

Abstract
In the regulatory setting, clinical pharmacology focuses on the impact of intrinsic and extrinsic factors on inter-patient and intra-subject variability in drug exposure and response. This translational science contributes to the understanding of the benefit-risk profile in individual patients and the development of relevant therapeutic monitoring and management strategies. Clinical pharmacology also plays a major role in the development and qualification of drug development tools. This article presented some recent examples to illustrate the important roles of clinical pharmacology in drug development and evaluation. In addition, emerging trends in clinical pharmacology regulatory sciences were also discussed, including the Model-Informed Drug Development (MIDD) pilot program, the use of real-world data to generate real-world evidence, and leveraging advances in basic, biomedical, and clinical science into useful tools for drug development and evaluation. Continued advances in clinical pharmacology can be the basis of more rational and efficient drug development and improved access to new drug treatments that are tailored to the patient to achieve better efficacy and safety.
KEY WORDS: clinical pharmacology, regulatory science, precision medicine, drug development
THE MISSION OF CLINICAL PHARMACOLOGISTS IN THE REGULATORY SETTING
Despite its critical role in drug development and precision medicine, the definition of clinical pharmacology is not universally understood. The American Society of Clinical Pharmacology and Therapeutics defines this discipline as the “study of drugs in humans,” which is “underpinned by the basic science of pharmacology, with added focus on the application of pharmacological principles and methods in the real world” (1). In the regulatory setting, clinical pharmacology focuses on the impact of intrinsic and extrinsic factors on inter-patient and intra-subject variability in drug exposure and response. This translational science contributes to the understanding of the benefit-risk profile in individual patients and the development of relevant therapeutic monitoring and management strategies. Clinical pharmacology also plays a major role in the development and qualification of drug development tools.
At the US Food and Drug Administration (FDA), the clinical pharmacologists regulating drug development work in the Office of Clinical Pharmacology (OCP), a sub-office of the Office of Translational Sciences in the Center of Drug Evaluation and Research (CDER). Their mission is twofold: (1) advance development of innovative medicines by applying state-of-the-art scientific principles and (2) promote therapeutic optimization and individualization through best practices in research, policy development, and drug evaluation throughout the product lifecycle (2). This mission is also consistent with the mission of OTS, which is to drive advancements in human health through scientific and regulatory innovation. To achieve this mission, in 2019 OCP engaged in 138 research projects and published 158 manuscripts that describe its scientific findings or discuss critical questions, challenges, and opportunities that arise as it applies the principles and methods of clinical of pharmacology to advance development, evaluation, and use of new medical products (3).
THE ROLE OF CLINICAL PHARMACOLOGY IN DRUG DEVELOPMENT AND EVALUATION
Drug development proceeds through a planned sequence of studies that includes an initial pre-clinical phase and subsequent clinical studies. In this sequence, early in vitro, in vivo, and in silico investigations provide vital information to guide further studies. Clinical pharmacology studies in the course of drug development include both in vitro and in vivo studies and are focused on evaluating the drug’s pharmacokinetics (e.g., absorption, distribution, metabolism, and excretion (ADME)), its pharmacodynamics (both the desired drug activity and the adverse effects), and the impact of intrinsic factors (e.g., age, gender, race/ethnicity, genomics, and organ dysfunction) and extrinsic factors (e.g., food effect, smoking, and drug-drug interactions) on drug exposure and response. As compared to small-molecule drugs, biologics may require different studies. For example, the development programs for monoclonal antibodies usually do not include mass balance, food effect, or studies of a drug’s effect on the QT interval, but they require immunogenicity evaluation, which is usually not needed for small-molecule drugs. Starting with the Pre-Investigational New Drug Application (Pre-IND) meeting (which ensures that the drug development plan and future clinical trials are acceptable), clinical pharmacologists at OCP provide guidance to the drug developers throughout the development process, including recommendations and advice on what clinical pharmacology questions need to be addressed for each investigational drug and when and how to conduct the required studies. The advice is conveyed during FDA-sponsor meetings and/or in written responses. In addition, formal guidance documents are developed to assist drug developers. In 2019, OCP issued six clinical pharmacology guidances (4–9) and contributed to 24 multidisciplinary guidances on broad drug development topics. Each published guidance is the product of years of regulatory research and represents the agency’s current thinking on a particular subject. For example, the recently published guidances for industry In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry(10) and Clinical Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry (11) reflect over a decade of regulatory research and broad collaborations of FDA with industry, academia, and other regulatory agencies. In addition, the comments received during the public comment period when the draft guidance documents were published have been considered in making the final recommendations.
