

Abstract
Oncolytic virotherapy holds promise of effective immunotherapy against otherwise nonresponsive cancers such as glioblastoma. Our previous findings have shown that although oncolytic Semliki Forest virus (SFV) is effective against various mouse glioblastoma models, its therapeutic potency is hampered by type I interferon (IFN-I)-mediated antiviral signaling. In this study, we constructed a novel IFN-I-resistant SFV construct, SFV-AM6, and evaluated its therapeutic potency in vitro, ex vivo, and in vivo in the IFN-I competent mouse GL261 glioma model. In vitro analysis shows that SFV-AM6 causes immunogenic apoptosis in GL261 cells despite high IFN-I signaling. MicroRNA-124 de-targeted SFV-AM6-124T selectively replicates in glioma cells, and it can infect orthotopic GL261 gliomas when administered intraperitoneally. The combination of SFV-AM6-124T and anti-programmed death 1 (PD1) immunotherapy resulted in increased immune cell infiltration in GL261 gliomas, including an increased tumor-reactive CD8+ fraction. Our results show that SFV-AM6-124T can overcome hurdles of innate anti-viral signaling. Combination therapy with SFV-AM6-124T and anti-PD1 promotes the inflammatory response and improves the immune microenvironment in the GL261 glioma model.
Keywords: Semliki Forest virus, oncolytic virotherapy, glioblastoma, type I interferon, Alphavirus, cancer immunotherapy, anti-PD1, immune checkpoint inhibitors, anti-tumor T cells
Graphical abstract
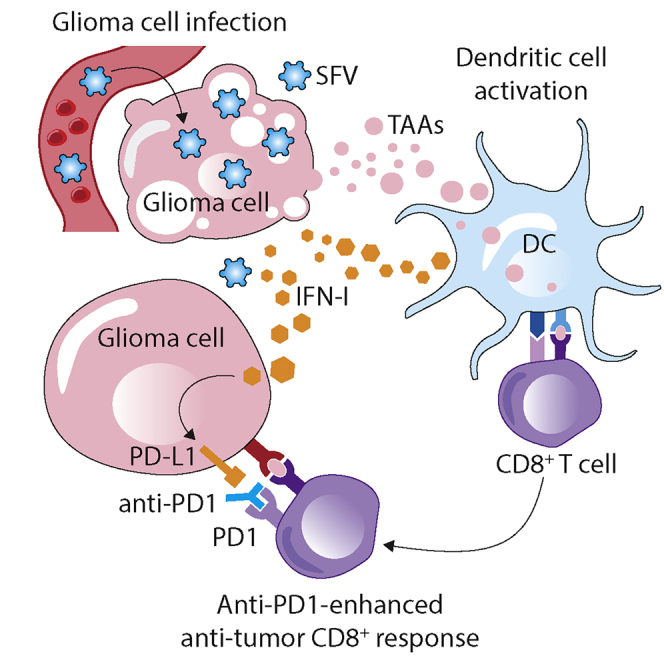
Glioblastoma is a highly malignant brain tumor with no curative therapy currently available. In this study, Miika Martikainen and colleagues have constructed a novel enhanced oncolytic Semliki Forest virus, and they show that it can be used as an immunotherapeutic agent to promote an anti-tumor immune response in the mouse glioblastoma model.
Introduction
Glioblastoma (GBM) is the most common and most severe form of primary brain tumor in adults.1 The current standard care for GBM consists of radiotherapy, chemotherapy with temozolomide, and surgical resection of the tumor (if possible). None of these interventions is curative, and the median survival remains short (14–15 months) due to relapse in virtually 100% of the cases.2,3
GBM thrives upon the tightly immune-controlled microenvironment in the brain and further skews it toward immunosuppression, making it a challenging target for immunotherapeutic approaches. Recent early phase clinical trials have, however, provided evidence that oncolytic viruses (OVs) can be used for effective GBM immunotherapy in some patients.4, 5, 6 Results from preclinical and clinical studies indicate that OV-mediated cancer cell killing and the innate inflammatory response can stimulate an adaptive anti-cancer immune response.7,8 Induction of tumor immunity de novo via virus-induced immunogenic oncolysis should be especially beneficial in treatment of “cold” tumors such as GBM.
Semliki Forest virus (SFV) is an alphavirus (group IV, positive-sense single-stranded RNA [+ssRNA] virus, family Togaviridae), which has a natural capability to penetrate the blood-brain barrier (BBB) in mice. This makes SFV an ideal candidate virus to target brain tumors, provided that viral replication can be restricted to tumor cells. We have previously studied modified oncolytic SFV in different syngeneic mouse glioma model results ranging between encouraging and underwhelming.9, 10, 11, 12 The main obstacle in these models is the type I interferon (IFN-I)-mediated innate antiviral response, which inhibits SFV replication in GBM tumors.
IFN-Is play a key role in mediating antiviral immune responses, especially for RNA viruses. Although IFN-I defects are frequent in cancer cells, they cannot be taken for granted in GBM.13,14 Accumulating evidence suggest that IFN-I signaling has major effects on cancer immunosurveillance.15 Therefore, despite its negative antiviral effect on OV replication and spread in the tumor tissue, fully functional IFN-I signaling in dendritic cells (DCs) is also needed for the full immunostimulatory therapeutic effect of oncolytic virotherapy. Viruses that can replicate and spread in the cancer cells despite IFN-I signaling would therefore be preferable.
In this study, we aimed to develop a novel oncolytic SFV construct with increased resistance to IFN-I to be used as a potent immunotherapeutic agent against GBM.
Results
SFV-AM6 shows enhanced oncolytic potency in GL261 cells in vitro despite a high IFN-I response
We constructed a novel recombinant SFV termed “SFV-AM6,” which carries 4 aa substitutions as compared to the parental SFV4, that is nsP3A70G, nsP3Y369F, nsP4R611K and E2K162E (Figure 1A). These mutations were expected to enhance replication and improve in vivo efficacy of the virus based on previous results by us and others (unpublished data).16, 17, 18
Figure 1.

