

Abstract
Following an acute myocardial infarction, reperfusion therapy is currently the most effective way to save the ischemic myocardium; however, restoring blood flow may lead to a myocardial ischemia-reperfusion injury (MIRI). Recent studies have confirmed that long-chain noncoding RNAs (LncRNAs) play important roles in the pathophysiology of MIRIs. These LncRNA-mediated roles include cardiomyocyte apoptosis, autophagy, necrosis, oxidative stress, inflammation, mitochondrial dysfunction, and calcium overload, which are regulated through the expression of target genes. Thus, LncRNAs may be used as clinical diagnostic markers and therapeutic targets to treat or prevent MIRI. This review evaluates the research on LncRNAs involved in MIRIs and provides new ideas for preventing and treating this type of injury.
1. Introduction
An acute myocardial infarction (AMI) is a type of cardiovascular event that seriously endangers human health [1, 2]. Timely reperfusion treatment following the onset of an AMI can restore blood flow, salvage viable myocardium, reduce the myocardial infarction size, and preserve left ventricular (LV) systolic function [3–5]. However, the process of myocardial reperfusion can further aggravate ischemic myocardium damage, leading to a myocardial ischemia-reperfusion injury (MIRI). Clinical presentations of a MIRI are reperfusion-induced arrhythmias, myocardial stunning, microvascular obstruction, and lethal myocardial reperfusion injury [6–8]. Protecting the heart from the detrimental effects of a MIRI is a therapeutic challenge for physicians. Therefore, elucidating the key mechanisms that prevent a MIRI would greatly aid in the development of an effective therapy for AMIs. Recent studies have shown that the mechanisms of a MIRI include cardiomyocyte apoptosis, autophagy, necrosis, oxidative stress, inflammation, mitochondrial dysfunction, and intracellular calcium overload, all of which involve a multitude of signaling pathways and molecular players [6, 7, 9].
Recently, long noncoding RNAs (LncRNAs) have been shown to modulate key interactions with molecular pathways associated with MIRI progression and potentially serve as novel therapeutic targets. LncRNAs are a class of noncoding RNAs longer than 200 nucleotides. LncRNAs can regulate gene expression at multiple levels, including regulation of epigenetic, transcriptional, and posttranscriptional processes. This review discusses recent studies that have examined the functional role of LncRNAs in MIRIs, their therapeutic potential and aspects that should be further evaluated in the future.
2. LncRNAs
LncRNAs are a class of noncoding RNAs that are more than 200 nucleotides in length that are typically synthesized via RNA polymerase II. Based on the position of the LncRNA and coding gene, these types of RNAs are divided into five categories: (1) sense LncRNA, (2) antisense LncRNA, (3) intron LncRNA, (4) bidirectional noncoding LncRNA, and (5) intergenic LncRNA. LncRNAs regulate gene expression on both chromatin and DNA at the transcriptional and posttranscriptional level, all of which can affect the occurrence and outcome of various diseases [10]. Some regulatory mechanisms exerted through LncRNAs may involve interfering with the translation of an encoded gene, inhibiting polymerase II activity, facilitating posttranscriptional modification, binding to functional proteins, or serving as a precursor for a small molecule RNA that binds to chromosomes to regulate signaling pathways [11, 12].
In addition to being directly involved in regulating gene expression, LncRNAs can also function as a competing endogenous RNA (ceRNA)—i.e., competes with other RNA transcripts for the same microRNA (miRNA), thereby affecting resulting interactions and subsequent signaling cascades. miRNAs are endogenous noncoding RNAs composed of 21 to 25 nucleotides that mainly facilitate the posttranscriptional regulation of target genes through binding to the 3′ untranslated region (UTR) of the target mRNA [13]. In a study on the interactions between RNA transcripts, Salmena et al. [14] first proposed a ceRNA-centered hypothesis as a novel model to regulate gene expression. In this model, transcripts such as LncRNA, pseudogene transcripts, or mRNAs can serve as ceRNAs through miRNA response elements (MREs), which can competitively bind to miRNAs with the same MRE to regulate gene expression levels, thereby affecting downstream cellular functions. Currently, functional studies on ceRNAs suggest that LncRNAs and miRNAs are mutually regulated via competitive binding to the corresponding MRE, thereby effectively controlling the subsequent posttranscriptional regulation of the miRNAs [15, 16].
3. The Role of LncRNAs in MIRIs
The relationship between LncRNAs and a MIRI has been reported in several studies. Recently, Wu et al. reported that 2,292 LncRNAs were upregulated and 1,848 LncRNAs were downregulated in patients with a MIRI [17]. Using gene chip analysis in a mouse model of ischemia-reperfusion (I/R), Liu et al. found that 64 LncRNAs were upregulated and 87 LncRNAs were downregulated in the region with the infarction [18]. Similarly, using gene chip analysis in a cell-based study, Huang et al. found that 309 LncRNAs were upregulated and 488 LncRNAs were downregulated following hypoxia/reoxygenation (H/R) in H9c2 cells [19]. Recent studies have provided credible evidence that LncRNAs play a vital role in the initiation and progression of a MIRI through their involvement in apoptosis, autophagy, and necrosis of cardiomyocytes, as well as by affecting oxidative stress, inflammation, mitochondrial dysfunction, and intracellular calcium overload (Figure 1 and Table 1).
Figure 1.
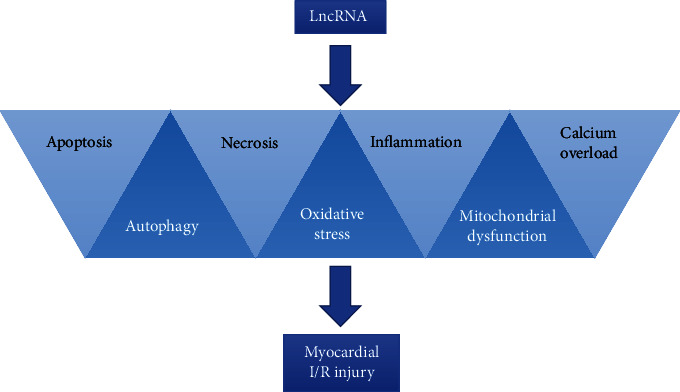
The role of LncRNAs in MIRI.
Table 1.
The role of LncRNAs in MIRI.
| Mechanisms | LncRNA | Mechanism | Species | Expression | Functions | Ref. |
|---|---|---|---|---|---|---|
| Apoptosis | MALAT1 | β-Catenin protein | Rat | Up | Knockdown of MALAT1 inhibits the cardiomyocyte apoptosis and improves cardiac function | [26] |
| miR-122/AKT/GSK-3β/β-catenin | RPCM/H9c2 | Up | Knockdown of MALAT1 inhibits the cardiomyocyte apoptosis | [27] | ||
| miR-200a-3p/PDCD4 | AC16/mice | Up | Promote cardiomyocyte apoptosis | [28] | ||
| miR-320/PTEN | NMCM/mice | Up | Promote cardiomyocyte apoptosis and aggravate myocardial injury | [29] | ||
| PFL | HSP-20/Bax/Bcl-2 | Rat | Up | Knockdown of PFL inhibits apoptosis and reduces the MI area | [30] | |
| HIF1A-AS1 | miR-204/SOCS2 | Mice | Up | Silence of HIF1A-AS1 inhibits cardiomyocyte apoptosis and alleviates ventricular remodeling | [31] | |
| NEAT1 | miR-520a | H9c2/rat | Up | Knockdown of NEAT1 inhibits cardiomyocyte apoptosis | [32] | |
| MAPK | H9c2/mice | Up | Promote cardiomyocyte apoptosis | [33] | ||
| Meg3 | miR-7-5p/PARP1 | H9c2/rat | Up | Promote cardiomyocyte apoptosis and myocardial damage | [34] | |
| MIAT | NF-κB/PUMA | H9c2/mice | Up | Silence of MIAT inhibits cardiomyocyte apoptosis | [35] | |
| XIST | miR-130a-3p/PDE4D | AC16 | Up | Knockdown of XIST inhibits apoptosis and promotes cell survival | [36] | |
| AK123484 | PARP and caspase-3 | H9c2/rat | Up | Knockdown of AK123483 inhibits apoptosis | [37] | |
| BDNF-AS | BDNF/VEGF/Akt | NMCM | Up | Promote cardiomyocyte apoptosis | [38] | |
| H19 | miR-877-3p/Bcl-2 | NMVC/mice | Down | H19 alleviates cardiomyocyte apoptosis | [39] | |
| UCA1 | p27 | RPCM/rat | Down | UCA1 inhibits cardiomyocyte apoptosis | [40] | |
| FAF | PI3K/AKT/FGF9/FGFR2 | NRCM/rat | Down | FAF inhibits cardiomyocyte apoptosis | [41] | |
| Autophagy | TUG1 | miR-142-3p/HMGB1 and Rac1 | NMCM/mice | Up | Knockdown of TUG1 inhibits excessive autophagy | [46] |
| XIST | miR-133a/SOCS2 | H9c2/mice | Up | Knockdown of XIST inhibits cardiomyocyte autophagy | [47] | |
| HRIM | LC3 | H9c2/rat | Up | Inhibition of HRIM reduces excessive autophagy | [19] | |
| AK139328 | miR-204-3p | Mice | Up | Silence of AK139328 inhibits cardiomyocyte autophagy | [48] | |
| AK088388 | miR-30a/Beclin-1/LC3-II | HL-1 | Up | Enhanced autophagy and promote cardiomyocyte injury | [49] | |
| FOXD3-AS | NF-κB/iNOS/COX2 | H9c2 | Up | Aggravate cardiomyocyte injury via promoting autophagy | [50] | |
| RMRP | miR-206/ATG3 | H9c2/rat | Up | Promote cardiomyocyte autophagy | [51] | |
| APF | miR-188-3p/ATG7 | NMCM/mice | Up | Increase autophagy and myocardial infarction | [52] | |
| Galont | miR-338/ATG5 | NMCM | Up | Promote autophagy and cell death | [53] | |
| 2810403D21Rik/Mirf | miR-26a/Usp15 | NMCM/mice | Up | Suppress autophagy and exacerbate myocardial injury | [54] | |
| CAIF | p53/Myocardin | NMCM/mice | Down | Suppress autophagy and attenuate myocardial infarction | [55] | |
| Necrosis | NRF | miR-873/RIPK1/RIPK3 | H9c2/mice | Up | Knockdown of NRF inhibits necrosis and reduces infarction size | [59] |
| H19 | miR-103/107/FADD | H9c2 | Down | Alleviate cardiomyocyte necrosis | [60] | |
| Oxidative stress | Gpr19 | miR-324-5p/Mtfr1 | Mice/NRCM | Up | Suppression of Gpr19 inhibits oxidative stress | [66] |
| ROR | p38/MAPK | H9c2/HCM | Up | Aggravate oxidative stress | [67] | |
| HOTAIR | miR-125/MMP2 | H9c2 | Down | Suppression of HOTAIR aggravates oxidative stress-induced cardiomyocyte injury | [68] | |
| FTX | miR-29b-1-5p/Bcl2l2 | NMCM/mice | Down | Overexpression of FTX inhibits oxidative stress | [69] | |
| Inflammation | KCNQ1OT1 | p38 MAPK/NF-κB | H9c2 | Up | Suppression of KCNQ1OT1 reduces inflammation | [75] |
| NEAT1 | miR-495-3p/MAPK6 | H9C2/mice | Up | Activating inflammation | [76] | |
| H19 | miR675/PPARα | NMCM/mice | Up | Knockdown of H19 inhibits inflammation, improves cardiac structure and function | [77] | |
| Mirt1 | NF-κB | NMCM/mice | Up | Knockdown of Mirt1 inhibits inflammation and reduces infarction size | [78] | |
| NF-κB | Rat | Up | Inhibition of Mirt1 relieves inflammatory injury | [79] | ||
| Gm2691 | PI3K/Ak | NRCM/rat | Down | Anti-inflammation | [80] | |
| Mitochondrial dysfunction | CARL | miR-539/PHB2 | NMCM/mice | Down | Attenuate mitochondrial fission and cell death | [87] |
| MDRL | miR-361/miR-484 | NMCM/mice | Down | Attenuate mitochondrial fission and apoptosis | [88] | |
| Calcium overload | ZFAS1 | SERCA2a | NMCM/mice | Up | Aggravate calcium overload | [94–96] |
3.1. LncRNAs and Apoptosis
Cardiomyocyte apoptosis is a key pathophysiological mechanism underlying a MIRI. Apoptosis is a type of programmed cell death induced by physiological or pathological factors. The process is characterized by cell shrinkage, chromatin condensation, and systematic DNA cleavage [20]. Apoptosis is a highly regulated cellular process, which can be divided into two classic pathways: the extrinsic or intrinsic pathway. The extrinsic pathway, also known as the death receptor-associated pathway, is activated via the death-inducing signal complex (DISC). The DISC activates caspase-8, which in turn triggers the downstream activation of executioner caspases, leading to apoptosis. The intrinsic pathway, also known as the mitochondrial pathway, is initiated by increased permeability of the outer mitochondrial membrane (OMM), leading to the translocation of proapoptotic molecules such as cytochrome C (cytC) and apoptosis-inducing factor (AIF) from the mitochondrial intermembrane space to the cytosol. This leads to the formation of the apoptosome complex and subsequent downstream activation of the caspase cascade. Members of the Bcl-2 family are major participants in the intrinsic pathway [20, 21]. In some circumstances, the intrinsic pathway is also initiated via the endoplasmic reticulum stress- (ERS-) induced apoptotic signaling pathway. It has been demonstrated that prolonged and/or excess ERS may lead to substantial apoptosis and be a major contributor to a MIRI. Herein, we expound on the relationships between different LncRNAs and their role during apoptosis in response to a MIRI [22–25].
