

Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor and a member of the PER-ARNT-SIM (PAS) superfamily of environmental sensors. The AHR is involved in a series of biological processes including adaptive metabolism of xenobiotics, toxicity of certain environmental pollutants, vascular development, fertility, and immune function. Mouse models, including the Ahr null and Ahr conditional null (Ahrfx) mice, are widely used for the study of AHR-mediated biology and toxicity. The Ahr conditional null mouse harbors the low-affinity Ahrd allele that exhibits approximately a 10-fold lower binding affinity for certain xenobiotic AHR ligands than the widely used C57BL/6 mouse that harbors the higher affinity Ahrb1 allele. Here, we report a novel mouse model that introduces a V375A polymorphism that converts the low-affinity allele into a high-affinity allele, offering a more sensitive conditional model. In the generation of this novel conditional allele, two additional mutants arose, including a 3-bp deletion in the PAS-B domain (AhrNG367R) and an early termination codon in the PAS-B domain (AhrTer383). The AhrNG367R allele presents as a phenocopy of the null and the AhrTer383 allele presents as an antimorph when assessing for the ductus venosus and liver lobe weight endpoints. These new models represent a series of tools that will be useful in further characterizing AHR biology.
Keywords: aryl hydrocarbon receptor, AHR, conditional allele, mouse model, dioxin, xenobiotics, receptors, CRISPR, PAS, dioxin receptor
Impact Statement
We describe the generation of 3 novel conditional mutations at the Ahr locus in mice. These include: a high affinity allele, a novel null allele, and an antimorphic allele. We propose these mutants will serve as powerful new tools for the study of the AHR pathway.
The aryl hydrocarbon receptor (AHR) is a ligand-activated basic helix-loop-helix (bHLH) transcription factor that is a member of the PER-ARNT-SIM (PAS) superfamily of environmental sensors (McIntosh et al., 2010). The AHR is highly conserved in Chordates, though variation in receptor activity, sequence, and size is common (Hahn, 2002; Poland et al., 1991). The naturally occurring Ahr polymorphisms in mice were essential tools in the discovery and characterization of the receptor (Ema et al., 1994; Nebert and Gelboin, 1969; Poland et al., 1976, 1994; Poland and Glover, 1975, 1980). For example, the differential response to polycyclic aromatic hydrocarbons across polymorphic strains contributed to the receptor’s discovery (Nebert et al., 1975; Poland et al., 1994; Poland and Glover, 1980, 1990). Subsequent characterization studies uncovered a prominent role for the AHR in upregulating genes, such as the cytochromes P450 that are responsible for metabolism of many of the receptor’s ligands (Dragin et al., 2008). More recent studies have unveiled a role for the AHR in proper development of the vasculature as well as maintenance of the immune response at barrier tissues (Baricza et al., 2016; Bhaumik and Basu, 2017; Haas et al., 2016; Harstad et al., 2006; Kiss et al., 2011; Lahvis et al., 2005; Metidji et al., 2018; Quintana et al., 2008; Veldhoen et al., 2008).
Decades of AHR research have yielded many different mouse models in efforts to answer important questions in AHR biology. Among these models is the conditional Ahr mouse (Ahrfx) that allows cell-specific deletion when crossed to transgenic lines expressing Cre recombinase in a cell-specific manner (Walisser et al., 2005). Although much has been learned from this model, the Ahrfx allele was derived from 129-SvJ embryonic stem cells (ESCs) which harbor the less sensitive Ahrd allele as compared to the Ahrb1 allele found in C57BL/6J (B6) mice (Poland et al., 1994). The resultant lack of sensitivity to many xenobiotic ligands arising from the Ahrd allele makes it difficult to characterize the cell-autonomous role of the AHR for certain endpoints using the Ahrfx model. For example, to induce endpoints of interest, study of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) toxicity requires significantly higher doses in Ahrd mice as compared with Ahrb mice (eg, hepatocellular toxicity, hepatocellular carcinoma, and cleft pallet) (Kennedy et al., 2014). To improve this model, we selectively modified the ligand-binding domain of the AHR in the existing Ahrfx model to convert the low-affinity V375 residue to the high-affinity A375 (generating the AhrV375A allele) (Ema et al., 1994; Poland et al., 1994; Xing et al., 2012). To this end, we employed an “Edited Recombinant Approach” (ERA) to introduce a modification into the existing Ahrfx allele using CRISPR-Cas9 (Ran et al., 2013; Sander and Joung, 2014). The ERA allows enhancement of previously generated conditional alleles while limiting the technical challenges associated with generating multiple independent mutations in series.
In addition to the high-affinity conditional allele (AhrV375A), we describe 2 additional alleles, a novel null allele and an antimorphic allele, generated serendipitously through the ERA that may also prove useful in our attempts to understand various aspects of AHR biology (AhrNG367R and AhrTer383). To characterize each allele and understand the consequences of each mutation on AHR biology, we examined their influence on characteristic AHR-dependent phenotypes (Table 1). These endpoints included the transcriptional responsiveness of these Ahr alleles to the AHR ligand beta-naphthoflavone (βNF), the prevalence of the ductus venosus (DV), liver lobe weights, and the response to the chemical colitigen dextran sodium sulfate (DSS) (Avilla et al., 2020; Díaz-Díaz et al., 2016; Furumatsu et al., 2011; Harstad et al., 2006; Lahvis et al., 2000, 2005; Nebert and Gelboin, 1969; Poland and Glover, 1975). Finally, we examined the cell autonomy of AHR signaling to determine if these mutant proteins function in the same cell-specific manner as the wild-type receptor (Bunger et al., 2003, 2008; Schmidt et al., 1996; Walisser et al., 2004a,b, 2005; ). We propose that each of these alleles has utility and are important new members of an allelic series at the Ahr locus.
Table 1.
Nomenclature for Ahr Alleles
| Mouse Strain | Formal Name | Informal Name | Technology | Description |
|---|---|---|---|---|
| Wild-type | C57BL/6J | Ahrb1 | n/a | Wild-type allele found in B6 mice |
| Conditional | Ahrtm3.1Bra | Ahrfx | Homologous recombination | Low-affinity conditional allele; generated from 129 Sv ESCs (Ahrd at the Ahr locus) |
| Null | Ahr.tm1Bra | Ahr−/− | Homologous recombination | Exon 2 removed; generated from 129 Sv ESCs cells (Ahrd at the Ahr locus) |
| High affinity conditional | Ahrem1Bra | AhrV375A | CRISPR-Cas9 | High-affinity conditional allele; Ahrd at the Ahr locus except for a single-nucleotide polymorphism at residue 375 introduced via homology directed repair |
| AhrNG367R | Ahrem2Bra | AhrNG367R | CRISPR-Cas9 | Three-base pair deletion near ligand-binding domain generated through nonhomologous end joining; conditional; Ahrd at the Ahr locus |
| AhrTer383 | Ahrem3Bra | AhrTer383 | CRISPR-Cas9 | Single-nucleotide insertion generated through nonhomologous end joining;Ahrd at the Ahr locus except for a fame shift at residue 371 resulting in 11 amino acid changes and early termination at residue 383; conditional |
Mouse strain refers to the name commonly used in manuscripts and scientific presentations. The formal locus names listed are defined by the International Committee on Standardized Genetic Nomenclature for Mice and assigned by Jackson Laboratory. The informal locus names of alleles defines the name used in this article. The technology column describes the technique used to generate each model, followed by a brief description of important allelic traits.
