, TPA-ace-Br, and 2TPA-ace. From our photophysical studies, we observe changing local and CT emission, delayed fluorescence, and phosphorescence emission in these molecules, even at room temperature. With a focus on CT excited states, a mechanistic approach has been attempted to establish the structure–property relationship in these molecules.
The syntheses of TPA-ace, 2TPA-ace, TPA-ace-Br, TPA-ace-CN, and TPA-ace-TRZ are outlined in Scheme 1. Each of the target materials has been characterized by a melting-point determination, 1H and 13C NMR spectroscopy, and mass spectrometry. Elemental analysis and HPLC analysis were used to evidence the purity of the emitters (Supporting Information).
Scheme 1. Synthesis Scheme for TPA-ace, 2TPA-ace, TPA-ace-Br, TPA-ace-CN, and TPA-ace-TRZ, Including X-ray Structure Diagrams Showing Intramolecular Interactions.

The solvent molecule and hydrogen atom are omitted for clarity.
Single crystals of TPA-ace, TPA-ace-CN, 2TPA-ace, and TPA-ace-TRZ were grown by the vapor diffusion of n-hexane into a saturated dichloromethane solution of the compound. The molecular structures were determined by single-crystal X-ray diffraction analysis and are shown in Scheme 1. Despite the design of enforced proximity between substituents of the acenaphthene, the flexibility of the ring system allowed sufficient splay to develop between substituents such that surprisingly few intramolecular interactions are observed. In TPA-ace-CN, there is the potential for a weak interaction between the cyano group and the bridging phenyl of the TPA [N···centroid distance of 3.598(2) Å], while in both TPA-ace-TRZ and 2TPA-ace intramolecular π···π interactions occur between the proximal phenyl rings only of the substituents [centroid···centroid distances of 3.5876(8) and 3.6881(12) Å, respectively]. Three different patterns of intermolecular interactions are seen in the structures. In TPA-ace, head-to-head dimers are formed via pairs of weak CH···π interactions between two methylene hydrogens and the naphthalene (H···centroid distances of 2.87 and 2.94 Å). In both 2TPA-ace and TPA-ace-CN, three-dimensional networks are formed from different combinations of weak interactions. In 2TPA-ace, the network arises from four sets of CH···π interactions involving TPA-phenyl hydrogens and either naphthalene or TPA-phenyl π-systems (H···centroid distances of 2.69 to 2.90 Å). In TPA-ace-CN, the network is formed from two different CH···π interactions between phenyl hydrogens and the terminal phenyls of the TPA (H···centroid distances of 2.91 and 2.99 Å) as well as CH···N hydrogen bonds between one methylene hydrogen and the cyano nitrogen [H···N 2.54 Å, C···N 3.443(3) Å]. In TPA-ace-TRZ, there are three pairs of interactions: π···π interactions between the triazine and one of the TRZ-phenyl groups of an adjacent molecule [centroid···centroid distance of 3.361(8) Å] and two CH···π interactions involving both methylene and naphthalene hydrogens and either naphthalene or TRZ-phenyl π-systems (H···centroid distances of 2.83 and 2.91 Å). The combination of these leads to two-dimensional sheets in the ac-plane.
The ground-state geometries of each of the acenaphthene compounds were optimized in the gas phase at the PBE0/6-31G(d,p) level starting from the geometry obtained from the single-crystal X-ray diffraction analysis; that of TPA-ace-Br was optimized starting from an initial geometry drawn in GaussView. Time-dependent DFT calculations were performed within the Tamm–Dancoff approximation (TDA)17 using the ground-state optimized geometries. The energies and electron density distributions of the highest occupied and lowest unoccupied molecular orbitals (HOMO/LUMO) and the energies of the S1 and T1 states are shown in Figure 1. In TPA-ace-TRZ, TPA-ace-CN, and TPA-ace-Br, the HOMO is localized on the TPA moiety while the HOMO is delocalized over both the TPA and acenaphthene bridge in TPA-ace and 2TPA-ace. In TPA-ace-TRZ, the LUMO is localized mainly on the TRZ unit while in TPA-ace-CN and TPA-ace-Br the LUMO is localized on the CN- or Br-substituted acenaphthene moiety. In the case of TPA-ace and 2TPA-ace, the LUMO is delocalized across both the phenyl ring of the TPA and the acenaphthene. A large electron density overlap exists in TPA-ace and 2TPA-ace, which is reflected in their large calculated ΔEST values; the ΔEST for TPA-ace-Br is also large due to a change in the nature of the T1 state, which is mainly described by a HOMO – 1 to LUMO transition that is of a localized excited (LE) character on the acenaphthene. Much smaller ΔEST values were observed for TPA-ace-TRZ and TPA-ace-CN, which is reflective of the significantly greater spatial separation of the electron density distributions of the HOMO and LUMO. Importantly, for TPA-ace-TRZ there is no predicted through-bond communication, and instead we hypothesize that there is a through-space electronic communication directly between the TPA and TRZ moieties. The predicted energy of the HOMO level for each of the emitters is around −5.00 eV, a reflection of a HOMO localized in all compounds on the TPA moiety, coupled with poor conjugation to the acenaphthene bridge, except for TPA-ace-TRZ (−4.87 eV) where there is significant stabilization. On the other hand, the energy of the LUMO level varies significantly depending upon the nature of the electron acceptor. There is a cluster of compounds, TPA-ace, 2TPA-ace, and TPA-ace-Br, with associated LUMO values of between −0.85 and −0.99 eV, and then there are the two emitters, TPA-ace-CN and TPA-ace-TRZ, containing strong electron-withdrawing acceptors, that have LUMOs that are significantly stabilized at −1.56 and −1.68 eV, respectively. This same analysis is true for the S1 energy level as well where TPA-ace, 2TPA-ace, and TPA-ace-Br possess high S1 energies ranging from 3.41 to 3.65 eV while TPA-ace-CN and TPA-ace-TRZ have significantly lower S1 energies of 2.87 and 2.51 eV, respectively. The triplet energies cluster over a narrow range between 2.65 and 2.82 eV while there is greater divergence in the singlet-state energies. This thus leads to two groups among these compounds where TPA-ace, 2TPA-ace, and TPA-ace-Br have large ΔEST values and TPA-ace-TRZ and TPA-ace-CN possess significantly smaller ΔEST values.
