

Abstract
The bio.tools registry is a main catalogue of computational tools in the life sciences. More than 17 000 tools have been registered by the international bioinformatics community. The bio.tools metadata schema includes semantic annotations of tool functions, that is, formal descriptions of tools’ data types, formats, and operations with terms from the EDAM bioinformatics ontology. Such annotations enable the automated composition of tools into multistep pipelines or workflows. In this Technical Note, we revisit a previous case study on the automated composition of proteomics workflows. We use the same four workflow scenarios but instead of using a small set of tools with carefully handcrafted annotations, we explore workflows directly on bio.tools. We use the Automated Pipeline Explorer (APE), a reimplementation and extension of the workflow composition method previously used. Moving “into the wild” opens up an unprecedented wealth of tools and a huge number of alternative workflows. Automated composition tools can be used to explore this space of possibilities systematically. Inevitably, the mixed quality of semantic annotations in bio.tools leads to unintended or erroneous tool combinations. However, our results also show that additional control mechanisms (tool filters, configuration options, and workflow constraints) can effectively guide the exploration toward smaller sets of more meaningful workflows.
Keywords: proteomics, scientific workflows, computational pipelines, workflow exploration, automated workflow composition, semantic tool annotation
Introduction
Converting experimental biological data into interpretable results increasingly involves the combination of multiple, diverse computational tools into pipelines or workflows performing specific sequences of operations.1−3 Working out which tools and combinations are applicable and scientifically meaningful in practice is often hard, in particular, when the tools were not developed by the same research group, consortium, or company. The idea of automated workflow exploration and composition is to let an algorithm perform or assist in this process. Different approaches to this idea have been proposed in the past,4−10 the applicability and success of which depend on rich and consistent semantic tool annotations.
In 2018, we published the case study “Automated Workflow Composition in Mass Spectrometry-Based Proteomics”.11 There, we demonstrated based on four selected proteomics use cases how the thorough semantic annotation of tools with regard to operations, data types, and formats enables their automated composition into tentatively viable workflows as permutations of a data analysis plan. As a proof-of-concept study, the study used a relatively small, well-defined set of tools, with carefully handcrafted semantic annotations as a basis. In this Technical Note, we outline the potential of the approach when applied to larger collections of semantically annotated, complex tools with multiple inputs and outputs.
Since our initial case study, much has happened: The bio.tools registry12 has grown and matured substantially13 and currently covers more than 17 000 bioinformatics tools, including more than 750 tools in the specially curated proteomics subset (https://proteomics.bio.tools).14,15 Furthermore, bio.tools now has a REST application programming interface (API) that allows for programmatic access to the registry and, in particular, to the tools’ metadata through their unique bio.tools identifiers. Along with the registration and annotation of proteomics tools in bio.tools, the coverage of proteomics terms in the EDAM ontology of bioinformatics operations, types of data, data identifiers, data formats, and topics (http://edamontology.org/)16 has evolved and matured. Finally, the PROPHETS framework17,18 used for automated workflow exploration in the previous study has been discontinued. With the Automated Pipeline Explorer (APE),19,20 we have developed a new framework that implements essentially the same workflow composition method but is generally better tailored to the work with scientific workflows.
In this Technical Note, we revisit the four proteomics use cases from our previous study that describe typical scenarios for the analysis of proteomics data. We use APE for the workflow exploration and move “into the wild”: Instead of using a small, handcrafted set of tools and annotations, we work with tools and annotations directly from the bio.tools registry. This opens up an unprecedented wealth of tools and, accordingly, a huge number of possible alternative workflows. With APE, we systematically explore the space of possibilities; however, as the quality of semantic annotations in bio.tools varies, deviations in the quality of the automatically explored workflows are to be expected. Therefore, we focus here on evaluating the quality of the suggested workflows, in particular, checking for impossible solutions and discussing possible ways of preventing their occurrence.
The remainder of this Technical Note is structured as follows. In the next section, we describe the experimental setup. Then, we present and discuss the results from the workflow exploration experiments before concluding with a short summary and perspectives for future work. The data and code for running this study, along with the workflow exploration results and evaluation data, are available online at https://github.com/sanctuuary/Proteomics_domain_setup and in the Supporting Information.
Experimental Section
Here we describe the setup of the study, summarized in Figure 1. This includes a short introduction to APE as the workflow exploration tool used, the process of fetching and filtering the semantic tool annotations from the bio.tools registry, the workflow use cases and corresponding workflow specifications, the parameters and configurations of the different workflow exploration runs, and the workflow evaluation process.