3.1.1. LncRNA MALAT1
The expression of the LncRNA MALAT1 is significantly increased in the myocardial tissue of the rat I/R model. Knockdown of MALAT1 can markedly improve I/R-induced myocardial contractile dysfunction and inhibit cardiomyocyte apoptosis in the rat I/R injury model by upregulating the expression of β-catenin [26]. LncRNA MALAT1 was prominently elevated in oxygen-glucose deprivation/reoxygenation- (OGD/R-) induced H9c2 cells. MALAT1 knockdown inhibited OGD/R-induced cardiomyocyte apoptosis by activating the AKT/GSK-3β/β-catenin signaling pathway via downregulation of miR-122 [27]. LncRNA MALAT1 expression was upregulated in H/R-treated AC16 cells. Moreover, MALAT1 regulated hypoxia-induced myocardial cell apoptosis by sponging miR-200a-3p to upregulate PDCD4 [28]. The expression of LncRNA MALAT1 and phosphatase and tensin homolog deleted on chromosome 10 (Pten) was upregulated, whereas that of miR-320 was downregulated in myocardial tissues from the AMI mouse model. MALAT1 functions as a ceRNA for miR-320 to regulate Pten in mouse cardiomyocytes. Overexpression of MALAT1 aggravated cardiomyocyte apoptosis in vitro, and knockdown of MALAT1 improved cardiac function, resulting from AMI [29].
3.1.2. LncRNA PFL
The expression level of the LncRNA PFL was markedly improved in the myocardial tissues, whereas the level of HSP-20 was markedly decreased in the myocardium of the I/R rat model. HSP-20 can form a stable complex with Bax and to in turn regulate the ratio of Bax/Bcl-2, thereby maintaining mitochondrial integrity and inhibiting both cytC release and caspase-3 activation. Knocking down PFL can inhibit MIRI-mediated apoptosis by upregulating HSP-20 expression [30].
3.1.3. LncRNA HIF1A-AS1
LncRNA HIF1A-AS1 expression was upregulated in the mouse model of I/R. Interestingly, LncRNA HIF1A-AS1 functions as a ceRNA when it binds with miR-204, thereby suppressing its expression and upregulating suppressor of cytokine2 (SOCS2) expression. Members of the SOCS protein family were initially described as cytokine-induced JAK/STAT signaling feedback inhibitors. Silencing HIF1A-AS1 can inhibit I/R-induced apoptosis in cardiomyocytes to alleviate ventricular remodeling following an AMI in mice [31].
3.1.4. LncRNA Nuclear Paraspeckle Assembly Transcript 1 (NEAT1)
LncRNA NEAT1 expression was upregulated in H/R-treated H9c2 cells. LncRNA NEAT1 can bind to miR-520a to suppress its expression. Additionally, NEAT1 knockdown can elevate Bax expression levels, cleave caspase-3, and reduce the expression level of Bcl-2. Knocking down NEAT1 can inhibit cardiomyocyte apoptosis induced via H/R [32]. Another study also reported that NEAT1 plays a crucial role in exacerbating the myocardial injury in MIRI mouse models. Moreover, NEAT1 could prompt cardiomyocyte apoptosis induced via I/R by activating the MAPK signaling pathway [33].
3.1.5. LncRNA Meg3
LncRNA Meg3 expression was markedly upregulated in I/R-treated H9c2 cells and in the I/R rat model. Meg3 can promote cell apoptosis, inhibit cell proliferation, and promote myocardial damage by directly binding to miR-5-7p. PARP1, the downstream target gene of miR-7-5p, plays an important role in cardiomyocyte apoptosis. LncRNA Meg3 regulates cardiomyocyte apoptosis in MIRI via the miR-7-5p/PARP1 signaling pathway [34].
3.1.6. LncRNA Myocardial Infarction-Associated Transcript (MIAT)
The expression of MIAT, another LncRNA, significantly increased in H9c2 cells when H/R stimulated as well as in the heart of mice in response to the I/R-induced injury. MIAT can regulate the expression of PUMA via NF-κB activation in H/R-induced H9c2 cells. Subsequently, PUMA plays an important role in inducing cell apoptosis. Silencing LncRNA MIAT could inhibit H/R-induced cardiomyocyte apoptosis and injury by suppressing the NF-κB and PUMA signaling pathways [35].
3.1.7. LncRNA X-Inactive Specific Transcript (XIST)
In H/R-treated AC16 cells, LncRNA XIST expression was upregulated, whereas miR-130a-3p was downregulated. Additionally, XIST can negatively regulate miR-130a-3p expression. PDE4D is a direct target gene of miR-130a-3p that promotes cardiomyocyte apoptosis. Depleting LncRNA XIST suppressed apoptosis and promoted cell survival in H/R-treated AC16 cells via the miR-130a-3p/PDE4D signaling pathway [36].
3.1.8. LncRNA AK123484
The expression of LncRNA AK123484 was upregulated in H9c2 cells treated with anoxia/reoxygenation (A/R). LncRNA AK123483 positively correlated with the expression of PARP. Consequently, LncRNA AK123483 knockdown protects cardiomyocytes against A/R-induced apoptosis by inhibiting PARP expression [37].
3.1.9. LncRNA BDNF-AS
LncRNA BDNF-AS expression was upregulated in H/R-treated cardiomyocytes. Knocking down BDNF-AS can inhibit apoptosis and promote cardiomyocyte viability under H/R conditions by upregulating BDNF expression. Therefore, BDNF upregulation exerts its protective effect against MIRIs by activating survival-signaling pathways, including upregulating VEGF and phosphorylating, i.e., activating, AKT [38].
3.1.10. LncRNA H19
H19 is a conserved LncRNA that is transcribed from the imprinted H19-insulin growth factor 2 (IGF2) locus; moreover, it is located in both the nucleus and the cytoplasm. LncRNA H19 was downregulated in the mouse model of I/R and H2O2-treated cardiomyocytes. Further, H19 acts as a ceRNA to inhibit miR-877-3p expression and suppress Bcl-2 expression. Therefore, expression of miR-877-3p exacerbates H2O2-induced injuries in cardiomyocytes as well as MIRIs via Bcl-2 through its mediated mitochondrial apoptotic pathway. These effects suggest that miR-877-3p and Bcl-2 serve as downstream mediators of H19 under apoptotic conditions. Further, LncRNA H19 alleviated cardiomyocyte apoptosis by suppressing the miR-877-3p/Bcl-2-mediated mitochondrial apoptotic pathway [39].
3.1.11. LncRNA UCA1
LncRNA UCA1 expression was significantly downregulated, and reactive oxygen species (ROS) levels were significantly enhanced in the rat model of I/R. UCA1 can directly inhibit p27 at the protein level. Overexpression of p27 can activate caspase-3 by promoting its cleavage; moreover, it can trigger H2O2-induced cardiomyocyte apoptosis. Suppressing UCA1 has a proapoptotic function by regulating p27 protein levels [40].
3.1.12. LncRNA FAF
LncRNA FAF expression is downregulated in cardiomyocytes treated with ischemia-hypoxia as well as the myocardium of AMI rats. Overexpressing LncRNA FAF can inhibit cardiomyocyte apoptosis induced via ischemia and hypoxia. LncRNA FAF positively regulates FGF9 expression. Moreover, FGF9 can specifically activate FGFR2, which is associated with the PI3K/AKT pathway. Knocking down FGF9 can promote cardiomyocyte apoptosis induced by ischemia and hypoxia. We theorize that LncRNA FAF plays a protective role in inhibiting cardiomyocyte apoptosis by upregulation FGF9/FGFR2 via the PI3K/AKT signaling pathway [41].
