

Abstract
Head and neck squamous cell carcinoma (HNSCC) is the most frequent type of head and neck cancer that usually arises from the mucosal surfaces of several organs including nasal cavity, paranasal sinuses, oral cavity, tongue, pharynx, and larynx. The Wnt signaling pathway is a crucial mechanism for cellular maintenance and development. It regulates cell cycle progression, apoptosis, proliferation, migration, and differentiation. Dysregulation of this pathway correlates with oncogenesis in various tissues including breast, colon, pancreatic as well as head and neck cancers. The present study aims to assess the gene alterations in the Wnt family of genes so as to derive an association with HNSCC. Computational approaches have been utilized for the identification of gene alterations in the Wnt family of genes. Several databases such as cBioportal, STRING, and UALCAN were used for the purpose. The frequency of alteration was high in case of Wnt family member 11 (5%). Gene amplification, deep deletions, missense and truncating mutations were observed in HNSCC patients. There was a marked difference in the gene expression profile of WNT11 between grades as well as normal samples. The survival probability measured using the Kaplan-Meier curve also presented with a significant difference among male and female subjects experiencing a low/medium level expression. The female patients showed less survival probability when compared to the male subjects. This provides the prognostic significance of the WNT11 gene in HNSCC. Taken together, the present study provides clues on the possible association of WNT11 gene alterations with HNSCC, which has to be further validated using experimental approaches.
Keywords: gene amplification, genetic variants, head and neck squamous cell carcinoma, in silico, Wnt pathway
Introduction
The head and neck squamous cell carcinoma (HNSCC) represents a heterogeneous group of cancer affecting the mucosal surfaces of several organs including nasal cavity, paranasal sinuses, oral cavity, tongue, pharynx, and larynx [1]. HNSCC accounts for more than 330,000 deaths worldwide with more than 650,000 cases of HNSCCs reported annually [2]. The development of HNSCC is strongly associated with long-term tobacco use, excessive consumption of strong alcohols or, especially in the case of oropharyngeal tumors, the infection with human papillomavirus (HPV), usually HPV type 16 or 18 [3]. The incidence of HNSCC is high in males when compared to females, especially in eastern Europe and India with over 20 males affected per 100,000 individuals [4]. Despite relatively easy access for clinical inspection, these tumors are frequently detected at a late stage, when therapeutic options are less effective in curing patients, who are then at a greater risk of the development of recurrent tumors or metastasis [5]. Thus, the overall survival rates in this group of patients remain relatively low (~50%), especially when patients are diagnosed with advanced stages of the disease [6]. There is a need for novel biomarkers which could improve the clinical management of HNSCC, including better prognosis and disease monitoring. Moreover, the development of new therapeutic options is also necessary for the improvement of treatment outcomes.
Wnt signaling is vital for a plethora of cellular function ranging from homeostasis to the development of mature tissue. Embryonic development also requires Wnt-mediated canonical signaling [7-9]. Moreover, Wnt signaling is inevitable for regulating cellular proliferation, apoptosis, metastasis, and migration of cells [10]. Of note, Wnt operates through either canonical or non-canonical pathways which are differentiated by β-catenin involvement [11]. Cell cycle progression, differentiation, fate determination, and migration are generally orchestrated by canonical Wnt signaling. Altered Wnt/β-catenin signaling has been considered a promoting event for various types of cancers and the oncogenic potential of Wnt signaling has been discussed in numerous cancer types, including breast, pancreatic, colon as well as head and neck [12]. The present study investigates the genetic alteration within the Wnt family genes employing in silico approach. The study is first of its kind that reports frequency and type of mutation in genes of the Wnt signaling pathway which provides a clue on the putative association of these genes with HNSCC.
Methods
Sample data set
The data is obtained from the cBioportal database which is a web source for obtaining, analyzing and exploring genomes. It contains the description of patients from different cohorts and provides information on the genetic alterations among various samples and genes. The Cancer Genome Atlas, TCGA (Firehose Legacy) data set consists of a total of 528 cases of head and neck squamous cell carcinoma, of which 504 samples had sequencing and copy number alteration data. The complete profile of mutated, amplified, deleted genes for each sample has been recorded in the database. Table 1 contains the demographic data of the patients analyzed in the study. Oncoprint data was obtained on submitting user-defined queries on 19 genes of the Wnt family, which was further analyzed for gene expression profile.
