

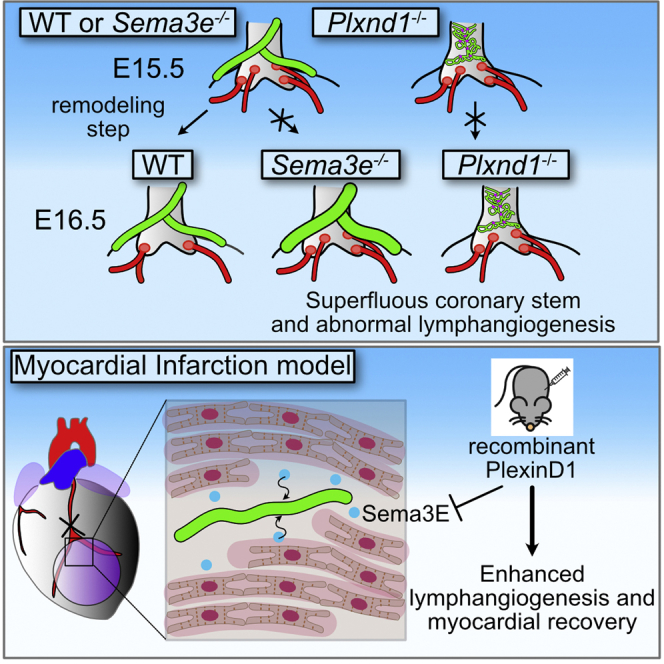
Summary
Blood and lymphatic vessels surrounding the heart develop through orchestrated processes from cells of different origins. In particular, cells around the outflow tract which constitute a primordial transient vasculature, referred to as aortic subepicardial vessels, are crucial for the establishment of coronary artery stems and cardiac lymphatic vessels. Here, we revealed that the epicardium and pericardium-derived Semaphorin 3E (Sema3E) and its receptor, PlexinD1, play a role in the development of the coronary stem, as well as cardiac lymphatic vessels. In vitro analyses demonstrated that Sema3E may demarcate areas to repel PlexinD1-expressing lymphatic endothelial cells, resulting in proper coronary and lymphatic vessel formation. Furthermore, inactivation of Sema3E-PlexinD1 signaling improved the recovery of cardiac function by increasing reactive lymphangiogenesis in an adult mouse model of myocardial infarction. These findings may lead to therapeutic strategies that target Sema3E-PlexinD1 signaling in coronary artery diseases.
Subject Areas: Biological Sciences, Cell Biology, Developmental Biology
Graphical abstract
Highlights
-
•
Sema3E-PlexinD1 signaling regulates coronary and cardiac lymphatic vessel development
-
•
Sema3E demarcate areas to repel PlexinD1-expressing lymphatic endothelial cells
-
•
Inhibition of Sema3E-PlexinD1 signaling improves the recovery after myocardial injury
Biological Sciences; Cell Biology; Developmental Biology
Introduction
The cardiac vascular system is composed of coronary and lymphatic vessel networks. The coronary arteries (CAs) are the first branches of the aorta and supply blood to the heart muscle. During mammalian CA development, endothelial cells derived from a variety of sources, including the proepicardium (Katz et al., 2012; Mikawa and Gourdie, 1996; Pérez-Pomares et al., 2002), endocardium (Wu et al., 2012), and sinus venosus (Red-Horse et al., 2010; Tian et al., 2013b), constitute an initial vascular plexus. Subsequent remodeling leads to maturation of the coronary vasculature and production of proper stems that are connected to the aorta through two ostia originating from the aortic sinuses (Chen et al., 2014a, 2014b). These connections are formed by the process in which endothelial cells from circumferential capillaries surrounding the outflow tract (i.e., the peritruncal vessels) penetrate the aortic wall at the level of the aortic sinuses (Ando et al., 2004; Bogers et al., 1989; Poelmann et al., 1993; Tian et al., 2013a; Waldo et al., 1990). Then, a bilateral pair of ostia is selected from the multiple connections between the peritruncal vessels and the aorta, resulting in the formation of normal left and right CA (Chen et al., 2014a, 2014b). Congenital anomalies that affect the coronary ostia and stems may cause myocardial ischemia and often lead to sudden death (Angelini, 2007; Hauser, 2005). Despite the clinical importance of CA stem formation, the mechanisms underlying this process remain largely unknown.
Recent studies have implicated transient embryonic microvessels surrounding the aorta, termed aortic subepicardial vessels (ASVs), in CA stem formation (Chen et al., 2014a, 2014b). ASVs are thought to be the primary components of peritruncal vessels, later remodeled into CA stems. In contrast, a subset of early ASV endothelial cells express prospero homeobox transcription protein 1 (Prox1), a master regulator of lymphangiogenesis (Chen et al., 2014a, 2014b). ASVs regress at later embryonic stages, whereas lymphatic vessels expressing lymphatic endothelial markers, vascular endothelial growth factor receptor 3 (VEGFR3) and lymphatic vessel endothelial hyaluronic acid receptor 1 (LYVE1), subsequently appear around the aorta (Klotz et al., 2015; Maruyama et al., 2019). However, no detailed analysis has described the possible relationship between ASVs and lymphatic vessels.
Semaphorin 3E (Sema3E) and its receptor PlexinD1 are members of the Sema-Plexin system, originally identified as an axon guidance regulator but currently recognized as a signaling axis regulating diverse (patho)physiological processes, including the morphogenesis and homeostasis of various organs (Corà et al., 2014; Epstein et al., 2015; Kruger et al., 2005; Sandireddy et al., 2019). Although other Sema members, such as Sema3A, 3C, and 4A, interact with PlexinA1 and PlexinD1 via Neuropilin (Nrp)-dependent mechanisms, only Sema3E directly binds to PlexinD1 (Corà et al., 2014; Epstein et al., 2015). Recent evidence has implicated this signaling axis in various vascular processes such as angiogenesis and pathological conditions (Casazza et al., 2010; Fukushima et al., 2011; Moriya et al., 2010; Shimizu et al., 2013). During development, Sema3e−/− and Plxnd1−/− mutant mice reportedly exhibit aberrant vascularization of the intersomitic vessels (Gu et al., 2005). In particular, Plxnd1 mutant mice show various cardiovascular anomalies, including high take-off CAs (Gitler et al., 2004; Zhang et al., 2009) and abnormal artery-lymph alignment with reduced lymphatic vessel branching in mouse embryonic back skin (Liu et al., 2016). Although Sema3c knockout is reported to cause a laterally dislocated connection to the aortic sinuses (Théveniau-Ruissy et al., 2008), the role of the Sema-Plexin system in coronary and lymphatic vessel formation remains largely unknown.
Here, we revealed a critical role of the Sema3E-PlexinD1 signaling axis in CA stem and cardiac lymphatic vessel development. Sema3e and Plxnd1 mutant mice exhibited excess CA ostia and abnormal cardiac lymphangiogenesis. On investigating the mechanism underlying these anomalies, a close relationship between CA and lymphatic vessel development was observed, suggesting the importance of the Sema3E-PlexinD1 signaling axis in ASV development. In addition, our in vitro studies revealed that Sema3E-PlexinD1 signaling was mediated via the actin cytoskeleton and focal adhesion rearrangement to elicit repulsive movement in lymphatic endothelial cells. Furthermore, the inhibition of Sema3E-PlexinD1 signaling significantly improved cardiac function after myocardial injury, possibly by altering the lymphangiogenic response. Our results could provide insights into therapeutic strategies for CA diseases, which remain the leading cause of death in most developed countries.
