

Summary
The acetylation of ATG9A within the endoplasmic reticulum (ER) lumen regulates the induction of reticulophagy. ER acetylation is ensured by AT-1/SLC33A1, a membrane transporter that maintains the cytosol-to-ER flux of acetyl-CoA. Defective AT-1 activity, as caused by heterozygous/homozygous mutations and gene duplication events, results in severe disease phenotypes. Here, we show that although the acetylation of ATG9A occurs in the ER lumen, the induction of reticulophagy requires ATG9A to engage FAM134B and SEC62 on the cytosolic side of the ER. To address this conundrum, we resolved the ATG9A interactome in two mouse models of AT-1 dysregulation: AT-1 sTg, a model of systemic AT-1 overexpression with hyperacetylation of ATG9A, and AT-1S113R/+, a model of AT-1 haploinsufficiency with hypoacetylation of ATG9A. We identified CALR and HSPB1 as two ATG9A partners that regulate the induction of reticulophagy as a function of ATG9A acetylation and discovered that ATG9A associates with several proteins that maintain ER proteostasis.
Subject areas: Molecular Biology, Cell Biology, Proteomics
Graphical abstract
Highlights
-
•
The ATG9A-FAM134B and ATG9A-SEC62 interaction requires specific structural features
-
•
Opposite Ca++-binding EF hands regulate ATG9A-FAM134B interaction
-
•
HSBP1 and CALR regulate ATG9A-mediated induction of reticulophagy
-
•
Many of the proteins that ensure ER proteostasis display spatial vicinity/cross talk
Molecular Biology; Cell Biology; Proteomics
Introduction
Nε-lysine acetylation in the endoplasmic reticulum (ER) maintains protein homeostasis (proteostasis) within the organelle and secretory pathway (reviewed in Farrugia and Puglielli, 2018). In particular, the acetylation status of the autophagy protein ATG9A regulates the induction of ER-specific autophagy (also referred to as reticulophagy or ER-phagy) and the disposal of toxic protein aggregates that form in the lumen of the organelle (Pehar et al., 2012; Peng et al., 2014, 2016, 2018). ER acetylation is ensured by AT-1 (also referred to as SLC33A1), a membrane transporter that translocates acetyl-CoA from the cytosol to the ER lumen, and ATase1 (also referred to as NAT8B) and ATase2 (also referred to as NAT8), two ER-bound type II membrane proteins that acetylate ER-resident and -transiting proteins (Jonas et al., 2010; Ko and Puglielli, 2009).
In humans, defective ER acetylation, as caused by loss-of-function mutations or gene duplication events of AT-1/SLC33A1, is linked to hereditary spastic paraplegia (heterozygous mutation), developmental delay and premature death (homozygous mutations), and autism spectrum disorder with intellectual disability and progeria-like dysmorphism (gene duplication) (Chiplunkar et al., 2016; Huppke et al., 2012a; Lin et al., 2008; Prasad et al., 2012; Sanders et al., 2011). These disease phenotypes are efficiently mimicked by mouse models of reduced or increased AT-1 activity (Hullinger et al., 2016; Peng et al., 2014, 2018). Importantly, reduced ER acetylation in AT-1S113R/+ hypomorphic mice causes increased (aberrant) induction of reticulophagy, whereas increased ER acetylation in AT-1-overexpressing (AT-1 sTg) mice has the opposite effect (Hullinger et al., 2016; Peng et al., 2014, 2018). Furthermore, inhibition of ATase1 and ATase2, downstream of AT-1, can restore reticulophagy and disposal of protein aggregates from the ER lumen in AT-1 overexpressors (Peng et al., 2016, 2018).
We recently reported that the acetylation status of ATG9A regulates its ability to interact with FAM134B (also referred to as RETREG1) and SEC62 (Peng et al., 2018), two ER membrane-bound proteins that can act as receptors for LC3B through their LC3B-interacting region (LIR) (Fumagalli et al., 2016; Khaminets et al., 2015; Lennemann and Coyne, 2017; Mochida et al., 2015). Importantly, the acetylation of ATG9A occurs on two lysine residues (K359 and K363) that face the ER lumen, whereas the LIR of FAM134B and SEC62 is on the C terminus of both proteins, exposed to the cytosolic side of the ER membrane (Fumagalli et al., 2016; Khaminets et al., 2015; Lennemann and Coyne, 2017; Mochida et al., 2015; Pehar et al., 2012). This topological conundrum is further complicated by the fact that FAM134B appears to assemble within the ER membrane with a reticulon-like conformation and has no available domain exposed to the ER lumen that could engage the acetylated loop of ATG9A (Bhaskara et al., 2019).
In this study, we dissected the structural aspects of the ATG9A-FAM134B and ATG9A-SEC62 interaction, resolved the ATG9A interactome, and took advantage of AT-1S113R/+ and AT-1 sTg mice to discover two ATG9A-interacting proteins—calreticulin (CALR) and heat shock protein beta 1 (HSPB1)—which engage from the lumenal and cytosolic sides of the ER membrane, respectively, to regulate the induction of reticulophagy. Collectively, our results disclose a previously unidentified aspect of the complex mechanisms that allow the ER acetylation machinery to maintain proteostasis within the ER and secretory pathway. Our findings also disclose pathogenic events underlying the disease phenotypes caused by dysfunctional ER acetylation.
Results
FAM134B and SEC62 engage ATG9A on the ER membrane with their cytosolic exposed C terminus
Membrane modeling suggests that FAM134B assembles with a reticulon-like conformation within the ER membrane to force curvature of the lipid bilayer (Figure 1A) (Bhaskara et al., 2019). However, different topology and disorder-prediction platforms suggest the presence of an ER lumen-exposed loop between amino acid (aa) 140 and 208 with two disordered segments of about 10 aa each (aa156–169 and aa187–198). Therefore, it is theoretically possible that disordered segments of FAM134B could move out of the ER membrane to interact with ATG9A within the ER lumen. To test this possibility, we introduced the Asn-Gly-Thr consensus sequence for N-glycosylation within the aa150–200 region of FAM134B. We reasoned that, if these segments were indeed exposed to the ER lumen, we would observe a migration shift caused by N-glycosylation of the Asn residue. Specifically, we introduced the Asn-Gly-Thr sequence (Dempski and Imperiali, 2002; Petrescu et al., 2004) at six independent locations. We also mutated N177 to neutralize a potential N-glycosylation motif within the aa140–208 segment of FAM134B. We only observed a migration shift of FAM134B when the Asn-Gly-Thr sequence was placed at aa200 (Figures 1B and 1C). Furthermore, no migration shift occurred with the N177D substitution (Figure 1B). Collectively, these results indicate that the aa140–208 region of FAM134B has limited movement and, therefore, is unlikely to engage ATG9A from the luminal side of the ER. These results also support the “hairpin” topology model proposed by Bhaskara et al. (2019).
Figure 1.

FAM134B engages ATG9A on the ER membrane.
(A) Schematic view of the predicted topology of FAM134B.
(B and C) (B) Insertion of the consensus sequence for N-glycosylation within the aa140–208 region of FAM134B did not consistently result in glycosylation. (C) Inset in (B). N-glyco, N-glycosylated FAM134B; no-glyco, non-glycosylated FAM134B.
(D and E) Immunoprecipitation of FAM134B pulled down only the immature and ER-based form of ATG9A. (E) Inset in (D). m, mature/N-glycosylated ATG9A; im, immature ATG9A.
(F) Imaris reconstruction of SIM showing interaction of Atg9a (cyan) and Fam134b (green). The ER (red) was labeled with ER3-mCherry. Interacting Atg9a and Fam134b are shown in gray.
FAM134B is solely found in the ER, whereas ATG9A can be found on different cellular locations, which highlights its complex biological functions (Imai et al., 2016; Lee and Finkel, 2009; Nishimura et al., 2017; Ohashi and Munro, 2010; Tamura et al., 2010; Webber et al., 2007; Young et al., 2006). As ATG9A is acetylated by ATase1 and ATase2 within the ER lumen (Pehar et al., 2012; Peng et al., 2018), it reasonable to assume that only the immature and ER-based form of ATG9A can engage FAM134B. To test this hypothesis, we transfected HEK293 cells with both ATG9A-GFP and FAM134B-DDK. Immunoprecipitation of FAM134B only pulled down the ER-based and immature form of ATG9A, thus supporting our hypothesis (Figures 1D and 1E). This was further confirmed by the visualization of ATG9A-FAM134B interaction on the ER surface with high-definition structured illumination microscopy (SIM) (Figure 1F).