3.2. LncRNAs and Autophagy
Autophagy is an intracellular protective mechanism under physiological conditions that can degrade damaged or unnecessary proteins, as well as organelles, to maintain cellular homeostasis. When a MIRI occurs, autophagy is activated in response to oxidative stress, ERS, and energy deprivation. Recent studies have demonstrated that autophagy can be either protective or detrimental to cardiac tissue during a MIRI event [42, 43]. Autophagy activated during the initial phase of ischemia has protective effects on cardiomyocytes. For instance, during the myocardial ischemia stage, AMPK is activated in response to the decreasing level of ATP, which occurs when a high ratio of AMP/ATP owing to the deprivation of nutrients and oxygen. Therefore, AMPK is an essential molecule to initiate autophagy in cardiac ischemia. AMPK could launch autophagy through the AMPK/mTORC1/ULK1 signaling pathway or by directly activating ULK1. Autophagy promotes cardiomyocyte survival by generating the amino acids and fatty acids required for maintaining cellular energy levels, as well as activating mitophagy, which can prevent damaged mitochondria from releasing their cytotoxic substances. However, uncontrolled excessive autophagy during the reperfusion stage exerts detrimental effects on cardiomyocytes. During the reperfusion phase, increased ROS generation prompts the overexpression of Beclin-1, which is a crucial mediator of autophagy during this phase. Consequently, overexpressing Beclin-1 can enhance autophagic activity during reperfusion. Excessive autophagy degrades essential proteins or organelles, thereby leading to autophagy-mediated cell death. Additionally, apoptosis is caused by detrimental effects of excessive autophagy in MIRI [44, 45].
3.2.1. LncRNA TUG1
LncRNA TUG1 expression was upregulated in I/R-injured heart tissues and H2O2-challenged cardiomyocytes. TUG1 was identified as a miRNA sponge that targets miR-142-3p, which affects the activity of downstream target molecules, e.g., HMGB1 and Rac1, that contribute to autophagic cell death. Depleting TUG1 could inhibit excessive autophagy-induced cell death and reduce I/R-induced infarction size [46].
3.2.2. LncRNA XIST
LncRNA XIST expression was upregulated in H/R-treated H9c2 cells. Subsequently, XIST increases SOCS2 expression by negatively regulating miR-133a, which results in excessive autophagy in cardiomyocytes. Further, XIST inhibition exerted a myocardial protective effect during a MIRI by suppressing autophagy and regulating the miR-133a/SOCS2 axis [47].
3.2.3. LncRNA-HRIM
The LncRNA-HRIM is located on chromosome 20p12, which includes three exons, and is a total of 1,470 bp. Notably, LncRNA-HRIM is upregulated in H/R-treated H9c2 cells as well as the rat model of I/R. Inhibition of LncRNA-HRIM expression can markedly increase myocardial cell survival by reducing excessive autophagy during H/R [19].
3.2.4. LncRNA AK139328
LncRNA AK139328 expression was evidently upregulated in diabetic mice with a MIRI. LncRNA AK139328 directly modulates miR-204-3p expression. Silencing LncRNA AK139328 can inhibit the expression of autophagy-related proteins (e.g., LC3-I/LC3-II, ATG5, and ATG7) in response to a MIRI. Knocking down AK139328 exerts a protective effect in diabetic mice with a MIRI by inhibiting cardiomyocyte autophagy [48].
3.2.5. LncRNA AK088388
LncRNA AK088388 expression is markedly upregulated in H/R-treated HL-1 cells. AK088388 can regulate LC3-II expression via the miR-30a/Beclin-1 pathway. The upregulated expression of AK088388 activates autophagy and promotes cardiomyocyte injury in response to H/R. Thus, AK088388 knockdown could promote cell viability and prevent extensive damage in cardiomyocytes [49].
3.2.6. LncRNA FOXD3-AS
LncRNA FOXD3-AS expression is upregulated in OGD/R-treated H9c2 cells. Overexpression of FOXD3-AS1 can increase the expression of autophagy-associated proteins (LC3II, Beclin-1, and ATG5) and reduce the expression of p62. Moreover, FOXD3-AS1 overexpression also augments the expression of phosphorylated (p)-NF-κB, p65, COX-2, and INOS. FOXD3-AS1 aggravates the I/R-induced injury in cardiomyocytes through excessive autophagy that is regulated by activating the NF-κB/iNOS/COX2 signaling pathway [50].
3.2.7. LncRNA RMRP
The expression of LncRNA RMRP is increased in hypoxia-treated H9c2 cells as well as in the rat model of I/R. RMRP negatively regulates the expression of miR-206; however, RMRP overexpression can activate the autophagic response and aggravate subsequent cell damage by downregulating miR-206. Moreover, overexpression of miR-206 has a protective effect on hypoxia-induced injuries by targeting the regulation of ATG3 expression. Overexpression of RMRP activates the PI3K/AKT/mTOR signaling pathway, which is reversed following miR-206 overexpression. The RMPR/miR-206/ATG3 axis can mediate cell autophagy by regulating the activation of PI3K/AKT/mTOR signaling in response to a MIRI [51].
3.2.8. LncRNA AK079427
The expression of the LncRNA AK079427, also known as autophagy-promoting factor (APF), was significantly upregulated in A/R-treated cardiomyocytes and the mouse model of I/R. APF specifically sponges miR-188-3p to regulate its activity. Overexpression of miR-188-3p can inhibit both autophagy and myocardial infarction by downregulating its downstream target, ATG7. ATG7 is a key autophagy promoting gene that encodes an E1-like enzyme, a member of the autophagy system. APF knockdown can attenuate cardiomyocyte autophagy and reduce myocardial infarction sizes in response to a I/R injury by targeting the miR-188-3p/ATG7 axis. Thus, the APF/miR-188-3p/ATG7 axis plays a key role in regulating autophagic cell death in cardiovascular tissues; moreover, it may be a potential target for novel therapeutic strategies for treating myocardial infarctions [52].
3.2.9. LncRNA Galont
LncRNA Galont expression was significantly upregulated in A/R-treated cardiomyocytes. Overexpression of Galont can aggravate cardiomyocyte autophagy and increase cell death in response to A/R stimuli, whereas Galont knockdown had the opposite effect. Galont was confirmed to directly bind with miR-338, a novel suppressor of autophagy. ATG5 is a gene targeted by miR-338, and Galont is positively correlated to ATG5 expression. Thus, the existing evidence suggests that the Galont/miR-338/ATG5 axis plays an important role in promoting A/R-induced autophagy in cardiomyocytes [53].
3.2.10. LncRNA 2810403D21Rik/Mirf
LncRNA 2810403D21Rik/Mirf expression was significantly upregulated in cardiomyocytes treated with H2O2 and in the heart tissue from the mouse model of MI. LncRNA 2810403D21Rik/Mirf acts as a ceRNA that competitively binds to and affects the activity of miR-26a. miR-26a also modifies the activity of the downstream target Usp15, which can inhibit mitochondrial autophagy by activating the PINK1/PRKN pathway. Silencing 2810403D21Rik/Mirf and overexpressing miR-26a enhanced cell viability and mitigated myocardial injury by increasing the expression of autophagy-related proteins. Further, overexpression of 2810403D21Rik/Mirf suppresses autophagy and exacerbates myocardial injury by limiting the signaling capacity of the miR-26a/Usp15 axis. Therefore, modulating the impact of the 2810403D21Rik/Mirf–miR-26a/Usp15 axis on autophagy can promote a cardioprotective effect in response to a MIRI [54].
3.2.11. LncRNA CAIF
LncRNA CAIF expression was significantly downregulated in H2O2-treated cardiomyocytes as well as the mouse model of I/R. CAIF can directly bind to the p53 protein and block p53-mediated transcription of MYOCD, which encodes myocardin. Myocardin contributes to the induction of the autophagic process and death of cardiomyocytes in the heart during the I/R-induced injury. Knockdown of CAIF can aggravate H2O2-induced cardiomyocyte autophagy, whereas myocardin overexpression augments the size affected from a MIRI. Moreover, CAIF inhibits autophagy and attenuates myocardial injury in the heart by targeting the p53/myocardin-dependent autophagy pathway [55].
3.3. LncRNAs and Necrosis
Cardiomyocyte necrosis is a major pathological event in an AMI. Defining features of necrosis include an increase in cell size from cytoplasmic and mitochondrial swelling, karyolysis, loss of ribosomes from the endoplasmic reticulum, and plasma membrane disruption, all of which initiate inflammation [24, 56]. Mitochondria play an important role in the execution of programmed necrotic cell death, especially in cardiomyocytes, through two mechanisms: mitochondrial permeability transition- (MPT-) dependent necrosis and death receptor-dependent necrosis (necroptosis). Necrosis usually destroys the inner mitochondrial membrane (IMM), thereby inducing the opening of mitochondrial permeability transition pores (MPTPs), which depletes ATP and the plasma membrane loses its integrity [57, 58]. Necroptosis can be induced by triggering several molecules, primarily through death receptors—e.g., TNFR, FasR, and TNF-related apoptosis-inducing ligand receptor (TRAIL-R)—that are stimulated by cytokines from the TNF family and Toll-like receptors (TLRs) [24, 56].
3.3.1. LncRNA NRF
LncRNA NRF expression was significantly upregulated in H2O2-treated H9c2 cells and the mouse model of I/R. LncRNA NRF directly binds to miR-873, which targets RIPK1 and RIPK3, thereby inhibiting necrosis in cardiomyocytes. Knocking down NRF inhibits necrosis in cardiomyocytes and reduces the infarct size in the I/R mouse model. p53 regulates NRF expression at the transcriptional level and is involved in cardiomyocyte necrosis via miR-873. These results demonstrate that p53 targets NRF–miR-873 and the RIPK1/RIPK3 axis, which play key roles in regulating cardiomyocyte necrosis during a MIRI [59].
3.3.2. LncRNA H19
LncRNA H19 expression decreased in H2O2-treated H9c2 cells. LncRNA H19 can directly bind to miR-103/107 to promote the expression of Fas-associated protein with death domain (FADD). FADD binds to RIPK1, thereby inhibiting cardiomyocyte necrosis by disrupting the formation of the RIPK1/RIPK3 complex. Inducing H19 expression can inhibit cardiomyocyte necrosis; moreover, H2O2 induces FADD downregulation. Collectively, these results reveal possible links among H19, miR-103/107, FADD, and RIPK1/RIPK3, which may collectively regulate cardiomyocyte necrosis during a MIRI [60].