Figure 1.

DFT-calculated ground-state (PBE0/6-31g(d,p)) and TDA-calculated excited-state energies, oscillator strengths, and electron density distributions (ISO value = 0.02) of the frontier molecular orbitals of the acenaphthene emitter derivatives.
The electrochemical properties of these compounds were investigated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Anodic scans reveal a reversible oxidation that is centered on the TPA (Figure S35), whereas the reduction waves were irreversible and inconclusive for all and were therefore omitted. The oxidation potential of TPA has been previously estimated at 0.87 V vs SCE.18 After grafting the acenaphthene unit, the oxidation potentials obtained from the DPVs were found to be much lower than that of TPA and were calculated at 0.26, 0.37, 0.38, 0.44, and 0.47 V for TPA-ace, 2TPA-ace, TPA-ace-TRZ, TPA-ace-Br, and TPA-ace-CN, respectively, which correspond to HOMO levels of −5.06, −5.17, −5.18, −5.24, and −5.27 eV, respectively. The addition of electron-withdrawing groups to the acenaphthene bridge stabilizes the oxidation potentials and reflects a certain degree of electronic coupling between the TPA and the acenaphthene bridge. The energies of the HOMO level determined from DPV are broadly in agreement with that predicted by DFT calculations.
Steady-state photophysical analysis was initially performed in dilute solution. Figure 2a shows the normalized optical absorption and emission spectra of TPA-ace in three different solvents. The TPA-ace absorption spectrum presents two peaks: a band centered at around 310 nm related to the acenaphthene scaffold16,19 and a second peak, at lower energy, associated with the π → π* transition of the delocalized TPA-ace system (Figure S36). With increasing solvent polarity, a minor red shift appears in the latter transition, whereas photoluminescence (PL) spectra exhibit a weak bathochromic shift and a change from a structured emission band to a Gaussian-shaped emission band. The small red shift could indicate that the bridge (ace) is not electronically decoupled and forms a weak through-bond charge-transfer (TBCT) state with the donor unit (TPA). This we propose as a highly mixed 1LE/1CT state with low CT character because there is conjugation between D and A since they are not orthogonally disposed, as found by DFT. In Figure 2b, we see that the blue-edge absorption feature in TPA-ace-Br is blue-shifted compared to TPA-ace, potentially indicating weaker conjugation between the TPA and ace-Br units. The PL spectrum of TPA-ace-Br yields dual emission in higher-polarity DCM. The high-energy band red shifts slightly with increasing solvent polarity, a behavior that matches very well with the mixed 1LE/1CT state observed in TPA-ace. The second, more intense, emission band at around 540 nm observed in DCM is assigned to a state with stronger CT character (stabilized by the high-polarity solvent), in line with the stronger acceptor strength of the ace-Br unit. With the addition of a second TPA donor unit, 2TPA-ace, changing from a donor–bridge (D–B) to a donor–bridge–donor (D–B–D) structure, the absorption and emission spectra remain similar to those of TPA-ace (Figure 2c). This similarity of the optical behavior is expected because the new TPA unit does not increase the strength of the donor, and the bridge effectively acts as a very weak acceptor unit and hence a similar ICT state forms. Careful inspection of the spectra, however, shows that the emission onset is red shifted by 822 cm–1 (in methylcyclohexane, MCH) and the band shape loses structure compared to the structured TPA-ace spectra. This we identify as the effect of an interaction between the two TPA units (i.e., a weak intramolecular dimer state), as supported by the X-ray structure. Concentration-dependent measurements (Figure S37) show that this is a purely monomolecular property, fully supporting the assumption that this is a cofacial intramolecular interaction between the two weakly overlapping TPA units.
Figure 2.

Normalized UV–vis absorption (dashed lines) and PL spectra (solid lines) of (a) TPA-ace, (b) TPA-ace-Br, and (c) 2TPA-ace molecules in different solvents at a concentration of 20 μM (λexc = 330 nm for TPA-ace and TPA-ace-Br; λexc = 340 nm for 2TPA-ace).