When a new drug application (NDA) or biologics license application (BLA) is submitted to FDA, OCP’s clinical pharmacologists work closely with colleagues from other disciplines such as medical reviewers, statisticians, chemists, pharmacologists, toxicologists, and regulatory project managers to evaluate the safety and effectiveness of the drug and to make the decision as to whether the application should be approved. The clinical pharmacology review is issue-driven, and the information in the application for a new drug or biologic is assessed to address important issues of dose selection and optimization, therapeutic individualization, and benefit-risk (including application in subpopulations) (12). If the drug is approved, the important clinical pharmacology information must be captured in the product labeling, especially factors that influence pharmacokinetics, pharmacodynamics, effectiveness, and safety, which may require dose adjustment (Fig. 1). When appropriate, OCP clinical pharmacologists may request post-marketing requirement (PMR) or post-marketing commitment (PMC) studies to address issues that have not been adequately addressed. For example, subpopulations of patients, such as patients with different stages of hepatic impairment, may not have been studied, and PMRs are needed to provide dosing instructions for these patients to prevent serious adverse events when treated with the new drug. In the case of remdesivir for the treatment of COVID-19, which was approved 76 days after submission, the approval letter required clinical pharmacology post-marketing studies in children and patients with renal and hepatic impairment, as well as evaluation of the drug’s potential to prolong the QT interval (13). The approval letter also included a PMC study in pregnant women. The FDA granted this application Fast Track and Priority Review designations (14, 15).
Fig. 1.

Clinical pharmacology information in the package insert. QTc, drug effect on corrected QT interval; PK, pharmacokinetics; DDI, drug-drug information; PK/PD, pharmacokinetics/pharmacodynamics
In these unprecedented and challenging times, it is especially critical to leverage the power of clinical pharmacology, because it provides unique tools to integrate different kinds of data and translate them into informed interpretations and predictions. A good example of the discipline’s importance in regulatory decision-making comes from the current pandemic, in which OCP clinical pharmacologists provided advice on the critical step of translating in vitro antiviral activity of potential drugs to treat the disease to appropriate clinical dosing regimens (16). Their analysis of the available data, which indicated that the oral administration of hydroxychloroquine under a safe dosing regimen is unlikely to produce lung concentrations that are effective against SARS-CoV-2, supported FDA’s decision to revoke its Emergency Use Authorization for oral formulations of chloroquine phosphate and hydroxychloroquine sulfate to treat COVID-19 (17).
EMERGING TRENDS IN CLINICAL PHARMACOLOGY REGULATORY SCIENCES
The Model-Informed Drug Development Pilot Program
The Model-Informed Drug Development (MIDD), in which a wide range of quantitative models are used to facilitate new drug development and healthcare, has been evolving rapidly in recent decades. These models, which provide a platform to integrate current understanding of a disease, patient characteristics, and pharmacology, have been used in dosage optimization and selection, the design of clinical program and trials, identification of supportive evidence of efficacy, and new policy development (Fig. 2) (18, 19).
Fig. 2.

Model-Informed Drug Development (MIDD). A wide range of quantitative models can be applied to integrate the information on patient characteristics, diseases, and drug effect, to inform drug development and regulatory decision-making. ADME, absorption, distribution, metabolism, excretion; E-R, exposure-response, the relationship between drug exposure (e.g., systemic drug concentrations) and response (e.g., biomarkers or clinical endpoints related to either effectiveness or safety); PBPK, physiologically based pharmacokinetic modeling; PK/PD, pharmacokinetic/pharmacodynamic; PopPK, population pharmacokinetics; QSP, quantitative systems pharmacology; QSAR, quantitative structure–activity relationship; QSPR, quantitative structure-property relationships
Under the sixth iteration of the Prescription Drug User Fee Act (PDUFA VI), FDA committed to developing MIDD-related guidance updates, holding public workshops on these approaches, and establishing a standard operating procedure for reviews of MIDD-related submissions. In 2018, FDA also established the MIDD Pilot Program, which affords sponsors or applicants the opportunity to meet with agency staff to discuss modeling approaches in medical product development (20). Jointly administered by CDER and the Center of Biologics Evaluation and Research, with OCP as the point of contact, the program provides a regulatory platform that engages all stake holders early in the development of a new drug and bridges multidisciplinary efforts.