MATERIALS AND METHODS
Guide RNA preparation
Two guide RNAs were designed for the generation of the AhrV375A conditional model to target the ligand-binding domain in exon-7 of the Ahr gene. Guide DNA oligonucleotides were synthesized into a forward primer (5′ GAAATTAATACGACTCACTATAGGN18-20GTTTTAGAGCTAGAAATAGC 3′), which includes the desired target site and overlap (designated in bold) with the reverse primer (5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAC-3′), which contains the sequence for the sgRNA stem loop structure necessary for incorporation into Cas9 (IDT, Coralville, Iowa) (Mali et al., 2013). The underlined nucleotides denote region where specific guide sequences are incorporated. Two guide sequences were used in the generation of these mice. Guide no.1 sequence: CCAagtcacgcttgatttacagaaatg; guide sequence no. 2: CCAgattacatcatcgtcactca where the bolded nucleotides define the PAM sequence. Guide DNA was generated through PCR using Phusion High-Fidelity DNA Polymerase (New England BioLabs, Ipswich, Massachusetts). Guide DNA was run on a 1% agarose gel (124 bp) and purified (Macherey-Nagel, Duren, Germany). Approximately 100 ng of purified PCR product was used as a template for sgRNA synthesis using the T7 Megashortscript kit according to manufacture protocol (Ambion, Austin, Texas). Following an in vitro transcription reaction, guide RNA was purified using MegaClear (Ambion, Austin, Texas) and resuspended in injection buffer (Transgenic Animal Facility, UW-Madison, Madison, Wisconsin) at 100 ng/μl.
Generation of the conditional AhrV375A mouse model using CRISPR gene editing
To generate mice conditional for a high-affinity ligand-binding domain, 3-week-old Ahrfx/fx dams (congenic on the B6 background for over 20 generations) were superovulated using Pregnant Mare Serum Gonadotropin (ProSpec Bio, East Brunswick, New Jersey) and human chorionic gonadotropin (Sigma-Aldrich, St. Louis, Missouri). Dams were then mated to Ahrfx/fx males to create homozygous Ahrfx/fx embryos. Day 0.5 embryos were collected by the UW Genome Editing and Animal Models core and microinjected with 50 ng/μl guide RNA, 50 ng/μl single-stranded oligodeoxynucleotide (ssODN), and 40 ng/μl Cas9 protein (PNA Bio, Newbury Park, California). Injected embryos were transplanted into pseudopregnant B6D2 F1 females. Pups were sequenced at weaning to screen for gene edits. The ssODN targeted the noncoding strand and encodeed the sequence is 5′-GTGTAAGAAGACTTATGAAAACACAAACGCAGGACATGTTCTTTTTCTGATGCTTACGTCAGTGGTCTCTGAGTGgCGATGATaTAATCTGGTCTTCCATTTCTGTAAATCAAGCGTGCATTGGACTGGACCCACCTCCAGCGACTGTGTTTTGCAAGA-3′ where the lowercase bolded letters denote induced point mutations. The first point mutation changed codon 375 from GAC (Valine) to GGC (Alanine), whereas the second point mutation is a silent mutation introduced to disrupt the targeted cut site to prevent subsequent cleavage and indel formation in the edited strand.
Screening for CRISPR events
To characterize gene editing events, genomic DNA from the tails of resultant mice were sequenced. Genomic DNA was PCR-amplified using OL8165 (5′-GTACTGTACTGTACTGATGG-3') and OL8166 (5′-CAGGTTCCATTGCCTTAGG-3′) which flank the region of Ahr targeted by the guide RNAs described above. The PCR products were run on a 1% agarose gel to ensure presence of product at approximately 300 bp. The PCR product was cleaned up using Exo-SAP IT according to manufacturer’s protocol (Applied Biosystems, Foster City, California). In order to determine the sequences of each allele, the PCR products were cloned into pGEM-T Easy vector (Promega, Madison, Wisconsin). Ten successfully transformed colonies (as indicated visually by the blue/white screen intrinsic to the pGEM-T Easy vector) were sequenced. Sanger Sequencing experiments were performed by the DNA Sequencing Core at UW-Madison. Results were analyzed using FinchTV (Digital World Biology, Seattle, Washington).
Animal care and treatment
Mice were housed in a selective pathogen-free facility in cages with corncob bedding. Mice had access to a chow diet and water ad libitum in accordance with the animal care protocol #M005987 approved by the University of Wisconsin Medical School’s Animal Care and Use Committee. The Ahr null mice (Ahr−/−) and Ahrfx mice used in these studies were previously backcrossed to B6 mice (Jackson Laboratories, Bar Harbor, Maine) for over 20 generations. The Ahrfx mouse was the parental strain for the CRISPR-mediated mutations. The 3 CRISPR strains, AhrV375A, AhrNG367R, and AhrTer383, were backcrossed onto B6 at least 5 times prior to use in experiments to eliminate potential off target events (Aryal et al., 2018). For induction experiments, mice were treated with 80 mg/kg body weight of βNF (Sigma Aldrich, St. Louis, Missouri) dissolved in corn oil by oral gavage. Control animals receive 1 ml/100 g of body weight of corn oil alone by oral gavage. Mice are euthanized 4 h following treatment via CO2 inhalation. A section of the left lobe of the liver was collected immediately following euthanasia and placed in RNAlater Stabilization Solution (Sigma Aldrich, St. Louis, Missouri). Mice used in induction studies were between 8 and 10 weeks old in age. Each group contains roughly equal number of males and females.
Western blot
Western blots were performed on cytosolic fractions prepared from freshly harvested adult mouse liver as described previously (LaPres et al., 2000). Protein samples were run on Mini-Protean TGX Stain-free gel (Bio-Rad Laboratories, Hercules, California) transferred to PVDF membrane, and incubated overnight with the BEAR-3 antibody (1μg/ml; raised against amino acids 1–402 of AHRb1) in 5% Blotto (nonfat dry milk in PBS), followed by 1 h incubation in goat anti-Rabbit secondary antibody (cat no. NB120-6024, Novus Biologics, Littleton, Colorado) in 5% Blotto. Blot was developed in alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate with nitroblue tetrazolium for 8 min. The BEAR3 antibody was generated as described previously (Pollenz et al., 1994).
Ductus venosus scoring and liver lobe weights
Prevalence of the DV was measured using gravity flow perfusion of the liver with trypan blue as described previously (Walisser et al., 2004a). Liver lobe weights were measured as previously described (Harstad et al., 2006). Briefly, livers were removed, and lobes were separated with scissors or razor blade. Each lobe (left, median, and rest) was weighed individually. The ratio of left: median lobe was calculated by dividing the weight of the left lobe by that of the median lobe. Mice examined for DV and LLW were between 8 and 12 weeks old in age. Each group contains roughly equal numbers of males and females. Statistical differences were calculated using 1-way ANOVA and deemed significant in cases where p < .05.
Dextran sodium sulfate treatment
Animals were given freshly prepared DSS (36 000–50 000 Da, colitis grade) diluted into acidified water from the animal facility at a concentration of 1% w/v (MP Biomedicals, Irvine, California) for 5 days as described previously (Díaz-Díaz et al., 2016). Mice were sacrificed on day 5 of DSS treatment. The colon was removed from each animal, cleaned with PBS, and measured from the distal end (following removal of the anus and rectum) to the proximal end. Whole colon was saved in RNA later as described below.
Cell-specific excision
The Ahrb1/V375A heterozygous mice or Ahrb1/Ter383 heterozygous mice were crossed to B6 mice harboring Cre recombinase transgenes under control of the Albumin (Alb; hepatocyte) or VE-Cadherin 5 (Cdh5; endothelial) promoter (CreAlb or CreCdh5, respectively) (Jackson Laboratory) (Alva et al., 2006; Postic et al., 1999; Walisser et al., 2005). For the genotyping of the Cre recombinase transgene and the low-affinity conditional Ahr allele, we used the PCR-based JAX protocols (Jackson Laboratory) (Walisser et al., 2005). For the detection of the CRISPR-mediated mutations, we employed the Sanger sequencing method described above. In all cases, Cre negative littermates were used as controls to Cre-positive animals. In some experiments, mice were genotyped using commercial services that employ quantitative real-time PCR (qPCR) (Transnetyx, Cordova, Tennessee).
RNA isolation and quantitative real-time PCR
Total RNA was isolated from liver and colon using Qiazol (Qiagen, Hilden, Germany) and the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The qPCR was performed as described previously (Baker et al., 2016). The reverse transcription reaction was completed using the High-Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol (Thermo Fisher, Waltham, Massachusetts) with 1 μg of RNA starting material as a template for this reaction. The qPCR reaction was completed using the Quantstudio 7 Flex Real-Time PCR System (Thermo Fisher, Waltham, Massachusetts). The probes used include Hprt (Mm03024075_m1), Cyp1a1 (Mm00487218_m1), Cyp1a2 (Mm00487224_m1), Cyp1b1 (Mm00487229_m1), Ahrr (Mm01352370_m1), and Il1β (Mm00434228_m1) (Thermo Fisher, Waltham, Massachusetts). Expression of target genes was calculated reletive to Hprt expression.