3.4. LncRNAs and Oxidative Stress
At the onset of reperfusion, the level of myocardial tissue oxygenation increases after restoring blood flow. However, when in a reduced state, respiratory chain complexes damage the mitochondrial electron transport chain, which generates a high level of ROS and leads to ischemia [61, 62]. ROS includes H2O2, hydroxyl radicals, and superoxide anions. The primary enzyme systems involved in ROS production during a MIRI are xanthine oxidase, NADPH oxidase, the mitochondrial electron transport chain, and uncoupled nitric oxide synthase [63]. Oxidative stress occurs when the antioxidant system is overwhelmed by ROS overproduction. ROS-mediated mitochondrial dysfunction and the sequence of biochemical events following reperfusion of the ischemic area are the key pathological features of a MIRI. ROS overproduction promotes the oxidation of DNA and proteins as well as and lipid peroxidation, thereby causing alterations in membrane permeability that structurally and functionally damage cells [62, 64, 65].
3.4.1. LncRNA Gpr19
The expression of the LncRNA Gpr19 is upregulated in the mouse model of AMI and in neonatal rat ventricular cardiomyocytes (NRCMs) exposed to OGD/R. Gpr19 targets miR-324-5p, a miRNA that negatively regulates Mtfr1 expression at the transcriptional and translational levels. The function of Mtfr1 is primarily related to the IMM. Overexpression of Mtfr1 aggravates oxidative stress in NRCMs induced via OGD/R. Further, suppression of Gpr19 inhibits oxidative stress and attenuates myocardial injury through the miR-324-5p/Mtfr1 axis [66].
3.4.2. LncRNA ROR
LncRNA ROR is highly expressed in I/R-treated H9c2 and HCM cells. Overexpression of ROR can aggravate oxidative stress in response to a MIRI by promoting NADPH oxidase activity, ROS production, and NOX2 protein levels. p38/MAPK inhibitors can rescue LncRNA-ROR-induced cell viability and reduce cell apoptosis in both H9c2 and HCM cells. Further, LncRNA ROR can activate oxidative stress pathways and induce myocardial apoptosis by regulating p38/MAPK signaling [67].
3.4.3. LncRNA HOTAIR
LncRNA HOTAIR expression was significantly downregulated in H2O2-treated H9c2 cells. HOTAIR modulates the expression of matrix metalloproteinases-2 (MMP2) by binding miR-125 in H9c2 cells when subjected to oxidative stress. Suppression of HOTAIR aggravates cardiomyocyte apoptosis induced by oxidative stress via the HOTAIR/miR-125/MMP2 axis [68].
3.4.4. LncRNA FTX
LncRNA FTX expression is significantly downregulated in the mouse model of I/R and in H2O2-treated cardiomyocytes. FTX sponges miR-29b-1-5p, which in turn regulates its activity. miR-29b-1-5p promotes H2O2-induced cardiomyocyte apoptosis by inhibiting the downstream target Bcl2l2. Overexpression of FTX inhibits H2O2-induced cardiomyocyte apoptosis by regulating the miR-29b-1-5p/Bcl2l2 axis [69].
3.5. LncRNAs and Inflammation
Inflammation is an important contributor to the pathophysiology of a MIRI. The I/R-induced injury can activate the inflammatory response by promoting the release of cytokines, chemokines, and other proinflammatory factors [70]. The extent of the inflammatory response to a MIRI can determine the infarct size and subsequent LV remodeling [71, 72]. During the postischemic phase, intracellular content released by necrotic cardiomyocytes initiates an intense inflammatory response by activating innate immune responses. During the early reperfusion phase, cardiomyocytes increase the expression of cytokines and adhesion molecules, which prompt the recruitment of neutrophils and other leukocytes to the infarcted myocardium [73]. As the adherent leukocytes transmigrate into these infarcted zones, they disrupt the microvascular barrier and cause hyperpermeability, leading to myocardium edema that increases the diffusion distance for oxygen and nutrients. Neutrophils are the most abundant among infiltrating inflammatory cells during the reperfusion stage. Notably, they impair endothelial function, activate platelets leading to coronary microembolization, and cause the myocardium no-reflow phenomenon. The infiltration of neutrophils induces a reperfusion injury that amplifies the cellular damage initiated by ischemia [71, 74].
3.5.1. LncRNA KCNQ1OT1
The expression of KCNQ1OT1 was significantly upregulated in OGD/R-induced H9C2 cells. Suppression of KCNQ1OT1 reduces the expression of inflammatory factors (e.g., TNF-α, IL-6, and IL-1b) by regulating adiponectin receptors, namely, AdipoR1, in H9C2 cells subjected to OGD/R treatment. Moreover, KCNQ1OT1 suppression also inhibits activating the p38/MAPK/NF-κB signaling pathway. Suppression of KCNQ1OT1 may exert protective effects against a MIRI by regulating AdipoR1, which in turn affects p38/MAPK/NF-κB signaling [75].
3.5.2. LncRNA NEAT1
LncRNA NEAT1 expression is upregulated in H2O2-treated H9C2 cells and in the mouse model of I/R. NEAT1 was identified as a functional sponge of miR-495-3p that negatively regulates its expression. miR-495-3p blocks the activation of the inflammation process induced via H2O2 treatment by downregulating its target, MAPK6. Thus, the NEAT1/miR-495-3p/MAPK6 axis plays a crucial role in activating inflammation and aggravating cardiomyocyte injury during a MIRI [76].
3.5.3. LncRNA H19
LncRNA H19—a precursor of miR-675 that regulates multifarious target genes posttranslationally and is involved in different biological processes—was upregulated in cardiomyocytes exposed to OGD/R. Silencing H19 reduces the production of proinflammatory cytokines and inhibits apoptosis in cardiomyocytes following exposure to OGD/R by downregulating the levels of p-IκB-α and p-p65. However, the effect of H19 inhibition was partially reversed when miR-675 was overexpressed. Moreover, miR-675 could directly suppress the expression of PPARα, as demonstrated with dual-luciferase reporter assays and software-based analysis. PPARα exerts an anti-inflammatory effect by suppressing the NF-κB signaling pathway. Therefore, H19 inhibition can markedly improve cardiac structure and function in response to a MIRI via the H19/miR-675/PPARα signaling axis [77].
3.5.4. LncRNA Mirt1
Mirt1, a LncRNA mainly expressed in cardiac fibroblasts, was upregulated in both the myocardium of AMI-induced mice and cardiac fibroblasts that were subjected to hypoxia. High Mirt1 levels contribute towards LV dysfunction in AMI-induced mice, whereas its knockdown could decrease the myocardial infarct size, improve cardiac functions, and potentially limit the inflammatory response. Knockdown of Mirt1 inhibits cardiomyocyte apoptosis and reduces the production of inflammatory cytokines by suppressing NF-κB signaling under hypoxic conditions in vitro. These data suggest that Mirt1 knockdown plays a protective role in response to an AMI by inhibiting NF-κB activation [78]. Another study reported that Mirt1 expression levels significantly increased in myocardial tissue of aged diabetic I/R rats. This study also found that the protective effects of Mirt1 inhibition in these rats may result from the inhibition of NF-κB activation by attenuating the inflammatory response, which would normally induce apoptosis and further aggravate the damage to cardiac tissues [79].
3.5.5. LncRNA Gm2691
LncRNA Gm2691 expression was markedly downregulated in the rat model of I/R and in hypoxia-treated NRCMs. Overexpression of Gm2691 significantly decreases IL-6, TNF-α, and IL-8 production in hypoxia-induced NRCMs. Hypoxia treatment in NRCMs can inhibit p-AKT expression, whereas the Gm2691 reverses this effect. Thus, LncRNA Gm2691 exerts antiapoptotic and anti-inflammatory effects by regulating the PI3K/AKT signaling pathway [80].
3.6. LncRNAs and Mitochondrial Dysfunction
Mitochondrial dysfunction is considered as the prominent pathological feature of a MIRI; therefore, it has become a key regulator of its inhibition. Mitochondria synthesize approximately 90% of ATP in cardiomyocytes [81, 82]. Mitochondrial dysfunction can cause the imbalance of calcium homeostasis, excess production of ROS, impaired cellular energy metabolism and ATP production, and opening of MPTPs, which can culminate in cell death [83]. Numerous studies have demonstrated that an imbalance between mitochondrial fission and fusion contributes towards mitochondrial dysfunction in response to a MIRI. During the early ischemic period, mitochondrial fusion leads to the generation of a disproportionately large amount of ATP, while sustained ischemia induces excessive mitochondrial fission. During the reperfusion period, excessive ROS and calcium overload exacerbate the activation of mitochondrial fission. Mitochondrial fission generates smaller mitochondrial fragments; however, mitophagy insufficiently eliminates damaged mitochondria. Thus, mitochondrial fission results in a higher susceptibility of opening MPTPs, activating apoptotic pathways by releasing cytC, and activating caspases, all of which result in cell death following myocardial reperfusion [84–86].
3.6.1. LncRNA CARL
LncRNA CARL is expressed in both the nucleus and the cytoplasm. The expression of CARL was downregulated in cardiomyocytes following anoxia treatment. CARL acts as an endogenous sponge of miR-539, thereby negatively regulating its levels and activity. miR-539 targets and regulates PHB2 expression, which plays an important role in regulating mitochondrial morphology. Overexpression of CARL attenuates mitochondrial fission and cell death during a MIRI by sponging miR-539 as well as upregulating PHB2 expression in vitro and in vivo. Combined, these data suggest that the CARL/miR-539/PHB2 signaling is crucial in affecting mitochondrial dynamics and cell apoptosis during a MIRI [87].
3.6.2. LncRNA MDRL
LncRNA MDRL is 1,039 nucleotides and is expressed in both the nucleus and the cytoplasm. MDRL expression was downregulated in A/R-treated cardiomyocytes. The bioinformatics program RNAhybrid revealed that MDRL carries a target site for miR-361. miR-361 promotes mitochondrial fission and apoptosis following A/R treatment by inhibiting the processing of pri-miR-484 by Drosha into pre-miR-484. MDRL acts as an endogenous sponge for miR-361 to regulate mature miR-484 levels. In an animal model, MDRL knockdown attenuated mitochondrial fission, cardiomyocyte death, and myocardial infarction size in response to I/R. These findings suggest that MDRL targets miR-361/miR-484 in the mitochondrial fission cascade and apoptosis during a MIRI, thereby revealing potential targets for treating cardiac disease [88].