As with TPA-ace-Br, the absorption of TPA-ace-CN (Figure 3a) also has a weak lowest-energy transition, ascribed to the delocalized TPA-ace unit, that is much less intense than that observed in TPA-ace and 2TPA-ace. The TPA-ace-TRZ absorption spectra (Figure 3b) reflect the introduction of the TRZ group with the appearance of a strong TRZ absorption band at 270 nm20 and, as with the other members of the series, a lower-energy band at 340 nm from the TPA-ace system. Furthermore, unlike the other compounds in this study, we observe a low-intensity tail/band stretching from 390 to 440 nm, characteristic of a direct CT transition21 and strongly indicative of a ground state through-space interaction between the TRZ and TPA units16 and fully in line with the large calculated permanent dipole moment compared to the other four materials. With the introduction of stronger acceptor groups, cyano and TRZ, a broader emission band is observed, even in nonpolar MCH (Figure 3a), and a larger red shift was obtained with increasing solvent polarity, which indicates that the presence of the electron-accepting ace-CN unit contributes to the formation of a stronger CT character excited state. DFT calculations reveal that this is indicative of a through-bond CT state between a stronger D–A pair. The emission spectra of TPA-ace-TRZ across the range of solvents show a red-shifted maximum and much stronger positive solvatochromism, even in MCH, which indicates that the TRZ unit has a much stronger acceptor character than the bridge (manifesto). This different behavior strongly suggests that this is a TSCT state between the TRZ···TPA units that has ground-state electronic coupling.
Figure 3.

Normalized UV–vis absorption (dashed lines) and PL spectra (solid lines) of (a) TPA-ace-CN and (b) TPA-ace-TRZ molecules in different solvents (λexc = 340 nm) at a concentration of 20 μM.
Turning to time-resolved PL spectra, we investigated the dynamics of the excited states in this family of CT materials dissolved in toluene. Figure 4a shows the time-resolved normalized emission spectra of TPA-ace in toluene at room temperature. In the first few nanoseconds, an emission band centered at 425 nm related to a short-lived LE state of the TPA donor unit mixed with a small amount of CT character is observed. This local emission decays rapidly with a broader band at around 500 nm growing in, indicating a weak, prompt ICT state (Figure S38). This CT state is transient and rapidly decays within 30 ns. This behavior is characteristic of a TICT state22 between the TPA and ace units, where the phenyl ring inking the TPA and ace units rotates from 48° to nearly orthogonal, fully breaking the conjugation of the two to stabilize the CT state for a short time. Analogous to the steady-state PL measurement, 2TPA-ace shows a time-resolved spectrum very similar to that of TPA-ace (Figure 4c), with a minor enhancement of the CT contribution potentially due to the through-space interaction of the two D units. Figure 4d shows the decay curve of these molecules in degassed toluene (λexc = 355 nm). In the nanosecond regime, TPA-ace and 2TPA-ace present fast emission decay from the mixed LE/CT state contribution, with τPL = 1.57 and 2.34 ns, respectively, showing the predominant LE character of this state. For TPA-ace-Br, at early times, two emission bands are observed simultaneously: an LE/CT emission at 425 nm (observed only in the first time window) analogous to that observed for TPA-ace and 2TPA-ace (Figure S38) as well as a prompt low-energy CT emission at 525 nm, with τPL = 9.4 ns (Table 1), but again it is a transient state. This CT band is much more intense than the transient species in TPA-ace and 2TPA-ace and has a longer lifetime. This we interpret as ace-Br being a sufficiently strong acceptor to permit the formation of a stable and more red-shifted through-bond CT state; the steric hindrance of the Br atom introduces a large torsion angle of 81° between TPA and ace-Br, corroborated by our DFT calculations. However, because we observed this state at about the same energy in toluene and DCM for all of these materials, we surmise that it must have weak CT character (i.e., high LE character), so again we assign this band to a mixed LE/CT state, which is red-shifted via the additional charge decoupling coming through the increased orthogonality between effective D (TPA) and A (ace-Br) groups. After an interval without emission (below the noise floor of the iCCD), at times after 50 μs, a delayed fluorescence (DF) emission having the same onset energy as the prompt CT band can be observed. In addition, there is a second weaker band centered at around 590 nm that we ascribe to phosphorescence. Because the energy gap between the triplet and singlet states (ΔEST) is large, ca. 260 meV, as described later, any RISC would be very weak and slow. This is why we observe dual emission in the form of TADF and room-temperature phosphorescence (RTP) in this case. We note that this dual emission behavior is in part observable because of the heavy atom effect of the Br, which increases the ISC, yielding a large triplet population, which in turn allows weak RISC to be observed. Again, through enhanced spin–orbit coupling, the phosphorescence radiative decay increases such that we can simultaneously observe RTP from the large triplet population.
Figure 4.

Time-resolved normalized PL spectra of (a) TPA-ace, (b) TPA-ace-Br, and (c) 2TPA-ace in toluene solution at a concentration of 20 μM. (d) Time-resolved PL decay curves in the entire region of analysis. λexc = 355 nm. (b) The spectrum recorded for TPA-ace-Br at 66 ns is very weak, and the feature between 380 and 460 nm is the dark signal from the iCCD, not LE emission from TPA-ace-Br.)
Table 1. Photophysical Properties of the Molecules in Toluene Solution.
|
1CT |
|||||
|---|---|---|---|---|---|
| emitter | Eg/eVa | /nm | /eV | ΦPLd | τPL/nse |
| TPA-ace | 3.29 | 495b | 2.50 | 0.67 | 1.57 |
| TPA-ace-Br | 3.33 | 522c | 2.37 | 0.02 | 9.49 |
| 2TPA-ace | 3.26 | 513b | 2.41 | 0.51 | 2.34 |
| TPA-ace-CN | 3.09 | 470c | 2.63 | 0.67 | 5.96 |
| TPA-ace-TRZ | 3.22 | 518c | 2.39 | 0.17 | 9.6 (72.6%) and 51.0 (27.4%) |
Optical band gap estimated from the absorption spectra onset of the first main (exciton) absorption band.