SFV-AM6 shows enhanced oncolytic potency in GL261 cells in vitro
(A) Schematic presentation of the SFV-AM6 genome. (B) Representative images of plaques produced by SFV4 and SFV-AM6 in the BHK-21 and GL261 cell lines. (C) Analysis of cell attachment (cell index) with the xCELLigence system (mean ± SD). GL261 cells infected with SFV4 (blue), SFV-AM6 (red) at an MOI of 0.01, or left uninfected (black) (n = 5) are shown. The time point of infection is marked with a black arrowhead. (D) Cell viability of infected GL261 cells was measured with an MTS assay (mean ± SD) 72 h after infection using various MOIs. (E) Caspase-3/7 activity (mean ± SD) in GL261 cells 16 h after infection at an MOI of 1. (F) Virus titers measured (mean ± SD) by plaque titration 24 h after infection at an MOI of 0.01. (G) qRT-PCR analysis of interferon (IFN)-β and indicated IFN-stimulated genes (ISGs) in GL261 cells, 16 h after infection at an MOI of 1 with SFV4 (blue) or SFV-AM6 (red). Data are plotted as mean ± SD. IFIT1, IFN-induced protein with tetratricopeptide repeats 1; TNF, tumor necrosis factor; CXCL10, C-X-C motif chemokine 10; PD-L1; programmed death-ligand 1. Statistical analysis was performed by a Student’s two-tailed, unpaired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The plaque phenotype of SFV-AM6 is larger in BHK-21 cells, indicating generally faster replication and spread of the virus (Figure 1B). Notably, SFV-AM6 produces plaques also in GL261 cells, whereas SFV4 does not (Figure 1B). SFV-AM6 showed enhanced GL261 killing potency as quantified using the xCELLigence system (Figure 1C) and the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay (Figure 1D). SFV-AM6 also induced higher caspase-3/7 activation (Figure 1E). An approximately 100-fold higher final titer of SFV-AM6 was detected in GL261 cell culture medium collected after an MOI of 0.01 infection (Figure 1F), which further supports that SFV-AM6 replicates better in GL261 cells than does the parental SFV4.
Both SFV4 and SFV-AM6 promoted expression of IFN-β, IFN-stimulated genes (ISGs) ISG15, ISG20, and IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), as well as tumor necrosis factor (TNF)-α, C-X-C motif chemokine 10 (CXCL10), and programmed death-ligand 1 (PD-L1), as compared to uninfected cells (Figure 1G). The increased oncolytic potency of SFV-AM6 in GL261 cells is therefore not due to lower/inhibited antiviral signaling.
In line with our earlier results, the mouse glioma cell line CT-2A, which lacks a strong antiviral response and produces lower levels of IFN-I,9,10 was readily killed by low MOI SFV4 (Figure S1A), and there was no obvious benefit in using SFV-AM6. However, SFV-AM6 had an increased cytopathic effect (CPE) in the mouse NXS2 neuroblastoma cell line (Figure S1B), suggesting that the potential benefit of using SFV-AM6 is not limited to the GL261 model.
SFV-AM6-infected GL261 cells promote activation and maturation of co-cultured DCs
We have recently published that SFV can induce immunogenic cell death in cancer cells, leading to activation of DCs in vitro,19 and we wanted to analyze whether this occurs also after SFV-AM6 infection in GL261 cells.
Phagocytic activity of mouse immature bone marrow-derived DCs was enhanced when co-cultured with SFV-AM6-infected GL261 cells, as evidenced by increased uptake of pHrodo reagent (Figure 2A) as well as GFP from infected GL261-GFP cells (Figure 2B). Co-culturing with infected GL261 cells also upregulated DC maturation markers CD40, CD80, CD86, and major histocompatibility complex (MHC) class II (Figures 2C–2F). In the case of CD80, CD86, and MHC class II, upregulation was also observed when DCs were cultured with SFV-AM6 virus alone (MOI of 10), indicating a direct DC activating potency of the virus.
Figure 2.

SFV-AM6 infection of GL261 cells triggers phagocytosis and maturation in co-cultured dendritic cells (DCs)
(A and B) Flow cytometry analysis of DC phagocytic activity. (A) Percentage of pHrodo reagent-positive DCs (CD11c+) after co-culture with noninfected or SFV-AM6-infected GL261 cells. (B) Analysis of direct GL261-GFP cell phagocytosis by DCs. Percentage of GFP+CD11c+ DCs after co-culture with noninfected or SFV-AM6-infected GL261-GFP cells. (C–F) Mean fluorescence intensity (MFI) of DC maturation markers, CD80 (C), CD86 (D), CD40 (E), and MHC class II (F) on DCs (CD11chigh/CD11b+ cells) co-cultured with SFV-AM6-infected GL261 cells. Noninfected GL261 cells or SFV-AM6 virus alone was used as a control. Data are plotted as mean ± SD. Statistical analysis was done by a two-tailed, unpaired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
MicroRNA-124 (miR-124) de-targeted SFV-AM6 selectively replicates in GL261 tumors ex vivo and in vivo
To alleviate the neurotoxicity of SFV-AM6 in mice, we constructed a miR-124 de-targeted version of the virus. The design (Figure 3A) was based on our earlier published work.11,20 SFV-AM6-124T displayed a large plaque phenotype, similar to SFV-AM6, in BHK-21 and GL261 cells (Figures S2A and S2B; compare with Figure 1), and it displayed SFV-AM6-like oncolytic potency in GL261 cells (Figure S2C), indicating that the viral replication in target tumor cells was not reduced.
Figure 3.