3.7. LncRNAs and Calcium Overload
Calcium homeostasis is disturbed during myocardial ischemia-reperfusion. Excessive accumulation and leakage of intracellular calcium lead to calcium overload, which exacerbates a MIRI [89, 90]. Cytosolic and mitochondrial calcium overload begins at ischemia and is aggravated during reperfusion. At the onset of ischemia, the Na+/H+ exchanger (NHE) is activated and transports H+ out of the cytosol in exchange for Na+. During reperfusion, NHE activity is accelerated, leading to additional accumulation of intracellular Na+. As a result, a high Na+ concentration activates the Na+/calcium exchanger (NCX), thereby leading to intracellular calcium overload [91]. Alternatively, more calcium influx through L-type calcium channels and the sarcoplasmic reticulum uptake of calcium from the cytoplasm is inhibited, which further exacerbates the calcium overload in response to the I/R [92]. During reperfusion, disruption of the mitochondrial membrane potential accelerates energy depletion, thereby resulting in mitochondrial calcium overload and ROS overproduction. In addition, disrupting the mitochondrial membrane potential also results in ATP synthase behaving as an ATPase, thereby accelerating energy depletion in response to the ischemic insult. Large amounts of calcium accumulated in cells inhibit ATP synthesis in mitochondria, leading to the dysfunctional processing of energy metabolism. These processes can trigger MPTPs to open, leading to the breakdown of respiratory chain coupling, mitochondrial membrane potential disorder, and eventually the induction of either necrotic- or apoptotic-mediated cell death [93].
3.7.1. LncRNA ZFAS1
LncRNA ZFAS1, an antisense LncRNA to the 5′ end of the protein-coding gene Znfx1, has been identified as an independent predictor of an AMI [94]. Calcium homeostasis is closely related to cardiac contractility, and abnormal intracellular calcium handling might contribute to the impairment of cardiac contractile function caused by ZFAS1 in a MIRI. Sarcoplasmic reticulum calcium-ATPase 2a (SERCA2a) is a key protein involved in the reuptake of calcium into the sarcoplasmic reticulum during diastole, thereby regulating calcium homeostasis in cardiomyocytes. ZFAS1 expression is markedly elevated in the myocardium of mice with an AMI. Knockdown of endogenous ZFAS1 improved cardiac function as the ejection fraction (EF), and fractional shortening (FS) resumed to almost normal levels in the mouse model of myocardial infarction. ZFAS1 overexpression in normal mice could impair cardiac function as EF and FS both decreased, thereby promoting an enlarged LV internal dimension at end-diastole (LVIDd) and LV internal dimension at systole (LVIDs), which is similar to the pathological process that occurs in response to a MIRI. Additionally, ZFAS1 conferred a negative effect on SERCA2a by repressing its expression at the transcriptional level. ZFAS1 deleteriously impacted the dynamic influx of calcium, thereby leading to intracellular calcium overload in cardiomyocytes, which might be the underlying mechanism of its proapoptotic property. Moreover, it was found that NFATc2 served as a transactivator to promote ZFAS1 expression [95]. Another study found that ZFAS1 activated the mitochondrial apoptosis pathway in the mouse model of myocardial infarction. Additionally, ZFAS1 knockdown relieved mitochondrial swelling and decreased mitochondrial membrane potential in hypoxia-treated cardiomyocytes. ZFAS1 induced cardiomyocyte apoptosis by inhibiting SERCA2a, thereby causing intracellular calcium overload. Therefore, knocking down ZFAS1 protects cardiac tissues against MIRI-induced dysfunction and cardiomyocytes against hypoxia treatment-induced apoptosis [96].
4. Clinical Utility of LncRNAs
4.1. Biomarkers
Due to the high mortality rate of patients with an AMI, early detection and treatment can considerably improve the prognosis of these patients. Finding a myocardial biomarker with high specificity and sensitivity has always been a hotspot in the cardiovascular research field. LncRNAs have a high degree of tissue specificity and are relatively stable secondary structures present within samples of plasma, serum, urine, and other body fluids. Thus, LncRNAs can be novel biomarkers to diagnose and predict the prognosis of several diseases [97, 98] (Table 2).
Table 2.
Clinical utility of LncRNA.
| Clinical utility | Model | Expression | LncRNA | Ref. |
|---|---|---|---|---|
| Biomarkers | AMI | Up | RP11-68I3.11, AC068831.6 | [99] |
| Up | LOC145474, LOC100129518, BRE-AS1, MIR22HG, MIR3945HG, ATP2B1-AS1, CATIPAS1, LINC00528 | [100] | ||
| Up | LIPCAR | [102] | ||
| Up | UCA1 (72–96 h after onset of AMI) | [103] | ||
| Up | KCNQ1OT1 | [104] | ||
| Up | CHAST | [105] | ||
| Up | H19, MIAT, MALAT1 | [106] | ||
| Up | CDR1AS | [94] | ||
| Down | RP11-133L14.5, PAX8-AS1, RP11-259K15.2, RP11-203M5.8, LINC01254 | [99] | ||
| Down | WDR86-AS1, A2M-AS1, and LINC00612 | [100] | ||
| Down | HOTAIR | [101] | ||
| Down | UCA1 (early stage of AMI) | [103] | ||
| Down | ANRIL | [104] | ||
| Down | ZFAS1 | [94] | ||
| CAD | Up | H19 | [107–108] | |
| Up | LIPCAR | [107] | ||
| Up | ENST00000444488.1 | [109] | ||
| Up | OTTHUMT00000387022 | [110] | ||
| Down | uc010yfd.1 | [109] | ||
| Therapy | Mice | Up | MDRL | [88] |
| Up | CARL | [87] | ||
| Down | APF | [52] | ||
| Down | Meg3 | [112] | ||
| Down | NEAT1 | [33] | ||
| Down | H19 | [77] | ||
| Down | MALAT1 | [29] | ||
| Down | NRF | [59] | ||
| Down | Mirt1 | [78] | ||
| Down | ZFAS1 | [95] | ||
| Rat | Up | HOTAIR | [113] | |
| Down | Mirt1 | [79] | ||
| Down | ZFAS1 | [114] |
An increasing number of clinical studies have measured changes in circulating LncRNAs of AMI patients and have investigated their roles as novel diagnostic biomarkers for AMIs. Lu et al. discovered seven LncRNAs by analyzing LncRNA expression in genomes from 52 acute coronary syndrome patients; moreover, they compared the LncRNA expression pattern between patients with a myocardial infarction or an unstable angina. The overall classification exhibited 90.38% accuracy, 100% sensitivity, and 68.75% specificity. These results suggest that these novel, functional LncRNAs may be candidate diagnostic biomarkers and potential therapeutic targets [99]. Li et al. reported LncRNA expression patterns using two microarray datasets from patients with AMI and healthy samples from the Gene Expression Omnibus (GEO) database. They found that 11 differentially expressed LncRNAs had a diagnostic value in AMI patients and may be used as potential biomarkers for early diagnosis of AMIs [100].
Gao et al. found that HOTAIR expression levels in the serum of AMI patients were significantly decreased compared with that of healthy controls. Thus, the LncRNA HOTAIR may serve as a sensitive predictor for AMI diagnosis [101]. Li et al. found that the LncRNA LIPCAR may serve as a potential biomarker for ST-segment elevation myocardial infarction. The extent of LncRNA elevation also could reflect the severity of a coronary stenosis. Multivariate Cox regression analysis revealed that the Gensini score and LIPCAR levels were independent predictors of major adverse cardiac events following an AMI [102]. Yan et al. found that LncRNA UCA1 levels decreased in plasma samples from patients with early stage AMI and increased in 72–96 h following AMI onset. They proposed that the circulating concentration of LncRNA UCA1 may serve as a potential biomarker for diagnosing AMI [103]. Vausort et al. reported that LncRNAs ANRIL and KCNQ1OT1 within the peripheral blood of patients with AMI can be used as predictors of LV dysfunction following a MIRI [104]. Wang et al. found that LncRNA CHAST expression was upregulated in patients with AMI at 24 h but substantially decreased 3 d following AMI onset, indicating that CHAST may serve as a potential biomarker for diagnosing AMI. Moreover, circulating CHAST levels within 24 h can be used to predict cardiac contractile function in patients with AMI independently [105]. Wang et al. found that the LncRNAs H19, MIAT, and MALAT1 indicated an adverse outcome from an AMI. Peripheral blood mononuclear cell- (PBMC-) derived levels of the LncRNAs H19, MIAT, and MALAT1 were increased in AMI patients suggesting that they may be a useful diagnostic biomarker for AMI [106]. Zhang et al. found that LncRNA ZFAS1 expression was downregulated, but LncRNA CDR1AS expression was upregulated in the peripheral blood of patients with AMI. They also found that either ZFAS1 or CDR1AS correlated with an AMI; however, the combination of the two or their reciprocal changes elicited greater sensitivity and specificity to accurately predict, thereby indicating that they are superior biomarkers for AMI [94].
In addition, there are several studies demonstrating that LncRNAs also serve as novel diagnostic biomarkers of coronary artery disease (CAD). Zhang et al. found that increased plasma levels of H19 and LIPCAR were associated with an increased risk of CAD and may be considered as novel biomarkers for diagnosing CAD [107]. Interestingly, the polymorphisms of H19 are associated with the risk and severity of CAD in a Chinese population, which might provide new markers for the prevention and early intervention of CAD [108]. Li et al. identified aberrantly expressed LncRNAs in CAD patients by analyzing LncRNA expression in the transcriptomes of 93 CAD patients and 48 healthy controls. The authors identified ENST00000444488.1 and uc010yfd.1 as novel LncRNA biomarkers for diagnosing CAD [109]. A recent study found that the LncRNA OTTHUMT00000387022 in PBMCs can be recognized as a novel biomarker due to its high sensitivity and specificity for diagnosing CAD [110].
Although the prospect of using LncRNAs as new diagnostic and prognostic biomarkers in cardiovascular disease is advancing, there are some limitations. For example, some LncRNAs need to be revalidated in a multicenter clinical study with a large sample size to ensure their diagnostic potential as a biomarker. Additionally, some previous clinical studies and samples related lack follow-up information; therefore, the predictive value of the LncRNA cannot be fully assessed. Recently, an in-depth study on the epigenetic mechanisms of differentially expressed LncRNAs in response to a MIRI is in progress; however, the standardized protocol of the test operation has not been confirmed. For instance, from acquiring biological fluids to recovering a small amount of nucleic acids, the quantification of these samples also presents a challenging technical query that should be further explored.
4.2. Therapy
With the recent advancing research on MIRI mechanisms, new therapeutic strategies have been developed to help limit the myocardium injury following an AMI. To reduce the infarct size, preserve the LV function, and improve clinical outcomes of AMI patients, LncRNAs have emerged as important regulatory molecules in the pathological progression of MIRIs [111]. Recent studies have reported that upregulating or downregulating the expression level of LncRNA can relieve a MIRI in mice. These trials also indicated multiple LncRNAs are likely to emerge as novel therapeutic candidates to target and treat AMI (Table 2).