Figure 1.

Experimental setup of the study.
Automated Pipeline Explorer (APE)
APE19,20 is a command line tool and Java API for the automated exploration of possible computational pipelines (scientific workflows) from large collections of computational tools. We use it as a standalone tool in this study, but in its capacity as an API, it could also be used as a component for workflow exploration within a workflow management system such as KNIME21 or Galaxy.22 Furthermore, APE can export the obtained workflows in Common Workflow Language (CWL, https://w3id.org/cwl/) format to facilitate integration with other systems. APE is open-source software and is available on GitHub (https://github.com/sanctuuary/APE).
The workflow exploration algorithm used by APE is a variation and extension of the Semantic Linear Time Logic (SLTL)-based process synthesis approach originally introduced by Steffen et al.23 Operating on input/output-annotated tools in combination with type and operation taxonomies, this approach aligns perfectly with the information that is available through bio.tools and EDAM. Therefore, we chose to follow this approach instead of potentially more powerful synthesis or planning techniques. In particular, planning approaches based on Description Logic24 have been demonstrated to be applicable to the automated workflow composition problem in the past.7,25 They do, however, require more comprehensive domain models (sophisticated ontologies), which are not readily available.
Concretely, the semantic domain model we provided to APE consists of operation, type, and format taxonomies as controlled vocabularies for the description of computational tools (directly derived from EDAM) and functional tool annotations (inputs, outputs, operations performed) using terms from these taxonomies (directly derived from bio.tools). The domain setup process is described in further detail as follows. The workflow specifications we provided to APE comprised the available inputs and intended outputs (data type and format, again in EDAM terms) and, in some cases, additional constraints (provided through open text templates that internally translate to SLTL formulas).
On this basis, APE computes possible workflows. Intuitively, this is a two-step process:
-
1.
The tools’ input/output annotations imply a network of technically possible tool sequences, which constitutes the search space for the workflow exploration.
-
2.
In this search space, APE looks for paths that match the concrete workflow specification (from workflow inputs to outputs, while respecting additional constraints).
Technically, this process is implemented as temporal logic-based program synthesis with iterative deepening; that is, the algorithm explores the workflows requiring the least amount of steps to solve the given problem first. APE translates the domain knowledge and workflow specification into propositional logical formulas and feeds them to an automated constraint solver to compute satisfying instances. These solutions are then translated into actual candidate workflows and sorted by length. For a detailed description of the implementation, we refer to Kasalica et al.20
Domain Models
In our previous study,11 the domain model comprised 26 tools (mostly but not exclusively from the ms-utils.org collection of mass-spectrometry data analysis tools) that we annotated in a CSV file with input and output data type and formats as well as operations using terms from the EDAM ontology. In this study, we fetched the corresponding tool annotations directly from the bio.tools registry via its REST API. The bio.tools annotation schema also uses the EDAM ontology as reference vocabulary, but in contrast with our previous tabular annotation format, it allows for the annotation of multiple inputs and outputs per tool. Thus we now work with significantly more comprehensive descriptions of the available tools’ functionality.
For this study, we worked with three different domain models:
-
1.
The original set of tools (comet, enrichnet, genetrail, genetrail2, gprofile_r, idconvert, isobar, libra, msconvert, mzXMLplot, Pep3D, PeptideProphet, peptideshaker, proteinprophet, protk, PTMProphet, ssrcalc, searchgui, Tandem2XML, rt, xml2tsv, xtandem, extract_protein_names), comprising the tools from the previous study that are available in bio.tools.
-
2.
An extended set of proteomics tools, corresponding to the labeled proteomics domain in bio.tools (https://proteomics.bio.tools), which extends the original set of tools.
-
3.
The full bio.tools set, containing all of the tools currently available in the registry.
To create the three domain models, we (1) used the bio.tools REST API to fetch the JSON files containing the respective bio.tools native annotations (which can contain multiple function annotations per tool), (2) cleaned the set of annotations by keeping only well-annotated functions (details follow), and (3) transformed the annotations to the APE annotation format. The quality of the domain model determines the quality of the workflows obtained through automated exploration. bio.tools contains thousands of tools, annotated by a diverse group of contributors. Not surprisingly, this leads to mixed levels of annotation quality. In particular, many of the tool annotations lack input or output definitions, specify them vaguely or incompletely, or use outdated EDAM references. Therefore, we discarded annotations that:
do not specify an input, as these tools can be used at any step of the workflow and typically introduce unnecessary new data
do not specify an output (typically these are interactive tools), as they do not contribute to solving a data analysis problem
miss a data type or format specification, as these are incomplete annotations
reference deprecated EDAM data type or format terms, as they are not part of the domain taxonomy anymore
Note that there are also many tool annotations that reference deprecated EDAM operation terms; however, we classify those as noncritical annotation errors and allow such tools. The only way that such domain model could produce wrong results is by restricting usage of the missing tool types through explicit constraints, which we rarely do in the studied use cases.