Results
Sema3e−/− and Plxnd1−/− mice exhibit severe CA defects
We performed a phenotypic analysis of Sema3e−/− and Plxnd1−/− embryos to obtain clues regarding the mechanism underlying CA stem development. As almost all Plxnd1−/− mice died by embryonic day 16.5 (E16.5) to E17.0, as previously reported (Zhang et al., 2009), owing to severe systemic edema and hemorrhage, accompanied by systemic vascular malformations (data not shown), we performed immunostaining with the endothelial marker PECAM (platelet endothelial cell adhesion molecule) and confocal imaging on Sema3e and Plxnd1 mutant hearts at E16.5. Although most all of the control hearts exhibited two coronary ostia at the aortic sinuses as expected, the number of coronary ostia was significantly increased in Sema3e−/− or Plxnd1−/− homozygous hearts (Figures 1A–1D). Occasionally, un-remodeled coronary vessels (CVs) were directly connected to the persistent truncus arteriosus (PTA) wall in Plxnd1−/− hearts (Figure 1C). Then, we analyzed postnatal Sema3e−/− animals to exclude the possibility that CA phenotypes were induced by a developmental delay. At postnatal day 7 (P7), Sema3e−/− hearts still showed an increased number of coronary ostia (Figures 1E and 1F). The supernumerary CA ostia were positioned higher or lower in aortic sinuses but not in the pulmonary artery sinus. Thus, Sema3E-PlexinD1 signaling may control the number of ostia and CA patterns.
Figure 1.

Sema3e−/− and Plxnd1−/− mice show CA defects
(A, C, and E) Whole-mount confocal images of hearts labeled for PECAM. (A) Images of control and Sema3e−/− hearts at E16.5. (C) Images of control and Plxnd1−/− hearts at E16.5. Plxnd1−/− mice showed PTA. (E) Images of control and Sema3e−/− hearts at P7. Control hearts exhibited two ostia at the aortic sinuses (red arrowheads). Sema3e−/− and Plxnd1−/− hearts showed an increased number of CA ostia and displaced coronary ostia (red arrowheads). Numbers on each schema corresponded to numbers on each figure.
(B, D, and F) Numbers of coronary ostia in control, Sema3e−/−, and Plxnd1−/− hearts were quantified. Ao, aorta; PA, pulmonary artery; PTA, persistent truncus arteriosus; CA, coronary artery. All results are expressed as the means ± standard error of the mean (SEM), and statistical analyses were performed using a non-parametric Mann-Whitney's U test. Each dot represents a value obtained from one sample. ∗P < 0.05, ∗∗P < 0.01. Scale bars, 100 μm (A, C, and E).
Sema3E-PlexinD1 signaling is required for coronary artery remodeling
We performed a sequential analysis of CA development in wild-type mice from E11.5 to E18.5 using PECAM staining to identify the mechanism through which supernumerary CA ostia are formed in mutant mice (Figures S1A and S1B). During normal development, the peritruncal vessels running through the dorsal side of the aorta first emerged at E11.5. These vessels, which were previously described as ASVs (Chen et al., 2014a, 2014b), extended toward the base of the aorta and reached future ostial sites at E12.5, where they communicated with CVs extending from the ventricular wall and connected to the aortic lumen at multiple sites by E13.5. At E15.0, CVs penetrated the aortic sinus at multiple sites in a reticular pattern. At E15.5, the CVs merged to form multiple large mature CAs. These multiple CAs had been remodeled into two mature ostia by E16.5. In Sema3e−/− and Plxnd1−/− mice, multiple CA ostia had typically formed by E15.5 (Figures 2A and 2B). Thus, the process of CA ostium formation involves penetration of the ASVs into the aortic lumen at E12.5 to E13.5, the connection of the CVs to the aortic sinuses at E13.5 to E15.5, and remodeling into two mature connections at normal ostial sites at E15.5 to E16.5. Sema3E-PlexinD1 signaling likely contributes to the last step of remodeling to produce two coronary stems at the proper positions (Figure 2C).
Figure 2.

The remodeling step in CA ostium development is disturbed in Sema3e−/− and Plxnd1−/− mice
(A) Whole-mount confocal images of control, Sema3e−/−, and Plxnd1−/− hearts at E13.0 and E15.5. Orange asterisks represent connections formed between ASVs and the aorta at E13.0. Red arrowheads represent large connections formed between the aorta and CVs at E15.5.
(B) Numbers of coronary ostia in control, Sema3e−/−, and Plxnd1−/− hearts at E15.5 were quantified.
(C) Schematic representation of CA ostium development in control, Sema3e−/−, and Plxnd1−/− mice. Ao, aorta; PA, pulmonary artery; PTA, persistent truncus arteriosus; CV, coronary vessel; CA, coronary artery; ASVs, aortic subepicardial vessels. All results are expressed as the means ± standard error of the mean (SEM), and statistical analyses were performed using a one-way analysis of variance (ANOVA) between groups and post hoc multiple comparisons using Bonferroni's test. Each dot represents a value obtained from one sample. ns, P ≥ 0.05. Scale bars, 50 μm (A, upper row), 100 μm (A, lower row).
Sema3E is expressed in the epicardium, whereas PlexinD1 is expressed in the endothelium
To explore the involvement of Sema3E-PlexinD1 signaling in CA development, we examined Sema3E and PlexinD1 expression patterns in the developing mouse heart through in situ hybridization and immunohistochemistry. From the embryonic (E10.5) to postnatal (P7) stages, PlexinD1 was broadly expressed in the endocardium and endothelium of large and small vessels, including CAs (Figure S2A). PlexinD1 was also expressed in lymphatic endothelial cells (LECs) at E16.5 (Figure S2C). These PlexinD1 signals were abolished in Plxnd1−/− hearts at E16.5, confirming their specificity in blood and lymphatic endothelial cells (Figures S2B and S2C). Subsequently, we analyzed Sema3e expression using in situ hybridization. Sema3e was expressed in the bronchial and esophageal epithelium, as well as somites at E11.5, E13.5, and E15.5 (Figures 3A, 3E, and 3I). In the heart, Sema3e was expressed in the mesenchyme at the base of the outflow tract, the pericardium, and the epicardium at E11.5 (Figures 3A–3D). Although mesenchymal expression was not observed at E13.5 and E15.5, pericardial and epicardial expressions were well maintained (Figures 3E–3L). At E13.5 and E15.5, a stronger expression was observed in the aortic epicardium and sinus venosus (Figures 3F, 3H, 3J, and 3L). These Sema3e signals were abolished in the Sema3e−/− hearts at E14.5, confirming their specificity (Figures 3M–3P).
Figure 3.

Sema3e is expressed in the developing heart
(A–P) Sagittal section in situ hybridization for Sema3e mRNA. (A–L) At E11.5, E13.5, and E15.5, Sema3e was expressed in the somites, nerves, and esophageal and bronchial epithelium. In the heart, Sema3e was expressed in the pericardium (black arrow heads), epicardium (black arrows), aortic epicardium (red arrows), and sinus venosus (red arrowheads). At E11.5, Sema3e was expressed at the base of the outflow tracts (green arrow). (M–P) Sema3e expression was absent in Sema3e−/− embryos at E14.5.