In the I/R mouse model, the intracoronary delivery of adenovirus-expressed LncRNAs MDRL [88] or CARL [87] notably inhibited mitochondrial fission, myocardial cell death, and myocardial infarction sizes and preserved cardiac function. Moreover, silencing any of the following LncRNAs, namely, APF [52], Meg3 [112], NEAT1 [33], H19 [77], MALAT1 [29], NRF [59], Mirt1 [78], or ZFAS1 [95], can reduce myocardial infarction size and preserve cardiac function.
In the rat I/R model, HOTAIR overexpression could suppress inflammation and myocardial cell apoptosis, as well as reduce myocardial infarction sizes [113]. Additionally, inhibiting either Mirt1 [79] or ZFAS1 [114] suppressed myocardial cell apoptosis, reduced myocardial infarction sizes, and improved cardiac function following an I/R-induced injury.
Although the results of using LncRNA therapeutics in animal experimental studies are promising, several limitations persist. Interestingly, owing to their therapeutic potential in preventing and treating a MIRI, gene therapy has become a promising approach to effectively target LncRNAs. However, our present understanding is limited to the animal models, and the safety and effectiveness of its clinical application require further research.
5. Conclusions and Perspectives
We have begun the era of “noncoding.” We look forward to seeing more reports of noncoding RNAs, particularly LncRNAs, that regulate various basic biological processes including cardiovascular biology and related diseases. LncRNAs have the potential to be biomarkers for clinical diagnoses and targets for gene therapy. However, LncRNA-based research in MIRIs is still in its infancy, and further investigation is required to fully elucidate the role of LncRNAs in MIRIs. For example, the majority of available evidence linking LncRNAs and MIRIs is limited in vitro data; moreover, nonconservative sequences and limitations of some experimental methods hinder the progress of in vivo experiments. Whether modifying LncRNAs could fully regulate the cellular response to a MIRI, there are relatively few studies on LncRNAs in this field. These hurdles must be overcome before LncRNAs can be used in clinical trials. With the innovation and advancement of LncRNA-based research, their mechanistic role in MIRIs is expected to be elucidated. Therefore, LncRNAs would be ideal and novel targets for the diagnosis and treatment of patients with AMI.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81570250) and the National Clinical Key Specialty Project Foundation.
Contributor Information
Hongyu Yu, Email: yuhongyudoctor@126.com.
Jichang Zhang, Email: jichang@jlu.edu.cn.
Conflicts of Interest
The authors declare that they have no conflict of interests.
Authors' Contributions
Zhuo Zhao and Wei Sun contributed equally to this work.
References
- 1.Sandoval Y., Thygesen K., Jaffe A. S. The universal definition of myocardial infarction: present and future. Circulation. 2020;141(18):1434–1436. doi: 10.1161/CIRCULATIONAHA.120.045708. [DOI] [PubMed] [Google Scholar]
- 2.Roger V. L., Sidney S., Fairchild A. L., et al. Recommendations for cardiovascular health and disease surveillance for 2030 and beyond: a policy statement from the American Heart Association. Circulation. 2020;141(9):e104–e119. doi: 10.1161/CIR.0000000000000756. [DOI] [PubMed] [Google Scholar]
- 3.Dehmer G. J., Badhwar V., Bermudez E. A., et al. 2020 AHA/ACC key data elements and definitions for coronary revascularization: a report of the American College of Cardiology/American Heart Association task force on clinical data standards (writing committee to develop clinical data standards for coronary revascularization) Journal of the American College of Cardiology. 2020;75(16):1975–2088. doi: 10.1016/j.jacc.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Bogaty P., Brophy J. M. Complete revascularization with multivessel PCI for myocardial infarction. New England Journal of Medicine. 2020;382(16) doi: 10.1056/NEJMc2000278. [DOI] [PubMed] [Google Scholar]
- 5.Neumann F. J., Sousa-Uva M., Ahlsson A., et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. EuroIntervention. 2019;14(14):1435–1534. doi: 10.4244/EIJY19M01_01. [DOI] [PubMed] [Google Scholar]
- 6.Davidson S. M., Ferdinandy P., Andreadou I., et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: Journal of the American College of Cardiology. 2019;73(1):89–99. doi: 10.1016/j.jacc.2018.09.086. [DOI] [PubMed] [Google Scholar]
- 7.Kulek A. R., Anzell A., Wider J. M., Sanderson T. H., Przyklenk K. Mitochondrial quality control: role in cardiac models of lethal ischemia-reperfusion injury. Cells. 2020;9(1):p. 214. doi: 10.3390/cells9010214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibanez B., Heusch G., Ovize M., Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. Journal of the American College of Cardiology. 2015;65(14):1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 9.Jennings R. B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circulation Research. 2013;113(4):428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y., Zhang R., Ying K. Long noncoding RNAs: novel links in respiratory diseases (review) Molecular Medicine Reports. 2015;11(6):4025–4031. doi: 10.3892/mmr.2015.3290. [DOI] [PubMed] [Google Scholar]
- 11.Ponting C. P., Oliver P. L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Kung J. T., Colognori D., Lee J. T. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Z., Sun W., Guo Z., Zhang J., Yu H., Liu B. Mechanisms of lncRNA/microRNA interactions in angiogenesis. Life Sciences. 2020;254:p. 116900. doi: 10.1016/j.lfs.2019.116900. [DOI] [PubMed] [Google Scholar]
- 14.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karreth F. A., Tay Y., Perna D., et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147(2):382–395. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tay Y., Kats L., Salmena L., et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147(2):344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X., Zhu H., Zhu S., Hao M., Li Q. lncRNA expression character associated with ischemic reperfusion injury. Molecular Medicine Reports. 2017;16(4):3745–3752. doi: 10.3892/mmr.2017.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y., Li G., Lu H., et al. Expression profiling and ontology analysis of long noncoding RNAs in post- ischemic heart and their implied roles in ischemia/reperfusion injury. Gene. 2014;543(1):15–21. doi: 10.1016/j.gene.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Huang Z., Ye B., Wang Z., et al. Inhibition of LncRNA-HRIM increases cell viability by regulating autophagy levels during hypoxia/reoxygenation in myocytes. Cellular Physiology and Biochemistry. 2018;46(4):1341–1351. doi: 10.1159/000489149. [DOI] [PubMed] [Google Scholar]
- 20.Bock F. J., Tait S. W. G. Mitochondria as multifaceted regulators of cell death. Nature Reviews. Molecular Cell Biology. 2020;21(2):85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 21.Elmore S. Apoptosis: a review of programmed cell death. Toxicologic Pathology. 2016;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thind G. S., Agrawal P. R., Hirsh B., et al. Mechanisms of myocardial ischemia-reperfusion injury and the cytoprotective role of minocycline: scope and limitations. Future Cardiology. 2015;11(1):61–76. doi: 10.2217/fca.14.76. [DOI] [PubMed] [Google Scholar]
- 23.Fang S. J., Li P. Y., Wang C. M., et al. Inhibition of endoplasmic reticulum stress by neuregulin-1 protects against myocardial ischemia/reperfusion injury. Peptides. 2017;88:196–207. doi: 10.1016/j.peptides.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Mishra P. K., Adameova A., Hill J. A., et al. Guidelines for evaluating myocardial cell death. American Journal of Physiology. Heart and Circulatory Physiology. 2019;317(5):H891–H922. doi: 10.1152/ajpheart.00259.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crow M. T., Mani K., Nam Y. J., Kitsis R. N. The mitochondrial death pathway and cardiac myocyte apoptosis. Circulation Research. 2004;95(10):957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 26.Xu X. Z., Luo B., Xiao Y., Zheng W. Q. Effects of lncRNA MALAT1-mediated β-catenin signaling pathway on myocardial cell apoptosis in rats with myocardial ischemia/reperfusion injury. European Review for Medical and Pharmacological Sciences. 2019;23(21):9557–9565. doi: 10.26355/eurrev_201911_19450. [DOI] [PubMed] [Google Scholar]
- 27.Gong L., Chang H., Xu H. LncRNA MALAT1 knockdown alleviates oxygen-glucose deprivation and reperfusion induced cardiomyocyte apoptotic death by regulating miR-122. Experimental and Molecular Pathology. 2019;111:p. 104325. doi: 10.1016/j.yexmp.2019.104325. [DOI] [PubMed] [Google Scholar]
- 28.Sun R., Zhang L. Long non-coding RNA MALAT1 regulates cardiomyocytes apoptosis after hypoxia/reperfusion injury via modulating miR-200a-3p/PDCD4 axis. Biomedicine & Pharmacotherapy. 2019;111:1036–1045. doi: 10.1016/j.biopha.2018.12.122. [DOI] [PubMed] [Google Scholar]
- 29.Hu H., Wu J., Li D., Zhou J., Yu H., Ma L. Knockdown of lncRNA MALAT1 attenuates acute myocardial infarction through miR-320-Pten axis. Biomedicine & Pharmacotherapy. 2018;106:738–746. doi: 10.1016/j.biopha.2018.06.122. [DOI] [PubMed] [Google Scholar]
- 30.Elmore S. Knocking down PFL can improve myocardial ischemia/reperfusion injury in rats by up-regulating heat shock protein-20. European Review for Medical and Pharmacological Sciences. 2019;23(17):7619–7627. doi: 10.26355/eurrev_201909_18885. [DOI] [PubMed] [Google Scholar]
- 31.Xue X., Luo L. LncRNA HIF1A-AS1 contributes to ventricular remodeling after myocardial ischemia/reperfusion injury by adsorption of microRNA-204 to regulating SOCS2 expression. Cell Cycle. 2019;18(19):2465–2480. doi: 10.1080/15384101.2019.1648960. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Wu H. J., Tang G. M., Shao P. Y., et al. Long non-coding RNA NEAT1 modulates hypoxia/reoxygenation-induced cardiomyocyte injury via targeting microRNA-520a. Experimental and Therapeutic Medicine. 2019;18(3):2199–2206. doi: 10.3892/etm.2019.7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X.‐. J., Wei J., Tian D., et al. NEAT1 promotes myocardial ischemia-reperfusion injury via activating the MAPK signaling pathway. Journal of Cellular Physiology. 2019;234(10):18773–18780. doi: 10.1002/jcp.28516. [DOI] [PubMed] [Google Scholar]