Values obtained from the peak of the time-resolved PL spectra at a 11 ns delay, after subtracting the pure LE spectra (Figure S37).
Values obtained from the peak of the time-resolved spectra at a 14 ns delay, after stabilization of the CT state (Figure S39).
Photoluminescence quantum yield in degassed solution at room temperature. (Standard: quinine sulfate in 0.1 M H2SO4, ΦPL = 0.54.)
Lifetimes associated with the monoexponential or biexponential decay fitting (Figure S38).
Turning to the time-resolved PL spectra of TPA-ace-CN, there is only one emission band over the observed time window originating from the prompt CT state (Figures 5a and S36b). Despite this CT state having a higher energy than the transient CT state seen in TPA-ace, the introduction of the CN unit resulted in a stronger, longer-lived CT state with fast electron transfer so that we observe no LE emission within our time resolution. One possible reason for this could be that the CT state in TPA-ace-CN has a greater spatial charge separation resulting in a smaller Coulomb attraction energy and so a larger total amount of CT energy than the transient species in TPA-ace.14
Figure 5.

Time-resolved normalized emission spectra of (a) TPA-ace-CN and (b) TPA-ace-TRZ in toluene solution at a concentration of 20 μM. (c) Time-resolved PL decay curves in the entire region of analysis. λexc = 355 nm.
Figure 5b shows the time-resolved PL spectra of TPA-ace-TRZ. Initially, an emission band at around 500 nm is observed, associated with the same transient CT state as observed in TPA-ace. As the time delay increases, a small red shift is seen with an isoemissive point at 515 nm. This change in the CT states occurs over 35 ns. From the emission decay curve, we observe a clear biexponential decay with τPL = 9.6 and 51 ns (Figure 5c), indicating emission from two different CT states. We interpret this as the initial through-bond CT state (with moderate D–A dihedral angles of 48° between TPA and ace and 57° between ace and TRZ) decaying rapidly to leave a more stable TSCT state, as indicated by the observation of an isoemissive point at ca. 505 nm in the time-resolved spectra. Measurable delayed emission is seen over 100 μs, having the same emission spectrum as the prompt decay ascribed to DF. Because this emission is measured in dilute toluene solution, it is several orders of magnitude too fast and highly unlikely to be triplet-triplet annihilation (TTA). If TTA were efficient, then we would have expected to see it in all materials, which we clearly do not. Thus, we are convinced that the data provides compelling evidence that this is TADF coming from the TSCT observed only in TPA-ace-TRZ. Furthermore, the weak DF from very low RISC is attributed to the large ΔEST (vide infra). The decay curve of TPA-ace-CN (Figure 5c) is monoexponential, with a τPL of 6 ns, assigned to the single prompt CT emission.
Table 1 summarizes the photophysical properties of the five molecules in toluene solution. Optical band gaps remained almost constant regardless of structure, at around 3.3 eV, except for TPA-ace-CN, which presented a smaller gap of 3.09 eV, revealing the expected impact of the presence of the stronger electron-accepting cyano group in line with calculations. Despite this strong acceptor character, TPA-ace-CN showed weaker positive solvatochromism when compared to TPA-ace-TRZ due to a larger charge-separation distance and hence a smaller induced dipole moment. TPA-ace-Br and TPA-ace-TRZ presented smaller ΦPL values, supporting the increases in ISC and the triplet population in these materials.
To further understand the properties of this family of materials, we also investigated the three members with the CT-character excited states in high-polarity solutions of degassed DCM. In the case of TPA-ace-Br, we observe that the spectral shift going from toluene to DCM for the low-energy emission band is only some 20 nm, far smaller than for the other materials. This clearly calls into question whether this low-energy state has CT character. From the phosphorescence data given below, the ΔEST in DCM should close substantially. Figure S41 shows the oxygen-dependent steady-state emission from TPA-ace-CN, TPA-ace-Br, and TPA-ace-TRZ in DCM. Both TPA-ace-CN and TPA-ace-Br show very strong emission quenching in oxygen, whereas TPA-ace-TRZ shows a much smaller quenching, consistent with singlet quenching by oxygen in the latter compound. Figure S42 shows a comparison of the emission spectra decay kinetics in DCM and toluene. In all cases, we observe only a prompt decay in DCM (except for a very weak hint of delayed emission in TPA-ace-Br, Figure S42), with lifetimes very similar to those found in toluene for TPA-ace-Br and TPA-ace-TRZ (the latter having two components, the longest with a lifetime of 40–50 ns, Figure S40), whereas the lifetime of TPA-ace-CN increases nearly 3-fold (Figure S43). The large oxygen quenching observed in TPA-ace-CN and TPA-ace-Br is not due to the quenching of triplet states that would give rise to DF but may simply reflect efficient singlet-state quenching in solution for excited states with very long lifetimes.