Table 1.
Demographic details of patients analyzed in the present study (as obtained from the cBioportal site)
| Characteristic | No. |
|---|---|
| Sex | |
| Male | 386 |
| Female | 142 |
| Mutation count | 6‒3,181 |
| Diagnosis age (y) | 19-90 |
| Smoking status | |
| Smokers | 515 |
| Data not available | 12 |
| Unknown | 1 |
| Alcohol history | |
| Yes | 352 |
| No | 165 |
| Data not available | 11 |
| Neoplasm histologic grade | |
| Grade 1 | 63 |
| Grade 2 | 311 |
| Grade 3 | 125 |
| Grade 4 | 7 |
| Grade GX | 18 |
| Data not available | 4 |
| Race category | |
| White | 452 |
| African | 48 |
| Asian | 11 |
| American Indian or Alaska native | 2 |
| Data not available | 15 |
Oncoprint data analysis
Oncoprint analysis is the shortened and concise summation of the genetic alterations in graphical format. It provides data on gene alterations based on user-defined query on a specific gene or a gene family. The details on frequency distribution of variations in each of the genes, the variant allele frequency, gene deletions, amplifications, insertions, frameshift etc., were recorded (Fig. 1) [13,14]. These information were used as baseline to track mutations or variations, gene expression, and survival of patients based on the gene alterations using several other computational tools.
Fig. 1.
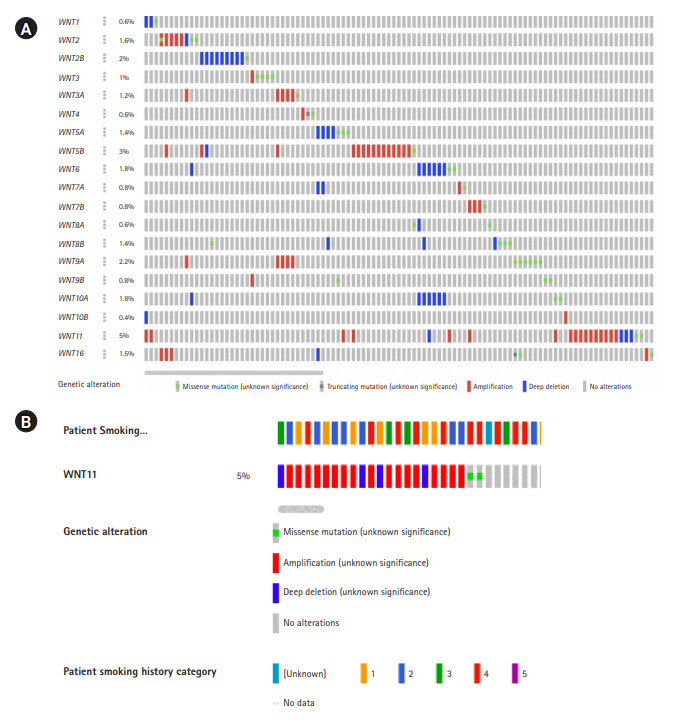
(A) Oncoprint analysis depicting gene alterations in the Wnt family of genes. Each of the grey bars represent patients with head and neck squamous cell carcinoma. (B) Frequency of gene amplification among different categories of smokers.
gnomAD analysis
The genome aggregation database (gnomAD) hosts information on 125,748 exome sequences and 15,708 whole genome sequences from unrelated individuals sequenced and deposited as part of various disease-specific or population genetic studies. The data source obtained from oncoprint was used to identify whether the variations observed in the present study were novel or reported elsewhere in any other population (Table 2). The exhaustive data source also provides information on minor allele frequencies which will provide a clue as whether the variant identified is a mutation or a polymorphism [15].
Table 2.