(Q) Schematic view of Sema3e expression. The boxed areas are magnified in the right images. OFT, outflow tract; V, ventricle; Ao, aorta; PA, pulmonary artery; LV, left ventricle; LB, lung bud; SV, sinus venosus; EP, epicardium; P, pericardium; E, esophagus; MB, main bronchus; L, lung; RA, right atrium; IVC, inferior vena cava. Scale bars, 500 μm (A, E, I, M), 100 μm (B, F, J, N)
Disruption of Sema3E-PlexinD1 signaling leads to cardiac lymphatic vessel malformation
Next, we examined whether lymphatic vessel development was affected in mice lacking Sema3E-PlexinD1 signaling by employing vascular and lymphatic endothelial markers. We first tested PECAM, Prox1, and VEGFR3 expression patterns. Consistent with our previous work (Maruyama et al., 2019), PECAM+/Prox1+/VEGFR3+ lymphatic vessels were distributed around the aorta and dorsal side of ventricles in control and Sema3e−/− embryos at E16.5 (Figures 4A and 4B). In Sema3e−/− embryos, lymphatic vessels were dilated and hyperplastic when compared with control vessels (Figures 4A, 4B, 4D, and 4F). Conversely, Plxnd1−/− embryos showed a fine plexus of lymphatic vessels with increased branch points surrounding the PTA wall (Figures 4C–4F). On the dorsal side of ventricles, Plxnd1−/− embryos also exhibited fine lymphatic vessels without large remodeled lymphatic vessels (Figures 4C, 4D, and 4F). We further tested LYVE1 expression patterns in control, Sema3e−/− and Plxnd1−/− embryos. Consistent with VEGFR3 expression patterns, in Sema3e−/− hearts, LYVE1+ lymphatic vessels were abnormally thickened when compared with control hearts (Figures 5A and 5B). By contrast, LYVE1+ areas were relatively sparse and discontinued without forming a reticulated structure around the PTA wall of Plxnd1−/− embryos (Figure 5C). In addition, only a small part of PECAM+/Prox1+ cells expressed LYVE1 (Figure 5C). Consistently, the vessel area of LYVE1+ lymphatic vessels was increased in Sema3e−/− embryos at E16.5, whereas mature lymphatic vessel formation appeared defective in Plxnd1−/− embryos (Figures 5D and 5E). These phenotypes were also observed in the dorsal ventricular wall (Figures S3A–S3C).
Figure 4.

Sema3E-PlexinD1 signaling contributes to cardiac lymphatic vessel development
(A–C) Whole-mount confocal images of control, Sema3e−/−, or Plxnd1−/− hearts labeled for PECAM, Prox1, and VEGFR3.
(D–F) Quantification of vessel diameter (D), number of branch points (E), and vessel area of ventral and dorsal side of the ventricles (F) (five points were counted in each sample). VEGFR3+ vessel areas were calculated using AngioTool. Ao, aorta; PA, pulmonary artery; PTA, persistent truncus arteriosus. All results are expressed as the means ± standard error of the mean (SEM), and statistical analyses were performed using a one-way analysis of variance (ANOVA) between groups and post hoc multiple comparisons using Bonferroni's test. Each dot represents a value obtained from one sample. ∗P < 0.05, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001. Scale bars, 100 μm (A-C).
Figure 5.

LYVE1 expression patterns in control, Sema3e−/−, and Plxnd1−/− hearts
(A–C) Whole-mount confocal images of control, Sema3e−/−, or Plxnd1−/− hearts labeled for PECAM, Prox1, and LYVE1 at E16.5. (A and B) In control and Sema3e−/− hearts, PECAM, Prox1, and LYVE1 were co-expressed in lymphatic vessels (red arrow heads). (C) In Plxnd1−/− hearts, hyperplastic PECAM+ capillaries were recognized in the PTA wall (white arrowheads). Fewer PECAM+/Prox1+/LYVE1+ lymphatic vessels (red arrowheads) were observed on the PTA wall.
(D) Confocal images of control, Sema3e−/−, or Plxnd1−/− hearts at E16.5 captured for AngioTool analyses. The LYVE1+ vessels (red) and branch points (blue) were depicted, which enabled the quantitative assessment of vessel parameters.
(E) Quantification of the LYVE1+ vessel area. Ao, aorta; PA, pulmonary artery; PTA, persistent truncus arteriosus. All results are expressed as the means ± standard error of the mean (SEM), and statistical analyses were performed using a one-way analysis of variance (ANOVA) between groups and post hoc multiple comparisons using Bonferroni's test. Each dot represents a value obtained from one sample. ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001. Scale bars, 100 μm (A-C)
To compare peripheral and cardiac lymphatic vessel development, we analyzed lymphatic vessel development in Sema3e−/− and Plxnd1−/− embryos using dorsal skin. In Sema3e−/− and Plxnd1−/− embryos, the distance between from each migration front to the midline was shorter than control (Figures S4A and S4C). Sema3e−/− and Plxnd1−/− embryos displayed dilated lymphatic vessels and reduced lymphatic vessel branching (Figures S4A, S4D, and S4E), as previously reported (Liu et al., 2016). Excessive sprouts were observed in Sema3e−/− and Plxnd1−/− embryos (Figures S4B and S4F). These results indicate the importance of Sema3E and PlexinD1 in cardiac and peripheral lymphatic vessel development; however, these appear to exert opposite effects in the heart. The phenotypic discrepancy between Sema3e−/− and Plxnd1−/− mice may be attributed to other semaphorin members, such as Sema3A and Sema3C (see the Discussion).
Sema3E-PlexinD1 signaling induces the repulsion and cytoskeletal collapse of LECs
Reportedly, Sema3E-PlexinD1 signaling negatively regulates angiogenesis in blood endothelial cells by inhibiting VEGF-A-VEGFR2 downstream signaling (Fukushima et al., 2011; Moriya et al., 2010). However, there have been no reports on the effect of Sema3E-PlexinD1 signaling on LECs. To investigate the molecular mechanism underlying the observed biological effects of Sema3E-PlexinD1 signaling on LECs, we performed in vitro studies using cultured human primary LECs (HLECs). To investigate the effect of Sema3E-PlexinD1 signaling on cell motility, we first performed scratch-wound-healing assays using recombinant Sema3E protein. Compared with control cells, wound healing was significantly impaired in Sema3E-treated HLECs (Figures S5A and S5B). Wound healing involves both migration and proliferation; accordingly, we analyzed the effect of Sema3E on cell proliferation using the MTT assay. Sema3E inhibited the proliferation of HLECs (Figure S5C), suggesting that decreased proliferation may partly explain the Sema3E-induced impairment of wound healing. In LECs, VEGFR3 activation by VEGF-C induces proliferation, migration, and survival, partly through extracellular signal-regulated kinase 1/2 (ERK1/2) and AKT phosphorylation (Adams and Alitalo, 2007). However, Sema3E stimulation did not affect VEGF-C-induced phosphorylation of ERK1/2 and AKT (Figure S5D). We further analyzed the effect of Sema3E on HLEC motility using HEK-293T cells expressing Sema3E. When Sema3E-expressing HEK-293T cells were seeded onto subconfluent HLECs, they repelled HLECs, whereas HEK-293T cells containing empty expression vector did not (Figures 6A–6C) (referred to as LEC repulsive assay). We also verified that Sema3E specifically acted through PlexinD1 in HLECs by using small interfering RNA (siRNA). First, we tested the efficiency of siRNA by western blotting against PlexinD1 and confirmed the downregulation of PlexinD1 protein levels (Figures S5E and S5F). When Sema3E-expressing HEK-293T cells were seeded onto subconfluent siControl transfected HLECs, they repelled HLECs, whereas control HEK-293T cells containing empty expression vector did not demonstrate similar behavior (Figures 6D–6F). These repulsive activities were nullified by siPlexinD1 (Figures 6E and 6F).
Figure 6.