- 34.Zou L., Ma X., Lin S., Wu B., Chen Y., Peng C. Long noncoding RNA-MEG3 contributes to myocardial ischemia-reperfusion injury through suppression of miR-7-5p expression. Bioscience Reports. 2019;39(8) doi: 10.1042/BSR20190210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L., Zhang D., Yu L., Dong H. Targeting MIAT reduces apoptosis of cardiomyocytes after ischemia/reperfusion injury. Bioengineered. 2019;10(1):121–132. doi: 10.1080/21655979.2019.1605812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou T., Qin G., Yang L., Xiang D., Li S. LncRNA XIST regulates myocardial infarction by targeting miR-130a-3p. Journal of Cellular Physiology. 2018;234(6):8659–8667. doi: 10.1002/jcp.26327. [DOI] [PubMed] [Google Scholar]
- 37.Zheng C., Wu Z., Tian L., et al. Long noncoding RNA AK12348 is involved in the regulation of myocardial ischaemia-reperfusion injury by targeting PARP and caspase-3. Heart, Lung & Circulation. 2018;27(5):e51–e58. doi: 10.1016/j.hlc.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 38.Zhao R., Wang X., Wang H., et al. Inhibition of long noncoding RNA BDNF-AS rescues cell death and apoptosis in hypoxia/reoxygenation damaged murine cardiomyocyte. Biochimie. 2017;138:43–49. doi: 10.1016/j.biochi.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 39.Li X., Luo S., Zhang J., et al. lncRNA H19 alleviated myocardial I/RI via suppressing miR-877-3p/Bcl-2-mediated mitochondrial apoptosis. Mol Ther Nucleic Acids. 2019;17:297–309. doi: 10.1016/j.omtn.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y., Zhou D., Li G., et al. Long non coding RNA-UCA1 contributes to cardiomyocyte apoptosis by suppression of p27 expression. Cellular Physiology and Biochemistry. 2015;35(5):1986–1998. doi: 10.1159/000374006. [DOI] [PubMed] [Google Scholar]
- 41.Shi H. J., Wang M. W., Sun J. T., et al. A novel long noncoding RNA FAF inhibits apoptosis via upregulating FGF9 through PI3K/AKT signaling pathway in ischemia-hypoxia cardiomyocytes. Journal of Cellular Physiology. 2019;234(12):21973–21987. doi: 10.1002/jcp.28760. [DOI] [PubMed] [Google Scholar]
- 42.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian J., Popal M. S., Zhao Y., Liu Y., Chen K., Liu Y. Interplay between exosomes and autophagy in cardiovascular diseases: novel promising target for diagnostic and therapeutic application. Aging and Disease. 2019;10(6):1302–1310. doi: 10.14336/AD.2018.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma S., Wang Y., Chen Y., Cao F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochimica et Biophysica Acta. 2015;1852(2):271–276. doi: 10.1016/j.bbadis.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Lin X. L., Xiao W. J., Xiao L. L., Liu M. H. Molecular mechanisms of autophagy in cardiac ischemia/reperfusion injury (review) Molecular Medicine Reports. 2018;18(1):675–683. doi: 10.3892/mmr.2018.9028. [DOI] [PubMed] [Google Scholar]
- 46.Su Q., Liu Y., Lv X. W., et al. Inhibition of lncRNA TUG1 upregulates miR-142-3p to ameliorate myocardial injury during ischemia and reperfusion via targeting HMGB1- and Rac1-induced autophagy. Journal of Molecular and Cellular Cardiology. 2019;133:12–25. doi: 10.1016/j.yjmcc.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 47.Li Z., Zhang Y., Ding N., et al. Inhibition of lncRNA XIST improves myocardial I/R injury by targeting miR-133a through inhibition of autophagy and regulation of SOCS2. Mol Ther Nucleic Acids. 2019;18:764–773. doi: 10.1016/j.omtn.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu S. Y., Dong B., Fang Z. F., Hu X. Q., Tang L., Zhou S. H. Knockdown of lncRNA AK139328 alleviates myocardial ischaemia/reperfusion injury in diabetic mice via modulating miR-204-3p and inhibiting autophagy. Journal of Cellular and Molecular Medicine. 2018;22(10):4886–4898. doi: 10.1111/jcmm.13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J. J., Bie Z. D., Sun C. F. Long noncoding RNA AK088388 regulates autophagy through miR-30a to affect cardiomyocyte injury. Journal of Cellular Biochemistry. 2019;120(6):10155–10163. doi: 10.1002/jcb.28300. [DOI] [PubMed] [Google Scholar]
- 50.Tong G., Wang Y., Xu C., et al. Long non-coding RNA FOXD3-AS1 aggravates ischemia/reperfusion injury of cardiomyocytes through promoting autophagy. American Journal of Translational Research. 2019;11(9):5634–5644. [PMC free article] [PubMed] [Google Scholar]
- 51.Kong F., Jin J., Lv X., et al. Long noncoding RNA RMRP upregulation aggravates myocardial ischemia- reperfusion injury by sponging miR-206 to target ATG3 expression. Biomedicine & Pharmacotherapy. 2019;109:716–725. doi: 10.1016/j.biopha.2018.10.079. [DOI] [PubMed] [Google Scholar]
- 52.Wang K., Liu C.-Y., Zhou L.-Y., et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nature Communications. 2015;6(1):p. 6779. doi: 10.1038/ncomms7779. [DOI] [PubMed] [Google Scholar]
- 53.Yin G., Yang X., Li Q., Guo Z. GATA1 activated lncRNA (Galont) promotes anoxia/reoxygenation-induced autophagy and cell death in cardiomyocytes by sponging miR-338. Journal of Cellular Biochemistry. 2018;119(5):4161–4169. doi: 10.1002/jcb.26623. [DOI] [PubMed] [Google Scholar]
- 54.Liang H., Su X., Wu Q., et al. LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy. 2020;16(6):1077–1091. doi: 10.1080/15548627.2019.1659610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C.-Y., Zhang Y.-H., Li R.-B., et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nature Communications. 2018;9(1) doi: 10.1038/s41467-017-02280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhe-Wei S., Li-Sha G., Yue-Chun L. The role of necroptosis in cardiovascular disease. Frontiers in Pharmacology. 2018;9:p. 721. doi: 10.3389/fphar.2018.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J., Liu D., Zhang M., Zhang Y. Programmed necrosis in cardiomyocytes: mitochondria, death receptors and beyond. British Journal of Pharmacology. 2019;176(22):4319–4339. doi: 10.1111/bph.14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feoktistova M., Leverkus M. Programmed necrosis and necroptosis signalling. The FEBS Journal. 2015;282(1):19–31. doi: 10.1111/febs.13120. [DOI] [PubMed] [Google Scholar]
- 59.Wang K., Liu F., Liu C. Y., et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death and Differentiation. 2016;23(8):1394–1405. doi: 10.1038/cdd.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J. X., Zhang X. J., Li Q., et al. MicroRNA-103/107 regulate programmed necrosis and myocardial ischemia/reperfusion injury through targeting FADD. Circulation Research. 2015;117(4):352–363. doi: 10.1161/CIRCRESAHA.117.305781. [DOI] [PubMed] [Google Scholar]
- 61.Widgerow A. D. Ischemia-reperfusion injury: influencing the microcirculatory and cellular environment. Annals of Plastic Surgery. 2014;72(2):253–260. doi: 10.1097/SAP.0b013e31825c089c. [DOI] [PubMed] [Google Scholar]
- 62.Kurian G. A., Rajagopal R., Vedantham S., Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited. Oxidative Medicine and Cellular Longevity. 2016;2016 doi: 10.1155/2016/1656450.1656450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Granger D. N., Kvietys P. R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biology. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ham P. B., 3rd, Raju R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Progress in Neurobiology. 2017;157:92–116. doi: 10.1016/j.pneurobio.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez-Montero J., Brito R., Gajardo A. I., Rodrigo R. Myocardial reperfusion injury and oxidative stress: therapeutic opportunities. World Journal of Cardiology. 2018;10(9):74–86. doi: 10.4330/wjc.v10.i9.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang L., Guo B., Liu S., Miao C., Li Y. Inhibition of the LncRNA Gpr19 attenuates ischemia-reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR-324-5p/Mtfr1 axis. IUBMB Life. 2020;72(3):373–383. doi: 10.1002/iub.2187. [DOI] [PubMed] [Google Scholar]
- 67.Zhang W., Li Y., Wang P. Long non-coding RNA-ROR aggravates myocardial ischemia/reperfusion injury. Brazilian Journal of Medical and Biological Research. 2018;51(6):p. e6555. doi: 10.1590/1414-431x20186555. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Li L., Zhang M., Chen W., et al. LncRNA-HOTAIR inhibition aggravates oxidative stress-induced H9c2 cells injury through suppression of MMP2 by miR-125. Acta Biochim Biophys Sin (Shanghai) 2018;50(10):996–1006. doi: 10.1093/abbs/gmy102. [DOI] [PubMed] [Google Scholar]
- 69.Long B., Li N., Xu X. X., et al. Long noncoding RNA FTX regulates cardiomyocyte apoptosis by targeting miR-29b-1-5p and Bcl2l2. Biochemical and Biophysical Research Communications. 2018;495(1):312–318. doi: 10.1016/j.bbrc.2017.11.030. [DOI] [PubMed] [Google Scholar]
- 70.Tong G., von Garlen N. N. A., Wowro S. J., et al. Post-TTM rebound pyrexia after ischemia-reperfusion injury results in sterile inflammation and apoptosis in cardiomyocytes. Mediators of Inflammation. 2019;2019 doi: 10.1155/2019/6431957.6431957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ong S.-B., Hernández-Reséndiz S., Crespo-Avilan G. E., et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacology & Therapeutics. 2018;186:73–87. doi: 10.1016/j.pharmthera.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marchant D. J., Boyd J. H., Lin D. C., Granville D. J., Garmaroudi F. S., McManus B. M. Inflammation in myocardial diseases. Circulation Research. 2012;110(1):126–144. doi: 10.1161/CIRCRESAHA.111.243170. [DOI] [PubMed] [Google Scholar]
- 73.Steffens S., Montecucco F., Mach F. The inflammatory response as a target to reduce myocardial ischaemia and reperfusion injury. Thrombosis and Haemostasis. 2017;102(2):240–247. doi: 10.1160/TH08-12-0837. [DOI] [PubMed] [Google Scholar]
- 74.Li J., Zhang H., Zhang C. Role of inflammation in the regulation of coronary blood flow in ischemia and reperfusion: mechanisms and therapeutic implications. Journal of Molecular and Cellular Cardiology. 2012;52(4):865–872. doi: 10.1016/j.yjmcc.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li X., Dai Y., Yan S., et al. Down-regulation of lncRNA KCNQ1OT1 protects against myocardial ischemia/reperfusion injury following acute myocardial infarction. Biochemical and Biophysical Research Communications. 2017;491(4):1026–1033. doi: 10.1016/j.bbrc.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 76.Luo M., Sun Q., Zhao H., Tao J., Yan D. Long noncoding RNA NEAT1 sponges miR-495-3p to enhance myocardial ischemia-reperfusion injury via MAPK6 activation. Journal of Cellular Physiology. 2019;235(1):105–113. doi: 10.1002/jcp.28791. [DOI] [PubMed] [Google Scholar]