The list of genes, proteins encoded, genetic alterations, loci, frequency of alteration, and variant allele frequency in genes of the Wnt signaling pathway
| Gene | Protein | Alteration | Loci | % of alteration | Variant allele frequency in tumor sample | gnomAD data |
|---|---|---|---|---|---|---|
| WNT1 | Wnt family member 1 | Deep deletion | 12q13.12 | 0.6 | ||
| D49N | 0.16 | Novel | ||||
| WNT2 | Wnt family member 2 | Gene amplification | 7q31.2 | 1.6 | ||
| Deep deletion | ||||||
| S158R | 0.42 | Novel | ||||
| T315P | 0.23 | Novel | ||||
| T315N | 0.57 | Novel | ||||
| WNT2B | Wnt family member 2B | Deep deletion | 1p13.2 | 2 | ||
| R16L | 0.63 | Novel | ||||
| WNT3 | Wnt family member 3 | Gene amplification | 17q21.31‒q21.32 | 1 | ||
| R85Q | 0.68 | rs1483494147 | ||||
| E2Q | 0.18 | Novel | ||||
| A261P | 0.46 | Novel | ||||
| G318D | 0.21 | Novel | ||||
| WNT3A | Wnt family member 3A | Gene amplification | 1q42.13 | 1.2 | ||
| P283L | 0.33 | Novel | ||||
| WNT4 | Wnt family member 4 | Gene amplification | 1p36.12 | 0.6 | ||
| E76Q | 0.26 | Novel | ||||
| S176Efs*12 | 0.13 | Novel | ||||
| WNT5A | Wnt family member 5A | Deep deletion | 3p14.3 | 1.4 | ||
| I93V | 0.28 | rs750646727 | ||||
| G341S | 0.20 | rs1370251695 | ||||
| R215C | 0.57 | Novel | ||||
| WNT5B | Wnt family member 5B | Gene amplification | 12p13.33 | 3 | ||
| Deep deletion | ||||||
| G242W | 0.36 | Novel | ||||
| WNT6 | Wnt family member 6 | Deep deletion | 2q35 | 1.8 | ||
| R46W | 0.29 | rs766635655 | ||||
| T105M | 0.23 | rs759013954 | ||||
| WNT7A | Wnt family member 7A | Gene amplification | 3p25.1 | 0.8 | ||
| Deep deletion | ||||||
| R90C | 0.28 | rs751362548 | ||||
| WNT7B | Wnt family member 7B | Gene amplification | 22q13.31 | 0.8 | ||
| Q261E | 0.10 | Novel | ||||
| WNT8A | Wnt family member 8A | Deep deletion | 5q31.2 | 0.6 | ||
| G12A | 0.25 | Novel | ||||
| L346V | 0.32 | Novel | ||||
| WNT8B | Wnt family member 8B | Deep deletion | 10q24.31 | 1.4 | ||
| W63C | 0.36 | Novel | ||||
| R157Q | 0.34 | Novel | ||||
| A59S | 0.12 | Novel | ||||
| A94S | 0.02 | Novel | ||||
| WNT9A | Wnt family member 9A | Gene amplification | 1q42.13 | 2.2 | ||
| R333Q | 0.17 | Novel | ||||
| D156N | 0.25 | Novel | ||||
| E37K | 0.41 | Novel | ||||
| V290M | 0.18 | Novel | ||||
| R61W | 0.14 | Novel | ||||
| A264V | 0.28 | Novel | ||||
| WNT9B | Wnt family member 9B | Gene amplification | 17q21.32 | 0.8 | ||
| P35A | 0.20 | Novel | ||||
| Y353H | 0.15 | Novel | ||||
| L102P | 0.18 | Novel | ||||
| WNT10A | Wnt family member 10A | Deep deletion | 2q35 | 1.8 | ||
| A240V | 0.16 | rs201578578 | ||||
| G88D | 0.28 | Novel | ||||
| WNT10B | Wnt family member 10B | Gene amplification | 12q13.12 | 0.4 | - | - |
| Deep deletion | ||||||
| WNT11 | Wnt family member 11 | Gene amplification | 11q13.5 | 5 | ||
| Deep deletion | ||||||
| L65P | 0.61 | rs1173549137 | ||||
| R202H | 0.07 | rs374455490 | ||||
| WNT16 | Wnt family member 16 | Gene amplification | 7q31.31 | 1.6 | ||
| Deep deletion | ||||||
| E271Q | 0.20 | Novel | ||||
| S218* | 0.18 | Novel | ||||
| G304W | 0.09 | Novel |
Protein protein interaction network analysis
The STRING database is a collection of known and predicted protein-protein interactions. These interactions could either be direct (physical) and indirect (functional) associations. They are derived from computational predictions and text mining of protein interactions in different organisms and information aggregated from several other primary databases [16]. The gene exhibiting the highest frequency of gene alteration was selected from the entire family and investigated for gene expression and derivation of expression based survival curves for different combinations of parameters such as sex, ethnicity, tumor grade