Protein modeling
Predicted AHR structures were visualized through PyMOL v 4.60. The PAS-B domain of AHRb1 shown was modeled and artificially colored in PyMOL using the protein model described previously (Xing et al., 2012).
Statistical analysis
Statistics were performed using GraphPad Prism version 8.3.1 for Windows (GraphPad Software, La Jolla, California). For gene induction data, groups were compared using a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). For DV and LLW analysis, groups were compared using Chi-squared (Fisher’s exact test) or 1-way ANOVA, respectively. The differences between groups were considered statistically significant when the p-value was < .05. In figures, groups sharing a superscript are not statistically different. Statistically significant differences are marked by different superscripts. All experiments contain both male and female mice in each group.
RESULTS
Generation of an Allelic Series at Ahr
To generate mice harboring a high-affinity conditional allele, Ahrfx dams were mated to males of the same genotype to generate homozygous embryos. Embryos were harvested from pregnant females and microinjected with a guide RNA targeting the ligand-binding domain in exon-7, Cas9 protein, and an ssODN containing the desired mutation. The injected embryos were then transferred to a pseudo-pregnant female and pups were screened for CRISPR events at weaning. Use of guide no. 1 yielded one CRISPR event in 1 mouse, resulting in a 3-bp deletion that spans codons 367 and 368 (AhrNG367R) (Figs. 1A and 2B). Use of guide no. 2 yielded 2 CRISPR events in 1 mouse on separate chromosomes (Figure 1B). The first event was a single adenine insertion at residue 371 resulting in a frame shift and early termination at residue 383 (AhrTer383) (Figure 2C). The second event incorporates the ssODN to encode the high-affinity alanine at residue 375 and a silent mutation 7 bp downstream (AhrV375A) (Figure 2A). To separate these two events into distinct lines, the female founder was crossed to a B6 male. To confirm the segregation of alleles, F1s were sequenced. Each F1 mouse harbored the Ahrb1 allele (the B6 paternal allele) and either the V375A allele or the Ter383 allele (the CRISPR’ed maternal allele). To eliminate potential off target CRISPR events, each line was backcrossed to B6 at least 5 times before being employed in an experiment (Aryal et al., 2018).
Figure 1.

Gene editing strategy for CRISPR-generated mutant mice. Alleles are shown in chronological order for which they were generated. The AhrNG367R allele was generated in an independent experiment using guide RNA no. 1, whereas AhrV375A and AhrTer383 alleles were generated on separate chromosomes in the same mouse during a second experiment using guide RNA no. 2. A, Proposed mechanism generating AhrNG367R allele. The Cas9 endonuclease incorporated guide RNA no. 1, directing the Cas9 protein to the Ahr locus in Ahrfx embryos. The Cas9 cleaved the DNA at the PAM sequence, inducing a double-stranded break. The cell recognized the double-stranded break and repaired the break using nonhomologous end joining (NHEJ). In this case, 3 bp were deleted resulting in the AhrNG367R mutation. B, Proposed mechanism generating the AhrV375A and AhrTer383 alleles. Guide RNA no. 2 directed Cas9 endonuclease in Ahrfx embryos. The Cas9 cleaved the DNA at the PAM site, inducing a double-stranded break. On 1 chromosome (left), the break was repaired through NHEJ, resulting in a single nucleotide insertion at the Ahr locus and the AhrTer383 mutation. On the second chromosome (right), the cell recognized homology with the ssODN. Homology directed repair (HDR) resulted in incorporation of the ssODN sequence at the Ahr locus, to yield the AhrV375A mutation. Both mutations occurred in one mouse, but each mutation occurred on a separate chromosome. The alleles were separated into distinct lines through breeding with wild-type B6 mice.
Figure 2.

Gene structure and protein map of induced mutations. Green boxes represent exons corresponding to amino acids in the bHLH domain, responsible for DNA binding and nuclear translocation. Blue boxes represent the PAS domain where dimarization with Ah Receptor Nuclear Translocator (ARNT) and ligand binding occur. Yellow boxes represents the transactivation domain. Red arrows represent IoxP sites flanking exon-2. Boxes above each allele show the DNA sequence; boxes below each allele describe the amino acid sequence. A, Protein map for the V375A allele. The gene encoded a point mutation at residue 375, changing GTC (Valine) to GCC (Alanine). The point mutation and silent mutation are shown bolded and underlined. B, Protein map for the NG367R allele. At the Ahr locus, 3 bp [ATG] were deleted, resulting in an amino acid deletion and substitution (NG367R), yielding a mutated PAS-B and ligand-binding domain. The deleted nucleotides are designated by brackets in the DNA sequence. The amino acid change is bolded and underlined. C, Protein map for the Ter383 allele based on genomic mutations. An adenine insertion at the codon for residue 371 in the gene resulted in a frame shift and early termination at residue 383. The dark blue box in the protien map represents amino acids following 371 that have been altered as a result of the frame shift. The adenine insertion and mutated amino acids are bolded and underlined.
Protein Expression in AhrV375A, AhrNG367R, and AhrTer383 Mice
The 3 mutant alleles generated protein of the appropriate predicted size as recognized by an antibody (BEAR-3) raised against amino acids 1–402 of AHRb1 (Figure 3). Because proteins of different sizes transfer at different rates from polyacrylamide gel upon electrotransfer to PVDF membranes, the proteins were not quantified. Rather, the Western blot was employed as a qualitative measure of protein size and its similarity to that predicted by the corresponding mutations described above.
Figure 3.

Expression of mutant proteins. A, The epitope for the BEAR-3 antibody recognizes amino acids 1–402 of AHRb1. The protein products from the four recombinant Ahr alleles are shown relative to AHRb1. The asterisk (*) denotes single nucleotide polymorphisms in AHRfx relative to AHRb1. The # denotes the V375A mutation in AHRV375A. The ^ marks the NG367R mutation in AHRNG367R resulting from a 3-bp deletion spanning codons 367 and 368, yielding a single amino acid deletion and a change in residue 367. Expected protein size is shown next to each predicted protein map. The orange box on the C-terminus of AHRTer383 represents the mutated amino acids resulting from the frame shift, beginning at 371 spanning until a premature stop codon at 383. B, Western blot shows 63 μg of protein products using BEAR-3 antibody. Cytosols were isolated fresh from 2 adult mice per genotype homozygous for each genotype. Image is representative of 2 experiments.
Ductus Venosus Phenotype of AhrV375A, AhrNG367R, and AhrTer383 Mice
Wild-type B6 mice (Ahrb1/b1) and Ahrfx/fx mice developed with normal DV closure by early adulthood (n = 0/8 and n = 0/12, respectively) (Table 2A). Similarly, mice heterozygous for the Ahrb1 allele and the high-affinity conditional allele (ie, Ahrb1/V375A) and mice homozygous for the high-affinity conditional allele (AhrV375A/V375A), also did not exhibit patent DV (n = 0/9 and n = 0/10, respectively) (Table 2A). In mice homozygous for the 3-bp deletion allele (AhrNG367R/NG367R), the DV was patent at a prevalence of 100% (n = 13/13) (Table 2B). Mice heterozygous for this allele ( Ahrb1/NG367R) exhibited normal DV closure (n = 0/11) (Table 2B). For comparison, mice homozygous for the Ahrb1 (Ahrb1/b1) allele did not display patent DV (n = 0/6) whereas mice homozygous for the null allele (Ahr−/−) consistently exhibited patent DV (n = 15/15) (Table 2B). Mice homozygous for the truncated allele (AhrTer383/Ter383) also exhibited 100% DV patency (n = 9/9) (Table 2C). Interestingly, mice heterozygous for the b1 and Ter383 allele (Ahrb1/Ter383) exhibited patent DV with 84% penetrance (n = 16/19) compared with 100% of Ahr null animals (n = 15/15) (Table 2C). Haploinsufficient mice (Ahrb1/-), as well as homozygous Ahrb1/b1 mice, did not exhibited patent DV (n = 0/13 and n = 0/10, respectively) (Table 2C).