As of October 2020, FDA had received 27 meeting requests and granted 19 submissions. These submissions covered many aspects of drug development (e.g., dose selection in a first-in-human study, post-approval dose optimization, and design of a phase 3 clinical trial) and a wide range of approaches, including population pharmacokinetics, exposure-response modeling, quantitative systems biology, and physiologically based pharmacokinetic (PBPK) modeling. Under this pilot program, approval of two regulatory submissions, a supplemental New Drug Application (sNDA) and a supplemental Biological License Application (sBLA), was facilitated by discussions between FDA and sponsors. Figure 3 shows the increasing use of PBPK for drug development in various areas (e.g., drug-drug interactions involving metabolizing enzymes and transporters; pediatrics, assessment of drug exposure in patients with renal impairment) (21, 22). Another emerging trend that needs attention is the use of hybrid modeling strategies, where the model types shown in Fig. 2 are used in sequence or simultaneously (e.g., combining mechanistic modeling with machine learning for response prediction) (23).
Fig. 3.
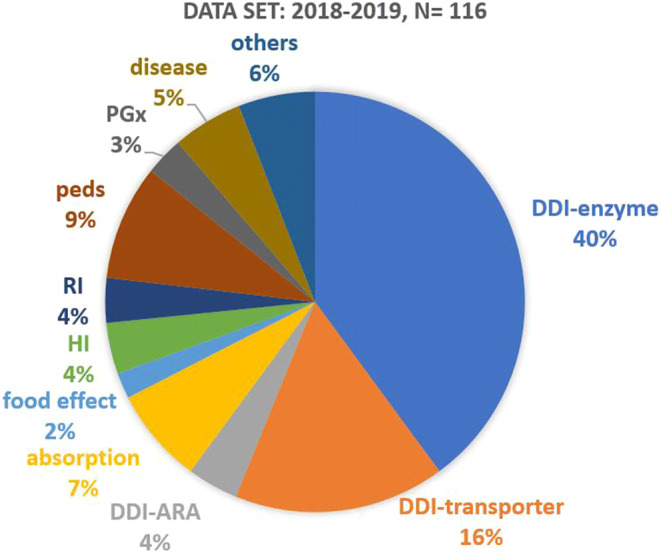
Distribution of PBPK submissions by application areas (2018-2019). DDI-enzyme, enzyme-mediated DDI; DDI-transporter, transporter-mediated DDI; DDI-ARA, acid reducing agent-mediated DDI; HI, hepatic impairment; RI, renal impairment; peds, pediatrics; PGx, pharmacogenomics. Figure Source: Zhang XY, Yang Y, Grimstein M et al. Application of PBPK Modeling and Simulation for Regulatory Decision-Making and Its Impact on the US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol. 2020 Oct;60 Suppl 1:S160-S178. doi: 10.1002/jcph.1767. PMID: 33205429
The Use of Real-World Data to Generate Real-World Evidence
In the past decade, electronic health record systems have been widely adopted, and smart phones and wearables make it possible for patients to generate vast amounts of health data on a continual basis. The use of digital health technology and telemedicine has increased due to the COVID19 pandemic, and the digital ecosystem for healthcare is rapidly evolving. We have an opportunity (and an obligation) to strive to derive real-world evidence (RWE) from real-world data (RWD) to promote public health. Although RWD and RWE have been traditionally used for safety surveillance by FDA, the 21st Century Cures Act places additional focus on their use to support regulatory decisions, including to help support the approval of new indications for approved drugs and to help support or satisfy post-marketing study requirements (24). Congress defined RWE as data regarding the usage, or the potential benefits or risks, of a drug derived from sources other than traditional clinical trials. FDA has expanded on the definition of RWE as the clinical evidence regarding the usage and potential benefits or risks of a medical product derived from analysis of RWD, while defining RWD as data relating to patient health status and/or the delivery of healthcare routinely collected from a variety of sources including, but not limited to, electronic health records (EHRs), claims and billing activities, product and disease registries, patient-generated data (in home-use or other settings), and data gathered from other sources like mobile devices (25).
The 21st Century Cures Act mandates that FDA develop a framework to evaluate the use of RWD/RWE in regulatory decisions. This framework, issued in December 2018 (26), includes the following considerations: (1) Are the RWD fit for use? (2) Does the trial or study design used to generate RWE provide adequate scientific evidence to answer or help answer the regulatory question? (3) Does the conduct of the study meet FDA regulatory requirements?