SFV-AM6-124T infects GL261 tumors ex vivo and in vivo
(A) Schematic presentation of the SFV-AM6-124T genome. (B and C) 50,000 PFUs of SFV-AM6-124T virus was added on top of brain slice cultures with GL261 tumors. Samples were fixed 2 days after infection and stained with anti-SFV antibody (green), anti-CD31 (red), and Hoechst (blue). (C) Magnified image of the infected tumor area. (D–G) Intraperitoneally administered SFV-AM6-124T infects GL261 brain tumor in vivo. Representative images from oncolytic regions in orthotopic GL261 tumors from mice treated with SFV-AM6-124T. (D) Staining for SFV proteins in GL261 sample collected from SFV-AM6-124T-treated mice 2 days after last virus dose. Nuclei were stained with Hoechst (blue). (E) Staining for cleaved caspase-3 in the same sample as in (D). (F and G) Sample collected from SFV-AM6-124T-treated mice at the endpoint. (F) Staining for SFV proteins (green) and CD11c (magenta). (G) Staining for cleaved caspase-3 (green) and CD11c (magenta).
To evaluate the infectivity of SFV-AM6-124T in a relevant ex vivo model, we prepared organotypic coronal brain slice cultures from GL261-bearing mice and infected them with SFV-AM6-124T. As seen in Figures 3B and 3C, viral proteins were detected in the GL261 tumor, indicating that SFV-AM6-124T can infect GL261 cells also under ex vivo conditions.
Next, we treated orthotopic GL261 glioma-bearing mice with four intraperitoneal (i.p.) 1 × 107 plaque-forming unit (PFU) doses of the virus on days 2, 5, 8, and 11 after tumor induction. Immunofluorescence staining revealed tumor regions that were positive both for viral proteins and cleaved caspase-3, 2 days after the last virus injection (Figures 3D and 3E) as well as at the experiment end point (Figures 3F and 3G). Notably, apoptotic tumor areas in SFV-AM6-124T-treated mice were infiltrated by CD11c+ cells (Figures 3F and 3G), which gives evidence of immunogenic cell death in vivo.
SFV-AM6-124T in combination with anti-PD1 therapy increases immune cell infiltration in GL261 gliomas
To evaluate the therapeutic potential of SFV-AM6-124T, we analyzed the survival of GL261-bearing mice and performed an extensive flow cytometry analysis of immune cell infiltrates in GL261 tumors after therapy with SFV-AM6-124T, anti-PD1, and a combination of both (Figure 4A). SFV-AM6-124T alone did not improve survival of mice (Figure 4B).
Figure 4.

Intraperitoneally injected SFV-AM6-124T increases immune cell infiltration in orthotopic GL261 gliomas
(A) Mouse treatment scheme. (B) Kaplan-Meier plot of GL261-bearing mice either treated with SFV-AM6-124T (n = 26), anti-PD1 (n = 7), SFV-AM6-124T + anti-PD1 combination (n = 16), or left untreated (n = 24). (C–F) Flow cytometry analysis of CD45+ cells that were isolated from the brains of GL261-bearing mice after different treatments (n = 6). Absolute counts of CD45high cells (C), CD3+ cells (D), CD8+ T cells (E), CD4+ T cells (F), CD3+CD4−CD8− double-negative (DN) T cells (G), B cells (H), DCs (I), granulocytes (J), CD11b+ myeloid cells (K), and NK cells (L). Data are plotted as mean ± SD. Statistical analysis was done by one-way ANOVA with Tukey’s post hoc test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
However, combination therapy improved survival when compared to the control group or virus alone, but it failed when compared to anti-PD1 monotherapy (Figure 4B). Interestingly, flow cytometry analysis did show that the combination therapy significantly increased absolute numbers of all tumor-infiltrating immune cells (CD45high) (Figure 4C). Detailed analysis revealed a significant increase in CD3+ T cells (Figure 4D), including CD4+, CD8+, and double-negative (CD3+, CD4−, CD8−) T cell subsets (Figures 4E–4G), B cells (Figure 4H), and DCs (Figure 4H) in the combination therapy. In addition, there was a trend to increase in infiltrating granulocytes, CD11b+ cells, and natural killer (NK) cells in the combination therapy (Figures 4J–4L). In summary, the combination of SFV-AM6-124T and PD1 therapies synergize to improve intra-tumoral immune cell infiltration and inflammation.
SFV-AM6-124T promotes a distinct CD8+ T cell phenotype in the GL261 microenvironment
Encouraged by the significant increase in infiltration of cytotoxic T cells after combination therapy, we characterized them in detail by flow cytometry (Table S2). SFV-AM6-124T monotherapy modestly upregulated the activation markers CD69 and CD25 and the proliferation marker Ki67, but it led to a more distinct upregulation of CD127 and the senescence marker KLRG1 on tumor-infiltrating CD8+ T cells as compared to controls (Figure 5A), as reflected in higher numbers of KLRG1+CD127− and KLRG1+CD127+ CD8+ T cell populations in the virus treatment groups (Figures 5B–5D). Anti-PD1 as monotherapy induced strong expression of Ki67 and activation markers CD69, CD25, CD44, and the homing receptor CD62L on tumor-infiltrating CD8+ T cells, suggesting an effector memory phenotype (Figure 5A). In addition, only anti-PD1 monotherapy induced upregulation of CX3CR1 (Figure 5A; Figures S3A–S3C), a subset of CD8+ T cells known to be responsive to PD1 therapy.21 After combination therapy, tumor-infiltrating CD8+ T cells had an activated (CD69+CD25+), effector phenotype (CD44+CD62L−) but were in a state of low proliferation (Ki67low) (Figure 5A; Figures S3D–S3F). Combination therapy also markedly increased co-expression of multiple T cell activation/exhaustion markers PD1, TM3, and TIM3 (Figures 5A and 5E). In addition, there was an increase in intra-tumoral regulatory T cells (Tregs) (CD4+CD25+FOXP3+CD127−) in the combination treatment group (Figures S3G and S3H). The above results indicate that SFV-AM6-124T treatment induced a distinct effector CD8 T cell phenotype with high KLRG1 expression, and when combined with anti-PD1, it was associated with elevated PD1 and TIM3 expression.
Figure 5.