The solid-state photophysical properties of the emitters were analyzed in a ZEONEX matrix, a low-polarity neutral polymer host. Films were fabricated by drop-casting method at a concentration of 1 wt % of emitter to host. As observed in the solution measurements, TPA-ace had a short-lifetime 1LE/1CT emission at earlier times, which decays rapidly to leave a residual broad 1CT band centered at around 480 nm, which itself decays within 20 ns (Figure 6a). Despite 2TPA-ace presenting results similar to those of TPA-ace, the time-resolved spectra (Figure 6c) showed a narrowed line width mixed LE/CT emission, which again decays rapidly to leave a 1CT band having higher intensity that is red-shifted by ca. 15 nm compared to that of TPA-ace. This corroborates our view that the interaction between the two TPA units enhances the probability of emission from the CT state. The time-resolved PL spectra of TPA-ace-CN (Figure 6b) shows an initial 1CT emission that red shifts with increasing time delay, indicating the apparent relaxation of 1CT emission due to a distribution of different molecular conformations with different decay times.21,23 This is typical behavior for a TADF molecule where the CT state D–A dihedral angle takes a range of values. Phosphorescence spectra (black lines) were recorded at a long time delay (80 ms), at 80 K, where the onsets of the spectra were used to obtain the 3LE energy of the molecules (Table 2). In all cases, large singlet–triplet energy gaps were found. The PL kinetic decay curves (Figure 6d) show that these molecules present a rapid decay, giving only prompt emission at room temperature, and no long-lived DF was found, analogous to the behavior in solution.
Figure 6.

Time-resolved normalized emission spectra of (a) TPA-ace, (b) TPA-ace-CN, and (c) 2TPA-ace in 1 wt % ZEONEX films. λexc = 355 nm. (d) Time-resolved PL kinetic decay curves in the entire region of analysis. The curves were obtained with 355 nm excitation. Note that the TPA-ace and TPA-ace-CN spectra after 10 and 20 ns, respectively, are very weak, and clear oscillatory dark noise from the iCCD can be seen. This is not intrinsic structure of the CT emission bands.
Table 2. Photophysical Properties of the Molecules in 1 wt % ZEONEX Films.
| emitter | 1CT/eVa | 3LE/eVb | ΔEST/eVc | ΦPLd | τp/nse | eτd/mse |
|---|---|---|---|---|---|---|
| TPA-ace | 2.99 | 2.53 | 0.46 | 0.37 | ||
| TPA-ace-Br | 2.77 | 2.51 | 0.26 | 8.05 | 0.33 (90.6%) | |
| 2.05 (9.4%) | ||||||
| 2TPA-ace | 2.95 | 2.55 | 0.40 | 0.36 | 1.93 (92.1%) | |
| 4.39 (7.9%) | ||||||
| TPA-ace-CN | 2.99 | 2.47 | 0.52 | 0.20 | 2.55 (97.0%) | |
| 7.50 (3.0%) | ||||||
| TPA-ace-TRZ | 2.94 | 2.46 | 0.48 | 0.12 | 6.9 (73%) | |
| 24.7 (27%) |
Values estimated from the onset of time-resolved PL spectra after the stabilization of 1CT or deconvoluted in order to obtain only the 1CT onset (Figure S42).
Values estimated from the onset of phosphorescence spectra. Spectra were collected with an 80 ms time delay and measured at 80 K.
ΔEST = 1CT – 3LE.
ΦPL measured for spin-coated films with a concentration of 1 wt % in the ZEONEX matrix using an integrating sphere connected to a Fluorolog-3.
Lifetimes estimated from monoexponential and biexponential decay fitting of the prompt and delayed regimes, respectively (Figure S46).
The time-resolved PL spectrum of TPA-ace-Br in the ZEONEX matrix is presented in Figure 7a, and its behavior was found to be very similar to that observed in solution. At early times, there is a very short-lived band at around 400 nm, and a highly red-shifted band at 500 nm emits until 65 ns. In the first time window, where clear dual emission is observed, the low-energy band has a pronounced blue-edge component and is structured. This suggests that it has mixed LE/CT character and a pure CT state, which we observe within our time resolution even at 80 K. The (relaxed) CT band possesses the same onset energy in ZEONEX as in toluene and is also very similar to that in DCM, indicating that it has a very small transition dipole moment, which aligns with the calculated TDM of 1.63 D. Moreover, after 100 μs, room-temperature phosphorescence grows in, contrary to solution measurement where mixed DF and phosphorescence emissions are seen at these times. This would point to greater stabilization of the CT state in toluene through the solvatochromic stabilization effect. Figure 7b shows the time-resolved PL spectra of TPA-ace-TRZ. Initially, during the prompt fluorescence regime, a relaxation of the 1CT band is observed, which decays after a few hundred nanoseconds. The onset of the CT emission is blue-shifted by 20 meV in ZEONEX compared to in toluene solution (Figure S44); we again observe an isoemissive point at 505 nm (Figure S45e). Thus, the CT emission observed in ZEONEX is predominantly from the through-bond state, with the formation of the (lower-energy) TSCT state hindered by the host matrix. This would indicate that reorganization of the TPA and TRZ moieties of the TPA-ace-TRZ must occur to enable the TSCT state to be accessed, which is hindered in ZEONEX. At later times, a very weak emission appears with an energy onset similar to that of phosphorescence, observed at late delay times at 80 K, indicative of inefficient RTP and not DF.
Figure 7.

Time-resolved normalized emission spectra of (a) TPA-ace-Br and (b) TPA-ace-TRZ in 1 wt % ZEONEX films. (c) Time-resolved PL kinetic decay curves. λexc = 355 nm.
Different regimes were observed in the decay curves (Figure 7c), assigned to prompt 1CT emission and a delayed emission component. While the delayed emission component of TPA-ace-TRZ is observable only at very long times (tens of milliseconds) and has a very weak contribution ascribed to phosphorescence, TPA-ace-Br makes a strong RTP contribution, confirming that attaching a heavy atom such as Br to the molecule significantly enhances the intersystem crossing (ISC) in this system.