Sema3E-expressing cells repel lymphatic endothelial cells
(A) Time schedule for the LEC repulsive assays.
(B and C) (B) Control HEK-293T cells transfected with pFLAG-T2A-EGFP vectors (control) or HEK-293T cells transfected with pFLAG-Sema3E-T2A-EGFP (Sema3E) were seeded on top of HLECs. Shown are merged phase-contrast and fluorescence images taken after 48 hr. Cell-free area is marked with a dotted black line and quantified in (C) (n = 4, biological replicates in all conditions. Three areas were counted in each condition).
(D) Time schedule for the siRNA transfected LEC repulsive assays.
(E and F) (E) HLECs were transfected with control siRNA or siPlexinD1. Control HEK-293T cells transfected with pFLAG-T2A-EGFP vectors (control) or HEK-293T cells transfected with pFLAG-Sema3E-T2A-EGFP (Sema3E) were seeded on top of siRNA transfected HLECs. Cell-free area is marked with a dotted white line and the boxed area is magnified in the right box. Cell-free area was quantified in (F) (n = 3, biological replicates in all conditions. Three areas were counted in each condition).
(G) Time schedule for the LEC spheroid sprouting assay.
(H) Spheroids and control or Sema3E-expressing HEK-293T cells were embedded in type I collagen gel and incubated in EGM2 medium. After 48 hr, spheroids and HEK-293T cells were stained for F-actin, GFP, and DAPI. The blue box represented spheroid branches facing to the GFP-expressing HEK-293T cell, and the green box represented control branches. Boxed area was magnified in lower boxes.
(I and J) Spheroids surrounded by 1 or 2 GFP-expressing cells were subjected to quantification. For analyzing the branch number and branch length, spheroids were divided into two parts (GFP+ cells containing side and the opposite side), depicted in the schema. For statistical analysis, spheroids located near a single GFP-expressing cell were divided by a line orthogonally crossed with a straight line traveling a GFP-expressing cell and the spheroid core. Spheroids surrounded by two GFP-expressing cells were divided by a line through the spheroid core parallel to a line connecting the two GFP-expressing cells. (n = 5, biological replicates in all conditions. Four spheroids were quantified in each condition). All results are expressed as the means ± standard error of the mean (SEM), and statistical analyses were performed using a one-way analysis of variance (ANOVA) between groups, post hoc multiple comparisons, Bonferroni's test (F), or non-parametric Mann-Whitney's U test (C, I, J). ∗∗P < 0.01, ∗P < 0.05. Scale bar, 100 μm (B, E, H)
To mimic the in vivo repulsive activity of Sema3E-PlexinD1 signaling against lymphatic vessel growth and patterning, we employed aggregates of HLECs (LEC spheroid sprouting assay) with control- or Sema3E-expressing HEK-293T cells (Figures 6G–6J). We first attempted to establish aggregates with siPlexinD1 transfected HLECs; however, we were unable to obtain proper spheroids to evaluate the repulsive activity of Sema3E-PlexinD1 as both the number and branch length of sprouting were dramatically decreased when compared with controls (data not shown). Thus, we used untreated HLECs to form spheroids. In spheroids co-cultured with Sema3E-expressing HEK-293T cells, branch elongation was hindered in the vicinity of Sema3E-expressing cells, as reflected by a significant decrease in the number and length of sprouts in the direction of GFP+ (Sema3E-expressing) cells compared to the opposite direction (Figures 6H–6J), leading to cell-free area formation.
Live imaging revealed that Sema3E-expressing cells caused a remarkable change in the shape of HLECs, rapidly repelling HLECs (Figure S6A and Video S1). A compilation of the tracks corresponding to individual HLEC migration paths showed that Sema3E-expressing HEK-293T cells repelled HLECs, which migrated with significantly increased velocity when compared with cells far from Sema3E-expressing cells (Figures S6B, S6C, and Video S2). Moreover, the velocity appeared to correlate with the distance from Sema3E-expressing cells (Figure S6D). Notably, HLECs avoided areas where Sema3E-expressing cells passed, even though they did not contact each other directly. Considering that Sema3E is a secreted protein, it may demarcate areas that repel the formation of lymphatic vessels in a dose-dependent manner.
Semaphorins were characterized in part based on their ability to drastically alter actin cytoskeletal dynamics in neuronal and vascular processes (Hamm et al., 2016; Hung et al., 2010). Furthermore, recent evidence has shown that Sema3E signaling through PlxnD1 regulates vascular patterning by modulating the cytoskeleton and focal adhesion structures (Aghajanian et al., 2014; Sakurai et al., 2010). To elucidate the effect of Sema3E-PlexinD1 signaling on morphological changes in HLECs, we evaluated evoked cytoskeletal remodeling and focal adhesion in HLECs by staining actin fibers with fluorescence-labeled phalloidin and focal adhesion plaques with immunostaining for vinculin. Sema3E treatment inhibited actin stress fiber formation and induced contraction in HLECs when compared with the control (Figure S5G). The cell percentage demonstrating a loss of dense stress fibers increased significantly in the Sema3E-treated groups when compared with the control (Figure S5H). Concurrently, focal adhesion plaques visualized by vinculin staining also decreased in number following Sema3E exposure (Figures S5G and S5I). The loss of actin stress fibers and focal adhesion plaques after Sema3E treatment was abolished by siPlexinD1 (Figures S5J–S5L), indicating that Sema3E specifically acted through PlexinD1 to induce detachment and collapses of the actin cytoskeleton, resulting in increased cell motility.
Collectively, these results suggest that Sema3E, acting through PlexinD1, suppressed HLEC proliferation and repelled them by demarking HLEC-free areas, possibly by regulating the actin cytoskeleton and focal adhesion, which may contribute to proper positioning of lymphatic vessels.