SFV-AM6-124T promotes a distinct CD8+ T cell phenotype in GL261 gliomas
(A) MFI heatmap of Ki67, CD69, CD25, CD44, CD62L, CD127, CX3CR1, KLRG1, PD1, TIM3, and LAG3 expression in CD8+ T cells from control and treated tumors. (B) Flow cytometry gating strategy for quantification of KLRG1 and CD127-expressing CD8+ T cells. (C and D) Quantification of KLRG1+/CD127− (C) and KLRG1+/CD127+ (D) cells. (E) Quantification of PD-1, TIM3, and LAG3 co-expression on CD8 T cells with SPICE software. Colors of the pie arcs depict the expression of individual inhibitory receptors, while the pie depicts the average proportion of co-expressed inhibitory receptors. (F) Bivariate plots showing staining for tetramer (Kb-restricted peptide amino acids 604–611 of p15E protein [KSPWFTTL]) on tumor-infiltrating CD8 T cells, and (G) the quantification of tumor-infiltrating tetramer+ CD8 T cells in each treatment group. (H and I) Quantification of KLRG1+/CD127− (H) and KLRG1+/CD127+ (I) tetramer+ CD8 T cell subpopulation. (J) Quantification of PD-1, TIM3, and LAG3 co-expression on tetramer+ CD8 T cells with SPICE software. Panels C, D, G, H and I are plotted as mean ± SD. Statistical analysis was done with one-way ANOVA with Tukey’s post hoc test (C, D, H and I) or Kruskal-Wallis with Dunn's post hoc test (G). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We next analyzed CD8+ T cells targeting GL261 tumor, which is of importance in respect to cancer therapy. The GL261 cells overexpress endogenous retrovirus-based tumor antigens, and T cells against this endogenous antigen are detectable using tetramers against the murine leukemia virus (MuLV) emv2-env epitope (KSPWFTTL).22 We observed an increased infiltration of KSPWFTTL-tetramer (Tet)+ tumor-specific T cells in anti-PD1 monotherapy and in combination therapy compared to controls (Figures 5F and 5G). More importantly, there was no difference in the percentage of KLRG1+CD127+ anti-tumor CD8+ T cells (Figure 5H) and lower levels of terminally differentiated KLRG1+CD127− anti-tumor CD8+ T cells (Figure 5I) in anti-PD1 and combination treatment compared to SFV-AM6-124T monotherapy. In addition, there were no differences in the co-expression of multiple T cell activation/exhaustion markers PD1, LAG3, and TIM3 (Figure 5J) among the different treatment groups. In conclusion, our therapies induced anti-tumor-specific CD8+ T cells, and especially combination therapy rescued the terminally differentiated KLRG1+ T cell phenotype.
Discussion
Given the important role of IFN-I in promoting cancer immunity and evidence of functional IFN-I signaling in human GBM samples,13,14,23 it seems likely that IFN-I resistant/tolerant viruses would be favorable as oncolytic immunotherapy agents. Consistent with this, the clinically relevant poliovirus PVS-RIPO has recently been reported to be tolerant to IFN-I in cancer cells.24 In the present study, we report a novel SFV-AM6 virus that shows enhanced replication and cytotoxicity in a highly IFN-I-competent GL261 mouse glioma cell line (Figure 1). Importantly, the increase in replication was not due to a dampened IFN-I response (Figure 1G), indicating a clearly improved, IFN-I tolerant phenotype of the SFV-AM6 over wild-type SFV4. A plaque assay in particular gave a strong indication of its ability to spread in restrictive tumor microenvironment-like conditions (Figure 1B). In contrast to regular non-immunogenic apoptosis, SFV-AM6 infection triggers DC activation (Figure 2). This is in line with our previously reported findings of SFV-induced pathogen- and damage-associated molecular pattern (PAMP and DAMP)-driven immunogenic cell death,19 and it indicates the immunotherapeutic potential of the SFV-AM6 construct.
While the molecular mechanistic explanation for the increased oncolytic potency of SFV-AM6 is unclear, some reasonable speculations can be made. The NsP3A70G substitution is located in the conserved nsP3 macrodomain and is thus possibly linked to viral counteraction of IFN-stimulated poly(ADP-ribose) polymerases.16,17 The NsP3Y369F mutation has been recently shown to inhibit viral hyperactivation of the phosphatidylinositol 3-kinase (PI3K-AKT-mTOR) pathway and reduce replication in vivo.25 The PI3K-AKT pathway is, however, overactivated in many cancers, including GBM, due to frequent loss of the negative regulator PTEN.26 The NsP3Y369F mutation could therefore selectively increase virus replication in cancer cells with oncogenic over-activation in the PI3K-AKT pathway, such as PTEN-defective GL261 cells.27
The NsP4R611K substitution affects the penultimate residue of nsP4 and the overlapping 26S sub-genomic RNA promoter region, and most probably acts by enhancing replication speed and/or promoting synthesis of 26S RNA (unpublished data). Finally, a Lys to Glu switch at residue 162 in E2 was included in our construct due to its positive effect on induction of viremia as well as BBB penetration.18
miR-124 de-targeting11,20 did not interfere with GL261 infectivity in vitro or ex vivo (Figure S2; Figure 3B and 3C). Our previously published work indicates that although miR-124 de-targeting significantly attenuates neurovirulence, it does not result in complete protection when the virus is administered intracranially.20 Because of this, as well as the advantages of systemic delivery, we decided to study SFV-AM6-124T as an i.p. administered oncolytic agent. In the case of SFV, we consider also that infection/activation of brain endothelial cells through systemic delivery should favorably increase immune cell infiltration. Impressively, systemically administered SFV-AM6-124T can reach orthotopic GL261 tumors, resulting in apoptotic/dead regions surrounded by SFV-infected cells (Figures 3D and 3E). The infected regions were infiltrated by CD11c+ cells (Figures 3F and 3G), which are likely peripheral DCs and serve as an indication of in vivo immunogenic oncolysis. Evidence of immunogenic oncolysis has been reported previously in the GL261 model with Newcastle disease virus28 as well as in a clinical trial with parvovirus (ParvOryx).29
In the context of immunotherapy, we analyzed the effect of SFV-AM6-124T to the immune microenvironment in GL261 gliomas both as monotherapy and when combined with an anti-PD1 antibody, as anti-PD1 therapy has been shown to be effective against GL261 gliomas.30 Anti-PD1 combination was considered rational also due to SFV-induced PD-L1 expression in GL261 cells (Figure 1G), likely via IFN-I signaling.31,32 In the present study, combination therapy with virus and anti-PD1 improved survival when compared to the control group or virus alone, but it failed to improve survival when compared to anti-PD1 alone (Figure 4A). The combination therapy group actually showed somewhat reduced survival when compared to anti-PD1. The lack of statistical significance between these two groups can be explained by the lower number of mice in the anti-PD1 group. Despite lacking clear survival benefit, the combination therapy, however, increased general numbers of GL261-infiltrating T cells, B cells, DCs (Figure 4), and intra-tumoral inflammation, suggesting potential pseudo-progression of the tumors, a phenomenon frequently observed after OV therapy of cancers.33
Detailed analysis of the tumor-infiltrating cytotoxic CD8+ T cells revealed that SFV-AM6-124T promoted a low-proliferating and exhausted KLRG1+ effector phenotype (Figure 5), which is likely associated with the cumulative T cell receptor and inflammatory stimulus.34 Although KLRG1+CD8+ cells have been shown to differentiate into “exKLRG1” cells (KLRG1−CD127+) with potent anti-viral and anti-tumor effects,34 it is reasonable to speculate that their presence is a therapeutically unfavorable response. The KLRG1+CD127− population was also increased in the GL261-targeting CD8+ T cells in response to virus alone (Figure 5I). The combination with anti-PD1, however, reduced the population to anti-PD1 monotherapy levels (Figure 5I), indicating reversion of the SFV-AM6-124T-driven negative effect in anti-tumor T cells.