RWD/RWE can advance the mission of clinical pharmacology by filling the knowledge gaps between clinical trials and real-world clinical practice (Fig. 4) (27). For example, there is a need to understand drug usage and clinical outcomes in certain patients who may be underrepresented or absent in the clinical trials conducted in drug development programs, such as those experiencing organ dysfunction or receiving concomitant medications, pregnant women, individuals in whom metabolism of the drug is different, or older adults. Based on RWD, we may be able to better understand the benefit-risk profile of the drug in these patient subgroups and develop appropriate treatment monitoring and management strategies. RWD may also be used to help select the optimal treatment and dosing regimens for different patient populations. In addition, RWD can be used to facilitate clinical trials and drug development. For example, RWD can be used to guide the eligibility criteria of the clinical trials. Other areas of interest include the use of RWD for disease modeling and the development of biomarkers and clinical endpoints. However, there are significant challenges in deriving RWE from RWD: The data can be of suboptimal quality and incomplete; patient information may exist in multiple systems that are non-interoperable due to incompatible formats and standards, which may compromise data aggregation and subsequent analyses; the unstructured formats of much of the data make analyses time-consuming; and due to a lack of randomization, analyses may be subject to confounding and bias. More regulatory research is needed to learn where RWD can be most helpful and to develop best practices.
Fig. 4.

Real-world data can augment the clinical trial data to address clinical pharmacology issues
Through internal and external collaborations, FDA scientists have been working on some demonstration projects to explore and advance the use of RWE including the following:
In a collaborative effort involving FDA, a real-world data company, and a health system, investigators studied the incidence of on-treatment pneumonitis in patients with non-small cell lung cancer (NSCLC) that were treated with immune checkpoint inhibitors (ICI) or chemotherapy using both randomized clinical trial data and a cohort derived from RWD. For the clinical trial data, the FDA scientists pooled data from approximately 6500 patients from 8 randomized trials comparing ICI treatment (with/without chemotherapy) to the chemotherapy control arm. The real-world data sample consisted of approximately 1200 patients from the health system. This study showed a higher incidence of treatment-associated pneumonitis among ICI (+/-chemotherapy)-treated NSCLC patients compared to those receiving chemotherapy alone in both clinical trial and the RWD cohorts. The data also showed a consistent numerical increase of treatment-associated pneumonitis in patient with a past medical history of pneumonitis as compared to patients without this prior history. This trend was observed in both ICI and chemotherapy treated groups, and in both clinical trials and RWD (28).
-
To develop clinical practice guidelines for accurate prescribing of opioids to treat acute pain indications that can decrease opioid misuse, the Acute Pain Pathways (APP) study has gathered real-world data on opioids prescribed and dispensed, and actual opioid use from patients, providers, and pharmacies for diverse patient populations.
On an average day in the USA, more than 463,000 opioid prescriptions are dispensed (29), and 4402 individuals, age 12 or older, initiate nonmedical use of prescription opioids (30). Leftover prescription opioids can be a source of misuse. In a 2019 National Survey of Drug Use and Health report, the source for misused prescription opioids was mainly from a friend or relative. There has been considerable literature published since the review documented in Fig. 5 on the excess prescription opioid supply, variation in amounts used by medical procedures, and variations in use across patients getting the same procedure. Leftover pills may result in misuse and related outcomes such as addiction.
FDA is collaborating with the Yale-Mayo Center of Excellence in Regulatory Science and Innovation (CERSI) on the APP study in order to inform evidence-based individualized clinical practice guidelines for prescribing opioids for acute pain to ensure appropriate access to opioids for pain management while limiting leftover supply that can contribute to misuse, diversion, and accidental poisonings. A total of 1550 patients who will be prescribed opioids will be recruited from multiple healthcare systems, primary care/urgent care, emergency department, and dental practices. The sites selected in 9 states are geographically and racially diverse. The data sources include pharmacies, electronic health records, and personal device apps which will collect patient-reported outcome measures on fills/refills, pain, patient satisfaction, health status, and health outcomes such as adverse events and functional status. This will support FDA’s goal of decreasing exposure and preventing new addiction while providing effective individualized pain management.
-
The Reagan-Udall Foundation (RUF) launched the COVID-19 Evidence Accelerator Collaborative initiative for the FDA in collaboration with Friends of Cancer Research (Friends) to provide a unique venue for major data organizations, government and academic researchers, and health systems to gather and design quick-turn-around queries and share their results. As part of the COVID-19 Evidence Accelerator Collaborative initiative, FDA harmonized a set of COVID-19 data elements identified with several widely used common data models. The common data models refer to agreed-upon formats into which participating healthcare sites transform their data and standards (Table I). This COVID-19 data set includes a sub-set of data elements from EHRs, administrative claims, and other sources of RWD. Data elements included in this project include demographics, medical history, medication, travel and social history, vital signs, laboratory and imaging, diagnosis, intervention, and clinical outcome.
Figure 6 illustrates the steps taken for this harmonization process. Following the detailed review of the COVID-19 data elements by the subject matter experts at the FDA, the data elements were harmonized to several common data models and standards. Figure 7 illustrates an example of several COVID-19 data elements mapped to various common data models and data standards.