Inhibition of Sema3E improves cardiac function and alters lymphatic phenotypes following cardiac injury
Lymphangiogenesis has recently been implicated in post-cardiac injury syndrome. The stimulation of lymphangiogenesis by VEGF-C improves cardiac function after myocardial infarction (MI) (Henri et al., 2016; Klotz et al., 2015). Sema3e−/− mice presented hyperplastic cardiac lymphatic vessels (Figures 4 and 5). Additionally, our in vitro studies indicated that Sema3E-PlexinD1 signaling affected lymphatic endothelial cell migration and proliferation (Figures 6, S5 and S6). Therefore, we hypothesized that the inhibition of Sema3E-PlexinD1 signaling might improve cardiac function after MI via a lymphangiogenic mechanism. First, we evaluated the protein levels of Sema3E and VEGFR3 in the infarcted area of the left ventricle. Sema3E protein levels increased rapidly one week post-MI. VEGFR3 protein levels also increased following infarction (Figure 7A). Sema3E was produced in the intact myocardium of the infarcted area (Figure 7B). In contrast, PlexinD1 was produced in the LYVE1+/VEGFR3+ lymphatic vessels and capillaries positive for endomucin (EMCN), a marker of endothelial cells, in and/or around the infarcted area (Figure 7C). We performed immunohistochemistry for PECAM and LYVE1 to observe phenotypic changes in the vasculature of MI hearts. On day 28 post-MI, we identified the presence of a boundary between infarcted and non-infarcted areas (Figures S7A and S7C). LYVE1 signals were also colocalized with Iba1, a marker for macrophages and microglia, indicating macrophage infiltration (Figure S7B). In non-infarcted areas, dense capillaries and CVs were observed in regular patterns, along with reactive LYVE1+ lymphangiogenesis (Figure S7C). In infarcted areas, curved, distended abnormal vessels and a few thin LYVE1+ lymphatic vessels were present (Figure S7D). We then injected Phosphate-buffered saline (PBS) or the recombinant human PlexinD1 protein on days 0, 2, 3, 4, and 6 post-MI to inhibit Sema3E signaling. The use of human PlexinD1 protein to antagonize the effect of mouse Sema3E has been validated in previous studies (Casazza et al., 2010; Tata et al., 2014); additionally, we confirmed the inhibitory effect of human PlexinD1 protein against Sema3E using the LEC repulsive assay (Figures S8A and S8B). On day 28 post-MI, stronger lymphangiogenic responses were observed in regions surrounding injured areas in PlexinD1-treated mice when compared with control mice (Figures 7D and S8C). Distended lymphatic vessels were well developed and covered infarcted areas in PlexinD1-treated mice. Conversely, the newly formed lymphatic vasculature in PBS-treated mice covered relatively smaller areas (Figure S8C). We statistically analyzed EMCN, LYVE1, and VEGFR3 staining in vessels in the infarcted area. The number of EMCN+ and LYVE1+ vessels was significantly increased in PlexinD1-treated mice (Figure 7E). We further analyzed the lymphatic phenotypes on day 28 post-MI. PlexinD1-treated mice demonstrated a significant increase in the number of large LYVE1+ and VEGFR3+ vessels (Figure 7E). Notably, PlexinD1-treated mice exhibited significant improvements in cardiac function, as evidenced by the decreased left ventricular dimensions in diastole and systole (Figure 7F), accompanied by an increased ejection fraction and fractional shortening (Figure 7G). Significantly smaller fibrotic areas and left ventricle wall thinning were observed in PlexinD1-treated mice on day 28 post-MI when compared with controls (Figures 7H and S8E). Furthermore, Sema3E-PlexinD1 signaling is important for immunomodulation and neuronal development. To determine whether recombinant PlexinD1 affected the immune response, we evaluated the number of macrophages in the infarcted area with macrophage marker Iba1. However, we observed no difference in the number of Ib11+ macrophages (Figures S8F–S8H). We evaluated cardiac innervation in the infarcted area using the neuronal marker Tuj1 antibody. However, we failed to observe any difference in the number of Tuj1+ neurons (data not shown). In summary, the inhibition of Sema3E signaling in MI hearts increased the number of capillaries and large lymphatic vessels when compared with control mice, thus improving cardiac function.
Figure 7.

Inhibition of Sema3E-PlexinD1 signaling stimulates lymphangiogenesis and improves cardiac function after myocardial infarction
(A) Western blot analysis of Sema3E and VEGFR3 expression levels. Samples were collected from injured left ventricles of MI hearts (n = 3 animals analyzed per time point and n = 3, biological replicates in all conditions).
(B–D) Serial sections of 4-μm of MI hearts were labeled for Sema3E, PlexinD1, EMCN, LYVE1, VEGFR3, or Masson's trichrome solution. (B) Images of immunolabeling for Sema3E and Masson's trichrome staining in MI hearts. Sema3E could be observed in the intact myocardium of the infarcted area (black dotted lines). Boxed areas are magnified in the lower images. (C) Images of Masson's trichrome staining and immunostaining for EMCN, LYVE1, VEGFR3, and PlexinD1. PlexinD1 was expressed in EMCN+ capillaries (black arrowheads) and LYVE1+/VEGFR3+ lymphatic vessels in the infarcted area (black arrows). (D) Paraffin section images of PBS- and recombinant PlexinD1-treated (PlexinD1) MI hearts labeled for Masson's trichrome staining and EMCN, LYVE1, and VEGFR3. Infarcted areas are magnified in the right images. Blue dotted areas represent infarcted regions (C, D).
(E) The numbers of EMCN, LYVE1, and VEGFR3+ vessels in the infarcted area were quantified. Large vessels were defined as vessels with an area >0.003 mm2. n = 11 (PBS) and n = 8 (PlexinD1).
(F) Evaluation of LV cavity dimensions in end-diastole and end-systole of PBS- or recombinant PlexinD1-treated (PlexinD1) MI mice by echocardiography at the indicated times.
(G) Evaluation of the ejection fraction (EF) and fractional shortening (FS) of PBS- or recombinant PlexinD1-treated (PlexinD1) MI mice by echocardiography at the indicated times.
(H) Evaluation of the fibrotic area of PBS- or recombinant PlexinD1-treated mice (PlexinD1) at day 28 post-MI. LV, left ventricle; RV, right ventricle; MI, myocardial infarction. All results are expressed as the means ± standard error of the mean (SEM), and statistical analyses were performed using a one-way analysis of variance (ANOVA) between groups and post hoc multiple comparisons using Bonferroni's test (A) or non-parametric Mann-Whitney's U test (E, F, G, H). Each dot represents a value obtained from one sample. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001. Scale bars, 100 μm (B, C, and D, right side images), 500 μm (D, left side image).
Discussion
The present study reveals a role for Sema3E-PlexinD1 signaling in CA stem development that involves remodeling to produce the two coronary ostia at proper positions in the aortic sinuses. Sema3E-PlexinD1 signaling is essential for the proper formation of cardiac lymphatic vessels. This study further revealed the beneficial effect of Sema3E-PlexinD1 signal inhibition on post-MI cardiac dysfunction in adult mice by modulating angiogenic and lymphangiogenic activities in injured hearts.
CA stems were recently reported to be patterned in two steps: the establishment of peritruncal vessels, which are composed of ASVs and immature CVs derived from the proepicardium (Katz et al., 2012; Mikawa and Gourdie, 1996; Pérez-Pomares et al., 2002), endocardium (Wu et al., 2012), and sinus venosus (Chen et al., 2014a, 2014b; Red-Horse et al., 2010). These peritruncal vessels were remodeled and interconnected with the aortic endothelium (Chen et al., 2014a, 2014b; Ivins et al., 2015; Théveniau-Ruissy et al., 2016). The first step requires several molecules including VEGF-C (Chen et al., 2014a, 2014b), C-X-C motif chemokine ligand 12 (Ivins et al., 2015), and the collagen- and calcium-binding EGF-like domains 1 protein (Bonet et al., 2018). Deletion of these signals results in a lack of ASVs and hypoplastic peritruncal CVs, leading to a decrease in the number of CA stems (Chen et al., 2014a, 2014b; Ivins et al., 2015; Théveniau-Ruissy et al., 2016). Cardiomyocytes along the wall of the aorta contribute to the next step by guiding CA stem formation to aortic sinuses (Chen et al., 2014a, 2014b; Théveniau-Ruissy et al., 2016). In the present study, subsequent remodeling of multiple connections into the two CA stems requires Sema3E-PlexinD1 signaling, and the absence of this signaling induces the formation of excess CA ostia positioned randomly at the aortic root.