Table 1 summarizes the effects of cleaning the annotation sets while creating the three domain models. The annotation quality of the small, original tool set is again good, and only three tools were discarded. In the larger, community-curated domain of proteomics, we find around a third of the tools to be in a well-annotated format, whereas on the global level, <10% of bio.tools are directly suitable for automated exploration. Still, these new domain models with 271 and 1642 tools, respectively, provide a next-level challenge for automated workflow exploration and resemble the variety of tools in a real-world setting better than the original set.
Table 1. Effects of Cleaning the Tool Annotation Sets.
| original | extended | full bio.tools | |
|---|---|---|---|
| number of tools in bio.tools | 24 | 751 | 17 369 |
| number of functions annotated in the tool set | 24 | 858 | 18 408 |
| number of discarded functions | 3 | 587 | 16 778 |
| number of resulting APE annotations | 21 | 271 | 1642 |
Workflow Specification Use Cases
We reuse the four proteomics data analysis use cases from our previous study,11 which require workflows of increasing complexity:
-
1.
Extraction of the Amino acid index (hydropathy) from peptides in biological sample measured by liquid chromatography MS.
-
2.
Protein identification and pathway enrichment analysis of MS spectra.
-
3.
The identification and localization of post-translational modifications in a phosphoproteomic study.
-
4.
Protein quantitation of multiple biological samples labeled by iTRAQ.
Table 2 summarizes the corresponding workflow specifications, which are the input for APE. Because EDAM and bio.tools have evolved since the previous case study, we revised the workflow specifications to match the now available terms. Concretely, we generalized the output specifications in the first and the fourth use cases from the expected data types to their parent classes, as none of the annotated tools used the originally specified types as input/output anymore. In the first use case, we substituted Amino acid index (hydropathy) with Amino acid property, whereas in the fourth use case, we replaced Gene expression profile by Expression data. Similarly, we updated some of the workflow constraints. The term validation of peptide-spectrum matches became obsolete with EDAM 1.19, so instead, we opted for the similar Target-Decoy term. Furthermore, we updated the term gene-set enrichment analysis to enrichment analysis. Although it is not deprecated, it is not used in any of the tool annotations.
Table 2. Workflow Specification for the Four Use Cases.
| use case | inputs | outputs | constraints |
|---|---|---|---|
| #1 | Mass spectra in Thermo RAW format | Amino acid property in any format | (i) Use operation peptide identification; (ii) Use operation Target-Decoy; (iii) Use operation retention time prediction; (iv) Do not use operation Protein identification |
| #2 | Mass spectra in Thermo RAW format | Pathway or network in any format | (i) Use operation peptide identification operation; (ii) Use operation enrichment analysis; (iii) Use operation enrichment analysis only after peptide identification operation; (iv) Use tool ProteinProphet only after PeptideProphet |
| #3 | Mass spectra in Thermo RAW format | Protein identification in any format | (i) Use operation PTM identification; (ii) Use operation PTM identification only after operation Target-Decoy; (iii) Use operation Target-Decoy only after operation peptide database search; (iv) Do not use operation Target-Decoy more than once |
| #4 | Mass spectra in Thermo RAW format | Expression data in any format | (i) Use operation iTRAQ; (ii) Use operation iTRAQ only after operation Target-Decoy |
Workflow Exploration Runs
As illustrated in Figure 1 (top), this study comprised 24 different workflow exploration runs in total. For each of the three domain models (Original, Extended, and Full bio.tools), we let APE explore possible workflows for all four use cases. Furthermore, we apply two different versions of the workflow specifications: desired input/output only (I/O) and I/O with additional constraints (I/O+C). This distinction allows us to evaluate the effects of these additional constraints in comparison with I/O specification only when exploring workflows in large collections of tools. In the previous case study, even with a small set of tools, constraints were crucial to guide the exploration toward the intended workflows, as an I/O specification alone did not provide sufficient information. We expected this to be even more needed with the increased size of the domain model.