This project is critical to advance research on COVID-19 across healthcare networks, all of which use different models and standards for their data.
Fig. 5.

Prevalence of unused opioids prescribed after surgery. Figure Source: Bicket MC, Long JJ, Pronovost PJ, Alexander GC, Wu CL. Prescription Opioid Analgesics Commonly Unused After Surgery: A Systematic Review. JAMA Surg. 2017 Nov 1;152(11):1066-1071
Table I.
Common Data Models and Data Standards
| Common data models (CDMs) | i2b2/ACT | Informatics for Integrating Biology & the Bedside / Accrual to Clinical Trials |
|---|---|---|
| OMOP | Observational Medical Outcomes Partnership | |
| PCORnet | Patient-Centered Outcomes Research Network | |
| Sentinel | ||
| Standards | CDISC SDTM | Clinical Data Interchange Standards Consortium, Study Data Tabulation Model |
| HL7 FHIR r4 | Health Level Seven Fast Healthcare Interoperability Resources, Release 4 |
Fig. 6.

COVID-19 common data elements harmonization process
Fig. 7.

Three representative COVID-19 data elements (i.e., treatment setting, on ventilation, and COVID 19 medication dosing regimen) on the left mapped to the data elements of multiple common data networks in different colored boxes*. *The depicted common data networks include the FDA Sentinel Initiative, PCORnet (Patient-Centered Outcomes Research Network), i2b2/ACT (Informatics for Integrating Biology and the Bedside / Accrual to Clinical Trials), OMOP (Observational Medical Outcomes Partnership from Observational Health Data Sciences and Informatics), CDISC SDTM (Study Data Tabulation Model from Clinical Data Interchange Standards Consortium), USCDI HL7 FHIR R4 (The U.S. Core Data for Interoperability Health Level Seven Fast Healthcare Interoperability Resources Revision 4), and VA EHR-S (Veterans Affairs - Electronic Health Record System)
These demonstration projects provide us with valuable insights on when and how to use RWD/RWE to address clinical pharmacology questions. The lessons learned will help OCP and FDA prepare for what is expected to be an increasing number of regulatory submissions with RWD/RWE components.
Other new types of medical data which are gaining attention include medical imaging data and microbiome data. Radiomics, an emerging science that uses algorithms to extract features from medical images, has the potential to uncover disease features and characteristics that cannot be appreciated by the naked eye (31, 32). Combined with advanced machine learning algorithms, radiomics can improve disease detection, characterization, staging, as well as assessment and prediction of treatment response (33). A related field, radiogenomics, assesses the relationship between imaging features and gene expression (34, 35). The OCP clinical pharmacologists are exploring the opportunities to apply machine learning on medical imaging data to advance precision medicine (36). The microbiome research has evolved rapidly and has gained significant traction in recent years (37). The microbiome can be defined as a characteristic microbial community occupying a reasonable well-defined habitat which has distinct physio-chemical properties. It has been discovered that the microbiome can have an impact on both the metabolism and the clinical outcome of the drug treatment (38, 39). Consequently, destruction of the microbiome (e.g., use of antibiotics) may be expected to have an impact of the treatment outcome of certain other drugs. In addition, live microbiome-based products could be developed to prevent, treat, or cure a disease or condition in humans, which may cause a new type of interaction potential with the drugs (40). We can expect that the microbiome data will be considered in precision medicine in the future and that microbiome-related research will be incorporated into the regulatory science research.
Leveraging Advances in Basic, Biomedical, and Clinical Science into Useful Tools for Drug Development and Evaluation
Under 21st Century Cures, new language on the qualification of drug development tools (DDTs) was added to the Federal Food, Drug, and Cosmetic Act (24). DDTs are methods, materials, or measures that can potentially facilitate drug development (41). DDTs include, but are not limited to, a biomarker used for clinical trial enrichment, a clinical outcome assessment used to evaluate clinical benefit, and an animal model used for efficacy testing of medical countermeasures under the regulations commonly referred to as the Animal Rule. FDA has established qualification programs (42) to support DDT development and a multi-step process for DDT qualification (43). Clinical pharmacologists in OCP play an integral part in this program by participating in the review of the submissions to the DDT qualification program.