Our previous report shows that Prox1+/VEGFR3+ mesh-like structures appear around the outflow tract of embryonic mouse hearts around E12.5. These structures are remodeled into Prox1+/VEGFR3+ thickened lymphatic vessels around E15.0 to E16.5 and thereafter extend to the ventral side of the heart. LYVE1 expression in cardiac lymphatic vessels is also recognized at this stage (Maruyama et al., 2019). Conversely, ASVs appeared on the outflow tract around E11.5 and then expanded. These vessels form channels to the aortic lumen and contain blood cells, indicating them to be blood vessels; however, a subset of early ASV endothelial cells is reported to express Prox1 (Chen et al., 2014a, 2014b). Thus, the Prox1+ and/or VEGFR3+ mesh-like structures on the outflow tract wall described in our previous work seem to overlap with ASVs. Taken together with these findings, ASVs and cardiac lymphatic vessels may share a common Prox1+ cell origin. Furthermore, our recent report also shows that some cardiac lymphatic vessels are derived from the Isl1-expressing pharyngeal mesoderm (Maruyama et al., 2019). Moreover, Théveniau-Ruissy et al., using genetic lineage tracing with Cx40-CreERT2 and avian homo-transplantation, have reported that endothelial cells of the CA stem may have different origins from ventricular CAs. They are likely to arise from mesenchyme distal to the outflow tract (Théveniau-Ruissy et al., 2016). These results suggest that ASVs may appear as a primitive mesenchymal plexus which gives rise to blood vessels including CA stems and lymphatic vessels surrounding the outflow tract. During these developmental processes, Sema3E-PlexinD1 signaling may contribute to CA stem remodeling and normal lymphatic development.
Both Sema3e−/− and Plxnd1−/− mice exhibit excess CA stem formation, but the phenotype related to lymphatic vessel formation is different. In the heart, Sema3e−/− mice showed distended lymphatic vessels, whereas Plxnd1−/− mice exhibited fine lymphatic plexus positive for VEGFR3 with aberrant LYVE1 expression. In the dorsal skin, both Sema3e−/− and Plxnd1−/− mice showed an increased lymphatic vessel diameter and reduced branch numbers. This dissimilarity may be attributed to the involvement of the possible Sema3E antagonism to other semaphorin ligands or co-receptors, such as Sema3A, Sema3C, Sema4A, or Nrp1/2, which may stimulate lymphatic vessel formation upon PlexinD1 activation. In fact, Sema-Plexin signaling shows various redundancies (Gu et al., 2003; Toyofuku et al., 2008). Sema3a−/− mice present right-sided cardiac hypertrophy, sinus bradycardia owing to defective sympathetic patterning and lymphatic valve defects (Behar et al., 1996; Bouvrée et al., 2012; Ieda et al., 2007). Sema3c−/− mice exhibit PTA and an interrupted aortic arch (Feiner et al., 2001). Sema4A has also been shown to interact with PlexinD1 via Nrp-dependent mechanisms, but Sema4A deletion in mice does not lead to cardiovascular phenotypes (Toyofuku et al., 2007). These findings indicate that PlexinD1 is involved in different aspects of cardiovascular development with various ligand requirements. Additionally, a recent finding suggests a possible role for PlexinD1, forming a complex with VEGFR2 and Nrp1, as a shear stress mechanoreceptor without ligand requirements (Mehta et al., 2020). Thus, the phenotypic discrepancy in cardiac lymphatic vessel formation in Sema3e−/− and Plxnd1−/− mice may be attributed to redundancy among other semaphorin ligands in the PlexinD1-mediated stimulation or ligand-independent PlexinD1 signaling.
Our in vitro studies in HLECs suggest that Sema3E acts on PlexinD1 to induce actin filament reorganization and focal adhesion disassembly, leading to retraction and repulsion of HLECs. Remarkably, Sema3E secreted from transfected HEK-293T cells demarcated areas that repel HLECs through PlexinD1. This, in turn, may contribute to the ability of Sema3E to block HLEC proliferation and migration, thereby displaying potent anti-lymphangiogenic activity in vivo. These mechanisms of endothelial repulsion are common in blood endothelial cells (Sakurai et al., 2010) but appear to be independent of ERK1/2 and AKT phosphorylation downstream of VEGF-C-VEGFR3 signaling in LECs. At the molecular level, Sema3E-PlexinD1 signaling in blood endothelial cells relies on the GTPase-activating protein activity of PlexinD1 to inactivate R-Ras and activate Arf6 to modulate the cytoskeleton and cellular adhesion (Sakurai et al., 2010). Recent findings have suggested that actin-depolymerizing factor cofilin and molecules interacting with CasLs are involved in the interaction between Sema-Plexin and the actin cytoskeleton (Hung and Terman, 2011). Further studies are warranted to determine the detailed molecular signaling involved in this process.
Our present study identified Sema3e expression in the pericardium and epicardium, especially around the aortic root. PlexinD1 is broadly expressed in the endothelium. Taken together with the phenotypes of the mutant mice, regional cross talk between the epicardium and endocardium via Sema3E-PlexinD1 signaling may regulate the formation and remodeling of vasculature around the aorta. However, it remains unknown how two specific orifices are selected from multiple connections into aortic sinuses through Sema3E-PlexinD1 signaling, necessitating further study. Sema3e is also expressed in the ventricular epicardium. Thus, Sema3E-PlexinD1 signaling may affect sinus venosus- and epicardium-derived CVs. To test this possibility, sinus venosus- and epicardium-restricted Sema3e mutant mice would be necessary.
The present study further revealed the possible involvement of Sema3E-PlexinD1 signaling in pathological lymphangiogenesis and functional outcomes in post-ischemic myocardial injury using human recombinant PlexinD1 protein. PlexinD1 binds to Sema3E and blocks its effect. Given that coronary endothelial cells also express PlexinD1, the beneficial effect of Sema3E inhibition in post-MI hearts may arise from lymphangiogenesis, as well as from angiogenesis. Recent findings suggest that although Sema3C mainly interacts with PlexinD1 in an Nrp-dependent manner, it could directly interact with PlexinD1, forming complexes with PlexinA4 (Smolkin et al., 2018). Therefore, the effect of PlexinD1 may arise in a part from interaction with Sema3C. Recently, lymphangiogenesis has been implicated in various pathophysiological processes, including micro-edema, fibrosis after MI, and heart failure. Indeed, the new cardiac lymphatic vessels formed after MI remove excess fluid, cytokines, and cell debris and regulate the immune reaction in the injured area, resulting in the stabilization of scar tissue and improvements in cardiac function (Goichberg, 2016; Henri et al., 2016). Sema3E-PlexinD1 signaling may modulate these lymphangiogenic responses by antagonizing other semaphorin ligands and/or regional lymphangiogenic factors, including VEGF-C (Klotz et al., 2015). Thus, the present study provides mechanistic insights into post-MI lymphangiogenesis by identifying Sema3E-PlexinD1 signaling as a key participant. Additionally, Sema3E is known to act on PlexinD1-expressing monocytes/macrophages and modulate immune responses (Shimizu et al., 2013), which may partially explain the effect of Sema3E-PlexinD1 inactivation. Future studies are needed to dissect the contributions of each protein and cell type to this beneficial effect. Nevertheless, the developmental role of Sema3E-PlexinD1 signaling in cardiac vessel formation was extrapolated to pathological processes after MI, and strategies that interfere with Sema3E-PlexinD1 signaling may represent a therapeutic target for improving lymphatic development coupled with immune regulation.
In summary, Sema3E-PlexinD1 signaling has important functions in CA development and pathological processes associated with MI through lymphangiogenic mechanisms. Further characterization of the molecular processes that control the activity of Sema3E-PlexinD1 signaling will provide a fundamental basis for future therapeutic interventions to regenerate the cardiac vasculature and promote effective cardiac repair.
Limitations of the study
The limitations of this study are mainly related to ambiguity of cell types responsible for Sema3E-PlexinD1 signaling. Although we speculate epicardium-derived Sema3E is responsible for remodeling of vasculature around the aorta, definite evidence should be obtained by conditional Sema3E deletion. It is also true for the role of Sema3E-PlexinD1 in adult myocardial infarction, in which the source of Sema3E is likely different from the case of embryonic coronary development. Thus, Sema3e conditional knockout mice crossed with cell type-specific Cre mice would be expected to dissect more specific roles of Sema3E-PlexinD1 signaling in lymphatic vessel and coronary artery development and diseases in the future. In addition, macrophage/monocyte-specific Cre mice crossed with Plexind1 conditional knockout mice would be beneficial to explore the possible involvement of immune components such as macrophages/monocytes in the pathophysiology of myocardial infarction.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kazuaki Maruyama (k.maruyama0608@gmail.com) or Hiroki Kurihara (kuri-tky@umin.net).