The C terminus of FAM134B has intrinsic disordered features, which make it a good candidate for protein-protein interactions. To dissect the mechanism that allows FAM134B to engage ATG9A, we generated truncated versions of FAM134B missing the large C-terminal region (aa235–495), as well as the predicted “lumen loop” (aa141–207). The results show that, indeed, truncation of the entire C-tail greatly reduced the interaction with ATG9A, whereas deletion of the aa141–207 loop did not (Figures 2A and 2B). Iterative Threading ASSEmbly Refinement (I-TASSER) modeling of aa235–495 domain predicts a long arm with a globular end (aa310–495) uniquely positioned to engage in protein-protein interactions (Figure 2C; the arm is shown in blue). This globular domain is predicted to assemble as three very similar sub-domains, each assembled as two opposing short α-helices connected by a coiled region (aa310–380, aa380–440, and aa440–495; see Figure 2D). Deletion of the arm (aa228–310) or sub-domains aa310–380 and aa380–440 increased, whereas deletion of the aa440–495 sub-domain reduced the FAM134B-ATG9A interaction (Figures 2E–2H). Interestingly, the LIR, which is necessary for FAM134B to recruit LC3B, is situated within the aa440–495 domain as a connecting loop between the two small α-helices of the sub-domain (Figure 2I; the LIR is in yellow). However, deletion of the LIR did not affect the ATG9A-FAM134B interaction, indicating that, although necessary for the engagement of LC3B, it is not required for interacting with ATG9A (Figure 2J).
Figure 2.

The globular C terminus of FAM134B is required for ATG9A interaction
(A and B) Deletion of aa235–495 of FAM134B prevented functional interaction with ATG9A. Representative western blot is shown in (A), whereas quantitative changes are shown in (B). Data are represented as mean ± SD. #p < 0.0005.
(C and D) I-TASSER model of the cytosolic C terminus of FAM134B. The entire C-terminus (C) and the globular domain (D) are shown. Different colors highlight the domains targeted in follow-up experiments.
(E–H) Deletion of different domains within the cytosolic C terminus of FAM134B affected the ATG9A-FAM134B interaction differently. Representative western blots are shown in (E and G), whereas quantitative changes are shown in (F and H). Data are represented as mean ± SD. ∗∗p < 0.005; #p < 0.0005.
(I) I-TASSER model of the aa440–495 region of FAM134B with the LIR domain (shown in yellow).
(J) Deletion of the LIR domain of FAM134B did not prevent functional interaction with ATG9A.
Next, we decided to target the ATG9A-SEC62 interaction. The topology of SEC62 is shown in Figure 3A. As with FAM134B, SEC62 was also found to interact with the ER-based immature form of ATG9A following co-immunoprecipitation (Figure 3B; see inset) as well as SIM-mediated visualization (Figure 3C). Truncation of the cytosolic C terminus of SEC62 greatly reduced the ATG9A-SEC62 interaction (Figures 3D and 3E). The C terminus of SEC62 is highly disordered and displays several flexible areas; as a result, we were unable to generate reliable structural models. However, even with SEC62, we observed that truncation of two different segments of the C terminus elicited opposing results, with the aa260–289 deletion stimulating the ATG9A-SEC62 interaction and the aa291–395 deletion (which included the LIR) reducing the ATG9A-SEC62 interaction (Figures 3F–3I).
Figure 3.

SEC62 engages ATG9A on the ER membrane with the cytosolic C terminus
(A) Schematic view of the predicted topology of SEC62.
(B) Immunoprecipitation of SEC62 pulled down only the immature and ER-based form of ATG9A. m, mature/N-glycosylated ATG9A; im, immature ATG9A.
(C) Imaris reconstruction of SIM showing interaction of Atg9a (cyan) and Sec62 (green). The ER (red) was labeled with ER3-mCherry. Interacting Atg9a and Sec62 are shown in gray.
(D and E) Deletion of the cytosolic domain of SEC62 reduced the ATG9A-SEC62 interaction. Representative western blots are shown in (D), whereas quantitative changes are in (E). Data are represented as mean ± SD. #p < 0.0005.
(F–I) Deletion of different domains within the cytosolic C terminus of SEC62 affected the ATG9A-SEC62 interaction differently. Representative western blots are shown in (F and H), whereas quantitative changes are in (G and I). Data are represented as mean ± SD. #p < 0.0005.
The aforementioned results suggest that FAM134B and SEC62 behave similarly in multiple aspects. First, they both engage ATG9A at the ER with their cytosolic C terminus. Second, they both engage ATG9A with the amino acid region that encompasses the LIR. Finally, in both cases, the interaction appears to require specific structural features rather than a simple amino acid sequence, thus suggesting that intrinsic dynamic events within the C terminus of FAM134B and SEC62 are necessary for functional engagement of ATG9A.
Engagement of ATG9A by FAM134B and SEC62 is accompanied by induction of reticulophagy
To determine the functional relevance of the above-mentioned findings, we generated HEK293 cells with stable expression of the ER autophagy tandem reporter (EATR) (Liang et al., 2018). The probe includes a tandem mCherry-eGFP reporter fused to RAMP4 (Figure S1A). As RAMP4 is an integral ER-membrane resident protein, EATR detects induction of reticulophagy (Liang et al., 2018). We transfected the aforementioned HEK293EATR cells with two opposing mutants of FAM134B and SEC62. The results show that deletion mutants that displayed reduced (Δ235–495 for FAM134B and Δ291–395 for SEC62) engagement of ATG9A were unable to stimulate reticulophagy, whereas deletion mutants that increased (Δ310–380 for FAM134B and Δ260–289 for SEC62) the engagement of ATG9A displayed increased induction of reticulophagy (Figures 4A and 4B).
Figure 4.

FAM134B and SEC62 mutants that engage ATG9A can induce reticulophagy
(A and B) Reticulophagy flux induced by different FAM134B (A) or SEC62 (B) deletion mutants. Data are represented as mean ± SD. #p < 0.0005.
(C and D) Sequence alignment of presumed EF hands in FAM134B and SEC62. Both EF1 (C) and EF2 (D) are shown. The Ca++-stabilizing amino acids that were mutated in the following experiments are indicated by arrowheads. The LIR domain is highlighted.
(E and F) EF1 and EF2 of FAM134B differentially affect reticulophagy flux. (E) Flux at 2hr and 4hr. (F) Time-course. Data are represented as mean ± SD. ∗p < 0.05; ∗∗p < 0.005; #p < 0.0005.
(G) I-TASSER model of the opposing EF1 and EF2 of FAM134B showing predicted interaction.
(H and I) EF1 and EF2 of SEC62 have a modest effect on the induction of reticulophagy. (H) Flux at 2hr and 4hr. (I) Time-course. Data are represented as mean ± SD. #p < 0.0005.
I-TASSER modeling of FAM134B predicts two opposing interacting Ca++-binding helix-loop-helix EF hands (Gifford et al., 2007; Kawasaki and Kretsinger, 2017). One of them (EF2) is situated within the aa440–495 loop that is required for binding to ATG9A, whereas the other (EF1) is situated on the opposing arm (Figures 4C and 4D; see also Figures 2C and 2D). The opposing and interacting arrangement of the two EF hands would suggest that intrinsic Ca++ dynamics can influence engagement of LC3B and induction of reticulophagy. To test whether this was indeed the case, we mutated the Ca++-stabilizing residues within each EF hand to Ala, specifically E270, D272, and E274 within EF1 and D464 and E467 within EF2 (Figures 4C and 4D). When transfected into HEK293EATR cells, the EF1 mutant increased, whereas the EF2 mutant decreased the induction of reticulophagy (Figures 4E and 4F). Importantly, the EF1 mutant version of FAM134B displayed a sustained reticulophagy flux that appeared independent of starvation (Figure 4F). In contrast, the EF2 mutant version of FAM134B was unable to respond at all to starvation (Figure 4F). Therefore, it is plausible to assume that Ca++ dynamics between EF1 and EF2 are key for the progression of reticulophagy, perhaps through an “opening” mechanism that allows the aa440–495 loop to engage LC3B (see Figure 4G).