Thin-film photophysical data are summarized in Table 2. Here, the energies of the S1 and T1 states were determined using the onset of the time-resolved spectra at room temperature (S1) and low temperature (80 K) (T1). The 3LE values of all molecules are slightly different but within error are around 2.53 eV for TPA-ace, TPA-ace-TPA, and TPA-ace-Br, implying that the structural modifications have a minimal influence on the energy and location of the local triplet state. For TPA-ace-CN and TPA-ace-TRZ, the triplet energy is lower by some 5–10 meV. At 80 K, the phosphorescence is structured for TPA-ace and 2TPA-ace and is assigned to the lowest-energy 3LE state of the ace unit;24 however, when acenaphthene is further substituted, the structure is lost. This identification is fully consistent with the large heavy-atom effects we observe in TPA-ace-Br enhancing this ace phosphorescence. The ΔEST values are higher than 0.4 eV in all cases except for TPA-ace-Br, where ΔEST is 0.26 eV; thus, it is not surprising that little or no DF is found in these compounds apart from TPA-ace-Br, where it is weak. The thin-film ΦPL values, measured under a N2 atmosphere, are higher for TPA-ace and TPA-ace-TPA than for the other compounds under study, which have emission mainly coming from a mixed 1LE/1CT state. For the molecules with stronger CT character, TPA-ace-CN and TPA-ace-TRZ, the ΦPL is significantly lower, indicating that the more stable CT states result in higher 3CT triplet state formation, which cannot be harvested by RISC. The transient PL decay curves were fitted by single or double exponentials; the very rapid decay of the emission of TPA-ace precluded the estimation of the prompt lifetime. 2TPA-ace presented two different lifetimes: a faster one with a larger contribution of around 2 ns from the mixed LE/CT state and a slower, smaller contribution term from the CT state, with a lifetime of around 4 ns. The prompt emission observed in TPA-ace-CN also has two components ascribed to the mixed LE/CT state and the relaxed 1CT. TPA-ace-TRZ has two characteristic lifetimes corresponding to the fast-decaying through-bond CT state and longer-lived TSCT state. On the other hand, TPA-ace-Br exhibited a single CT state lifetime in the prompt regime, while in the delayed regime it had two exponential lifetimes on the order of milliseconds and assigned to RTP.
Time-resolved PL decays of the ZEONEX films were also measured at 80 K (Figure S47). For TPA-ace, 2TPA-ace, and TPA-ace-CN, as at room temperature, the 490 nm mixed 1LE/1CT state decay is very rapid, leaving a transient CT state at 520 nm which itself has totally decayed within 100 ns. In particularly, TPA-ace-CN presented a weak, shorter-lifetime prompt CT state emission at 80 K, when compared with room-temperature measurements. This would indicate that a twisting motion is necessary, possible in the open ZEONEX polymer network, in order to stabilize the CT state in TPA-ace-CN, and as a result of steric hindrance caused by the CN group, this is thermally activated. Phosphorescence is observed in the millisecond time range. The CT emission from TPA-ace-Br at 80 K was found to be highly structured but at the same onset energy as at RT. This surprising observation suggests that this low-energy (weak) CT state is of mixed LE/CT character with a high LE contribution at 80 K.25 Finally, similar to the observed RT behavior, TPA-ace-TRZ showed initial CT emission, changing to a second low-energy CT state over tens of nanoseconds. An isoemissive point was observed, but the processes occur on a slower time scale, ca. 200 ns, indicative of a conformational reorganization of the TPA and TRZ moieties to stabilize the TSCT state, which is itself observed to live longer at low temperature. In all cases, well-resolved phosphorescence is observed at long times from which accurate 3LE energies are calculated.
To understand why we observe little or no TADF from the TSCT state in TPA-ace-TRZ, even in DCM where the ΔEST becomes rather small, we consider where the lowest-energy triplet state of the molecule resides. This is found to be the local triplet state of the ace bridge (thus a Br attached to the ace gives a heavy-atom enhancement to the ace phosphorescence), and in TPA-ace-TRZ, the ace unit is orthogonal and electronically decoupled from D and A, especially in the TSCT conformation. Hence, in this conformation 1CT and 3CT will be degenerate26 and triplet harvesting must occur through the vibronic-coupling spin–orbit coupling (SOC) mechanism4,5 as in D–A and exciplex27 TADF systems. However, in the case of TPA-ace-TRZ, the potentially mediating isoenergetic local triplet state is the ace bridge triplet, which is orthogonal to both D and A and thus cannot efficiently couple to the TSCT states. Therefore, even though TPA-ace-TRZ has a strong TSCT, it cannot produce TADF because of the lack of coupling to a mediating triplet state. From this we see that TSCT D–A pairs give rise to TADF through the vibronic coupling SOC mechanism, as do through-bond D–A systems and exciplex molecules.28
In summary, we report a TPA-ace-TRZ compound which we unambiguously demonstrate emits via a TSCT state. We compared the photophysical properties of this compound with several model systems: TPA-ace, TPA-ace-Br, 2TPA-ace, and TPA-ace-CN. In all five compounds, there is an emissive mixed through-bond LE/CT state with differing levels of LE and CT mixing: TPA-ace shows the strongest degree of LE character while TPA-ace-TRZ exhibits the strongest CT character. Time-resolved measurements showed that the introduction of a second TPA donor onto the ace unit in 2TPA-ace resulted in intramolecular dimer formation causing a relative enhancement of the overall ICT contribution, while the addition of the electron-accepting CN unit in TPA-ace-CN resulted in a pure-blue ICT emission. TPA-ace-Br also was shown to have a strong ICT state in the prompt region, the result of the nearly orthogonal conformation between the TPA and the ace-Br groups in the ground state. However, because of the relatively large ΔEST along with the existence of the heavy bromine atom, a dual TADF/RTP emission appeared weakly in solution, and with dominant RTP character in the solid state. The TSCT state, uniquely observed in TPA-ace-TRZ, exhibited a small delayed contribution in toluene solution, the result of a ΔEST being above 200 meV. Our study provides one of the few clear experimental demonstrations of the existence of a TSCT state, one that is corroborated by extensive DFT calculations. Our study also reveals the intimate interplay that the bridging ace group has on mediating both the through-bond ICT state and the TSCT state.