We limited the exploration runs to the first 100 (shortest) workflows. This choice was motivated by two observations: First, as workflow candidates get longer, they tend to simply extend already considered shorter solutions and introduce redundant steps. Second, users of automated workflow exploration tools are able to process and compare only a limited number of workflows, and in our experience, 100 is a reasonable bound in practice. Finally, because of resource limitation, we terminate the search if no solutions are found until length 20 (workflows with 20 operation steps).
The experiments were run using bio.tools annotations as of November 25, 2020, EDAM version 1.24, and APE version 1.1.2 on a i7-6500U CPU at 2.50 GHz and a 16 GB RAM machine running on Ubuntu 20.04. We did not measure the runtime behavior of the workflow exploration algorithm systematically in this study. For the smallest domain model, the runtime was a few seconds; exploration using the largest domain model was averaging ∼10 min, with the longest time of almost 1 h. In general, runtime performance increases with the size of the domain model and the length of the solutions.
Workflow Evaluation
To evaluate the quality of the suggested workflows, two domain experts (proteomics researchers with extensive tool and workflow experience) scored the first 20 workflow candidates of each exploration run on a scale from 0 to 3 according to the following criteria:
-
3:
Good workflow, have seen it or similar before, I know it will work
-
2:
Interesting suggestion, seems viable, could work, worth trying
-
1:
Might work, but does not seem very useful or has unnecessary steps
-
0:
I know that it will not work
The two experts scored the workflows independently. We calculated the averages of their scores for subsequent analysis. Further evaluation through implementation, execution, and benchmarking, like we performed in the previous study,11 was out of scope for this study and is left for future work.
Results and Discussion
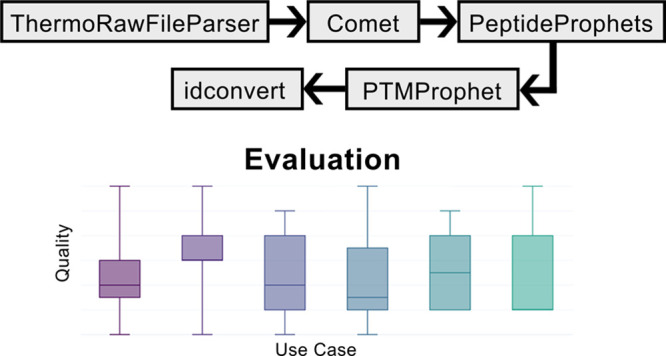
As follows, we summarize and discuss the workflows found in all exploration settings previously described. We use the number of obtained workflows and the scores they received from the domain experts (see Figure 2) as indicators of the workflow exploration comprehensiveness and the quality in the different setups.
Figure 2.

Workflow quality evaluation.
As general observations, the workflow-evaluating domain experts remarked that they found this to be an interesting and insightful exercise. On the one hand, they came across several interesting, sometimes even surprising, workflow suggestions that they would not have thought about themselves but that seem worth trying. On the other hand, for faulty or insensible workflow suggestions, they could usually see how the (flawed) annotations of the involved tools set the automated composer on a wrong track.
Use Case #1
The workflows we obtained for the first use case with the original tool set closely resemble the corresponding results from the previous study. First workflows for the specification are found at a length of three, which corresponds to workflows of three successive tools. As the examples in Figure 3 indicate, the main difference from the results of the previous study is that some tools that were previously used (e.g., X!Tandem, SSRCalc) are not included anymore. This is due to different annotations of the tools, which are both now annotated to expect two inputs instead of one.
Figure 3.

Example workflow candidates: Use Case #1.
The example workflows for the original tool set also show another frequently observed pattern: Short workflows with a conversion step (such as msConvert) often are contained again in longer workflows that simply repeat the conversion step. These redundant steps could be avoided by introducing constraints that prevent multiple conversion operations over the same data. Currently, this requires formulating such constraints per individual tool, so we are considering ways of adding this as a more convenient configuration option to APE.