Sequence alignment suggests the presence of two EF hands on the C terminus of SEC62 that follow the same pattern of FAM134B (Figures 4C and 4D). However, secondary structure prediction as well as I-TASSER failed to support this scenario. The inconsistency might be because the C terminus of SEC62 is highly dynamic. Indeed, previous studies have confirmed the Ca++-binding properties of one of the EF hands (labeled as EF1 in Figure 4C) (Linxweiler et al., 2013). Therefore, we decided to mutate predicted Ca++-stabilizing residues within each EF hand of SEC62 (Figures 4C and 4D) and test their functional role. In contrast to FAM134B, only the EF2 mutant version of SEC62 was able to significantly affect the induction of reticulophagy upon starvation (Figure 4H). However, this effect was not as dramatic and immediate as with FAM134B (Figure 4I). Therefore the biological significance of the EF structure and Ca++ dynamics with SEC62 remains unclear.
To validate the results obtained with the EATR system, we also used live imaging to visualize mCherry puncta as an indication of successful transition from the ER membrane to the lysosomes. The results were consistent with EATR for both FAM134B and SEC62 (Figures S1B and S1C). Indeed, we observed increased puncta with the Δ310–380 (for FAM134B) and Δ260–289 (for SEC62) deletion mutants, as well as with the EF1 mutants (Figures S1B and S1C). Next, we generated HEK293 cells with stable expression of an mCherry-RAMP4 fusion system (referred to as mCherry cleavage from ER [CCER]) (Liang et al., 2018). In this system, the mCherry is cleaved from RAMP4 during progression of reticulophagy, thus causing a migration shift on immunoblotting (Figure S1D) (Liang et al., 2018). Again, we observed concordance with EATR- and imaging-based approaches (Figures S1E and S1F).
When taken together, the array of modeling, structural biochemistry, and targeted mutagenesis indicates that engagement of ATG9A by the C terminus of FAM134B and SEC62 is accompanied by the induction of reticulophagy. Furthermore, at least with FAM134B, this process appears to depend on Ca++ dynamics that might influence the engagement of LC3B. The possible regulatory functions of Ca++ are supported by the fact that calcium-modulating agents, thapsigargin and BAPTA, were able to influence the induction of reticulophagy in vivo (Figure S2).
CALR and HSPB1 are ATG9A partners
Collectively, these results reveal previously unidentified mechanistic aspects of the ATG9A-FAM134B and ATG9A-SEC62 interaction. However, they do not resolve the topological conundrum that we discussed earlier. Indeed, the acetylation of ATG9A occurs on the luminal side of the ER, whereas the engagement of FAM134B or SEC62 occurs on the cytosolic side. A possible scenario is that additional ATG9A-interacting proteins might be required to connect a luminal (acetylation) to a cytosolic (FAM134B or SEC62 engagement) event. To address this, we overexpressed wild-type (WT) ATG9A with a myc tag at the C terminus in HEK293 cells and generated stable clones. Next, we purified ATG9A by affinity chromatography following cell permeabilization with either Triton X-100 or DDM, and analyzed the migration profile of ATG9A under native conditions. Depending on the different detergent used, we resolved two large complexes of approximately 720 and 900 kDa (Figure 5A) suggesting that ATG9A exists within a large supercomplex. Importantly, similar high-molecular-mass species have already been described in yeast, indicating that this is an intrinsic aspect of ATG9A biology (He et al., 2008).
Figure 5.

Identification and characterization of the ER-based ATG9A interactome
(A) Migration profile of the ATG9A supercomplex.
(B) ER-based ATG9A-interacting proteins are shown in clusters based on KEGG and STRING classification.
(C) Endogenous Hspb1 and Calr preferentially associate with Atg9a in AT-1 sTg mice. Co-immunoprecipitation was performed with intact native ER vesicles.
(D) Schematic view of the experimental conditions used in (C) and (E–G). Atg9a is shown in blue.
(E–G) Endogenous Hspb1 and Calr preferentially associate with Atg9a in AT-1 sTg mice. Co-immunoprecipitation was performed with permeabilized native ER vesicles. Representative western blots are shown in (E), whereas quantitative changes are shown in (F and G). Data are represented as mean ± SD. ∗∗p < 0.005; #p < 0.0005.
(H and I) Functional interaction of transgenic HSBP1 and ATG9A when expressed in HEK293 cells. (H) IP of HSBP1. (I) IP of ATG9A.
(J and K) Functional interaction of transgenic CALR and ATG9A when expressed in HEK293 cells. (J) IP of CALR. (K) IP of ATG9A.
(L) Imaris reconstruction of SIM showing interaction of Atg9a (cyan) and Hspb1 (green). The ER (red) was labeled with ER3-mCherry. Interacting Atg9a and Hspb1 are shown in gray.
(M) Imaris reconstruction of SIM showing interaction of Atg9a (cyan) and Calr (green). The ER (red) was labeled with ER3-mCherry. Interacting Atg9a and Calr are shown in gray.
Next, we prepared ER-enriched fractions and purified ATG9A-interacting proteins following Triton X-100 or DDM permeabilization and affinity chromatography. This ATG9A supercomplex was then resolved by mass spectrometry (MS). MS identified several proteins, which were broadly classified as cytosolic and ER associated based on our own interest in ER-specific events. The 35 ER-associated proteins included ER luminal proteins, integral ER membrane proteins, and proteins that can associate with the cytosolic side of the ER membrane (Figure 5B and Table S1). KEGG pathway and STRING analysis broadly defined three groups of proteins. (1) Folding/chaperones, which included a subset of 16 proteins responsible for folding of nascent polypeptides. Of note are PDIA3 and PDIA6, two members of the large protein disulfide isomerase family, which recognize free sulfhydryl groups and catalyze the formation of disulfide bonds of nascent glycoproteins. Also of note are UDP-glucose:glycoprotein glucosyltransferase (UGGT1), which is responsible for reglucosylation of unfolded glycoproteins as part of the calreticulin/calnexin cycle, and calreticulin (CALR) and calnexin (CANX) themselves, which are Ca++-binding chaperones responsible for quality control of nascent glycoproteins. (2) ERES/COP, which included nine proteins involved in the assembly of ER exit sites (ERES) as well as formation and trafficking of vesicles between the ER and the Golgi apparatus. Of note are SEC22B, SEC23B, SEC24C, and SEC31A, which are essential for the organization of the ERES and the packaging of correctly folded polypeptides for transport to the Golgi apparatus. Also of note are COPA, COPB1, COPB2, COPG1, and COPE, all members of the large coatomer superfamily. They are cytosolic proteins that are recruited on the surface of the ER and Golgi apparatus to mediate anterograde and retrograde ER-Golgi transport of cargo proteins. (3) Cytosolic chaperones, which included a group of six cytosolic members of the heat shock protein superfamily that can be recruited on the ER membrane as part of their chaperone-like activity.
To support the MS findings, we isolated ER membranes, immunoprecipitated ER-bound endogenous Atg9a, and probed for all target proteins identified in Figure 5B. We were able to confirm in vivo association with 32 of the proteins identified by MS (see Figures 5C and S3A). Only exceptions were a subset of three proteins (Sec24c, Sec31a, and Cdc42), which could not be detected in a consistent fashion. As mentioned above, we launched this MS study to identify possible ATG9A-interacting proteins that specifically regulate the ability of ATG9A to engage FAM134B and SEC62 as a function of ATG9A acetylation. Therefore, we took advantage of AT-1S113R/+ and AT-1 sTg mice, which display reduced and increased acetylation of Atg9a, respectively (Peng et al., 2014, 2018). We reasoned that ATG9A-interacting proteins that are relevant for our study should display different association with ATG9A as a function of ATG9A's own acetylation status. Among the 35 ER-associated proteins that were identified in the ATG9A interactome, only six displayed preferential association with endogenous Atg9a in one of our models: heat shock protein beta-1 (Hspb1; also referred to as Hsp27), Calr, protein disulfide-isomerase A3 (Pdia3), peptidyl-prolyl cis-trans isomerase B (Ppib), coatomer subunit alpha (Copa), and coatomer subunit gamma 1 (Copg1). Specifically, Hspb1, Calr, Ppib, and Copg1 appeared to preferentially associate with Atg9a in AT-1 sTg mice, which exhibit hyperacetylated Atg9a and reduced reticulophagy (Dieterich et al., 2019; Peng et al., 2018), whereas Copa appeared to preferentially associate with Atg9a in AT-1S113R/+ mice, which exhibit hypoacetylation of Atg9a and increased reticulophagy (Peng et al., 2014, 2016) (Figures 5C and S3A). This initial co-immunoprecipitation experiment was done by pulling down endogenous Atg9a from intact native ER membranes (Figure 5D). A limitation of this approach is that the ER vesicles are intact and, therefore, can include proteins that incorporate or associate with the vesicles but are not specifically bound to Atg9a (Figure 5D). To resolve this limitation, we permeabilized ER vesicles with 0.5% Triton X-100 before immunoprecipitation of endogenous Atg9a (Figure 5D). We found a striking divergence between AT-1S113R/+ and AT-1 sTg mice with Hspb1 and Calr, but not with Pdia3, Ppib, Copa, or Copg1 (Figures 5E and S3B). Interestingly, both Hspb1 and Calr were found to preferentially associate with ER-bound Atg9a of AT-1 sTg mice (Figures 5E–5G). Finally, we co-transfected human ATG9A-GFP with either HSPB1-DDK or CALR-DDK in HEK293 cells. Co-immunoprecipitation experiments confirmed that both HSPB1 and CALR can interact with ATG9A (Figures 5H–5K). Importantly, immunoprecipitation of CALR, an ER luminal protein, only pulled down the immature and ER-based form of ATG9A (Figure 5J), whereas immunoprecipitation of HSBP1, a cytosolic protein, pulled down both the immature and mature versions of ATG9A (Figure 5H). These results provide further significance to our findings. Finally, high-definition SIM confirmed that Atg9a-Hspb1 (Figure 5L) and Atg9a-Calr (Figure 5M) interaction can occur at the ER.