In addition, OCP has been actively conducting regulatory research to help develop DDTs. For example, a clinical study with implications for treatment of cardiac conditions like high cholesterol is currently being sponsored by the Division of Applied Regulatory Science/OCP to identify pharmacodynamic biomarkers to support development of biosimilars for PCSK9 inhibitors (44). This study will assess pharmacokinetics and pharmacodynamics of evolocumab and alirocumab across an appropriate dose range to inform clinical trial operating characteristics for future pharmacodynamics similarity studies. Another major research focus within the division is the in vitro Proarrhythmia Assay (CiPA) collaboration which brings together FDA, its foreign counterparts, non-profits, academia, and drug developers to improve our ability to predict the risk that a drug will cause the potentially fatal cardiac arrhythmia torsades de pointes (45).
OCP also plays a role in the use of in vitro pharmacology assays for precision medicine. Ivacaftor is a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator. It is indicated for the treatment of cystic fibrosis (CF) in patients age 4 months and older who have one mutation in the CFTR gene that is responsive to ivacaftor based on clinical and/or in vitro assay data. If the patient’s genotype is unknown, an FDA-cleared CF mutation test should be used to detect the presence of a CFTR mutation followed by verification with bi-directional sequencing when recommended by the mutation test instructions for use (46).
OCP scientists have also been scanning the scientific horizon (see the recent workshop on 3D Cell Culture Models for Drug Pharmacokinetics, Safety, and Efficacy Assessment co-sponsored by the University of Maryland Center of Excellence in Regulatory Science and Innovation and FDA) (47, 48) and researching emerging technologies.
CONCLUSION
Continued advances in clinical pharmacology can be the basis of more rational and efficient drug development and improved access to new drug treatments that are tailored to the patient to achieve better efficacy and safety. If we are to achieve this scenario, we need a vigorous, sustained, and forward-looking program of research that brings together diverse scientific talents in productive collaborations. FDA has a wide range of mechanisms to support these collaborations both nationally and globally, including working groups, consortia, Center of Excellence in Regulatory Science and Innovation (CERSI) (49), the Broad Agency Announcement Program (50), and collaborative agreements (e.g., Memoranda of Understanding (51) and Cooperative Research and Development Agreements (52)). OCP is strongly committed to working with all stakeholders to further the ever-expanding discipline of clinical pharmacology in support of the public health.
Acknowledgements
The authors thank Mark Geanacopoulos for critical review of the manuscript, Giang Ho and Kimberly Bergman for assisting in the production of Figs. 1, 2, 4, and 5, and Wei Chen for assisting in the production of Fig. 7.
Author’s Contribution
All authors made substantial contributions to the conception and design of the work, data interpretation, writing, and revising the manuscript and agreed to the manuscript content.
Declarations
Conflict of Interest
The authors declare no competing interests.
Disclaimer
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.What is Clinical Pharmacology? : The American Society for Clinical Pharmacology and Therapeutics (ASCPT); [Available from: https://www.ascpt.org/Resources/Knowledge-Center/What-is-Clinical-Pharmacology.
- 2.The Website of Office of Clinical Pharmacology: The Office of Clinical Pharmacology, Office of Translational Sciences, US FDA; [Available from: https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/office-clinical-pharmacology.
- 3.2019 Annual Report: The Office of Clinical Pharmacology, Office of Translational Sciences, US FDA; [Available from: https://www.fda.gov/media/134935/download.
- 4.Assessing the Effects of Food on Drugs in INDs and NDAs – Clinical Pharmacology Considerations: US FDA; 2019 [Available from: https://www.fda.gov/media/121313/download.
- 5.Bioavailability Studies Submitted in NDAs or INDs – General Considerations: US FDA; 2019 [Available from: https://www.fda.gov/media/121311/download.
- 6.General Clinical Pharmacology Considerations for Neonatal Studies for Drugs and Biological Products Guidance for Industry: US FDA; 2019 [Available from: https://www.fda.gov/media/129532/download.
- 7.Maximal Usage Trials for Topically Applied Active Ingredients Being Considered for Inclusion in an Over-The -Counter Monograph: Study Elements and Considerations: US FDA; 2019 [Available from: https://www.fda.gov/media/125080/download.
- 8.Population Pharmacokinetics: US FDA; 2019 [Available from: https://www.fda.gov/media/128793/download.
- 9.Bioanalytical Methods Templates: US FDA; 2019 [Available from: https://www.fda.gov/media/131425/download.
- 10.In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry: US FDA; 2020 [Available from: https://www.fda.gov/media/134582/download.
- 11.Clinical Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry: US FDA; 2020 [Available from: https://www.fda.gov/media/134581/download.
- 12.MANUAL OF POLICIES AND PROCEDURES: Good Review Practices: Clinical Pharmacology Review of New Molecular Entity (NME) New Drug Applications (NDAs) and Original Biologics License Applications (BLAs): US FDA; 2016 [Available from: https://www.fda.gov/media/71709/download.