When we explore workflows for the same specification but with the extended and full sets of tools, the number of results significantly increases. As Figure 3 indicates, the additional results comprise several new and sometimes surprising workflow suggestions, such as the combination of using MZmine before Mascot Server. However, there are also workflow suggestions that are less sensible. For example, some workflows start with the tool CompassXPort, although it cannot read Thermo RAW files. The reason is that CompassXPort is annotated in bio.tools to read Mass spectrometry data format files, which is the parent term for 30 specific file formats including Thermo RAW. CompassXPort cannot read Thermo RAW, but this cannot be inferred when using general annotations including parent terms. Ideally, CompassXPort would be annotated in bio.tools with a precise list of accepted input formats and not their parent term. To work around this with the current version of APE, a constraint can be added that, for example, excludes CompassXPort from the exploration. Alternatively, one could restrict the domain model to include only tools described by sufficiently specific file formats. However, given the current state of tool annotations, such a restriction is likely to strongly decrease the domain model size. To solve such problems more generally, we are currently investigating possibilities for extending APE with new configuration options or heuristics for data format handling.
Interestingly, we obtained almost the same results for the extended and full domain models. The only notable difference is the occurrence of a rather new parsing tool ThermoRawFileParser in the workflow solutions with the full domain model. ThermoRawFileParser is not yet included in the proteomics domain, as it has been recently added. With the exception of this tool, extending the domain model with tools from outside the bio.tools proteomics domain does not create new possibilities for this use case. This can be interpreted as evidence that the coverage of proteomics tools in the respective bio.tools domain is comprehensive.
A general observation from the evaluation is that the constraints do indeed have a considerable effect on the number and quality of workflows obtained. As Figure 2 shows, for all domain models, the domain experts gave higher scores to the workflows explored with constraints. This is especially important with the extended and full domain models. There, the number of solutions exceeds the threshold of 100 already at length 4 in the unconstrained case. To a large extent, these are workflows that are (presumably) implementable based on their input/output annotations but that do not perform the operations actually intended by the workflow developer. Constraints that specify these intentions thus better help to drastically decrease the number of unfeasible workflows. Furthermore, they allow for the exploration of longer and, at the same time, more meaningful workflows. For this use case, the hand-curated domain model appears to be more restrictive and effective for finding appropriate solutions than the constrained case over a larger domain model. We attribute this to the specific tailoring of the original domain model to this use case, an approach that does, however, hardly scale in practice. This reemphasizes the importance of using appropriate constraints when dealing with larger collections of tools and annotations from community repositories.
Use Case #2
When exploring workflows for the second use case with the original tool set, somewhat surprisingly, we did not find any. The reason is that the enrichment analysis tools (gProfiler, EnrichNet, etc.), which are needed to generate the specified workflow output, are annotated to expect a Gene ID type as one of the two obligatory inputs. However, there is no tool in the domain model that would generate a Gene ID as output, and hence there is a missing link that prevents the exploration from finding corresponding workflows. A “shim” tool that converts IDs into each other (e.g., UniProt protein accession into Gene IDs) could provide this missing link. It is an ongoing discussion, however, if such shims should be registered in bio.tools or if that would lead to an overload of tools with insignificant functionality. Many shims are, in fact, included in larger software suites or libraries but are not annotated as individual functions of these and thus are missed by automated exploration. A related issue is incomplete annotations due to large numbers of functions performed or formats supported. For example, some enrichment tools accept tens of different ID types but list only a few in their tool annotation. Thus some possible matches are missed. However, using a more abstract parent term in the annotation can cause erroneous matches, as previously described for CompassXPort.
Such missing links are typical risks of using small domain models. Interestingly, here the use of the larger sets of tools does not resolve the issue. The exploration with the extended domain model does return possible workflows, but these are questionable. As Figure 4 shows, the first suggestion is a single tool (ProCoNa) that matches the input/output specification. Like CompassXPort, it is annotated as using the general Mass spectrometry data format as input, whereas it actually accepts only some of these formats (a table containing peptide identifications), so the problem is similar to the one previously described. The other suggestions are then actually meaningless extensions of this first workflow. In the constrained case, no workflows are returned, as the constraints try to enforce the aforementioned enrichment analysis tools, which cannot be used due to the missing shims.
Figure 4.

Example workflow candidates: Use Case #2.
The unconstrained exploration with the full domain model yields the same results as that with the extended model, with the exception of the ThermoRawFileParser tool for the initial file conversion. As in the previous constrained cases, the constrained exploration with the full domain model resulted in no solutions. Unfortunately, in the case of the full model, we could not explore solutions up to length 20. Because of the exponential runtime complexity and memory requirements of the exploration algorithm, the composition for lengths longer than 8 exceeded the available memory, so the exploration process stopped there. However, on the basis of the results from the extended model, we assumed that no solutions would have been found among longer workflows either.