Collectively, the aforementioned results identified HSBP1 and CALR as previously unidentified ATG9A-interacting proteins. They also revealed that both HSBP1 and CALR preferentially associate with acetylated ATG9A, which is unable to engage FAM134B or SEC62 and trigger reticulophagy (Peng et al., 2018). Our MS-based studies also revealed a surprising association of ER-bound ATG9A with a series of proteins that are involved with essential aspects of ER biology (discussed later).
CALR and HSPB1 play different roles in the regulation of reticulophagy
To dissect the role of CALR in ATG9A-mediated induction of reticulophagy, we overexpressed human CALR in HEK293 cells and then determined levels of acetylated ATG9A. Surprisingly, we found a marked increase in the levels of acetylated ATG9A (Figures 6A and 6B), suggesting that CALR can either acetylate ATG9A or stabilize already acetylated ATG9A. To differentiate between these two possibilities, we analyzed the acetylation profile of the ER following overexpression of CALR, but did not observe significant differences (Figure 6C). We also determined acetyltransferase activity of affinity-purified CALR, but were not able to detect significant acetyl-CoA:lysine acetyltransferase activity in vitro (data not shown).
Figure 6.

CALR and HSPB1 differentially regulate the induction of reticulophagy
(A and B) Overexpression of CALR-stabilized acetylated ATG9A. Representative western blots are shown in (A), whereas quantitative changes are shown in (B). Data are represented as mean ± SD. ∗p < 0.05.
(C) Overexpression of CALR did not affect the acetylation profile of the ER.
(D) Overexpression of CALR did not affect the induction of reticulophagy. Data are represented as mean ± SD.
(E and F) siRNA-mediated downregulation of CALR stimulated the induction of reticulophagy. (E) Reticulophagy flux was measured at 2 and 4 h. (F) Western blot to document the downregulation of CALR. siNEG were used as negative siRNA controls. Data are represented as mean ± SD. #p < 0.0005.
(G–H) I-TASSER structure of dimeric (G) and monomeric (H) forms of HSPB1 with mutations and deletions targeted in (I–O).
(I) Overexpression of HSPB1 stimulated the induction of reticulophagy, whereas disease-associated mutants or downregulation of HSPB1 did not. Reticulophagy flux was measured at 2 and 4 h. Data are represented as mean ± SD. #p < 0.0005.
(J) Western blot to document the downregulation of HSPB1. siNEG were used as negative siRNA controls.
Next, we used the EATR system to determine whether CALR itself could influence the induction of reticulophagy. Interestingly, overexpression of CALR did not elicit any effect (Figure 6D), whereas small interfering RNA (siRNA)-mediated downregulation of CALR caused a significant increase in induction and progression of reticulophagy (Figures 6E and 6F).
Overall, the aforementioned results are consistent with a model where CALR recognizes—and perhaps stabilizes—already acetylated ATG9A, thus preventing the engagement of FAM134B and/or SEC62 by ATG9A (discussed later). The limiting element of this model would be the acetylation status of ATG9A. As a result, overexpression of CALR would not affect the induction of autophagy because the acetylated pool of endogenous ATG9A is already “sequestered” by endogenous CALR. However, downregulation of endogenous CALR would prevent “sequestration” of ATG9A, thus favoring engagement of FAM134B and/or SEC62 and induction/progression of reticulophagy.
To dissect the role of HSPB1, we took advantage of the already reported disease association with axonal Charcot-Marie-Tooth type 2F (CMT2F) and distal hereditary motor neuropathy 2B (HMN2B). Specifically, we targeted the R136W single mutation and the R140G/K141Q double mutation, as well as the N terminus (Almeida-Souza et al., 2010; Echaniz-Laguna et al., 2017; Ikeda et al., 2009; Muranova et al., 2015; Nefedova et al., 2013a, 2013b). It is worth mentioning that HSPB1 exists in equilibrium between different assembly states: monomers, dimers, and oligomers (Alderson et al., 2019; Arrigo, 2017). The R136W and R140G/K141Q mutations preclude the dimerization of HSPB1, whereas deletion of the N terminus prevents the formation of HSPB1 oligomers (Alderson et al., 2019; Arrigo, 2017). Therefore the mutations and deletion that we chose provide biological as well as disease-relevant information (see Figures 6G and 6H).
Expression of WT HSPB1 stimulated the induction and progression of reticulophagy as assessed by EATR. However, all the point mutations and terminal deletions that we used in this study appeared to be defective and unable to stimulate EATR flux at the same level of the WT protein (Figure 6I). Interestingly, siRNA-mediated downregulation of HSPB1 did not elicit any effect (Figures 6I and 6J), suggesting that although WT HSPB1 can stimulate the induction of reticulophagy, it is not essential for its normal progression.
When taken together, the aforementioned results suggest that CALR and HSPB1 are ATG9A-interacting proteins; they play opposing roles and act on the opposing sides of the ER membrane to regulate the ATG9A-mediated induction of reticulophagy.
Discussion
The ER acetylation machinery includes three essential components: an ER membrane transporter, AT-1, which translocates acetyl-CoA from the cytosol to the ER lumen, and two ER-bound acetyl-CoA:lysine acetyltransferases, ATase1 and ATase2, which acetylate ER-resident and -transiting proteins (Farrugia and Puglielli, 2018; Jonas et al., 2010; Ko and Puglielli, 2009). Loss-of-function mutations and gene duplication events that disrupt ER acetylation are associated with severe human diseases (Chiplunkar et al., 2016; Huppke et al., 2012a; Lin et al., 2008; Prasad et al., 2012; Sanders et al., 2011). Furthermore, mouse models of reduced or increased AT-1 activity develop severe phenotypes that mimic the associated human diseases (Hullinger et al., 2016; Peng et al., 2014, 2018). A combination of cell- and animal-based studies indicate that the ER acetylation machinery is necessary to maintain proteostasis within the ER and secretory pathway (Farrugia and Puglielli, 2018; Pehar et al., 2012; Peng et al., 2014, 2016, 2018).
The removal of toxic protein aggregates through reticulophagy is an essential aspect of the proteostatic functions of ER acetylation. Reduced ER acetylation in AT-1S113R/+ hypomorphic mice causes increased (aberrant) induction of reticulophagy, whereas increased ER acetylation in AT-1-overexpressing mice has the opposite effect (Hullinger et al., 2016; Peng et al., 2014, 2018). Furthermore, inhibition of ATase1 and ATase2, downstream of AT-1, can restore reticulophagy in AT-1 overexpressors (Peng et al., 2016, 2018). Mechanistically, the regulation of reticulophagy involves acetylation of ATG9A on two lysine residues (K359 and K363) that face the lumen of the ER (Pehar et al., 2012). The acetylation status of ATG9A, in turn, regulates the ability of ATG9A to interact with FAM134B and SEC62, which can then engage cytosolic LC3B through the LIR on their C terminus (Peng et al., 2018).