Acknowledgments
We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for analytical services. We thank UMICORE AG for the gift of catalysts.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.1c00265.
General experimental procedures, synthesis, melting point, NMR spectra, HRMS spectra, elemental analysis report, HPLC analysis report, crystallographic data (CCDC 2041945-2041948), molecular orbitals, and Cartesian coordinates of optimized ground states and additional photophysical data (PDF)
Author Contributions
S.K. and E.Z.-C. designed the materials. S.K. synthesized the materials and performed the electrochemistry study. L.G.F. and K.S. carried out the photophysics study. S.K. and E.C. performed the theoretical calculations and analyzed the results. D.B.C. and A.M.Z.S. solved the structure of the single crystals. S.K., L.G.F., D.B.C., A.P.M., and E.Z.-C. wrote the manuscript. A.P.M. and E.Z.-C. supervised the progress of the project.
We acknowledge the EU Horizon 2020 grant agreement no. 812872 (TADFlife) for funding. S.K. acknowledges financial support from the European Union’s Horizon 2020 research and innovation program under a Marie Skłodowska Curie Individual Fellowship (MCIF; agreement no. 748430-THF-OLED).
The authors declare no competing financial interest.
Notes
The research data supporting this publication can be accessed at https://doi.org/10.17630/0ae146df-b0f9-4590-9e7e-c30191cc4461.
Supplementary Material
References
- Goushi K.; Adachi C. Efficient Organic Light-Emitting Diodes through up-Conversion from Triplet to Singlet Excited States of Exciplexes. Appl. Phys. Lett. 2012, 101, 023306. 10.1063/1.4737006. [DOI] [Google Scholar]
- Uoyama H.; Goushi K.; Shizu K.; Nomura H.; Adachi C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Dias F. B.; Bourdakos K. N.; Jankus V.; Moss K. C.; Kamtekar K. T.; Bhalla V.; Santos J.; Bryce M. R.; Monkman A. P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. 10.1002/adma.201300753. [DOI] [PubMed] [Google Scholar]
- Etherington M. K.; Gibson J.; Higginbotham H. F.; Penfold T. J.; Monkman A. P. Revealing the Spin–Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence. Nat. Commun. 2016, 7, 13680. 10.1038/ncomms13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J.; Monkman A. P.; Penfold T. J. The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules. ChemPhysChem 2016, 17, 2956–2961. 10.1002/cphc.201600662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Song X.; Gillett A. J.; Drummond B. H.; Jones S. T. E.; Li G.; He H.; Cai M.; Credgington D.; Duan L. Efficient and Stable Deep-Blue Fluorescent Organic Light-Emitting Diodes Employing a Sensitizer with Fast Triplet Upconversion. Adv. Mater. 2020, 32, 1908355. 10.1002/adma.201908355. [DOI] [PubMed] [Google Scholar]
- Schwartz G.; Reineke S.; Rosenow T. C.; Walzer K.; Leo K. Triplet Harvesting in Hybrid White Organic Light-Emitting Diodes. Adv. Funct. Mater. 2009, 19, 1319–1333. 10.1002/adfm.200801503. [DOI] [Google Scholar]
- Im Y.; Kim M.; Cho Y. J.; Seo J.-A.; Yook K. S.; Lee J. Y. Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters. Chem. Mater. 2017, 29, 1946–1963. 10.1021/acs.chemmater.6b05324. [DOI] [Google Scholar]
- Goushi K.; Yoshida K.; Sato K.; Adachi C. Organic Light-Emitting Diodes Employing Efficient Reverse Intersystem Crossing for Triplet-to-Singlet State Conversion. Nat. Photonics 2012, 6, 253–258. 10.1038/nphoton.2012.31. [DOI] [Google Scholar]
- Sarma M.; Wong K.-T. Exciplex: An Intermolecular Charge-Transfer Approach for TADF. ACS Appl. Mater. Interfaces 2018, 10, 19279–19304. 10.1021/acsami.7b18318. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Zheng C.; Zhou Y.; Li C.; Ye J.; Du X.; Li W.; He Z.; Zhang M.; Lin H.; Tao S.; Zhang X. Novel Small-Molecule Electron Donor for Solution-Processed Ternary Exciplex with 24% External Quantum Efficiency in Organic Light-Emitting Diode. Mater. Horiz. 2019, 6, 1425–1432. 10.1039/C9MH00373H. [DOI] [Google Scholar]
- Jeon S. K.; Lee J. Y. Highly Efficient Exciplex Organic Light-Emitting Diodes by Exciplex Dispersion in the Thermally Activated Delayed Fluorescence Host. Org. Electron. 2020, 76, 105477. 10.1016/j.orgel.2019.105477. [DOI] [Google Scholar]