Use Case #3
For this use case, the workflows obtained with the original tool set largely again correspond to the results from the previous study. As Figure 2 shows, there is again a notable difference between the runs with and without constraints. In fact, for this use case, the constraints are crucial, as the input/output specification is quite general and does not provide sufficient information to actually solve the problem as intended. As Figure 5 shows, the workflows obtained with the unconstrained specification are, in principle, valid but lack the validation part of the reference workflow scenarios. Only the use of constraints ensures that Target-Decoy tools like PeptideProhphet and PTMProphet are included.
Figure 5.

Example workflow candidates: Use Case #3.
This observation also holds for the extended and full tool sets. Looking at the workflow candidates, we see that the slight difference in results between the extended and the full domain model is, in fact, again caused by the additional tool ThermoRawFileParser in the full domain. This aligns with our findings from Use Case #1.
Note that also for this use case we observe the spurious use of CompassXPort, T2D converter, and similar tools, for the same reasons as previously discussed.
Use Case #4
Similar to Use Cases #1 and #3, the workflows obtained for the fourth use case largely correspond to those from the previous study when exploring workflows for the original tool set (Figure 6). Furthermore, this scenario again affirms that the usage of additional constraints to specify the problem decreases the number and increases the quality of the workflows. (See Figure 2.) Interestingly, whereas the constrained solutions over the extended and the full domain differ in the usage of the aforementioned ThermoRawFileParser tool, the unconstrained solutions differ in two more tools, namely, TDimpute and pyQms. These two tools, similarly to ThermoRawFileParser, have not yet been added to the proteomics domain but in fact contain the EDAM topic Proteomics.
Figure 6.

Example workflow candidates: Use Case #4.
Conclusions
Annotating computational tools with ontologically defined terms describing their operations, data types, and formats enables their automated composition into tentatively viable workflows. Providing such annotations (using terms from EDAM ontology) is one of the main goals of the bio.tools registry, which has become a main catalogue of computational tools in the life sciences. For this Technical Note, we applied APE to the bio.tools registry and revisited workflow use cases from a previous proof-of-concept study in proteomics. Our results show that this combination can provide an effective means for searching for possible purpose-specific workflows in large collections of semantically annotated tools. We see this as another milestone on the long way toward a workflow exploration system that would ultimately be as comprehensive, reliable, and easy to use as contemporary route planners.
Naturally, the overall quality of the automatically obtained workflows highly depends on the quality of the semantic domain model, here the domain ontology (EDAM) and the tool annotations (bio.tools). Many tools in the community-driven bio.tools registry are not accurately annotated, presumably due to a lack of awareness and understanding among curators of what constitutes good annotations for the purpose of automated tool composition and workflow exploration. Even partially inadequate annotations can lead to wrong compositions or the exclusion of tools that suit a requested combination of input, output, and operation. Hence, to use bio.tools with APE, the relevant tool sets have to be preprocessed and filtered to sort out poorly annotated annotations. Conversely, small curated domains can yield high-quality results, but they tend to overfit, do not enable the exploration of new, possibly better performing tools and workflows, and furthermore, hardly scale, so they are not an ideal solution in the long term.
Accepting that domain models will never be perfect, APE’s ability to incrementally provide additional workflow constraints to the exploration algorithm gains importance. As our results show, such constraints are crucial for filtering out nonsensical and undesirable alternatives and for guiding the search toward actually desirable tool combinations in a still huge space of possibilities. The resulting workflows also contain valuable information that can be employed to improve tool annotations. A knowledgeable researcher can adapt the annotations of neglected tools and correct erroneous annotations in tools that were assigned to a workflow despite their inability to fulfill that particular task.
In this Technical Note, we presented a manual evaluation and comparison of automatically composed workflows for four different proteomics use cases. The evaluation relied mostly on our understanding of the domain and our experiences with the used tools. To be able to objectively evaluate each of the proposed workflows, the workflows should furthermore be checked for the actual compatibility of the involved tools, implemented, and benchmarked, like we did in our previous study.11 Indeed, our goal for the future is to develop a platform that supports the automated exploration, implementation, execution, and benchmarking in one coherent framework and thus assists the workflow developer in systematically exploring and evaluating possible workflows for a specific research question.