Materials availability
All data and materials associated with this study are available in the main text or the Supplemental Information.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We thank all the lab members for participating in helpful discussions. We also thank Tetsuro Watabe (Tokyo Medical and Dental University) and Yasuhiro Yoshimatsu (Niigata University) for advises for the in vitro experiments. We also thank Xin Geng, YenChun Ho, Boksik Cha, Md. Riaj Mahamud, Lijuan Chen, and R. Sathish Srinivasan (Oklahoma Medical Research Foundation) for advices for experimental procedures. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (19H01048 to H. K. and 20K17072 to K. M.); the Platform for Dynamic Approaches to Living System from the Ministry of Education, Culture, Sports, Science and Technology, Japan; and Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency (JST), Japan (JPMJCR13W2).
Author contribution
K.M., Y.A., S.M.-T., and H.K. conceived the study and designed the experiments. K.M. performed the majority of the experiments. K.N. implemented the cardiac injury model, and K.M. and H.N. performed the histological analysis. K.Y. performed echocardiography. A.U., Y.Y., and F.M. contributed to the mutant mice. Y.K. and Y.U. helped with data analyses. K.M., S.M.-T., M.K.S., and H.K. coordinated the experimental work, analyzed the data, and wrote the manuscript, with contributions from all authors.
Declaration of interests
The authors declare no competing interests.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102305.
Contributor Information
Kazuaki Maruyama, Email: k.maruyama0608@gmail.com.
Hiroki Kurihara, Email: kuri-tky@umin.net.
Supplemental information
References
- Adams R.H., Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- Aghajanian H., Choi C., Ho V.C., Gupta M., Singh M.K., Epstein J.A. Semaphorin 3d and semaphorin 3e direct endothelial motility through distinct molecular signaling pathways. J. Biol. Chem. 2014;289:17971–17979. doi: 10.1074/jbc.m113.544833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K., Nakajima Y., Yamagishi T., Yamamoto S., Nakamura H. Development of proximal coronary arteries in quail embryonic heart. Circ. Res. 2004;94:346–352. doi: 10.1161/01.res.0000112963.79064.09. [DOI] [PubMed] [Google Scholar]
- Angelini P. Coronary artery anomalies. Circulation. 2007;115:1296–1305. doi: 10.1161/circulationaha.106.618082. [DOI] [PubMed] [Google Scholar]
- Behar O., Golden J.A., Mashimo H., Schoen F.J., Fishman M.C. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature. 1996;383:525–528. doi: 10.1038/383525a0. [DOI] [PubMed] [Google Scholar]
- Bogers A.J.J.C., Groot A.C.G., Poelmann R.E., Péault B.M., Huysmans H.A. Development of the origin of the coronary arteries, a matter of ingrowth or outgrowth? Anat. Embryol. 1989;180:437–441. doi: 10.1007/bf00305118. [DOI] [PubMed] [Google Scholar]
- Bonet F., Pereira P.N.G., Bover O., Marques S., Inácio J.M., Belo J.A. CCBE1 is required for coronary vessel development and proper coronary artery stem formation in the mouse heart. Dev. Dyn. 2018;247:1135–1145. doi: 10.1002/dvdy.24670. [DOI] [PubMed] [Google Scholar]
- Bouvrée K., Brunet I., Toro R.D., Gordon E., Prahst C., Cristofaro B., Mathivet T., Xu Y., Soueid J., Fortuna V. Semaphorin3A, Neuropilin-1, and PlexinA1 are required for lymphatic valve formation. Circ. Res. 2012;111:437–445. doi: 10.1161/circresaha.112.269316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casazza A., Finisguerra V., Capparuccia L., Camperi A., Swiercz J.M., Rizzolio S., Rolny C., Christensen C., Bertotti A., Sarotto I. Sema3E-Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J. Clin. Invest. 2010;120:2684–2698. doi: 10.1172/jci42118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Heidi I., Poduri A., Numi H., Kivela R., Saharinen P., McKay A.S., Raftrey B., Churko J., Tian X., Zhou B. VEGF-C and aortic cardiomyocytes guide coronary artery stem development. J. Clin. Invest. 2014;124:4899–4914. doi: 10.1172/jci77483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Heidi I., Sharma B., Akerberg B.N., Numi H.J., Kivelä R., Saharinen P., Aghajanian H., McKay A.S., Bogard P.E., Chang A.H. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Dev. Camb. Engl. 2014;141:4500–4512. doi: 10.1242/dev.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corà D., Astanina E., Giraudo E., Bussolino F. Semaphorins in cardiovascular medicine. Trends Mol. Med. 2014;20:589–598. doi: 10.1016/j.molmed.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Epstein J.A., Aghajanian H., Singh M.K. Semaphorin signaling in cardiovascular development. Cell Metab. 2015;21:163–173. doi: 10.1016/j.cmet.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiner L., Webber A.L., Brown C.B., Lu M.M., Jia L., Feinstein P., Mombaerts P., Epstein J.A., Raper J.A. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Dev. Camb. Engl. 2001;128:3061–3070. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- Fukushima Y., Okada M., Kataoka H., Hirashima M., Yoshida Y., Mann F., Gomi F., Nishida K., Nishikawa S.-I., Uemura A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J. Clin. Invest. 2011;121:1974–1985. doi: 10.1172/jci44900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler A.D., Lu M.M., Epstein J.A. PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev. Cell. 2004;7:107–116. doi: 10.1016/j.devcel.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Goichberg P. Therapeutic lymphangiogenesis after myocardial infarction: vascular endothelial growth factor-C paves the way. J. Thorac. Dis. 2016;8:1904–1907. doi: 10.21037/jtd.2016.07.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C., Rodriguez E.R., Reimert D.V., Shu T., Fritzsch B., Richards L.J., Kolodkin A.L., Ginty D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell. 2003;5:45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C., Yoshida Y., Livet J., Reimert D.V., Mann F., Merte J., Henderson C.E., Jessell T.M., Kolodkin A.L., Ginty D.D. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science. 2005;307:265–268. doi: 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- Hamm M.J., Kirchmaier B.C., Herzog W. Sema3d controls collective endothelial cell migration by distinct mechanisms via Nrp1 and PlxnD1Sema3d regulates collective cell migration. J. Cell Biol. 2016;215:415–430. doi: 10.1083/jcb.201603100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser M. Congenital anomalies of the coronary arteries. Heart. 2005;91:1240–1245. doi: 10.1136/hrt.2004.057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henri O., Pouehe C., Houssari M., Galas L., Nicol L., Edwards-Lévy F., Henry J.-P., Dumesnil A., Boukhalfa I., Banquet S. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. 2016;133:1484–1497. doi: 10.1161/circulationaha.115.020143. [DOI] [PubMed] [Google Scholar]