Here, we report mechanistic aspects of the ATG9A-FAM134B and ATG9A-SEC62 interactions. Both FAM134B and SEC62 engage ATG9A on the cytosolic side of the ER through specific structural features that appear to involve changes in Ca++ dynamics. The modeling data obtained with FAM134B are particularly intriguing, as they support a scenario where interaction with ATG9A and changes in Ca++ binding through two opposing EF hands might allow structural rearrangement of the globular end of FAM134B that is necessary for the LIR to “open” and engage LC3B. The typical helix-loop-helix structure of the EF hand is stabilized by Ca++, which is engaged by the side chains of Glu and Asp residues. This provides geometric and spatial constraint to the otherwise very flexible turn-loop structure (Gifford et al., 2007; Kawasaki and Kretsinger, 2017). EF hands tend to occur in pairs, and cooperativity is often ensured by their opposing placement on the folded protein that generates a defined 11 Å distance between bound Ca++ ions (Biekofsky and Feeney, 1998; Biekofsky et al., 1998). However, the opposing EF hands tend to be slightly different. This difference causes asymmetric Ca++ binding, which ultimately ensures dynamic fluctuation of specific regions within a folded polypeptide (Kawasaki and Kretsinger, 2017; Lopez et al., 2002; Shaw et al., 1991). Consistent with the above, inactivation of the opposing EF1 and EF2 hands on the globular domain of FAM134B elicited opposing effects. Inactivation of EF1 increased the ability of FAM134B to induce reticulophagy, whereas inactivation of EF2 decreased the ability of FAM134B to induce reticulophagy, suggesting that structural fluctuations are indeed necessary for FAM134B to engage LC3B. This is somewhat reminiscent of the assembly-disassembly fluctuations described with circular dichroism and NMR studies of both typical and atypical EF hands (Gifford et al., 2007; Kawasaki and Kretsinger, 2017). The fact that selective deletions of opposing domains within the globular end of FAM134B positively and negatively influenced ATG9A-FAM134B interaction provides further structural support to the model delineated above.
A challenging aspect of our findings is that the acetylation of ATG9A occurs in the lumen of the ER, whereas the engagement of FAM134B or SEC62 occurs on the cytosolic side of the organelle. To resolve this, we used unbiased MS to resolve a crude “ATG9A interactome.” The analysis identified several proteins broadly divided into “cytosolic” and “ER-associated.” The former included cytoskeletal and cytoskeletal-associated proteins. The latter included ER-integral as well as ER-associated proteins. It is worth stressing that, although translated in the ER, ATG9A normally translocates out of the ER toward the Golgi apparatus, the plasma membrane, and LC3B-positive autophagosomes during the induction of autophagy (Bejarano et al., 2014; Ohashi and Munro, 2010; Puri et al., 2013; Saitoh et al., 2009; Tamura et al., 2010; Young et al., 2006). Also important is the fact that ATG9A can interact with ERES and COP structures (Graef et al., 2013; Kakuta et al., 2012; Lynch-Day et al., 2010; Suzuki et al., 2013), and that cross talk between the autophagy machinery and the ERES/COPII assembly machinery has been described (Sanchez-Wandelmer et al., 2015). Therefore, the ATG9A interactome that we report here appears to reflect essential aspects of ATG9A biology. It is also worth mentioning that both FAM134B and SEC62 are found on the rough ER, where the bulk of protein biosynthesis occurs (Fumagalli et al., 2016; Khaminets et al., 2015; Mochida et al., 2015; Rubinsztein, 2015; Schuck, 2016). Furthermore, SEC62 is part of the translocon, which allows insertion of newly synthesized proteins within the ER (Gemmer and Forster, 2020). It has been proposed that FAM134B and SEC62 couple the machinery that allows biosynthesis and insertion of newly synthesized proteins into the ER with the machinery that controls the disposal of unfolded/misfolded polypeptides, and maintains the size of the ER (Nakatogawa and Mochida, 2015; Schuck, 2016). The presence of integral components of the ER quality control machinery as well as ERES and COPII structural components within the ATG9A interactome seems to support the idea of vicinity and cross talk of the essential machineries that maintain proteostasis within the ER.
By using mouse models of decreased (AT-1S113R/+) and increased (AT-1 sTg) AT-1 activity, we were able to identify two ATG9A-interacting proteins, CALR and HSPB1, that preferentially bind acetylated ATG9A. Importantly, CALR is a non-classical chaperone involved in the quality control of newly synthesized glycoproteins (Hebert and Molinari, 2007; Trombetta and Parodi, 2003); being an ER lumenal protein, it can only engage ATG9A within the lumen of the organelle. Our findings suggest that CALR can recognize—and perhaps stabilize—acetylated ATG9A; therefore, CALR is the first confirmed ER lumenal ATG9A-interacting protein. Calnexin (CANX), the membrane-bound structural homolog of CALR, has recently emerged as a possible “facilitator” of ER-phagy/reticulophagy through its interaction with FAM134B (Fregno et al., 2018). Therefore the CANX/CALR system may collectively regulate the induction of ER-phagy/reticulophagy through FAM134B and ATG9A, respectively. Interestingly, in addition to CALR, the MS analysis of the ATG9A interactome also identified CANX, PDIA3, PDIA6, and UGGT1; these are all part of the classical CALR/CANX cycle that retrieves unfolded glycoproteins as part of the normal quality control activities of the ER. These findings further support the existence of a close interaction (and spatial vicinity) between the machinery that ensures quality control and the one that ensures reticulophagy within the ER.
In addition to CALR, the MS analysis of the ATG9A interactome identified the small heat shock protein HSPB1 as a cytosolic ATG9A-interacting protein. HSPB1 mutations have been linked to axonal forms of CMT disease and HMN (Almeida-Souza et al., 2010; Capponi et al., 2011; Echaniz-Laguna et al., 2017; Evgrafov et al., 2004; Holmgren et al., 2013; Ikeda et al., 2009; Irobi et al., 2010; Lin et al., 2011; Muranova et al., 2015; Nefedova et al., 2013a, 2013b), which belong to the general family of hereditary sensory autonomic and motor neuropathies (HSANs/HMNs). How these mutations affect HSPB1 activity is unknown, although studies suggest altered substrate-binding activity (Almeida-Souza et al., 2010; Capponi et al., 2011; Echaniz-Laguna et al., 2017; Evgrafov et al., 2004; Holmgren et al., 2013; Ikeda et al., 2009; Irobi et al., 2010; Lin et al., 2011; Muranova et al., 2015; Nefedova et al., 2013a, 2013b). However, the real substrate(s) is/are unknown. Our data indicate that CMT/HMN-associated mutant versions of HSPB1 inactivate the ability of HSPB1 to stimulate reticulophagy. Interestingly, AT-1 or FAM134B mutations are also associated with related forms of HSANs/HMNs (Almeida-Souza et al., 2010; Capponi et al., 2011; Davidson et al., 2012; Echaniz-Laguna et al., 2017; Evgrafov et al., 2004; Holmgren et al., 2013; Huppke et al., 2012a, 2012b; Ikeda et al., 2009; Ilgaz Aydinlar et al., 2014; Irobi et al., 2010; Kurth et al., 2009; Lin et al., 2008, 2011; Muranova et al., 2015; Murphy et al., 2012; Nefedova et al., 2013a, 2013b; Wakil et al., 2018). When taken together, these data support the possibility that defects in induction and progression of reticulophagy might underlie disease-associated mutations affecting AT-1, FAM134B, and HSBP1.
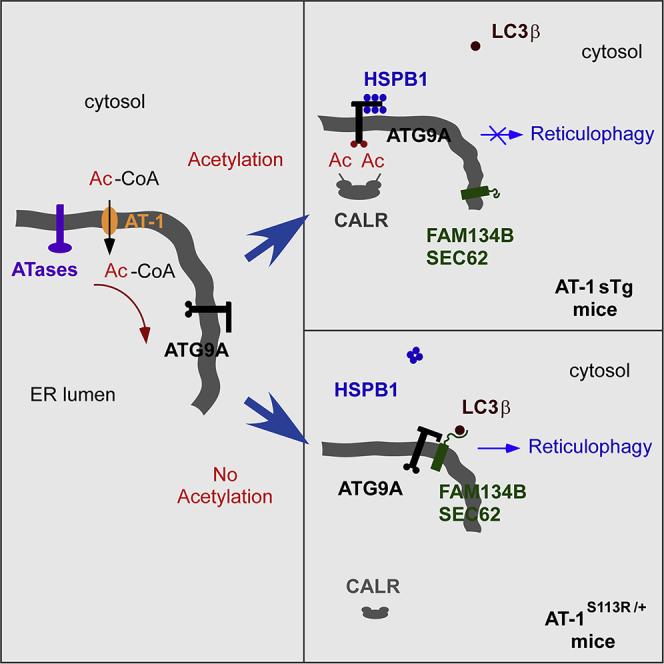
Based on our findings, we can envision a working model where increased influx of acetyl-CoA into the ER lumen caused by increased expression and activity of AT-1 in AT-1 sTg and AT-1 nTg mice leads to hyperacetylation of the ER pool of ATG9A, perhaps caused by allosteric activation of ATase1 (Rigby et al., 2020) (Figure 7A). Conversely, reduced influx of acetyl-CoA into the ER lumen caused by haploinsufficiency of AT-1 in AT-1S113R/+ mice or inhibition of the ATases leads to hypoacetylation of the ER pool of ATG9A (Figure 7B). When acetylated, ATG9A is recognized—and perhaps sequestered—by CALR, thus preventing interaction with FAM134B or SEC62, engagement of LC3B, and induction of reticulophagy (Figure 7A). Under this condition, the cytosolic-exposed domain(s) of ATG9A is occupied by HSPB1, probably in the monomeric form. In contrast, when non-acetylated, ATG9A is not recognized by CALR and is able to interact with FAM134B or SEC62. This might force displacement or rearrangement of HSPB1. The ATG9A-FAM134B and ATG9A-SEC62 interaction allows engagement of LC3B and induction of reticulophagy (Figure 7B). The structural arrangement of HSPB1 seems to be an important component, because disease-associated mutations that prevent oligomerization of HSPB1 appear to act as negative effectors. Although several aspects of this model are supported by the present study and by already published work (Hullinger et al., 2016; Pehar et al., 2012; Peng et al., 2014, 2016, 2018; Peng and Puglielli, 2016; Rigby et al., 2020), there are different components that remain to be fully dissected and, as such, we consider this only a working and incomplete model.