Future work on the way toward this vision requires addressing different conceptual as well as practical issues of automated workflow exploration. For example, constraint specification and tool annotation possibilities need to be extended and flexibilized to allow for “identity annotation” (i.e., for format conversion tools that change the data format but maintain the data type) and quantified constraints (such as constraints that should hold for all tools in a set individually, e.g., when using constraints to avoid the redundant use of tools). On the practical side, bio.tools entries need to be updated (or flagged) automatically when EDAM changes and the used annotation terms become obsolete. Another pragmatic next step could be the introduction of an “automated exploration badge” in bio.tools that marks tools with validated semantic tool annotations. Thinking further, it might be desirable for bio.tools to provide quantitative annotations that could be informative for automated workflow exploration tools to prioritize possible workflows over others. In addition to the technical monitoring information from OpenEBench26 that is already now available for some tools, this could include download numbers or marking of tools highly used in workflows (for example, derived from the WorkflowHub, https://workflowhub.eu/) as indicators of popularity and value to the community. Similarly, allowing the community to rate or mark tools as useful or well-maintained could be a basis for providing quality control on tools used within a workflow.
Acknowledgments
We thank Hans Ienasescu for his support with the bio.tools API.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00983.
Table S5: Spreadsheet containing general information about the exploration runs. Figure S1: Distribution of the workflow quality scores using box plots (PDF)
Table S1. List of up to 100 solutions generated for each run for Use Case 1 (XLSX)
Table S2. List of up to 100 solutions generated for each run for Use Case 2 (XLSX)
Table S3. List of up to 100 solutions generated for each run for Use Case 3 (XLSX)
Table S4. List of up to 100 solutions generated for each run for Use Case 4 (XLSX)
Table S6. Quality evaluation of the first 20 workflows per each run for Use Case 1 (XLSX)
Table S7. Quality evaluation of the first 20 workflows per each run for Use Case 2 (XLSX)
Table S8. Quality evaluation of the first 20 workflows per each run for Use Case 3 (XLSX)
Table S9. Quality evaluation of the first 20 workflows per each run for Use Case 4 (XLSX)
Table S10. Summary of the evaluation results (XLSX)
File S1: Tool annotations for the original domain (TXT)
File S2: Tool annotations for the extended domain (TXT)
File S3: Tool annotations for the full domain (TXT)
The authors declare no competing financial interest.
Supplementary Material
References
- Atkinson M.; Gesing S.; Montagnat J.; Taylor I. Scientific workflows: Past, present and future. Future Generation Computer Systems 2017, 75, 216–227. 10.1016/j.future.2017.05.041. [DOI] [Google Scholar]
- Garijo D. AI Buzzwords Explained: Scientific Workflows. AI Matters 2017, 3, 4–8. 10.1145/3067682.3067683. [DOI] [Google Scholar]
- Taylor I. J.; Deelman E.; Gannon D. B.; Shields M.. Workflows for e-Science: Scientific Workflows for Grids; Springer-Verlag, 2014. [Google Scholar]
- Lamprecht A.User-Level Workflow Design:A Bioinformatics Perspective; Lecture Notes in Computer Science; Springer, 2013; Vol. 8311. [Google Scholar]
- Wilkinson M. D.; McCarthy E. L.; Vandervalk B. P.; Withers D.; Kawas E. A.; Samadian S. SADI, SHARE, and the in silico scientific method. BMC Bioinf. 2010, 11, S7. 10.1186/1471-2105-11-S12-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos J.; Karlsson T. J. M.; Trelles O. Magallanes: a web services discovery and automatic workflow composition tool. BMC Bioinf. 2009, 10, 334. 10.1186/1471-2105-10-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žaková M.; Křemen P.; Železný F.; Lavrač N. Automating Knowledge Discovery Workflow Composition through Ontology-Based Planning. IEEE Transactions on Automation Science and Engineering 2011, 8, 253–264. 10.1109/TASE.2010.2070838. [DOI] [Google Scholar]