- Hung R.-J., Terman J.R. Extracellular inhibitors, repellents, and semaphorin/plexin/MICAL-mediated actin filament disassembly. Cytoskelet Hoboken N. J. 2011;68:415–433. doi: 10.1002/cm.20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R.-J., Yazdani U., Yoon J., Wu H., Yang T., Gupta N., Huang Z., Berkel W.J.H.van, Terman J.R. Mical links semaphorins to F-actin disassembly. Nature. 2010;463:823–827. doi: 10.1038/nature08724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M., Kanazawa H., Kimura K., Hattori F., Ieda Y., Taniguchi M., Lee J.-K., Matsumura K., Tomita Y., Miyoshi S. Sema3a maintains normal heart rhythm through sympathetic innervation patterning. Nat. Med. 2007;13:604–612. doi: 10.1038/nm1570. [DOI] [PubMed] [Google Scholar]
- Ivins S., Chappell J., Vernay B., Suntharalingham J., Martineau A., Mohun T.J., Scambler P.J. The CXCL12/CXCR4 Axis plays a critical role in coronary artery development. Dev. Cell. 2015;33:455–468. doi: 10.1016/j.devcel.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz T.C., Singh M.K., Degenhardt K., Rivera-Feliciano J., Johnson R.L., Epstein J.A., Tabin C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell. 2012;22:639–650. doi: 10.1016/j.devcel.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz L., Norman S., Vieira J.M., Masters M., Rohling M., Dubé K.N., Bollini S., Matsuzaki F., Carr C.A., Riley P.R. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522:62. doi: 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R.P., Aurandt J., Guan K.-L. Semaphorins command cells to move. Nat. Rev. Mol. Cell Biol. 2005;6:789–800. doi: 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- Liu X., Uemura A., Fukushima Y., Yoshida Y., Hirashima M. Semaphorin 3G provides a repulsive guidance cue to lymphatic endothelial cells via neuropilin-2/PlexinD1. Cell Rep. 2016;17:2299–2311. doi: 10.1016/j.celrep.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Maruyama K., Miyagawa-Tomita S., Mizukami K., Matsuzaki F., Kurihara H. Isl1-expressing non-venous cell lineage contributes to cardiac lymphatic vessel development. Dev. Biol. 2019;452:134–143. doi: 10.1016/j.ydbio.2019.05.002. [DOI] [PubMed] [Google Scholar]
- Mehta V., Pang K.-L., Rozbesky D., Nather K., Keen A., Lachowski D., Kong Y., Karia D., Ameismeier M., Huang J. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature. 2020;578:290–295. doi: 10.1038/s41586-020-1979-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikawa T., Gourdie R.G. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 1996;174:221–232. doi: 10.1006/dbio.1996.0068. [DOI] [PubMed] [Google Scholar]
- Moriya J., Minamino T., Tateno K., Okada S., Uemura A., Shimizu I., Yokoyama M., Nojima A., Okada M., Koga H., Komuro I. Inhibition of semaphorin as a novel strategy for therapeutic angiogenesis. Circ. Res. 2010;106:391–398. doi: 10.1161/circresaha.109.210815. [DOI] [PubMed] [Google Scholar]
- Pérez-Pomares J.-M., Carmona R., González-Iriarte M., Atencia G., Wessels A., Muñoz-Chápuli R. Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. Int. J. Dev. Biol. 2002;46:1005–1013. [PubMed] [Google Scholar]
- Poelmann R.E., Groot A.C.G., Mentink M.M., Bökenkamp R., Hogers B. Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ. Res. 1993;73:559–568. doi: 10.1161/01.res.73.3.559. [DOI] [PubMed] [Google Scholar]
- Red-Horse K., Ueno H., Weissman I.L., Krasnow M.A. Coronary arteries form by developmental reprogramming of venous cells. Nature. 2010;464:549–553. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A., Gavard J., Annas-Linhares Y., Basile J.R., Amornphimoltham P., Palmby T.R., Yagi H., Zhang F., Randazzo P.A., Li X. Semaphorin 3E initiates antiangiogenic signaling through plexin D1 by regulating Arf6 and R-ras. Mol. Cell Biol. 2010;30:3086–3098. doi: 10.1128/mcb.01652-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandireddy R., Cibi D.M., Gupta P., Singh A., Tee N., Uemura A., Epstein J.A., Singh M.K. Semaphorin 3E/PlexinD1 signaling is required for cardiac ventricular compaction. JCI Insight. 2019;4:e125908. doi: 10.1172/jci.insight.125908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I., Yoshida Y., Moriya J., Nojima A., Uemura A., Kobayashi Y., Minamino T. Semaphorin3E-Induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab. 2013;18:491–504. doi: 10.1016/j.cmet.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Smolkin T., Nir-Zvi I., Duvshani N., Mumblat Y., Kessler O., Neufeld G. Complexes of plexin-A4 and plexin-D1 convey semaphorin-3C signals to induce cytoskeletal collapse in the absence of neuropilins. J. Cell Sci. 2018;131:jcs208298. doi: 10.1242/jcs.208298. [DOI] [PubMed] [Google Scholar]
- Tata A., Stoppel D.C., Hong S., Ben-Zvi A., Xie T., Gu C. An image-based RNAi screen identifies SH3BP1 as a key effector of Semaphorin 3E-PlexinD1 signaling. J. Cell Biol. 2014;205:573–590. doi: 10.1083/jcb.201309004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théveniau-Ruissy M., Dandonneau M., Mesbah K., Ghez O., Mattei M.-G., Miquerol L., Kelly R.G. The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ. Res. 2008;103:142–148. doi: 10.1161/circresaha.108.172189. [DOI] [PubMed] [Google Scholar]
- Théveniau-Ruissy M., Pérez-Pomares J.-M., Parisot P., Baldini A., Miquerol L., Kelly R.G. Coronary stem development in wild-type and Tbx1 null mouse hearts. Dev. Dyn. 2016;245:445–459. doi: 10.1002/dvdy.24380. [DOI] [PubMed] [Google Scholar]
- Tian X., Hu T., He L., Zhang H., Huang X., Poelmann R.E., Liu W., Yang Z., Yan Y., Pu W.T., Zhou B. Peritruncal coronary endothelial cells contribute to proximal coronary artery stems and their aortic orifices in the mouse heart. PLoS One. 2013;8:e80857. doi: 10.1371/journal.pone.0080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X., Hu T., Zhang H., He Lingjuan, Huang X., Liu Q., Yu W., He Liang, Yang Zhongzhou, Zhang Z. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res. 2013;23:1075–1090. doi: 10.1038/cr.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T., Yabuki M., Kamei J., Kamei M., Makino N., Kumanogoh A., Hori M. Semaphorin-4A, an activator for T-cell-mediated immunity, suppresses angiogenesis via Plexin-D1. EMBO J. 2007;26:1373–1384. doi: 10.1038/sj.emboj.7601589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T., Yoshida J., Sugimoto T., Yamamoto M., Makino N., Takamatsu H., Takegahara N., Suto F., Hori M., Fujisawa H. Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev. Biol. 2008;321:251–262. doi: 10.1016/j.ydbio.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Waldo K.L., Willner W., Kirby M.L. Origin of the proximal coronary artery stems and a review of ventricular vascularization in the chick embryo. Am. J. Anat. 1990;188:109–120. doi: 10.1002/aja.1001880202. [DOI] [PubMed] [Google Scholar]
- Wu B., Zhang Z., Lui W., Chen X., Wang Y., Chamberlain A.A., Moreno-Rodriguez R.A., Markwald R.R., O’Rourke B.P., Sharp D.J. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Singh M.K., Degenhardt K.R., Lu M.M., Bennett J., Yoshida Y., Epstein J.A. Tie2Cre-mediated inactivation of plexinD1 results in congenital heart, vascular and skeletal defects. Dev. Biol. 2009;325:82–93. doi: 10.1016/j.ydbio.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.