It is not self-evident that OVs would synergize positively with an anti-PD1-boosted T cell response. In fact, it can be expected that the immunodominant viral antigens could also redirect the T cell response away from anti-tumor toward anti-virus and promote T cell exhaustion. A virus-induced IFN-I response can also negatively affect anti-tumor T cells, as reported recently by Evgin et al.35 using a combination of oncolytic vesicular stomatitis virus (VSV) and chimeric antigen receptor (CAR) T cell therapy in a mouse melanoma model. Herein, we report that systemically administered oncolytic SFV-AM6-124T in combination with anti-PD1 results in increased anti-tumor CD8+ infiltration in an orthotopic mouse GL261 model. Importantly, the GL261-reactive CD8+ T cells did not show a similar terminally differentiated phenotype that was observed in the total CD8+ population (Figure 5), indicating that the anti-tumor response is not negatively affected by the virus.
The lack of correlation to survival benefit in SFV-AM6-124T-treated mice makes interpretation of the results challenging. It is likely that the kinetics of the combination therapy-induced anti-tumor response is not sufficiently fast to prevent tumor growth within the short time frame of the GL261 model. The symptoms observed in the combination therapy group could be due to heavily increased inflammation in the tumors. Although some level of inflammation in the intracranial tumors is a favorable immunotherapeutic response, it can lead to severe symptoms in mice. We therefore advocate that monitoring mouse symptoms is not sufficient, and analysis of the immune cell infiltrates should be a key consideration when evaluating the therapeutic potency.
Taken together, our results support that the oncolytic potency of SFV-AM6-124T can be harnessed in combination with anti-PD1 to enhance the anti-tumor immune response. The results can be translated into development of effective oncolytic immunotherapy against GBM.
Materials and methods
Cell lines
GL261 mouse GBM cells (gift from Dr. G. Sáfrány, National Research Institute for Radiobiology and Radiohygiene, Budapest, Hungary) were cultured in Gibco DMEM GlutaMAX (Thermo Fischer Scientific, Waltham, MA, USA) supplemented with 10% Gibco heat-inactivated fetal bovine serum (FBS, Thermo Fischer Scientific), 10 U/mL Gibco penicillin-streptomycin (PEST, Thermo Fisher Scientific), and 1 mM Gibco sodium pyruvate (Thermo Fisher Scientific). Baby hamster kidney (BHK)-21 cells were cultured in Gibco Glasgow’s minimum essential medium (GMEM, Thermo Fisher Scientific) supplemented with 10% FBS, 2% tryptose phosphate broth (Teknova, Hollister, CA, USA), 20 mM HEPES, and 10 U/mL Gibco PEST (Thermo Fisher Scientific).
Viruses
All modifications/mutations were made into pCMV-SFV4 backbone plasmid36 either by in vitro mutagenesis and/or by replacement of fragments with the mutation-containing fragments from previously made constructs; the obtained construct was verified by sequencing and designated as pCMV-SFV-AM6. pCMV-SFV-AM6-124T was constructed by cloning the miR124T fragment from pSP6-SFV4-124T20 into pCMV-SFV-AM6. All SFV viruses were produced and titrated in BHK-21 cells as described in Supplemental materials and methods.
GL261 infection assays
The xCELLigence system (ACEA Biosciences, San Diego, CA, USA) was used to assess GL261 de-attachment following an MOI of 0.01 virus infection of 10,000 cells. The assay was performed as described in Supplemental materials and methods. For the cell viability assay, GL261 cells were seeded onto a 96-well plate (20,000 cells/well) and infected on the next day with virus (diluted in complete cell culture medium) at different MOIs. Cell viability was measured using a CellTiter 96 AQueous One solution cell proliferation assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions 72 h after infection. A Caspase-Glo 3/7 assay system (Promega) was used to measure caspase-3 and caspase-7 activity in GL261 cells (plated onto a 96-well plate as above) infected with an MOI of 1, 16 h after infection. A plaque assay in GL261 cells was performed according to a titration protocol (Supplemental materials and methods).
qRT-PCR
500,000 GL261 cells were infected at an MOI of 1. Infected and uninfected cells (three parallel samples each) were harvested 16 h after infection. RNA was extracted using an RNeasy mini kit (QIAGEN, Germany). cDNA was synthesized from pooled RNA samples with iScript cDNA synthesis (Bio-Rad, Hercules, CA, USA) from 1 μg of extracted RNA. iQ SYBR Green supermix (Bio-Rad) and the primers listed in Table S1. Each sample was run in duplicate, and the fold change of expression was quantified using the 2–ΔΔCt method.