- Jankus V.; Data P.; Graves D.; McGuinness C.; Santos J.; Bryce M. R.; Dias F. B.; Monkman A. P. Highly Efficient TADF OLEDs: How the Emitter–Host Interaction Controls Both the Excited State Species and Electrical Properties of the Devices to Achieve near 100% Triplet Harvesting and High Efficiency. Adv. Funct. Mater. 2014, 24, 6178–6186. 10.1002/adfm.201400948. [DOI] [Google Scholar]
- Colella M.; Danos A.; Monkman A. P. Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex. J. Phys. Chem. Lett. 2019, 10, 793–798. 10.1021/acs.jpclett.8b03646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Shen P.; Zhao Z.; Tang B. Z. Through-Space Conjugation: A Thriving Alternative for Optoelectronic Materials. CCS Chem. 2019, 1, 181–196. 10.31635/ccschem.019.20180020. [DOI] [Google Scholar]
- Woon K.-L.; Yi C.-L.; Pan K.-C.; Etherington M. K.; Wu C.-C.; Wong K.-T.; Monkman A. P. Intramolecular Dimerization Quenching of Delayed Emission in Asymmetric D–D′–a TADF Emitters. J. Phys. Chem. C 2019, 123, 12400–12410. 10.1021/acs.jpcc.9b01900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S.; Head-Gordon M. Time-Dependent Density Functional Theory within the Tamm–Dancoff Approximation. Chem. Phys. Lett. 1999, 314, 291–299. 10.1016/S0009-2614(99)01149-5. [DOI] [Google Scholar]
- Quinton C.; Alain-Rizzo V.; Dumas-Verdes C.; Miomandre F.; Clavier G.; Audebert P. Redox-Controlled Fluorescence Modulation (Electrofluorochromism) in Triphenylamine Derivatives. RSC Adv. 2014, 4, 34332–34342. 10.1039/C4RA02675F. [DOI] [Google Scholar]
- Herington E. F. G.; Jones J. I. Ultraviolet Absorption Spectra of Some Copolymers Containing Acenaphthylene, and of Related Compounds. J. Polym. Sci. 1949, 4, 725–733. 10.1002/pol.1949.120040605. [DOI] [Google Scholar]
- de Sa Pereira D.; Lee D. R.; Kukhta N. A.; Lee K. H.; Kim C. L.; Batsanov A. S.; Lee J. Y.; Monkman A. P. The Effect of a Heavy Atom on the Radiative Pathways of an Emitter with Dual Conformation, Thermally-Activated Delayed Fluorescence and Room Temperature Phosphorescence. J. Mater. Chem. C 2019, 7, 10481–10490. 10.1039/C9TC02477H. [DOI] [Google Scholar]
- Dias F. B.; Santos J.; Graves D. R.; Data P.; Nobuyasu R. S.; Fox M. A.; Batsanov A. S.; Palmeira T.; Berberan-Santos M. N.; Bryce M. R.; Monkman A. P. The Role of Local Triplet Excited States and D-a Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Adv. Sci. 2016, 3, 1600080. 10.1002/advs.201600080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariasse K. A. Comment on “Pseudo-Jahn–Teller and Tict-Models: A Photophysical Comparison of Meta-and para-Dmabn Derivatives” [Chem. Phys. Lett. 305 (1999) 8]: The Pict Model for Dual Fluorescence of Aminobenzonitriles. Chem. Phys. Lett. 2000, 320, 8–13. 10.1016/S0009-2614(00)00230-X. [DOI] [Google Scholar]
- Northey T.; Stacey J.; Penfold T. J. The Role of Solid State Solvation on the Charge Transfer State of a Thermally Activated Delayed Fluorescence Emitter. J. Mater. Chem. C 2017, 5, 11001–11009. 10.1039/C7TC04099G. [DOI] [Google Scholar]
- Stevens B.; Walker M. The Kinetics of Phosphorescence and Delayed Fluorescence Decay for Aromatic Hydrocarbons in Liquid Paraffin. Proc. Math. Phys. Eng. Sci. 1964, 281, 420–436. [Google Scholar]
- Aydemir M.; Haykır G.; Türksoy F.; Gümüş S.; Dias F. B.; Monkman A. P. Synthesis and Investigation of Intra-Molecular Charge Transfer State Properties of Novel Donor–Acceptor–Donor Pyridine Derivatives: The Effects of Temperature and Environment on Molecular Configurations and the Origin of Delayed Fluorescence. Phys. Chem. Chem. Phys. 2015, 17, 25572–25582. 10.1039/C5CP03937A. [DOI] [PubMed] [Google Scholar]
- Wada Y.; Nakagawa H.; Matsumoto S.; Wakisaka Y.; Kaji H. Organic Light Emitters Exhibiting Very Fast Reverse Intersystem Crossing. Nat. Photonics 2020, 14, 643–649. 10.1038/s41566-020-0667-0. [DOI] [Google Scholar]
- dos Santos P. L.; Dias F. B.; Monkman A. P. Investigation of the Mechanisms Giving Rise to TADF in Exciplex States. J. Phys. Chem. C 2016, 120, 18259–18267. 10.1021/acs.jpcc.6b05198. [DOI] [Google Scholar]
- Penfold T. J.; Dias F. B.; Monkman A. P. The Theory of Thermally Activated Delayed Fluorescence for Organic Light Emitting Diodes. Chem. Commun. 2018, 54, 3926–3935. 10.1039/C7CC09612G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.