Figure 7.

Working Model
Description is in the main text. Events preventing (A) or stimulating (B) reticulophagy are shown. Although several aspects of this working model are supported by Hullinger et al. (2016); Pehar et al. (2012); Peng et al. (2014, 2016, 2018); Peng and Puglielli (2016); and Rigby et al. (2020), there are different components that remain to be fully dissected. The most urgent questions include: (1) the structural mechanism that allows CALR to recognize the acetylated lysine residues and sequester ATG9A and (2) the specific role of HSPB1, as well as the structural requirements that underlie the apparent different behavior of monomeric and oligomeric HSPB1.
The specific role of K359/K359Ac and K363/K363Ac and how they influence ATG9A activity remains an unresolved aspect of our findings. All ATG9 proteins, including human ATG9A, are predicted to contain six transmembrane (TM) helices with three loops facing the luminal side. While this manuscript was under preparation and editorial process, four different ATG9 cryoelectron microscopic (cryo-EM) structures were reported (Guardia et al., 2020; Lai et al., 2020; Maeda et al., 2020; Matoba et al., 2020). Three of them (from yeast and human ATG9A) yielded a model with four spanning TM helices and two reentrant helices (Guardia et al., 2020; Maeda et al., 2020; Matoba et al., 2020), whereas one (from Arabidopsis thaliana) yielded a model with six spanning TM helices (Lai et al., 2020). In all cases, ATG9 appeared to organize as a trimer with a central open pore. ATG9A is acetylated on two lysine residues, K359 and K363, both residing in the second of the three intraluminal loop within the six TM predicted model (Pehar et al., 2012). The ER-based ATases, which acetylate ATG9A, are both type-II membrane proteins with the catalytic domain facing the ER lumen (Farrugia and Puglielli, 2018; Ko and Puglielli, 2009). Therefore, the predicted intraluminal location of K359 and K363 is consistent with the topology of the ATases. The six TM helices cryo-EM model of A. thaliana ATG9 would be consistent with the intraluminal K359/K363 acetylation model. In contrast, the four fully spanning and two reentrant helices cryo-EM model of human ATG9A would place K359 and K363 in the central pore of the trimeric complex. Although the central pore of the trimeric structure appears to be necessary for the autophagy-related functions of ATG9A, the dynamics of the trimeric assembly itself are currently unknown (Maeda et al., 2020; Matoba et al., 2020). Specifically, it is unknown when and how the ATG9A monomers interact to build the trimeric structure. As the acetylation status of ATG9A regulates its autophagy-related functions (Farrugia and Puglielli, 2018; Pehar et al., 2012; Peng et al., 2014, 2016, 2018), we can speculate that the acetylation/deacetylation of K359/K363 might play, either directly or indirectly, some role in the trimerization process of ATG9A or in the proposed scramblase/flippase activity of the open pore. However, this remains to be studied. The in vivo implication of the structural models also remains to be fully studied. However, ongoing studies with Atase1−/− and Atase2−/− mice in our laboratory might reveal additional mechanistic aspects related to ATG9A assembly and, perhaps, further clarify pathogenic events underlying the disease phenotypes caused by dysfunctional ER acetylation.
Limitations of the study
-
1.
The fact that ATG9A might work at the intersection between engagement of the secretory pathway and induction of reticulophagy is an intriguing new area of investigation. Two independent studies (one recently published [Dieterich et al., 2021] and one in press) from our laboratory using mouse models of increased (AT-1 sTg) and reduced (AT-1S113R/+, Atase1−/− and Atase2−/−) ER acetylation confirm this dual function of the ER acetylation machinery. However, how this balance is achieved remains to be dissected.
-
2.
This study revealed a previously unknown regulatory function of Ca++. Ca++ regulates the ATG9A-FAM134B and ATG9A-SEC62 interaction on the cytosolic side of the ER. Ca++ also regulates CALR activity within the ER lumen. Importantly, CALR itself normally functions as a high-affinity “storage system” for ER luminal Ca++. The precise mechanism(s) underlying the role of Ca++ release and—perhaps—transport across the ER membrane remains to be dissected.
-
3.
The assembly state of HSPB1 is crucial to the activity of the protein and seems to be important for the pathophysiology of HSPB1-associated HSANs/HMNs. However, why and how remains to be determined.
Resource availability
Lead contact
Further information, requests, and enquiries should be directed to and will be fulfilled by the lead contact and corresponding author, Prof. Luigi Puglielli (lp1@medicine.wisc.edu).
Materials availability
All unique/stable reagents generated in this study are available on request from the lead contact.
Data and code availability
The published article includes all data generated or analyzed during this study.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank Grzegorz Sabat at the Proteomics-Mass Spectrometry Facility of the University of Wisconsin-Madison for help with LC-MS and Dr. John Svaren for critical reading of an early version of this paper. This work was supported by the NIH (NS094154 and AG057408 to L.P.) and the Department of Veterans Affairs (I01 BX004202 to L.P.). This study was also supported in part by a core grant to the Waisman Center from the National Institute of Child Health and Human Development (U54 HD090256), and to the University of Wisconsin Carbone Cancer Center from the National Cancer Institute (P30 CA014520). SIM was performed at the University of Wisconsin-Madison Biochemistry Optical Core.
Author contributions
B.K.S., N.S.O., Y.P., and S.L.S. performed the experiments and analyzed the data. L.P. designed the overall study and wrote the paper with input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102315.