Infection of brain slice culture
Brains from GL261 tumor-bearing mice were collected in complete Hanks’ balanced salt solution (HBSS; 2.5 mM HEPES [pH 7.4], 30 mM d-glucose, 1 mM CaCl2, 1 mM MgSO4, 4 mM NaHCO3) and embedded in low-melting point agarose on ice. Agarose-embedded brains were cut into 200-μm coronal sections with a vibratome. Slices were transferred onto cell culture inserts (8-μm membrane, VWR International, Radnor, PA, USA) on 12-well plates with 350 μL of added culture medium (68% DMEM + l-glutamine, 5% FBS, 1% PEST in complete HBSS) on the bottom of wells. Slice cultures were incubated (37°C, 5% CO2) for 1 h, after which 5 μL of virus (50,000 PFU) was pipetted onto the tumor. After 2 days of incubation with the virus (37°C, 5% CO2), slices were fixed overnight with 4% paraformaldehyde (PFA) at 4°C. Fixed slices were transferred onto 12-well plates (without inserts) and stained using rabbit anti-SFV (kind gift from Ari Hinkkanen, University of Eastern Finland, Kuopio, Finland) and hamster anti-CD31 (Thermo Fisher Scientific) as primary antibodies. Life Technologies Alexa Fluor 488 (AF488)-conjugated donkey anti-rabbit (Thermo Fisher Scientific) and Alexa Fluor 647 (AF647)-conjugated goat anti-hamster (Thermo Fisher Scientific) were used as secondary antibodies. The background was stained with Invitrogen Hoechst 33342 (Thermo Fisher Scientific). For the staining protocol, see Supplemental materials and methods.
Animal experiments
2 × 104 GL261 cells were injected intracranially into 6- to 8-week-old female C57BL/6NRj mice (Janvier Labs, France), 1 mm anterior and 1.5 mm right from bregma at 3-mm depth using a Hamilton syringe and stereotactic equipment (AgnTho’s, Sweden). Mice treated with virus received a 1 × 107 PFU dose of SFV-AM6-124T i.p. at 2, 5, 8, and 11 days after tumor induction. Mouse anti-PD1 (clone RMP1-14) was administered at 200 μg/mouse i.p. at days 9 and 12 after tumor induction. Mice were sacrificed upon appearance of symptoms such as paralysis, hunchback, 20% loss of body weight, or notable distress. For flow cytometry analysis of immune cells, brains were collected at day 14 after tumor induction.
Immunofluorescence
Brains from sacrificed mice were cut in half (midsagittal), snap-frozen in 2-methylbutane on dry ice, embedded to Neg-50 (Thermo Fisher Scientific), and cut into 7-μm sections with a cryostat. The sections were fixed with −20°C methanol and stained with rabbit anti-SFV, rabbit anti-cleaved caspase-3 (Cell Signaling Technology, Danvers, MA, USA), and hamster anti-mouse CD11c (BD Biosciences, San Jose, CA, USA). Life Technologies donkey anti-rabbit-AF488 and goat anti-hamster-AF647 (Thermo Fisher Scientific) were used as secondary antibodies. The sections were imaged with an Eclipse Ti-S microscope (Nikon, Japan).
Flow cytometry
Single cells from the murine brain earing GL261 tumors were isolated using a mouse tumor dissociation kit (Miltenyi Biotec, Germany) and a gentleMACS Octo dissociator with heaters (Miltenyi Biotec). Myelin was depleted using 25% BSA density gradient centrifugation for 20 min at 2,600 rpm and subsequently removing the myelin rim on top of the BSA gradient. CD45+ cells were enriched using mouse CD45 MicroBeads (Miltenyi Biotec) following the manufacturer’s instructions. Unspecific Fc receptor binding in all single-cell suspensions was then blocked by using anti-mouse CD16/CD32 antibody (clone 93, BioLegend, San Diego, CA, USA), and were then stained using fluorochrome-conjugated antibodies (Table S2). All antibodies were diluted 1:100 from stock concentration in Brilliant Violet stain buffer (BD Biosciences). CountBright absolute counting beads (Thermo Fisher Scientific) were added to calculate absolute numbers of immune infiltrates, and samples were acquired in an LSRFortessa (BD Biosciences). Data were analyzed using FlowJo version 10.7 (FlowJo) or SPICE.37
DC phagocytosis and maturation assays
Bone marrow-derived DCs were isolated from the femur and tibia of female 7- to 8-week-old wild-type C57BL/6NRj mice (Janvier Labs, France) and cultured in Gibco Iscove’s modified Dulbecco’s medium (IMDM, Thermo Fischer Scientific) supplemented with 10% heat-inactivated FBS, 10 U/mL PEST, 10 mM HEPES, 50 mM β-mercaptoethanol, 20 ng/mL recombinant murine interleukin (IL)-4, and 20 ng/mL recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF, Nordic BioSite, Sweden). The medium was changed every 3 days. Non-adherent immature DCs were harvested for a co-culture assay on day 7.