- 13.NDA Approval Letter: VEKLURY (remdesivir) for injection: US FDA; 2020 [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2020/214787Orig1s000ltr.pdf.
- 14.FDA News Release: FDA Approves First Treatment for COVID-19: US FDA; 2020 [Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19.
- 15.Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics US FDA; 2014 [Available from: https://www.fda.gov/media/86377/download.
- 16.Fan J, Zhang X, Liu J, Yang Y, Zheng N, Liu Q, Bergman K, Reynolds K, Huang SM, Zhu H, Wang Y. Connecting hydroxychloroquine in vitro antiviral activity to in vivo concentration for prediction of antiviral effect: a critical step in treating COVID-19 patients. Clin Infect Dis. 2020;71:3232–3236. doi: 10.1093/cid/ciaa623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Letter revoking EUA for chloroquine phosphate and hydroxychloroquine sulfate: US FDA; 2020 [Available from: https://www.fda.gov/media/138945/download.
- 18.Wang Y, Zhu H, Madabushi R, Liu Q, Huang SM, Zineh I. Model-Informed Drug Development: current US regulatory practice and future considerations. Clin Pharmacol Ther. 2019;105(4):899–911. doi: 10.1002/cpt.1363. [DOI] [PubMed] [Google Scholar]
- 19.Madabushi R, Wang Y, Zineh I. A holistic and integrative approach for advancing Model-Informed Drug Development. CPT Pharmacometrics Syst Pharmacol. 2019;8(1):9–11. doi: 10.1002/psp4.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Model-Informed Drug Development Pilot Program: US FDA; 2020 [Available from: https://www.fda.gov/drugs/development-resources/model-informed-drug-development-pilot-program.
- 21.Grimstein M, Yang Y, Zhang X, Grillo J, Huang SM, Zineh I, Wang Y. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–25. doi: 10.1016/j.xphs.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X, Yang Y, Grimstein M, Fan J, Wang YH, Grillo J, et al. Application of PBPK modeling and simulation for regulatory decision-making and it’s impact on the US prescribing information: an update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. The Journal of Clinical Pharmacology. Submitted in 2020. [DOI] [PubMed]
- 23.Mentre F, Friberg LE, Duffull S, French J, Lauffenburger DA, Li L, et al. Pharmacometrics and systems pharmacology 2030. Clin Pharmacol Ther. 2020;107(1):76–78. doi: 10.1002/cpt.1683. [DOI] [PubMed] [Google Scholar]
- 24.PUBLIC LAW 114–255: US Congress; 2016 [Available from: https://www.congress.gov/114/plaws/publ255/PLAW-114publ255.pdf.
- 25.Real-World Evidence: US FDA; 2020 [Available from: https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence.
- 26.Framework for FDA’s Real-World Evidence Program: US FDA; 2018 [Available from: https://www.fda.gov/media/120060/download.
- 27.Liu Q, Ramamoorthy A, Huang SM. Real-world data and clinical pharmacology: a regulatory science perspective. Clin Pharmacol Ther. 2019;106(1):67–71. doi: 10.1002/cpt.1413. [DOI] [PubMed] [Google Scholar]
- 28.NSCLC: Pneumonitis, Immunotherapy, and Chemotherapy: The ASCO Post; 2020 [Available from: AACR annual meeting 2020; https://www.ascopost.com/videos/aacr-virtual-annual-meeting-2020/qi-liu-on-pneumonitis-immunotherapy-and-chemotherapy-in-nsclc/.
- 29.National Prescription Audit and static data 2006-2011. January 2006-December 2018. IQVIA.
- 30.2019 National Survey on Drug Use and Health (NSDUH) – 12 or older.