Table 2.
Patent DV frequencies in the Three Alleles
| Genotype | % Patent DV | N |
|---|---|---|
| A. V375A | ||
| Ahrb1/b1 | 0 | 0/8 |
| Ahrfx/fx | 0 | 0/12 |
| Ahrb1/V375A | 0 | 0/9 |
| AhrV375A/V375A | 0 | 0/10 |
|
B. NG367R
| ||
| Ahrb1/b1 | 0 | 0/6 |
| Ahrb1/NG367R | 0 | 0/11 |
| AhrNG367R/NG367R | 100 | 13/13 |
| Ahr−/− | 100 | 15/15 |
| C. Ter383 | ||
| Ahrb1/b1 | 0 | 0/10 |
| Ahrb1/− | 0 | 0/13 |
| Ahr−/− | 100 | 15/15 |
| Ahrb1/Ter383 | 84 | 16/19 |
| AhrTer383/Ter383 | 100 | 9/9 |
A. Penetrance of patent DV in AhrV375A mutants and controls. B. Penetrance of DV in AhrNG367R mutants and controls. C. Penetrance of DV in truncated AhrTer383 mutants and controls. The DV penetrance is measured by portal vein gravity flow liver perfusion. Roughly equal numbers of male and female mice were represented within each genotype. Statistical analysis using a Chi-squared (Fisher’s exact test) showed no statistically significant differences between AhrV375A/V375A and controls (Ahrb1/b1, Ahrfx/fx, and Ahrb1/V375A); statistically significant differences between AhrNG367R/NG367R and Ahr−/− versus wild-type and heterozygous controls (Ahrb1/b1 and Ahrb1/NG367R) (p < .0001); and a statistical difference between Ahrb1/Ter38, AhrTer383/Ter383, and Ahr−/− versus wild-type and haploinsufficient controls (Ahrb1/b1 and Ahrb1/−) (p < .0001).
Liver Lobe Weights of AhrV375A, AhrNG367R, and AhrTer383 Mice
To further characterize the consequences of each mutation on an additional AHR-mediated developmental phenotype, liver lobes were weighed. Wild-type B6 Ahrb1/b1 mice, Ahrfx/fx mice, and AhrV375A/V375A mice exhibited normal ratios of left to median lobe weights (Table 3). In Ahr−/−, AhrNG367R/NG367R, and Ahrb1/Ter383 mice, the left to median liver lobe weights were significantly decreased in comparison to wild-type B6 Ahrb1/b1, Ahrfx/fx , and AhrV375A/V375A counterparts (p < .0001) (Table 3).
Table 3.
Liver Lobe Weights in the 3 Mutants
| Genotype | Left: Median Lobe Weight (mean ± SD) | n |
|---|---|---|
| Ahrb1/b1 | 1.14 ± 0.13 | 19 |
| Ahrfx/fx | 1.06 ± 0.13 | 6 |
| Ahr−/- | 0.75 ± 0.04 | 6 |
| AhrV375A/V375A | 1.14 ± 0.12 | 9 |
| AhrNG367R/NG367R | 0.92 ± 0.14 | 10 |
| Ahrb1/Ter383 | 0.84 ± 0.18 | 12 |
Left: median liver lobe weight ratios from homozygous AhrV375A/V375A and AhrNG367R/NG367R mice, as well as heterozygous Ahrb1/Ter383 mice, relative to Ahrb1/b1, Ahrfx/fx, and Ahr−/− controls. Ratios of all the mice within a given genotype are represented as the mean weights of left to median lobes ± standard deviation. Roughly equal number of male and female mice were represented for each genotype. Statistical analysis using a 1-way ANOVA showed significant differences between the livers of Ahrb1/b1, Ahrfx/fx, and AhrV375A/V375A mice compared with Ahr−/−, AhrNG367R/NG367R and Ahrb1/Ter383 mice (p < .0001).
Transcription of AHR-Driven Genes in the Liver of AhrV375A, AhrNG367R, and AhrTer383 mice
To assess the consequences of the 3 mutations in the Ahr on expression of characteristic AHR-driven genes, we measured hepatic mRNA transcript levels of Cyp1a1, Cyp1a2, Cyp1b1, and Ahrr from mice following a 4-h treatment with the AHR agonist βNF or the corn oil vehicle control. Mice homozygous for the high-affinity conditional allele (AhrV375A/V375A) were compared with wild-type (Ahrb1/b1) and low-affinity conditional (Ahrfx/fx) counterparts (Figure 4). Mice with the high-affinity conditional allele (AhrV375A/V375A), which harbors A375, displayed a greater response (2.6–9.6-fold higher) for all transcripts compared with Ahrfx/fx, which harbors V375. For example, induction of Cyp1a1 was 9.6 times higher in AhrV375A/V375A than in Ahrfx/fx mice, with a p-value = .0002. Induction of Cyp1a2 and Cyp1b1 was 2.6 and 3.6 times higher, respectively, in AhrV375A/V375A than in Ahrfx/fx mice (p-value < .0001). Induction of Ahrr was 8 times higher in AhrV375A/V375A than in Ahrfx/fx mice (p < .0001). Mice homozygous for the 3-bp deletion (AhrNG367R/NG367R) were less responsive compared to their high-affinity conditional (AhrV375A/V375A) counterparts for induction of AHR target genes (Figure 5). The AhrNG376R/NG367R mice were significantly (p < .001) less inducible (198, 6, 5.5, and 7.2 times, respectively) for Cyp1a1, Cyp1a2, Cyp1b1, and Ahrr than AhrV375A mice. Mice heterozygous for the truncated allele (Ahrb1/Ter383) were less responsive to AHR-driven gene induction compared with wild-type (Ahrb1/b1) but comparable to haploinsufficient (Ahrb1/-) controls (Figure 6). The B6 wild-type controls (Ahrb1/b1) were slightly more inducible for Cyp1a1 (1.7 times), Cyp1a2 (1.2 times), and Cyp1b1 (1.6 times) than Ahrb1/Ter383 counterparts, but these differences did not attain significance. The B6 wild-type controls are 7.3 times more inducible for Ahrr expression than the Ahrb1/Ter383 mice. The haploinsufficient controls (Ahrb1/-) and the Ahrb1/Ter383 mice displayed similar induction of target genes.
Figure 4.

Induction of AHR-driven genes in homozygous high-affinity conditional mutant (AhrV375A) compared with mice homozygous for the low-affinity conditional allele (Ahrfx) and homozygous wild-type animals (Ahrb1). Mice are treated with βNF or corn oil vehicle control (CO) for 4 h. Liver mRNA expression of AHR-driven genes relative to Hprt. Groups sharing a superscript are not different at the level of statistical significance (p > .05) determined by a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). Each point represents the mean of 3 technical replicates of one biological replicate. Males are represented by a circle whereas females are represented by a square. Error bars represent standard error of the mean.
Figure 5.

Induction of AHR-driven genes in homozygous ligand-binding mutant (AhrNG367R) compared with mice homozygous for the high-affinity conditional allele (AhrV375A). Mice are treated with βNF or corn oil vehicle control (CO) for 4 h. Liver mRNA expression of AHR-driven genes relative to Hprt. Groups sharing a superscript are not different at the level of statistical significance (p > .05) determined by a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). Each point represents the mean of 3 technical replicates of 1 biological replicate. Males are represented by a circle while females are represented by a square. Error bars represent standard error of the mean.
Figure 6.

Induction of AHR-driven genes in heterozygous truncated mutants (Ahrb1/Ter383) compared with homozygous wild-type (Ahrb1/b1) and haploinsufficient (Ahrb1/-) controls. Mice are treated with βNF or corn oil vehicle control (CO) for 4 h. Liver mRNA expression of AHR-driven genes relative to Hprt. Groups sharing a superscript are not different at the level of statistical significance (p > .05) determined by a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). Each point represents the mean of 3 technical replicates of 1 biological replicate. Males are represented by a circle whereas females are represented by a square. Error bars represent standard error of the mean.