For measuring phagocytic activity, GL261 cells were infected with SFV-AM6 at an MOI of 10 for 48 h followed by co-culturing with DCs (1:1 ratio) and 100 μg of pHrodo Red Staphylococcus aureus Bioparticles conjugate for phagocytosis (Thermo Fisher Scientific) for 2 h. To measure phagocytosis of tumor cells directly, GL261-GFP cells were infected and co-cultured with DCs as above. Noninfected cells were used as a control. Cells were harvested and DCs were stained with anti-CD11c-allophycocyanin (APC)/Cy7 (BioLegend). Phagocytosis of pHrodo reagent was quantified as the percentage of CD11c+pHrodo+ cells. Similarly, the phagocytosis of dead GL261-GFP cells by DCs was quantified as the percentage of CD11c+GFP+ cells
For the DC maturation assay, GL261 cells were infected with SFV-AM6 (MOI of 10) for 48 h followed by co-culturing with immature DCs at a ratio 1:1 for 18 h. Noninfected GL261 cells were used as a negative control. Additionally, SFV-AM6 was added directly on DCs (MOI of 10). Maturation of DCs was analyzed by flow cytometry using anti-CD11c-APC/Cy7, anti-CD11b-peridinin chlorophyll protein (PerCP), anti-CD80-Pacific Blue, anti-CD86-phycoerythrin (PE)/Cy7, anti-CD40-APC, and anti-I-A/I-E-Brilliant Violet 510 (BV510, BD Biosciences) antibodies.
Statistical analysis
All statistical analysis was performed using GraphPad Prism software.
Ethics statement
The Swedish Work Environment Authority has approved the work with genetic modification of SFV (ID no. 202100-2932 v66a14 [laboratory] and v67a10 [mice]). All experiments regarding modified SFV were conducted under biosafety level 2. The local Animal Ethics Committee in Stockholm (N164/15, N170/13) approved the animal studies.
Acknowledgments
This work was supported by the Swedish Cancer Society (CAN 2016/318 and 19 0184 Pj); the Swedish Childhood Cancer Fund (PR2018-0127); the Swedish Research Council (2015-03688 and 20199-01326); the Knut and Alice Wallenberg Foundation (2019.0088); and by grant IUT20-27 from the Estonian Research Council. M.M. is a recipient of a Marie Curie fellowship from the EU (AVITAG, 707093). M.R. is a recipient of a postdoctoral fellowship from the Swedish Childhood Cancer Fund (TJ2017-0004). J.M. is supported by a fellowship from the Chinese Scholarship Council. We acknowledge Gerald McInerney (Karolinska Institutet, Stockholm, Sweden) for help with the design of the virus construct and for providing plasmids for the cloning. We also thank Alessandra Vaccaro (Uppsala University) and Tina Sarén (Uppsala University) for help with immune cell isolation.
Author contributions
M.M., M.R., R.L., A.D., D.Y., A.M., and M.E. designed the experiments. M.M., M.R., R.L., J.M., M.-M.M., and A.M. conducted the experiments. M.M., M.R., R.L., J.M., A.D., D.Y., A.M., and M.E. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.03.008.
Supplemental information
References
- 1.Louis D.N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P., Ellison D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous system: A summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Ostrom Q.T., Gittleman H., Liao P., Vecchione-Koval T., Wolinsky Y., Kruchko C., Barnholtz-Sloan J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-oncol. 2017;19(Suppl 5):v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desjardins A., Gromeier M., Herndon J.E., 2nd, Beaubier N., Bolognesi D.P., Friedman A.H., Friedman H.S., McSherry F., Muscat A.M., Nair S. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018;379:150–161. doi: 10.1056/NEJMoa1716435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang F.F., Conrad C., Gomez-Manzano C., Yung W.K.A., Sawaya R., Weinberg J.S., Prabhu S.S., Rao G., Fuller G.N., Aldape K.D. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018;36:1419–1427. doi: 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cloughesy T.F., Landolfi J., Vogelbaum M.A., Ostertag D., Elder J.B., Bloomfield S., Carter B., Chen C.C., Kalkanis S.N., Kesari S. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-oncol. 2018;20:1383–1392. doi: 10.1093/neuonc/noy075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aurelian L. Oncolytic viruses as immunotherapy: Progress and remaining challenges. OncoTargets Ther. 2016;9:2627–2637. doi: 10.2147/OTT.S63049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell S.J., Barber G.N. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. 2018;33:599–605. doi: 10.1016/j.ccell.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruotsalainen J., Martikainen M., Niittykoski M., Huhtala T., Aaltonen T., Heikkilä J., Bell J., Vähä-Koskela M., Hinkkanen A. Interferon-β sensitivity of tumor cells correlates with poor response to VA7 virotherapy in mouse glioma models. Mol. Ther. 2012;20:1529–1539. doi: 10.1038/mt.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramachandran M., Yu D., Dyczynski M., Baskaran S., Zhang L., Lulla A., Lulla V., Saul S., Nelander S., Dimberg A. Safe and effective treatment of experimental neuroblastoma and glioblastoma using systemically delivered triple microRNA-detargeted oncolytic Semliki Forest virus. Clin. Cancer Res. 2017;23:1519–1530. doi: 10.1158/1078-0432.CCR-16-0925. [DOI] [PubMed] [Google Scholar]
- 11.Martikainen M., Niittykoski M., von und zu Fraunberg M., Immonen A., Koponen S., van Geenen M., Vähä-Koskela M., Ylösmäki E., Jääskeläinen J.E., Saksela K., Hinkkanen A. MicroRNA-attenuated clone of virulent Semliki Forest Virus overcomes antiviral type I interferon in resistant mouse CT-2A glioma. J. Virol. 2015;89:10637–10647. doi: 10.1128/JVI.01868-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heikkilä J.E., Vähä-Koskela M.J.V., Ruotsalainen J.J., Martikainen M.W., Stanford M.M., McCart J.A., Bell J.C., Hinkkanen A.E. Intravenously administered alphavirus vector VA7 eradicates orthotopic human glioma xenografts in nude mice. PLoS ONE. 2010;5:e8603. doi: 10.1371/journal.pone.0008603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cosset É., Petty T.J., Dutoit V., Cordey S., Padioleau I., Otten-Hernandez P., Farinelli L., Kaiser L., Bruyère-Cerdan P., Tirefort D. Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral-like type I interferon gene response. Int. J. Cancer. 2014;135:1381–1389. doi: 10.1002/ijc.28670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duarte C.W., Willey C.D., Zhi D., Cui X., Harris J.J., Vaughan L.K., Mehta T., McCubrey R.O., Khodarev N.N., Weichselbaum R.R., Gillespie G.Y. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS ONE. 2012;7:e29653. doi: 10.1371/journal.pone.0029653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham R., Hauer D., McPherson R.L., Utt A., Kirby I.T., Cohen M.S., Merits A., Leung A.K.L., Griffin D.E. ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc. Natl. Acad. Sci. USA. 2018;115:E10457–E10466. doi: 10.1073/pnas.1812130115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung A.K.L., McPherson R.L., Griffin D.E. Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases. PLoS Pathog. 2018;14:e1006864. doi: 10.1371/journal.ppat.1006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson M.C., Saul S., Fragkoudis R., Weisheit S., Cox J., Patabendige A., Sherwood K., Watson M., Merits A., Fazakerley J.K. Ability of the encephalitic arbovirus Semliki Forest virus to cross the blood-brain barrier is determined by the charge of the E2 glycoprotein. J. Virol. 2015;89:7536–7549. doi: 10.1128/JVI.03645-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma J., Ramachandran M., Jin C., Quijano-Rubio C., Martikainen M., Yu D., Essand M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020;11:48. doi: 10.1038/s41419-020-2236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ylösmäki E., Martikainen M., Hinkkanen A., Saksela K. Attenuation of Semliki Forest virus neurovirulence by microRNA-mediated detargeting. J. Virol. 2013;87:335–344. doi: 10.1128/JVI.01940-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan Y., Cao S., Liu X., Harrington S.M., Bindeman W.E., Adjei A.A., Jang J.S., Jen J., Li Y., Chanana P. CX3CR1 identifies PD-1 therapy-responsive CD8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight. 2018;3:e97828. doi: 10.1172/jci.insight.97828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song E., Mao T., Dong H., Boisserand L.S.B., Antila S., Bosenberg M., Alitalo K., Thomas J.L., Iwasaki A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature. 2020;577:689–694. doi: 10.1038/s41586-019-1912-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alain T., Lun X., Martineau Y., Sean P., Pulendran B., Petroulakis E., Zemp F.J., Lemay C.G., Roy D., Bell J.C. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc. Natl. Acad. Sci. USA. 2010;107:1576–1581. doi: 10.1073/pnas.0912344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walton R.W., Brown M.C., Sacco M.T., Gromeier M. Engineered oncolytic poliovirus PVSRIPO subverts MDA5-dependent innate immune responses in cancer cells. J. Virol. 2018;92 doi: 10.1128/JVI.00879-18. e00879-e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzon M., Castro C., Thaa B., Liu L., Mutso M., Liu X., Mahalingam S., Griffin J.L., Marsh M., McInerney G.M. Alphavirus-induced hyperactivation of PI3K/AKT directs pro-viral metabolic changes. PLoS Pathog. 2018;14:e1006835. doi: 10.1371/journal.ppat.1006835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srividya M.R., Thota B., Shailaja B.C., Arivazhagan A., Thennarasu K., Chandramouli B.A., Hegde A.S., Santosh V. Homozygous 10q23/PTEN deletion and its impact on outcome in glioblastoma: a prospective translational study on a uniformly treated cohort of adult patients. Neuropathology. 2011;31:376–383. doi: 10.1111/j.1440-1789.2010.01178.x. [DOI] [PubMed] [Google Scholar]
- 27.Oh T., Fakurnejad S., Sayegh E.T., Clark A.J., Ivan M.E., Sun M.Z., Safaee M., Bloch O., James C.D., Parsa A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014;12:107. doi: 10.1186/1479-5876-12-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koks C.A., Garg A.D., Ehrhardt M., Riva M., Vandenberk L., Boon L., De Vleeschouwer S., Agostinis P., Graf N., Van Gool S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer. 2015;136:E313–E325. doi: 10.1002/ijc.29202. [DOI] [PubMed] [Google Scholar]
- 29.Geletneky K., Hajda J., Angelova A.L., Leuchs B., Capper D., Bartsch A.J., Neumann J.-O., Schöning T., Hüsing J., Beelte B. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 2017;25:2620–2634. doi: 10.1016/j.ymthe.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genoud V., Marinari E., Nikolaev S.I., Castle J.C., Bukur V., Dietrich P.-Y., Okada H., Walker P.R. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. OncoImmunology. 2018;7:e1501137. doi: 10.1080/2162402X.2018.1501137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao W., Klement J.D., Lu C., Ibrahim M.L., Liu K. IFNAR1 controls autocrine type I IFN regulation of PD-L1 expression in myeloid-derived suppressor cells. J. Immunol. 2018;201:264–277. doi: 10.4049/jimmunol.1800129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bazhin A.V., von Ahn K., Fritz J., Werner J., Karakhanova S. Interferon-α up-regulates the expression of PD-L1 molecules on immune cells through STAT3 and p38 signaling. Front. Immunol. 2018;9:2129. doi: 10.3389/fimmu.2018.02129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiyokawa J., Wakimoto H. Preclinical and clinical development of oncolytic adenovirus for the treatment of malignant glioma. Oncolytic Virother. 2019;8:27–37. doi: 10.2147/OV.S196403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herndler-Brandstetter D., Ishigame H., Shinnakasu R., Plajer V., Stecher C., Zhao J., Lietzenmayer M., Kroehling L., Takumi A., Kometani K. KLRG1+ effector CD8+ T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity. 2018;48:716–729.e8. doi: 10.1016/j.immuni.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evgin L., Huff A.L., Wongthida P., Thompson J., Kottke T., Tonne J., Schuelke M., Ayasoufi K., Driscoll C.B., Shim K.G. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat. Commun. 2020;11:3187. doi: 10.1038/s41467-020-17011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulper L., Sarand I., Rausalu K., Merits A. Construction, properties, and potential application of infectious plasmids containing Semliki Forest virus full-length cDNA with an inserted intron. J. Virol. Methods. 2008;148:265–270. doi: 10.1016/j.jviromet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roederer M., Nozzi J.L., Nason M.C. SPICE: Exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A. 2011;79:167–174. doi: 10.1002/cyto.a.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.