Supplemental information
References
- Alderson T.R., Roche J., Gastall H.Y., Dias D.M., Pritisanac I., Ying J., Bax A., Benesch J.L.P., Baldwin A.J. Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun. 2019;10:1068. doi: 10.1038/s41467-019-08557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida-Souza L., Goethals S., de Winter V., Dierick I., Gallardo R., Van Durme J., Irobi J., Gettemans J., Rousseau F., Schymkowitz J. Increased monomerization of mutant HSPB1 leads to protein hyperactivity in Charcot-Marie-Tooth neuropathy. J. Biol. Chem. 2010;285:12778–12786. doi: 10.1074/jbc.M109.082644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo A.P. Mammalian HspB1 (Hsp27) is a molecular sensor linked to the physiology and environment of the cell. Cell Stress Chaperones. 2017;22:517–529. doi: 10.1007/s12192-017-0765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano E., Yuste A., Patel B., Stout R.F., Jr., Spray D.C., Cuervo A.M. Connexins modulate autophagosome biogenesis. Nat. Cell Biol. 2014;16:401–414. doi: 10.1038/ncb2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara R.M., Grumati P., Garcia-Pardo J., Kalayil S., Covarrubias-Pinto A., Chen W., Kudryashev M., Dikic I., Hummer G. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 2019;10:2370. doi: 10.1038/s41467-019-10345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biekofsky R.R., Feeney J. Cooperative cyclic interactions involved in metal binding to pairs of sites in EF-hand proteins. FEBS Lett. 1998;439:101–106. doi: 10.1016/s0014-5793(98)01349-0. [DOI] [PubMed] [Google Scholar]
- Biekofsky R.R., Martin S.R., Browne J.P., Bayley P.M., Feeney J. Ca2+ coordination to backbone carbonyl oxygen atoms in calmodulin and other EF-hand proteins: 15N chemical shifts as probes for monitoring individual-site Ca2+ coordination. Biochemistry. 1998;37:7617–7629. doi: 10.1021/bi9800449. [DOI] [PubMed] [Google Scholar]
- Capponi S., Geroldi A., Fossa P., Grandis M., Ciotti P., Gulli R., Schenone A., Mandich P., Bellone E. HSPB1 and HSPB8 in inherited neuropathies: study of an Italian cohort of dHMN and CMT2 patients. J. Peripher. Nerv. Syst. 2011;16:287–294. doi: 10.1111/j.1529-8027.2011.00361.x. [DOI] [PubMed] [Google Scholar]
- Chiplunkar S., Bindu P.S., Nagappa M., Bineesh C., Govindaraj P., Gayathri N., Bharath M.M., Arvinda H.R., Mathuranath P.S., Sinha S. Huppke-Brendel syndrome in a seven months old boy with a novel 2-bp deletion in SLC33A1. Metab. Brain Dis. 2016;31:1195–1198. doi: 10.1007/s11011-016-9854-6. [DOI] [PubMed] [Google Scholar]
- Davidson G., Murphy S., Polke J., Laura M., Salih M., Muntoni F., Blake J., Brandner S., Davies N., Horvath R. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J. Neurol. 2012;259:1673–1685. doi: 10.1007/s00415-011-6397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempski R.E., Jr., Imperiali B. Oligosaccharyl transferase: gatekeeper to the secretory pathway. Curr. Opin. Chem. Biol. 2002;6:844–850. doi: 10.1016/s1367-5931(02)00390-3. [DOI] [PubMed] [Google Scholar]
- Dieterich I.A., Cui Y., Braun M.M., Lawton A.J., Robinson N.H., Peotter J.L., Yu Q., Casler J.C., Glick B.S., Audhya A. Acetyl-CoA flux from the cytosol to the ER regulates engagement and quality of the secretory pathway. Sci. Rep. 2021;11:2013. doi: 10.1038/s41598-021-81447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich I.A., Lawton A.J., Peng Y., Yu Q., Rhoads T.W., Overmyer K.A., Cui Y., Armstrong E.A., Howell P.R., Burhans M.S. Acetyl-CoA flux regulates the proteome and acetyl-proteome to maintain intracellular metabolic crosstalk. Nat. Commun. 2019;10:3929. doi: 10.1038/s41467-019-11945-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echaniz-Laguna A., Geuens T., Petiot P., Pereon Y., Adriaenssens E., Haidar M., Capponi S., Maisonobe T., Fournier E., Dubourg O. Axonal neuropathies due to mutations in small heat shock proteins: clinical, genetic, and functional insights into novel mutations. Hum. Mutat. 2017;38:556–568. doi: 10.1002/humu.23189. [DOI] [PubMed] [Google Scholar]
- Evgrafov O.V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C.L., Schagina O., Verpoorten N., Van Impe K., Fedotov V. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004;36:602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- Farrugia M.A., Puglielli L. Nepsilon-lysine acetylation in the endoplasmic reticulum - a novel cellular mechanism that regulates proteostasis and autophagy. J. Cell Sci. 2018;131:jcs221747. doi: 10.1242/jcs.221747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I., Fasana E., Bergmann T.J., Raimondi A., Loi M., Solda T., Galli C., D'Antuono R., Morone D., Danieli A. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018;37:e99259. doi: 10.15252/embj.201899259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F., Noack J., Bergmann T.J., Cebollero E., Pisoni G.B., Fasana E., Fregno I., Galli C., Loi M., Solda T. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016;18:1173–1184. doi: 10.1038/ncb3423. [DOI] [PubMed] [Google Scholar]
- Gemmer M., Forster F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020;133:jcs231340. doi: 10.1242/jcs.231340. [DOI] [PubMed] [Google Scholar]
- Gifford J.L., Walsh M.P., Vogel H.J. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- Graef M., Friedman J.R., Graham C., Babu M., Nunnari J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell. 2013;24:2918–2931. doi: 10.1091/mbc.E13-07-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia C.M., Tan X.F., Lian T., Rana M.S., Zhou W., Christenson E.T., Lowry A.J., Faraldo-Gomez J.D., Bonifacino J.S., Jiang J. Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep. 2020;31:107837. doi: 10.1016/j.celrep.2020.107837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C., Baba M., Cao Y., Klionsky D.J. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol. Biol. Cell. 2008;19:5506–5516. doi: 10.1091/mbc.E08-05-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert D.N., Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- Holmgren A., Bouhy D., De Winter V., Asselbergh B., Timmermans J.P., Irobi J., Timmerman V. Charcot-Marie-Tooth causing HSPB1 mutations increase Cdk5-mediated phosphorylation of neurofilaments. Acta Neuropathol. 2013;126:93–108. doi: 10.1007/s00401-013-1133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullinger R., Li M., Wang J., Peng Y., Dowell J.A., Bomba-Warczak E., Mitchell H.A., Burger C., Chapman E.R., Denu J.M. Increased expression of AT-1/SLC33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J. Exp. Med. 2016;213:1267–1284. doi: 10.1084/jem.20151776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppke P., Brendel C., Kalscheuer V., Korenke G.C., Marquardt I., Freisinger P., Christodoulou J., Hillebrand M., Pitelet G., Wilson C. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am. J. Hum. Genet. 2012;90:61–68. doi: 10.1016/j.ajhg.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppke P., Brendel C., Korenke G.C., Marquardt I., Donsante A., Yi L., Hicks J.D., Steinbach P.J., Wilson C., Elpeleg O. Molecular and biochemical characterization of a unique mutation in CCS, the human copper chaperone to superoxide dismutase. Hum. Mutat. 2012;33:1207–1215. doi: 10.1002/humu.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y., Abe A., Ishida C., Takahashi K., Hayasaka K., Yamada M. A clinical phenotype of distal hereditary motor neuronopathy type II with a novel HSPB1 mutation. J. Neurol. Sci. 2009;277:9–12. doi: 10.1016/j.jns.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Ilgaz Aydinlar E., Rolfs A., Serteser M., Parman Y. Mutation in FAM134B causing hereditary sensory neuropathy with spasticity in a Turkish family. Muscle Nerve. 2014;49:774–775. doi: 10.1002/mus.24145. [DOI] [PubMed] [Google Scholar]
- Imai K., Hao F., Fujita N., Tsuji Y., Oe Y., Araki Y., Hamasaki M., Noda T., Yoshimori T. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell Sci. 2016;129:3781–3791. doi: 10.1242/jcs.196196. [DOI] [PubMed] [Google Scholar]
- Irobi J., Almeida-Souza L., Asselbergh B., De Winter V., Goethals S., Dierick I., Krishnan J., Timmermans J.P., Robberecht W., De Jonghe P. Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum. Mol. Genet. 2010;19:3254–3265. doi: 10.1093/hmg/ddq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas M.C., Pehar M., Puglielli L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J. Cell Sci. 2010;123:3378–3388. doi: 10.1242/jcs.068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta S., Yamamoto H., Negishi L., Kondo-Kakuta C., Hayashi N., Ohsumi Y. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J. Biol. Chem. 2012;287:44261–44269. doi: 10.1074/jbc.M112.411454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H., Kretsinger R.H. Structural and functional diversity of EF-hand proteins: evolutionary perspectives. Protein Sci. 2017;26:1898–1920. doi: 10.1002/pro.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A., Heinrich T., Mari M., Grumati P., Huebner A.K., Akutsu M., Liebmann L., Stolz A., Nietzsche S., Koch N. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–358. doi: 10.1038/nature14498. [DOI] [PubMed] [Google Scholar]
- Ko M.H., Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 2009;284:2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth I., Pamminger T., Hennings J.C., Soehendra D., Huebner A.K., Rotthier A., Baets J., Senderek J., Topaloglu H., Farrell S.A. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 2009;41:1179–1181. doi: 10.1038/ng.464. [DOI] [PubMed] [Google Scholar]