Hepatocyte-Specific Excision of AhrV375A Reduced Expression of AHR-Driven Genes in Liver but Did Not Affect DV Closure
To ensure conditionality of the AhrV375A allele, we crossed mice harboring this allele with mice harboring CreAlb. Mice from the F2 generation homozygous for AhrV375A with and without CreAlb were treated with βNF. After 4 h, liver mRNA transcript levels of AHR-driven genes were measured. In AhrV375ACreAlb mice treated with βNF, gene expression of Cyp1a1 and Cyp1a2 were markedly reduced (p < .004) compared to AhrV375A counterparts (Figure 7). Expression of Ahrr and Cyp1b1 was not significantly reduced in AhrV375ACreAlb mice compared to AhrV375A mice. Excision of AhrV375A in hepatocytes (AhrV375ACreAlb) did not result in a patent DV (n = 0/10) (Table 4).
Figure 7.

Induction of AHR-driven genes in mice ubiquitously expressing the high-affinity conditional allele (AhrV375A) compared to mice with the AHR excised in hepatocytes (AhrV375ACreAlb). Mice are treated with βNF or corn oil vehicle control (CO) for 4 h. Liver mRNA expression of AHR-driven genes relative to Hprt. Groups sharing a superscript are not different at the level of statistical significance (p > .05) determined by a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). Each point represents the mean of 3 technical replicates of 1 biological replicate. Males are represented by a circle whereas females are represented by a square. Error bars represent standard error of the mean.
Table 4.
Penetrance of DV in V375A Mutants Crossed to Mice Harboring Hepatocyte (Alb) and Endothelial (Cdh5) Driven Cre Recombinase
| Ahr Genotype | Cre | % Patent DV | n |
|---|---|---|---|
| AhrV375A/V375A | (None) | 0 | 0/15 |
| AhrV375A/V375A | Alb | 0 | 0/10 |
| AhrV375A/V375A | Cdh5 | 100 | 10/10 |
Excision of the AhrV375A allele in hepatocytes (AhrV375ACreAlb) does not result in patent DV. Excision of the AhrV375A allele in endothelial cells (AhrV375ACreCdh5) results in patent DV. Roughly equal number of male and female mice are represented for each genotype. Statistical analysis using a Chi-squared (Fisher’s exact test) shows significant differences between AhrV375A and AhrV375ACreAlb versus AhrV375ACreCdh5 (p < .0001). There is no significant difference between AhrV375A/V375A and AhrV375A/V375ACreAlb.
Endothelial-Specific Deletion of AhrV375A Is a Phenocopy of the Ahr Null Mouse
To determine if the AhrV375A mouse displayed the same cell autonomy as the Ahrfx mouse characterized previously, we measured DV prevelence in AhrV375A mice crossed to mice harboring the endothelial specific Cdh5-driven Cre recombinase transgene (Alva et al., 2006; Walisser et al., 2005). Excision of AhrV375A in the endothelial compartment (AhrV375ACreCdh5) resulted in a patent DV (n = 10/10) (Table 4).
Endothelial Cell Autonomy of AhrTer383 Is a Phenocopy of the Ahr Null Mouse
Based upon the presence of patent DV in heterozygous AhrTer383 mice (Ahrb1/Ter383), we postulated this allele was an antimorph in endothelial cells. To test this idea, we deleted the AhrTer383 allele in endothelial cells of heterozygous mice through a cross to a mouse carrying CreCdh5. Prevalence of the patent DV was measured in mice globally heterozygous for the AhrTer383 construct but excised in Cdh5+ endothelial cells (Ahrb1/Ter383CreCdh5) (Table 5). In mice with the AhrTer383 construct removed in endothelial cells (Ahrb1/Ter383CreCdh5), we observed patent DV in 18% of mice (n = 2/11) compared with 86% patent DV in littermates that expressed AhrTer383 ubiquitously (Ahrb1/Ter383, but with no CreCdh5) (n = 19/22) (Table 5).
Table 5.
Penetrance of DV in Ter383 Mutants Crossed to Endothelial Specific (CreCdh5) Cre Recombinase
| Ahr Genotype | CreCdh5 Genotype | % Patent DV | n |
|---|---|---|---|
| Ahrb1/b1 | − | 0 | 0/10 |
| Ahrb1/b1 | + | 0 | 0/10 |
| Ahrb1/Ter383 | − | 86 | 19/22 |
| Ahrb1/Ter383 | + | 18 | 2/11 |
Excision of the AhrTer383 allele in endothelial cells rescues DV closure in heterozygous Ahrb1/Ter383 CreCdh5 animals. Roughly equal number of male and female mice are represented for each genotype. Statistical analysis using a Chi-squared (Fisher’s exact test) shows significant differences between Ahrb1/Ter383 versus Ahrb1/Ter383 CreCdh5 (p = .0002). There is no significant difference between Ahrb1/1b1, Ahrb1/b1CreCdh5, and Ahrb1/Ter383 CreCdh5.
Heterozygous Ahrb1/Ter383 Response to Chemically Induced Colitis
To determine if the AhrTer383 allele exhibited antimorphic properties for the chemically induced colitis endpoint as determined by colon length and induction of interleukin-1β (IL1β), mice were treated with DSS for 5 days. For both endpoints, the response of DSS treated Ahrb1/Ter383 mice was similar to that observed in Ahrb1/b1 wildtype mice, despite observing an enhanced response in Ahr−/− mice (Figure 8).
Figure 8.

Colitis phenotype in Ahrb1/Ter383 mice. A, Colon length of mice treated with 1% DSS in drinking water for 5 days. Differences between groups sharing a superscript are not statistically significant (p > .05) determined by a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). B, Relative expression of Il1β mRNA transcripts in whole colon of mice treated with 1% DSS in drinking water for 5 days. Differences between groups sharing a superscript are not statistically significant (p > .05) determined by a 2-way ANOVA with multiple comparisons of the means (Tukey’s multiple comparison test). Roughly equal number of males and females are represented in each genotype. Each point represents the mean of 3 technical replicates of 1 biological replicate for gene induction. Error bars represent standard error of the mean.
Structural Predictions
The PAS-B domain of the AHRb1 was modeled from that previously described (Figure 9) (Xing et al., 2012). Residues relevant for the 3 mutants were highlighted using Pymol (Xing et al., 2012).
Figure 9.
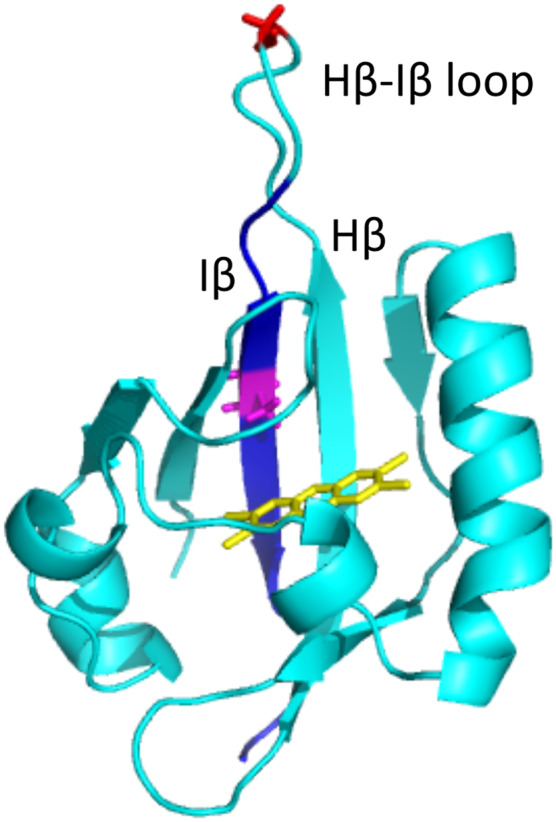
Structural rendition of PAS-B from AHRb1 showing location of amino acids relevant to described mutants. The structure of the PAS-B domain modeled from AHRb1 sequence is shown in light blue. A TCDD molecule is shown in the ligand-binding pocket in yellow. Residue 375 (Alanine) is highlighted in purple. The Glycine residue at 368 mutated in the AHRNG367R model is highlighted in red andlocated in the hairpin loop. The residues changed in AHRTer383 as a result of the frame shift mutation are highlighted in dark blue.