- 77.Luo H., Wang J., Liu D., et al. The lncRNA H19/miR-675 axis regulates myocardial ischemic and reperfusion injury by targeting PPARα. Molecular Immunology. 2019;105:46–54. doi: 10.1016/j.molimm.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 78.Li X., Zhou J., Huang K. Inhibition of the lncRNA Mirt1 attenuates acute myocardial infarction by suppressing NF-κB activation. Cellular Physiology and Biochemistry. 2017;42(3):1153–1164. doi: 10.1159/000478870. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y., Wang T., Zhang M., Chen P., Yu Y. Down-regulation of myocardial infarction associated transcript 1 improves myocardial ischemia-reperfusion injury in aged diabetic rats by inhibition of activation of NF-κB signaling pathway. Chemico-Biological Interactions. 2019;300:111–122. doi: 10.1016/j.cbi.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 80.Li T., Tian H., Li J., et al. Overexpression of lncRNA Gm2691 attenuates apoptosis and inflammatory response after myocardial infarction through PI3K/Akt signaling pathway. IUBMB Life. 2019;71(10):1561–1570. doi: 10.1002/iub.2081. [DOI] [PubMed] [Google Scholar]
- 81.Paradies G., Paradies V., Ruggiero F. M., Petrosillo G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. American Journal of Physiology. Heart and Circulatory Physiology. 2018;315(5):H1341–H1352. doi: 10.1152/ajpheart.00028.2018. [DOI] [PubMed] [Google Scholar]
- 82.Yang M., Linn B. S., Zhang Y., Ren J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2019;1865(9):2293–2302. doi: 10.1016/j.bbadis.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 83.Lesnefsky E. J., Chen Q., Tandler B., Hoppel C. L. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annual Review of Pharmacology and Toxicology. 2017;57(1):535–565. doi: 10.1146/annurev-pharmtox-010715-103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maneechote C., Palee S., Chattipakorn S. C., Chattipakorn N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. Journal of Cellular and Molecular Medicine. 2017;21(11):2643–2653. doi: 10.1111/jcmm.13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cooper H. A., Eguchi S. Inhibition of mitochondrial fission as a novel therapeutic strategy to reduce mortality upon myocardial infarction. Clinical Science (London, England) 2018;132(20):2163–2167. doi: 10.1042/CS20180671. [DOI] [PubMed] [Google Scholar]
- 86.Maneechote C., Palee S., Kerdphoo S., Jaiwongkam T., Chattipakorn S. C., Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clinical Science (London, England) 2018;132(15):1669–1683. doi: 10.1042/CS20180510. [DOI] [PubMed] [Google Scholar]
- 87.Wang K., Long B., Zhou L.-Y., et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nature Communications. 2014;5(1):p. 3596. doi: 10.1038/ncomms4596. [DOI] [PubMed] [Google Scholar]
- 88.Wang K., Sun T., Li N., et al. MDRL lncRNA regulates the processing of miR-484 primary transcript by targeting miR-361. PLoS Genet. 2014;10(7):p. e1004467. doi: 10.1371/journal.pgen.1004467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bagur R., Hajnoczky G. Intracellular Ca2+ sensing: its role in calcium homeostasis and signaling. Molecular Cell. 2017;66(6):780–788. doi: 10.1016/j.molcel.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mattiazzi A., Argenziano M., Aguilar-Sanchez Y., Mazzocchi G., Escobar A. L. Ca2 + sparks and Ca2 + waves are the subcellular events underlying Ca2 + overload during ischemia and reperfusion in perfused intact hearts. Journal of Molecular and Cellular Cardiology. 2015;79:69–78. doi: 10.1016/j.yjmcc.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pittas K., Vrachatis D. A., Angelidis C., Tsoucala S., Giannopoulos G., Deftereos S. The role of calcium handling mechanisms in reperfusion injury. Current Pharmaceutical Design. 2018;24(34):4077–4089. doi: 10.2174/1381612825666181120155953. [DOI] [PubMed] [Google Scholar]
- 92.Yang C. F. Clinical manifestations and basic mechanisms of myocardial ischemia/reperfusion injury. Ci Ji Yi Xue Za Zhi. 2018;30(4):209–215. doi: 10.4103/tcmj.tcmj_33_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gomez L., Li B., Mewton N., et al. Inhibition of mitochondrial permeability transition pore opening: translation to patients. Cardiovascular Research. 2009;83(2):226–233. doi: 10.1093/cvr/cvp063. [DOI] [PubMed] [Google Scholar]
- 94.Zhang Y., Sun L., Xuan L., et al. Reciprocal changes of circulating long non-coding RNAs ZFAS1 and CDR1AS predict acute myocardial infarction. Scientific Reports. 2016;6(1):p. 22384. doi: 10.1038/srep22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang Y., Jiao L., Sun L., et al. LncRNAZFAS1as a SERCA2a inhibitor to cause intracellular Ca2+ overload and contractile dysfunction in a mouse model of myocardial infarction. Circulation Research. 2018;122(10):1354–1368. doi: 10.1161/CIRCRESAHA.117.312117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiao L., Li M., Shao Y., et al. lncRNA- _ZFAS1_ induces mitochondria-mediated apoptosis by causing cytosolic Ca2+ overload in myocardial infarction mice model. Cell Death & Disease. 2019;10(12):p. 942. doi: 10.1038/s41419-019-2136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhong Z., Hou J., Zhang Q., et al. Differential expression of circulating long non-coding RNAs in patients with acute myocardial infarction. Medicine (Baltimore) 2018;97(51):p. e13066. doi: 10.1097/MD.0000000000013066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y., Zhang L., Wang Y., et al. MicroRNAs or long noncoding RNAs in diagnosis and prognosis of coronary artery disease. Aging and Disease. 2019;10(2):353–366. doi: 10.14336/AD.2018.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu Y., Meng X., Wang L., Wang X. Analysis of long non-coding RNA expression profiles identifies functional lncRNAs associated with the progression of acute coronary syndromes. Experimental and Therapeutic Medicine. 2018;15(2):1376–1384. doi: 10.3892/etm.2017.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li L., Cong Y., Gao X., Wang Y., Lin P. Differential expression profiles of long non-coding RNAs as potential biomarkers for the early diagnosis of acute myocardial infarction. Oncotarget. 2017;8(51):88613–88621. doi: 10.18632/oncotarget.20101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gao L., Liu Y., Guo S., et al. Circulating long noncoding RNA HOTAIR is an essential mediator of acute myocardial infarction. Cellular Physiology and Biochemistry. 2017;44(4):1497–1508. doi: 10.1159/000485588. [DOI] [PubMed] [Google Scholar]
- 102.Li M., Wang Y. F., Yang X. C., et al. Circulating long noncoding RNA LIPCAR acts as a novel biomarker in patients with ST-segment elevation myocardial infarction. Medical Science Monitor. 2018;24:5064–5070. doi: 10.12659/MSM.909348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yan Y., Zhang B., Liu N., et al. Circulating long noncoding RNA UCA1 as a novel biomarker of acute myocardial infarction. BioMed Research International. 2016;2016 doi: 10.1155/2016/8079372.8079372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vausort M., Wagner D. R., Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circulation Research. 2014;115(7):668–677. doi: 10.1161/CIRCRESAHA.115.303836. [DOI] [PubMed] [Google Scholar]
- 105.Wang X., Wang L., Ma Z., et al. Early expressed circulating long noncoding RNA CHAST is associated with cardiac contractile function in patients with acute myocardial infarction. International Journal of Cardiology. 2020;302:15–20. doi: 10.1016/j.ijcard.2019.12.058. [DOI] [PubMed] [Google Scholar]
- 106.Wang X. M., Li X. M., Song N., Zhai H., Gao X. M., Yang Y. N. Long non-coding RNAs H19, MALAT1 and MIAT as potential novel biomarkers for diagnosis of acute myocardial infarction. Biomedicine & Pharmacotherapy. 2019;118:p. 109208. doi: 10.1016/j.biopha.2019.109208. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Z., Gao W., Long Q.-Q., et al. Increased plasma levels of lncRNA H19 and LIPCAR are associated with increased risk of coronary artery disease in a Chinese population. Scientific Reports. 2017;7(1) doi: 10.1038/s41598-017-07611-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gao W., Zhu M., Wang H., et al. Association of polymorphisms in long non-coding RNA H19 with coronary artery disease risk in a Chinese population. Mutation Research. 2015;772:15–22. doi: 10.1016/j.mrfmmm.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 109.Li L., Wang L., Li H., et al. Characterization of LncRNA expression profile and identification of novel LncRNA biomarkers to diagnose coronary artery disease. Atherosclerosis. 2018;275:359–367. doi: 10.1016/j.atherosclerosis.2018.06.866. [DOI] [PubMed] [Google Scholar]
- 110.Cai Y., Yang Y., Chen X., et al. Circulating ‘lncRNA OTTHUMT00000387022’ from monocytes as a novel biomarker for coronary artery disease. Cardiovascular Research. 2016;112(3):714–724. doi: 10.1093/cvr/cvw022. [DOI] [PubMed] [Google Scholar]
- 111.van Zuylen V. L., den Haan M. C., Geutskens S. B., et al. Post-myocardial infarct inflammation and the potential role of cell therapy. Cardiovascular Drugs and Therapy. 2015;29(1):59–73. doi: 10.1007/s10557-014-6568-z. [DOI] [PubMed] [Google Scholar]
- 112.Wu H., Zhao Z. A., Liu J., et al. Long noncoding RNA Meg3 regulates cardiomyocyte apoptosis in myocardial infarction. Gene Therapy. 2018;25(8):511–523. doi: 10.1038/s41434-018-0045-4. [DOI] [PubMed] [Google Scholar]
- 113.Zhang D., Wang B., Ma M., Yu K., Zhang Q., Zhang X. lncRNA HOTAIR protects myocardial infarction rat by sponging miR-519d-3p. Journal of Cardiovascular Translational Research. 2019;12(3):171–183. doi: 10.1007/s12265-018-9839-4. [DOI] [PubMed] [Google Scholar]
- 114.Wu T., Wu D., Wu Q., et al. Knockdown of long non-coding RNA-ZFAS1 protects cardiomyocytes against acute myocardial infarction via anti-apoptosis by regulating miR-150/CRP. Journal of Cellular Biochemistry. 2017;118(10):3281–3289. doi: 10.1002/jcb.25979. [DOI] [PubMed] [Google Scholar]