- Lai L.T.F., Yu C., Wong J.S.K., Lo H.S., Benlekbir S., Jiang L., Lau W.C.Y. Subnanometer resolution cryo-EM structure of Arabidopsis thaliana ATG9. Autophagy. 2020;16:575–583. doi: 10.1080/15548627.2019.1639300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I.H., Finkel T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009;284:6322–6328. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennemann N.J., Coyne C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13:322–332. doi: 10.1080/15548627.2016.1265192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.R., Lingeman E., Ahmed S., Corn J.E. Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol. 2018;217:3354–3367. doi: 10.1083/jcb.201804185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K.P., Soong B.W., Yang C.C., Huang L.W., Chang M.H., Lee I.H., Antonellis A., Lee Y.C. The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One. 2011;6:e29393. doi: 10.1371/journal.pone.0029393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P., Li J., Liu Q., Mao F., Qiu R., Hu H., Song Y., Yang Y., Gao G., Yan C. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42) Am. J. Hum. Genet. 2008;83:752–759. doi: 10.1016/j.ajhg.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linxweiler M., Schorr S., Schauble N., Jung M., Linxweiler J., Langer F., Schafers H.J., Cavalie A., Zimmermann R., Greiner M. Targeting cell migration and the endoplasmic reticulum stress response with calmodulin antagonists: a clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer. 2013;13:574. doi: 10.1186/1471-2407-13-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M.M., Chin D.H., Baldwin R.L., Makhatadze G.I. The enthalpy of the alanine peptide helix measured by isothermal titration calorimetry using metal-binding to induce helix formation. Proc. Natl. Acad. Sci. U S A. 2002;99:1298–1302. doi: 10.1073/pnas.032665199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day M.A., Bhandari D., Menon S., Huang J., Cai H., Bartholomew C.R., Brumell J.H., Ferro-Novick S., Klionsky D.J. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc. Natl. Acad. Sci. U S A. 2010;107:7811–7816. doi: 10.1073/pnas.1000063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S., Yamamoto H., Kinch L.N., Garza C.M., Takahashi S., Otomo C., Grishin N.V., Forli S., Mizushima N., Otomo T. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol. 2020;27:1194–1201. doi: 10.1038/s41594-020-00520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba K., Kotani T., Tsutsumi A., Tsuji T., Mori T., Noshiro D., Sugita Y., Nomura N., Iwata S., Ohsumi Y. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020;27:1185–1193. doi: 10.1038/s41594-020-00518-w. [DOI] [PubMed] [Google Scholar]
- Mochida K., Oikawa Y., Kimura Y., Kirisako H., Hirano H., Ohsumi Y., Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522:359–362. doi: 10.1038/nature14506. [DOI] [PubMed] [Google Scholar]
- Muranova L.K., Weeks S.D., Strelkov S.V., Gusev N.B. Characterization of mutants of human small heat shock protein HspB1 carrying replacements in the N-terminal domain and associated with hereditary motor neuron diseases. PLoS One. 2015;10:e0126248. doi: 10.1371/journal.pone.0126248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S.M., Davidson G.L., Brandner S., Houlden H., Reilly M.M. Mutation in FAM134B causing severe hereditary sensory neuropathy. J. Neurol. Neurosurg. Psychiatry. 2012;83:119–120. doi: 10.1136/jnnp.2010.228965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H., Mochida K. Reticulophagy and nucleophagy: new findings and unsolved issues. Autophagy. 2015;11:2377–2378. doi: 10.1080/15548627.2015.1106665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefedova V.V., Datskevich P.N., Sudnitsyna M.V., Strelkov S.V., Gusev N.B. Physico-chemical properties of R140G and K141Q mutants of human small heat shock protein HspB1 associated with hereditary peripheral neuropathies. Biochimie. 2013;95:1582–1592. doi: 10.1016/j.biochi.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Nefedova V.V., Sudnitsyna M.V., Strelkov S.V., Gusev N.B. Structure and properties of G84R and L99M mutants of human small heat shock protein HspB1 correlating with motor neuropathy. Arch. Biochem. Biophys. 2013;538:16–24. doi: 10.1016/j.abb.2013.07.028. [DOI] [PubMed] [Google Scholar]
- Nishimura T., Tamura N., Kono N., Shimanaka Y., Arai H., Yamamoto H., Mizushima N. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017;36:1719–1735. doi: 10.15252/embj.201695189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y., Munro S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell. 2010;21:3998–4008. doi: 10.1091/mbc.E10-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M., Jonas M.C., Hare T.M., Puglielli L. SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J. Biol. Chem. 2012;287:29921–29930. doi: 10.1074/jbc.M112.363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Kim M.J., Hullinger R., O'Riordan K.J., Burger C., Pehar M., Puglielli L. Improved proteostasis in the secretory pathway rescues Alzheimer's disease in the mouse. Brain. 2016;139:937–952. doi: 10.1093/brain/awv385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Li M., Clarkson B.D., Pehar M., Lao P.J., Hillmer A.T., Barnhart T.E., Christian B.T., Mitchell H.A., Bendlin B.B. Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J. Neurosci. 2014;34:6772–6789. doi: 10.1523/JNEUROSCI.0077-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Puglielli L. N-lysine acetylation in the lumen of the endoplasmic reticulum: a way to regulate autophagy and maintain protein homeostasis in the secretory pathway. Autophagy. 2016;12:1051–1052. doi: 10.1080/15548627.2016.1164369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Shapiro S.L., Banduseela V.C., Dieterich I.A., Hewitt K.J., Bresnick E.H., Kong G., Zhang J., Schueler K.L., Keller M.P. Increased transport of acetyl-CoA into the endoplasmic reticulum causes a progeria-like phenotype. Aging Cell. 2018:e12820. doi: 10.1111/acel.12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrescu A.J., Milac A.L., Petrescu S.M., Dwek R.A., Wormald M.R. Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology. 2004;14:103–114. doi: 10.1093/glycob/cwh008. [DOI] [PubMed] [Google Scholar]
- Prasad A., Merico D., Thiruvahindrapuram B., Wei J., Lionel A.C., Sato D., Rickaby J., Lu C., Szatmari P., Roberts W. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G. 2012;2:1665–1685. doi: 10.1534/g3.112.004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri C., Renna M., Bento C.F., Moreau K., Rubinsztein D.C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell. 2013;154:1285–1299. doi: 10.1016/j.cell.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby M.J., Gomez T.M., Puglielli L. Glial cell-axonal growth cone interactions in neurodevelopment and regeneration. Front. Neurosci. 2020;14:203. doi: 10.3389/fnins.2020.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D.C. Cell biology: receptors for selective recycling. Nature. 2015;522:291–292. doi: 10.1038/nature14532. [DOI] [PubMed] [Google Scholar]
- Saitoh T., Fujita N., Hayashi T., Takahara K., Satoh T., Lee H., Matsunaga K., Kageyama S., Omori H., Noda T. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U S A. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Wandelmer J., Ktistakis N.T., Reggiori F. ERES: sites for autophagosome biogenesis and maturation? J. Cell Sci. 2015;128:185–192. doi: 10.1242/jcs.158758. [DOI] [PubMed] [Google Scholar]
- Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S. On keeping the right ER size. Nat. Cell Biol. 2016;18:1118–1119. doi: 10.1038/ncb3430. [DOI] [PubMed] [Google Scholar]
- Shaw G.S., Hodges R.S., Sykes B.D. Probing the relationship between alpha-helix formation and calcium affinity in troponin C: 1H NMR studies of calcium binding to synthetic and variant site III helix-loop-helix peptides. Biochemistry. 1991;30:8339–8347. doi: 10.1021/bi00098a009. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Akioka M., Kondo-Kakuta C., Yamamoto H., Ohsumi Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci. 2013;126:2534–2544. doi: 10.1242/jcs.122960. [DOI] [PubMed] [Google Scholar]
- Tamura H., Shibata M., Koike M., Sasaki M., Uchiyama Y. Atg9A protein, an autophagy-related membrane protein, is localized in the neurons of mouse brains. J. Histochem. Cytochem. 2010;58:443–453. doi: 10.1369/jhc.2010.955690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta E.S., Parodi A.J. Quality control and protein folding in the secretory pathway. Annu. Rev. Cell Dev. Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- Wakil S.M., Monies D., Hagos S., Al-Ajlan F., Finsterer J., Al Qahtani A., Ramzan K., Al Humaidy R., Al-Muhaizea M.A., Meyer B. Exome sequencing: mutilating sensory neuropathy with spastic paraplegia due to a mutation in FAM134B gene. Case Rep. Genet. 2018;2018:9468049. doi: 10.1155/2018/9468049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber J.L., Young A.R., Tooze S.A. Atg9 trafficking in Mammalian cells. Autophagy. 2007;3:54–56. doi: 10.4161/auto.3419. [DOI] [PubMed] [Google Scholar]
- Young A.R., Chan E.Y., Hu X.W., Kochl R., Crawshaw S.G., High S., Hailey D.W., Lippincott-Schwartz J., Tooze S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study.