DISCUSSION
The ERA described in this report employs the CRISPR-Cas9 technique to generate targeted mutations in a mouse line that has previously undergone a recombination event via gene targeting. This approach was initiated to improve the previously generated Ahrfx allele by increasing the ligand-binding affinity of the receptor product while maintaining the conditionality of the allele. To this end, we used CRISPR-Cas9 gene editing technology to generate the AhrV375A mouse by inducing the V375A codon change (Figure 1) (Adli, 2018; Ceccaldi et al., 2016; Ran et al., 2013; Sander and Joung, 2014; Singh et al., 2015). In this process, we observed neighboring recombination events in separate lines that were reflective of NHEJ. The apparent NHEJ mechanism, which can occur at a higher frequency than HDR, generated 2 additional mutations in codons near 375, yielding the AhrNG367R and AhrTer383 alleles (Figure 2). In this report, we provide an initial characterization of the 3 Ahr alleles in order to make them broadly available for developmental and toxicological studies.
The role of the AHR is highly context dependent, such that its biological outputs differ depending on the type of ligand, concentration of ligand, developmental stage, and cell type in which signaling occurs (Avilla et al., 2020; Cella and Colonna, 2015; Esser, 2016; Esser and Rannug, 2015; Harrill et al., 2013; Larigot et al., 2018; Nebert, 2017; Stevens et al., 2009). Previous studies have begun to unravel this complicated and differential biology using the conditional null model, commonly known as Ahrfx (Villa et al., 2017; Walisser et al., 2005). The importance of cell type can be clearly shown as TCDD-induced hepatocellular toxicity is dependent upon AHR expression in hepatocytes, whereas AHR expression in the endothelial compartment facilitates proper vascular development and DV closure (Walisser et al., 2005). More recently, the Ahrfx model has been used to show that the cell-specific expression of AHR plays a critical role in maintaining homeostasis at barrier tissues like the lung, skin, and gut (Esser and Rannug, 2015; Haas et al., 2016; Metidji et al., 2018). Use of the Ahrfx models has been essential in developing the hypothesis that AHR is an environmental and physiological sensor that monitors local chemistries to maintain homeostasis using a myriad of different mechanisms depending on cell type (Avilla et al., 2020; Esser and Rannug, 2015; Quintana et al., 2008; Stockinger et al., 2014; Veldhoen et al., 2008).
Although the Ahrfx model has been an essential tool in the field of AHR biology, it is a conditional Ahrd allele and thus has markedly reduced sensitivity to many classically defined ligands, as compared with Ahrb strains (B6, C3H, BALB, etc.). The shortcoming of the Ahrfx model is a consequence of the tools available at the time of its generation. That is, the allele was created by homologous recombination in 129SvJ-derived GS1 ESCs which harbor the low-affinity Ahrd allele (Poland et al., 1994; Walisser et al., 2005). Given that the low sensitivity of the Ahrfx models can reduce statistical power and limit experimental protocols (Kennedy et al., 2014), we employed the ERA to target residue V375 in the Ahrfx allele to create a model more sensitive to certain xenobiotic ligands (Poland et al., 1994; Walisser et al., 2005). Residue 375 is a polymorphism located in the ligand-binding pocket of AHR that is considered largely responsible for the differential ligand-binding affinities between Ahrd and Ahrb alleles (Ema et al., 1994; Poland et al., 1994; Xing et al., 2012). In keeping with this idea, the conversion of Ahrfx into AhrV375A improved the responsiveness to the xenobiotic AHR ligand βNF to levels comparable to those observed in B6 mice (Figure 4). Although the conditional AhrV375A mice harbor other polymorphisms that exist between Ahrd and Ahrb alleles, including an alternate stop codon that yields an additional 43 amino acids on the C-terminus (Figure 3) (Poland et al., 1994). Our results suggest that these polymorphisms do not impact the AHR-driven gene induction following treatment with βNF (Figure 4).
The increased sensitivity and responsiveness of the AhrV375A mouse allows the use of nontoxic xenobiotic ligands like βNF, as well as short-term, acute exposure protocols. In comparison, prior experiments employing the Ahrfx allele required chronic exposures to highly potent and toxic ligands, such as TCDD, to induce a significant upregulation of target gene expression. An example of the type of information gained from use of the AhrV375A model can be seen with the deletion of the AHR in hepatocytes (AhrV375ACreAlb), where induction of Cyp1a1 and Cyp1a2 are almost completely abrogated, whereas induction of the Cyp1b1 and Ahrr appear relatively unaffected following exposure to βNF (Figure 7). This cell-specific expression data is consistent with the idea that it is the AHR signaling in non-hepatocyte populations, eg, Kuepfer cells, biliary epithelial cells, and/or endothelial cells, that are responsible for the upregulation of Cyp1b1 and Ahrr in liver. This observation was previously unobservable due to the insensitive nature of the Ahrfx allele. Given the improved sensitivity of this model, it now seems possible to employ AhrV375A in future studies to examine the response of these gene targets in specific subpopulations. Importantly, use of AhrV375A supports the hypothesis that activation of the same cellular sensor, AHR, by the same ligand, βNF, is regulating different gene sets in different cell types within the same organ.
It is also important to note the unique genetics of AhrV375A. In addition to the conditional nature of the allele, it is also co-isogenic with the existing Ahrfx model from which it was derived. All residues are identical between the two encoded proteins with the singular exception of residue 375. This co-isogenicity offers the first in vivo opportunity to examine the contribution of residue 375 in isolation on AHR-mediated endpoints in a conditional mouse model. Although congenic lines like C57BL/6J-Ahrd and DBA/2-Ahrb1 have proven valuable, those strains differ at residue 375 in addition to residues 324, 471, 532, and 588, and employ different stop codons (Poland et al., 1994). Importantly, the AhrV375A allele reported here is not identical to any known allele found in nature, but most closely resembles the Ahrb2 allele found in C3H and BALB/cBy mouse strains. That is, the AhrV375A allele harbors an A375, conferring high affinity for ligand binding, and has the C-terminal extension of 43 amino acids common to Ahrb2 strains (in addition to the 4 other polymorphisms listed above).
It is known that CRISPR-Cas9 can yield unexpected mutations at or near the target site, often through the use of NHEJ rather than HDR to repair the induced double-stranded breaks (Sander and Joung, 2014). In this case, we identified two such mutations and examined their consequences on AHR biology. The first of these mutations was the AhrNG367R mutant. Early characterization of this mutation presented as a null for patent DV, liver lobe weights, and induction endpoints (Schmidt et al., 1996). Specifically, 100% of homozygous AhrNG367R and Ahr-/- mice exhibited a patent DV and were uninducible for target genes following ligand treatment (Tables 2B and 3; Figure 5). This phenotypic evidence supports the hypothesis that AhrNG367R is a novel null allele. Protein analysis revealed that this protein is expressed, suggesting the null phenotype is not the consequence of a lack of protein, but rather a nonfunctional protein (Figure 3). Modeling of the AHRNG367R protein suggests the mutation may significantly impact the tertiary structure of the PAS-B domain and could highlight the structural importance of the β hairpin in AHR structure (Figure 9). The mutation is located at the tip of the β hairpin, where G368 is deleted and N367 is changed to R367. As a result, the amino acid sequence around the tip of the hairpin is converted from RNGR to RRR. Glycine is energetically critical for the formation of the β-hairpin structure. In addition to the loss of the critical Glycine at position 368, this mutation creates a positive charged tip through the addition of Arginine, potentially disfavoring the formation of the β hairpin structure and resulting in significant changes in Hβ/Iβ, the 2 β sheets at the center of the “palm” of the ligand-binding pocket (Xing et al., 2012). The structural alteration caused by this mutant may significantly affect the structure and stability of the ligand-binding pocket and therefore impair ligand binding. This hypothesis is supported by the observation that homozygous mice harboring this mutation (AhrNG376R/NG367R) failed to respond to AHR ligand. Although this mutation warrants further characterization, this allele may have additional utility as a novel null allele that is co-isogenic with Ahrfx and AhrV375A.