- 31.Rizzo S, Botta F, Raimondi S, Origgi D, Fanciullo C, Morganti AG, Bellomi M. Radiomics: the facts and the challenges of image analysis. Eur Radiol Exp. 2018;2(1):36. doi: 10.1186/s41747-018-0068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Carvalho S, et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun. 2014;5:4006. doi: 10.1038/ncomms5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song J, Shi J, Dong D, Fang M, Zhong W, Wang K, Wu N, Huang Y, Liu Z, Cheng Y, Gan Y, Zhou Y, Zhou P, Chen B, Liang C, Liu Z, Li W, Tian J. A new approach to predict progression-free survival in stage IV EGFR-mutant NSCLC patients with EGFR-TKI therapy. Clin Cancer Res. 2018;24(15):3583–3592. doi: 10.1158/1078-0432.CCR-17-2507. [DOI] [PubMed] [Google Scholar]
- 34.Bodalal Z, Trebeschi S, Nguyen-Kim TDL, Schats W, Beets-Tan R. Radiogenomics: bridging imaging and genomics. Abdom Radiol (NY). 2019;44(6):1960–1984. doi: 10.1007/s00261-019-02028-w. [DOI] [PubMed] [Google Scholar]
- 35.Iwatate Y, Hoshino I, Yokota H, Ishige F, Itami M, Mori Y, Chiba S, Arimitsu H, Yanagibashi H, Nagase H, Takayama W. Radiogenomics for predicting p53 status, PD-L1 expression, and prognosis with machine learning in pancreatic cancer. Br J Cancer. 2020;123:1253–1261. doi: 10.1038/s41416-020-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Q, Zhu H, Liu C, Jean D, Huang SM, ElZarrad MK, et al. Application of machine learning in drug development and regulation: current status and future potential. Clin Pharmacol Ther. 2020;107(4):726–729. doi: 10.1002/cpt.1771. [DOI] [PubMed] [Google Scholar]
- 37.Berg G, Rybakova D, Fischer D, Cernava T, Verges MC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome. 2020;8(1):103. doi: 10.1186/s40168-020-00875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570(7762):462–467. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 40.Science and Regulation of Live Microbiome-Based Products Used to Prevent, Treat, or Cure Diseases in Humans: US FDA; 2018 [Available from: https://www.fda.gov/vaccines-blood-biologics/workshops-meetings-conferences-biologics/science-and-regulation-live-microbiome-based-products-used-prevent-treat-or-cure-diseases-humans.
- 41.Drug Development Tools | DDTs: US FDA; 2019 [Available from: https://www.fda.gov/drugs/development-approval-process-drugs/drug-development-tools-ddts.
- 42.Drug Development Tool (DDT) Qualification Programs: US FDA; 2020 [Available from: https://www.fda.gov/drugs/development-approval-process-drugs/drug-development-tool-ddt-qualification-programs#:~:text=The%20Cures%20Act%2C%20passed%20in,capabilities%20of%20a%20single%20entity.
- 43.Drug Development Tool (DDT) qualification process: US FDA; 2019 [Available from: https://www.fda.gov/drugs/drug-development-tool-ddt-qualification-programs/drug-development-tool-ddt-qualification-process.
- 44.Pharmacodynamic Biomarkers to Support Biosimilar Development: PCSK9 Inhibitors: The National Library of Medicine; 2019 [Available from: https://clinicaltrials.gov/ct2/show/NCT04189484?cond=biomarker&cntry=US&fund=1&draw=2&rank=10.
- 45.Impact Story: Improved Assessment of Cardiotoxic Risk in Drug Candidates: The Comprehensive in vitro Proarrhythmia Assay: US FDA; 2020 [Available from: https://www.fda.gov/drugs/regulatory-science-action/impact-story-improved-assessment-cardiotoxic-risk-drug-candidates-comprehensive-vitro-proarrhythmia.
- 46.US Package Insert of KALYDECO® (ivacaftor): US FDA; 2017 [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203188s019lbl.pdf.
- 47.3D Cell Culture Models for Drug PK, Safety, and Efficacy Assessment: University of Maryland CERSI; 2020 [Available from: https://www.pharmacy.umaryland.edu/media/SOP/wwwpharmacyumarylandedu/centers/cersievents/pdf/3d-cell-culture-agenda-vrs7.pdf.
- 48.Wang H, Brown P, Chow E, Ewart L, Ferguson S, Fitzpatrick S, et al. 3D cell culture models for drug pharmacokinetics, safety, and efficacy assessment. Clin Transl Sci. Submitted in. 2020. [DOI] [PMC free article] [PubMed]
- 49.Centers of Excellence in Regulatory Science and Innovation (CERSIs): US FDA; 2018 [Available from: https://www.fda.gov/science-research/advancing-regulatory-science/centers-excellence-regulatory-science-and-innovation-cersis.
- 50.Regulatory Science Extramural Research and Development Projects: US FDA; 2020 [Available from: https://www.fda.gov/science-research/advancing-regulatory-science/regulatory-science-extramural-research-and-development-projects.
- 51.FDA Memoranda of Understanding: US FDA; 2019 [Available from: https://www.fda.gov/about-fda/partnerships-enhancing-science-through-collaborations-fda/fda-memoranda-understanding.
- 52.Cooperative Research and Development Agreements (CRADAs): US FDA; 2018 [Available from: https://www.fda.gov/science-research/fda-technology-transfer-program/cooperative-research-and-development-agreements-cradas.