- DiBernardo M.; Pottinger R.; Wilkinson M. Semi-automatic web service composition for the life sciences using the BioMoby semantic web framework. J. Biomed. Inf. 2008, 41, 837–847. 10.1016/j.jbi.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Merelli E.; Armano G.; Cannata N.; Corradini F.; d’Inverno M.; Doms A.; Lord P.; Martin A.; Milanesi L.; Möller S.; Schroeder M.; Luck M. Agents in bioinformatics, computational and systems biology. Briefings Bioinf. 2006, 8, 45–59. 10.1093/bib/bbl014. [DOI] [PubMed] [Google Scholar]
- Gil Y.; Deelman E.; Blythe J.; Kesselman C.; Tangmunarunkit H. Artificial intelligence and grids: workflow planning and beyond. IEEE Intelligent Systems 2004, 19, 26–33. 10.1109/MIS.2004.1265882. [DOI] [Google Scholar]
- Palmblad M.; Lamprecht A.-L.; Ison J.; Schwämmle V. Automated workflow composition in mass spectrometry-based proteomics. Bioinformatics 2019, 35, 656–664. 10.1093/bioinformatics/bty646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison J.; Rapacki K.; Menager H.; Kalas M.; Rydza E.; Chmura P.; Anthon C.; Beard N.; Berka K.; Bolser D.; et al. Tools and data services registry: a community effort to document bioinformatics resources. Nucleic Acids Res. 2016, 44 (D1), D38–D47. 10.1093/nar/gkv1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison J. C.; Ménager H.; Brancotte B.; Jaaniso E.; Salumets A.; Racek T.; Lamprecht A.; Palmblad M.; Kalas M.; Chmura P.; Hancock J. M.; Schwämmle V.; Ienasescu H. Community curation of bioinformatics software and data resources. Briefings Bioinform. 2020, 21, 1697–1705. 10.1093/bib/bbz075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwämmle V.; Harrow J.; Ienasescu H. Proteomics Software in bio.tools: Coverage and Annotations. J. Proteome Res. 2021, 10.1021/acs.jproteome.0c00978. [DOI] [PubMed] [Google Scholar]
- Tsiamis V.; Ienasescu H.-I.; Gabrielaitis D.; Palmblad M.; Schwämmle V.; Ison J. One Thousand and One Software for Proteomics: Tales of the Toolmakers of Science. J. Proteome Res. 2019, 18, 3580–3585. 10.1021/acs.jproteome.9b00219. [DOI] [PubMed] [Google Scholar]
- Ison J. C.; Kalas M.; Jonassen I.; Bolser D. M.; Uludag M.; McWilliam H.; Malone J.; Lopez R.; Pettifer S.; Rice P. M. EDAM: an ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics 2013, 29, 1325–1332. 10.1093/bioinformatics/btt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokat S.; Lamprecht A.; Steffen B. Loose Programming with PROPHETS. FASE'12: Proceedings of the 15th international conference on Fundamental Approaches to Software Engineering 2012, 7212, 94–98. 10.1007/978-3-642-28872-2_7. [DOI] [Google Scholar]
- Lamprecht A.; Naujokat S.; Margaria T.; Steffen B. Synthesis-Based Loose Programming. Seventh International Conference on the Quality of Information and Communications Technology 2010, 262–267. 10.1109/QUATIC.2010.53. [DOI] [Google Scholar]
- Kasalica V.; Lamprecht A.-L. APE: A Command-Line Tool and API for Automated Workflow Composition. Computational Science – ICCS 2020 2020, 12143, 464–476. 10.1007/978-3-030-50436-6_34. [DOI] [Google Scholar]
- Kasalica V.; Lamprecht A.-L. Workflow Discovery with Semantic Constraints: The SAT-Based Implementation of APE. Electronic Communications of the EASST 2019, 78, 1–25. 10.14279/tuj.eceasst.78.1092. [DOI] [Google Scholar]
- Berthold M. R.; Cebron N.; Dill F.; Gabriel T. R.; Kötter T.; Meinl T.; Ohl P.; Thiel K.; Wiswedel B. KNIME - the Konstanz Information Miner: Version 2.0 and Beyond. SIGKDD Explor. Newsl 2009, 11, 26–31. 10.1145/1656274.1656280. [DOI] [Google Scholar]
- Afgan E.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen B.; Margaria T.; Freitag B.. Module Configuration by Minimal Model Construction; University of Passau, 1993.
- Gil Y. Description Logics and Planning. AI Mag. 2005, 26 (2), 73. 10.1609/aimag.v26i2.1814. [DOI] [Google Scholar]
- Ambite J. L.; Kapoor D.. Automatically Composing Data Workflows with Relational Descriptions and Shim Services; The Semantic Web: Berlin, 2007; pp 15–29. [Google Scholar]
- Capella-Gutierrez S.; Iglesia D. d. l.; Haas J.; Lourenco A.; Fernández J. M.; Repchevsky D.; Dessimoz C.; Schwede T.; Notredame C.; Gelpi J. L.; Valencia A. Lessons Learned: Recommendations for Establishing Critical Periodic Scientific Benchmarking. bioRxiv 2017, 181677. 10.1101/181677. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.