The other novel mutation that appears to have arisen through NHEJ is AhrTer383. This allele encodes an adenine insertion at codon 371 resulting in a frameshift and premature stop codon at 383. Based upon previous domain mapping studies and structural models, we predicted this protein would behave as an antimorph and designed experiments to test this idea. In support of this prediction, we observed mice heterozygous for the wild-type Ahrb1 and AhrTer383 alleles (Ahrb1/Ter383) display patent DV at a penetrance of 84%. This is compared with 0% patent DV in mice haploinsufficient for the Ahrb1 allele (Ahrb1/-) (Table 2). This partial penetrance of DV has been observed in other AHR-mutant mice, such as AHR and ARNT hypomorphs, as well as in mice haploinsufficient or hypomorphic for the AHR chaperone, Ara9 (Lin et al., 2008; Walisser et al. 2004a,b). Interestingly, while the AhrTer383 construct acts as a developmental antimorph (Tables 2C and 3), it does not appear to affect induction of target genes like Cyp1a1, Cyp1a2, Cyp1b1, and Ahrr (Figure 6) or sensitivity to DSS-induced colitis (Figure 8). The sequence of this mutant predicts the AHRTer383 mutant has amino acid replacement beginning at residue 371 following the point mutation, altering the identity of the following 11 amino acids of the PAS-B domain (ie, from YIIVTQRPLTD in Ahrfx mice to IHHRHSETTDG-stop in AhrTer383 mice). This mutant protein retains the bHLH domain and the PAS-A domain, which together have been shown to confer full activity in ARNT dimerization (Seok et al., 2017). Thus, this mutant is postulated to retain the ability to dimerize with ARNT and interact with the target DNA sequences but may not be able to activate the transcription of downstream genes or respond to ligand given the missing C-terminal transactivation domain and disrupted ligand binding pocket (Jain et al., 1994). Importantly, protein analysis reveals that AHRTer383 produces a protein of the predicted molecular weight around 37 kDa (Figure 3).
Although there is still considerable work to be done to determine the mechanism by which AhrTer383 invokes its antimorphic activity during hepatovascular development, these observations suggest the developmental pathway has different requirements for AHR and ARNT concentrations than the adaptive pathway or other physiological pathways such as that employed in intestinal barrier integrity. Although such ideas can be investigated in future studies, it is important to note that the AhrTer383 allele may have significant utility in AHR mechanism of action studies in vivo. It is not only antimorphic for the DV endpoint, but this activity is conditional, as the allele can be excised by cell-specific Cre recombinase (Table 5). When crossed to a mouse harboring the endothelial specific CreCdh5 transgene, normal DV closure proceeds, demonstrating that removal of AhrTer383 in endothelial cells rescues the DV phenotype. This is consistent with the observation that mice conditional for Ahrfx crossed to mice harboring the CreCdh5 transgene (Ahrfx/fxCreCdh5) exhibit a patent DV (Walisser et al., 2005).
The AhrV375A mouse was generated intentionally to solve the problem of xenobiotic sensitivity in the existing conditional model and serve as a useful tool in future cell autonomy, toxicology, drug metabolism, and immunology studies. Though generated serendipitously, the two other novel mutations described may also be useful in AHR biology studies. The AhrNG367R mouse is a potentially novel null allele that is co-isogenic with the other models in this series and provides a useful model in AHR protein structure studies. The AhrTer383 mouse model could prove useful in studies of patent DV due to the fact that the model may be used as a heterozygote, which helps to cut time and breeding requirements for experiments. Additionally, this allele may be optimized to generate an even more effective AHR antimorph and serve as a model for creating antimorphs in other PAS proteins. These studies remain on going, but our early characterization has shown that these animals may be useful to other laboratories working to better understand AHR biology.
ACKNOWLEDGMENTS
Thank you to Katie Stanley of the Arts and Media Solutions division at the University of Wisconsin-Madison School of Medicine and Public Health for contributing to figure design and digitalization.
FUNDING
This work was supported by the National Institutes of Health (R35-ES028377, T32-ES007015, T32-GM008692, T32-CA009135, and P30-CA014520). The UW SciMed GRS Program and The Morgridge Foundation.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Adli M. (2018). The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alva J. A., Zovein A. C., Monvoisin A., Murphy T., Salazar A., Harvey N. L., Carmeliet P., Iruela-Arispe M. L. (2006). VE-cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767. [DOI] [PubMed] [Google Scholar]
- Aryal N. K., Wasylishen A. R., Lozano G. (2018). Crispr/cas9 can mediate high-efficiency off-target mutations in mice in vivo. Cell Death Dis. 9, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilla M. N., Malecki K. M. C., Hahn M. E., Wilson R. H., Bradfield C. A. (2020). The Ah receptor: Adaptive metabolism, ligand diversity, and the xenokine model. Chem. Res. Toxicol. 33, 860–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker B. B., Yee J. S., Meyer D. N., Yang D., Baker T. R. (2016). Histological and transcriptomic changes in male zebrafish testes due to early life exposure to low level 2,3,7,8-tetrachlorodibenzo-p-dioxin. Zebrafish 13, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baricza E., Tamasi V., Marton N., Buzas E. I., Nagy G. (2016). The emerging role of aryl hydrocarbon receptor in the activation and differentiation of th17 cells. Cell. Mol. Life Sci. 73, 95–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik S., Basu R. (2017). Cellular and molecular dynamics of th17 differentiation and its developmental plasticity in the intestinal immune response. Front. Immunol. 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger M. K., Glover E., Moran S. M., Walisser J. A., Lahvis G. P., Hsu E. L., Bradfield C. A. (2008). Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 106, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger M. K., Moran S. M., Glover E., Thomae T. L., Lahvis G. P., Lin B. C., Bradfield C. A. (2003). Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J. Biol. Chem. 278, 17767–17774. [DOI] [PubMed] [Google Scholar]
- Ceccaldi R., Rondinelli B., D’Andrea A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M., Colonna M. (2015). Aryl hydrocarbon receptor: Linking environment to immunity. Semin. Immunol. 27, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragin N., Shi Z., Madan R., Karp C. L., Sartor M. A., Chen C., Gonzalez F. J., Nebert D. W. (2008). Phenotype of the cyp1a1/1a2/1b1-/- triple-knockout mouse. Mol. Pharmacol. 73, 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Díaz C. J., Ronnekleiv-Kelly S. M., Nukaya M., Geiger P. G., Balbo S., Dator R., Megna B. W., Carney P. R., Bradfield C. A., Kennedy G. D. (2016). The aryl hydrocarbon receptor is a repressor of inflammation-associated colorectal tumorigenesis in mouse. Ann. Surg. 264, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M., Ohe N., Suzuki M., Mimura J., Sogawa K., Ikawa S., Fujii-Kuriyama Y. (1994). Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J. Biol. Chem. 269, 27337–27343. [PubMed] [Google Scholar]
- Esser C. (2016). The aryl hydrocarbon receptor in immunity: Tools and potential. Methods Mol. Biol. 1371, 239–257. [DOI] [PubMed] [Google Scholar]
- Esser C., Rannug A. (2015). The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 67, 259–279. [DOI] [PubMed] [Google Scholar]
- Furumatsu K., Nishiumi S., Kawano Y., Ooi M., Yoshie T., Shiomi Y., Kutsumi H., Ashida H., Fujii-Kuriyama Y., Azuma T., et al. (2011). A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci. 56, 2532–2544. [DOI] [PubMed] [Google Scholar]
- Haas K., Weighardt H., Deenen R., Köhrer K., Clausen B., Zahner S., Boukamp P., Bloch W., Krutmann J., Esser C. (2016). Aryl hydrocarbon receptor in keratinocytes is essential for murine skin barrier integrity. J. Invest. Dermatol. 136, 2260–2269. [DOI] [PubMed] [Google Scholar]
- Hahn M. E. (2002). Aryl hydrocarbon receptors: Diversity and evolution. Chem. Biol. Interact. 141, 131–160. [DOI] [PubMed] [Google Scholar]
- Harrill J. A., Hukkanen R. R., Lawson M., Martin G., Gilger B., Soldatow V., Lecluyse E. L., Budinsky R. A., Rowlands J. C., Thomas R. S. (2013). Knockout of the aryl hydrocarbon receptor results in distinct hepatic and renal phenotypes in rats and mice. Toxicol. Appl. Pharmacol. 272, 503–518. [DOI] [PubMed] [Google Scholar]
- Harstad E. B., Guite C. A., Thomae T. L., Bradfield C. A. (2006). Liver deformation in Ahr-null mice: Evidence for aberrant hepatic perfusion in early development. Mol. Pharmacol. 69, 1534–1541. [DOI] [PubMed] [Google Scholar]
- Jain S., Dolwick K. M., Schmidt J. V., Bradfield C. A. (1994). Potent transactivation domains of the Ah receptor and the Ah receptor nuclear translocator map to their carboxyl termini. J. Biol. Chem. 269, 31518–31524. [PubMed] [Google Scholar]
- Kennedy G. D., Nukaya M., Moran S. M., Glover E., Weinberg S., Balbo S., Hecht S. S., Pitot H. C., Drinkwater N. R., Bradfield C. A. (2014). Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and tnf/il-1 receptors. Toxicol. Sci. 140, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss E. A., Vonarbourg C., Kopfmann S., Hobeika E., Finke D., Esser C., Diefenbach A. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565. [DOI] [PubMed] [Google Scholar]
- Lahvis G. P., Lindell S. L., Thomas R. S., McCuskey R. S., Murphy C., Glover E., Bentz M., Southard J., Bradfield C. A. (2000). Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 97, 10442–10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahvis G. P., Pyzalski R. W., Glover E., Pitot H. C., McElwee M. K., Bradfield C. A. (2005). The aryl hydrocarbon receptor is required for developmental closure of the Ductus venosus in the neonatal mouse. Mol. Pharmacol. 67, 714–720. [DOI] [PubMed] [Google Scholar]
- LaPres J. J., Glover E., Dunham E. E., Bunger M. K., Bradfield C. A. (2000). Ara9 modifies agonist signaling through an increase in cytosolic aryl hydrocarbon receptor. J. Biol. Chem. 275, 6153–6159. [DOI] [PubMed] [Google Scholar]
- Larigot L., Juricek L., Dairou J., Coumoul X. (2018). Ahr signaling pathways and regulatory functions. Biochim. Open 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B. C., Nguyen L. P., Walisser J. A., Bradfield C. A. (2008). A hypomorphic allele of aryl hydrocarbon receptor-associated protein-9 produces a phenocopy of the Ahr-null mouse. Mol. Pharmacol. 74, 1367–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh B. E., Hogenesch J. B., Bradfield C. A. (2010). Mammalian PER-ARNT-SIM proteins in environmental adaptation. Annu. Rev. Physiol. 72, 625–645. [DOI] [PubMed] [Google Scholar]
- Metidji A., Omenetti S., Crotta S., Li Y., Nye E., Ross E., Li V., Maradana M. R., Schiering C., Stockinger B. (2018). The environmental sensor Ahr protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert D. W. (2017). Aryl hydrocarbon receptor (Ahr): “Pioneer member” of the basic-helix/loop/helix PER-ARNT-SIM (BHLH/PAS) family of “sensors” of foreign and endogenous signals. Progr. Lipid Res. 67, 38–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert D. W., Gelboin H. V. (1969). The in vivo and in vitro induction of aryl hydrocarbon hydroxylase in mammalian cells of different species, tissues, strains, and developmental and hormonal states. Arch. Biochem. Biophys. 134, 76–89. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Robinson J. R., Niwa A., Kumari K., Poland A. P. (1975). Genetic expression of aryl hydrocarbon hydroxylase activity in the mouse. J. Cell. Physiol. 85, 393–414. [DOI] [PubMed] [Google Scholar]
- Poland A., Glover E. (1975). Genetic expression of aryl hydrocarbon hydroxylase by 2,3,7,8-tetrachlorodibenzo-p-dioxin: Evidence for a receptor mutation in genetically non-responsive mice. Mol. Pharmacol. 11, 389–398. [Google Scholar]
- Poland A., Glover E. (1980). 2,3,7,8,-tetrachlorodibenzo-p-dioxin: Segregation of toxocity with the ah locus. Mol. Pharmacol. 17, 86–94. [PubMed] [Google Scholar]
- Poland A., Glover E. (1990). Characterization and strain distribution pattern of the murine ah receptor specified by the Ahd and Ahb-3 alleles. Mol. Pharmacol. 38, 306–312. [PubMed] [Google Scholar]
- Poland A., Glover E., Bradfield C. A. (1991). Characterization of polyclonal antibodies to the Ah receptor prepared by immunization with a synthetic peptide hapten. Mol. Pharmacol. 39, 20–26. [PubMed] [Google Scholar]
- Poland A., Glover E., Kende A. S. (1976). Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 251, 4936–4946. [PubMed] [Google Scholar]
- Poland A., Palen D., Glover E. (1994). Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 46, 915–921. [PubMed] [Google Scholar]
- Pollenz R. S., Sattler C. A., Poland A. (1994). The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in hepa 1c1c7 cells by immunofluorescence microscopy. Mol. Pharmacol. 45, 428–438. [PubMed] [Google Scholar]
- Postic C., Shiota M., Niswender K. D., Jetton T. L., Chen Y., Moates J. M., Shelton K. D., Lindner J., Cherrington A. D., Magnuson M. A. (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315. [DOI] [PubMed] [Google Scholar]
- Quintana F. J., Basso A. S., Iglesias A. H., Korn T., Farez M. F., Bettelli E., Caccamo M., Oukka M., Weiner H. L. (2008). Control of t(reg) and t(h)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71. [DOI] [PubMed] [Google Scholar]
- Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F. (2013). Genome engineering using the crispr-cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander J. D., Joung J. K. (2014). Crispr-cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 32, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J. V., Su G. H., Reddy J. K., Simon M. C., Bradfield C. A. (1996). Characterization of a murine Ahr null allele: Involvement of the ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. U.S.A. 93, 6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok S. H., Lee W., Jiang L., Molugu K., Zheng A., Li Y., Park S., Bradfield C. A., Xing Y. (2017). Structural hierarchy controlling dimerization and target DNA recognition in the Ahr transcriptional complex. Proc. Natl. Acad. Sci. U.S.A. 114, 5431–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., Schimenti J. C., Bolcun-Filas E. (2015). A mouse geneticist's practical guide to CRISPR applications. Genetics 199, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens E. A., Mezrich J. D., Bradfield C. A. (2009). The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 127, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B., Meglio P. D., Gialitakis M., Duarte J. H. (2014). The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 32, 403–432. [DOI] [PubMed] [Google Scholar]
- Veldhoen M., Hirota K., Westendorf A. M., Buer J., Dumoutier L., Renauld J.-C., Stockinger B. (2008). The aryl hydrocarbon receptor links th17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109. [DOI] [PubMed] [Google Scholar]
- Villa M., Gialitakis M., Tolaini M., Ahlfors H., Henderson C. J., Wolf C. R., Brink R., Stockinger B. (2017). Aryl hydrocarbon receptor is required for optimal b‐cell proliferation. EMBO J. 36, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walisser J. A., Bunger M. K., Glover E., Bradfield C. A. (2004a). Gestational exposure of Ahr and ARNT hypomorphs to dioxin rescues vascular development. Proc. Natl. Acad. Sci. U.S.A. 101, 16677–16682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walisser J. A., Bunger M. K., Glover E., Harstad E. B., Bradfield C. A. (2004b). Patent Ductus venosus and dioxin resistance in mice harboring a hypomorphic ARNT allele. J. Biol. Chem. 279, 16326–16331. [DOI] [PubMed] [Google Scholar]
- Walisser J. A., Glover E., Pande K., Liss A. L., Bradfield C. A. (2005). Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc. Natl. Acad. Sci. U.S.A. 102, 17858–17863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y., Nukaya M., Satyshur K. A., Jiang L., Stanevich V., Korkmaz E. N., Burdette L., Kennedy G. D., Cui Q., Bradfield C. A. (2012). Identification of the ah-receptor structural determinants for ligand preferences. Toxicol. Sci. 129, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]