etc., Functional enrichment of the protein network and Kyoto Encyclopedia of Genes and Genomes pathway was derived from the protein protein interaction (PPI) network. The strength score is the ratio between the number of proteins annotated with a term interacting in the network and the number of proteins which is expected to be annotated with this term in a random protein network of the same size. The false discovery rate demonstrates the significance of the enrichment process. The false discovery rate is denoted by p-values which are corrected for multiple testing within each category using Benjamini-Hochberg procedure.
Gene expression and survival analysis
The expression of the gene presenting with highest frequency of gene alteration in HNSCC was analyzed using the UALCAN (http://ualcan.path.uab.edu/cgi-bin/TCGA-survival) database. Survival curve analysis based on the tumor grade and expression profile was performed to demonstrate the putative role of Wnt family of genes with HNSCC. Gene expression data is expressed as transcripts per million (TPM) which is a normalization method for RNA-sequencing data. The TPM values which were used for the generation of box-whisker plots were also used to determine the significant difference between the groups. The t test was performed using PERL script with the comprehensive Perl archive network (CPAN) module. Combined survival effect analysis of gene expression and other clinical parameters such as race, sex, tumor grade, and cancer subtypes were assessed using log-rank test that generated a p-value which was further used to indicate statistical significance of survival correlation between groups [17].
Results
Demographic data
The dataset (TCGA, Firehose Legacy) included in the present study had information on 528 HNSCC samples. The male:female ratio was found to be 2.7:1, with age groups ranging from 19 to 90 years. The number of individuals with the history of smoking and alcohol were roughly around 98% and 67%, respectively. There were five different groups of categories for smoking viz., 1-lifelong non-smoker, 2-current smoker, 3-current informed smoker for >15 years, 4-current reformed smoker ≤15 years, 5-current reformed smoker, duration not specified. The dataset had samples from patients of American (85.6%), African (9.1%), Asian (2.1%), and American Indian (0.4%) descent. The distribution of patients based on the histologic grade of neoplasm is given in Table 1, of which 59% of patients had grade 2 tumor.
Oncoprint data analysis
The oncoprint data analysis revealed alterations in 19 genes, of which WNT11 (5%) harbored the highest frequency of gene amplification and deep deletion. When the pattern of amplification was assessed in different groups of smokers a greater frequency of gene amplification was observed in current smokers (n=6) when compared to other categories (Table 2, Fig. 1B).
gnomAD analysis
The gnomAD analysis revealed several novel and reported variants as demonstrated by the oncoprint data. The variations in WNT3 (R85Q), WNT5A (I93V, G341S), WNT6 (R46W, T105M), WNT7A (R90C), WNT10A (A240V), and WNT11 (L65P, R202H) genes were reported. Other missense mutations were found to be novel (Table 2). Further investigations are warranted to identify the consequences and association of these variations with HNSCC.
Protein network analysis
The protein interaction network reveals the major interactions of WNT11 with genes such as DVL1, DVL2, DVL3, FZD1, FZD2, FZD3, FZD4, FZD6, FZD7, FZD8 which play key roles in governing cell polarity, embryonic development, formation of neural synapses, cell proliferation, and many other processes in developing and adult organisms (Fig. 2). Majority of genes interacting with WNT11 exhibit significant upregulation of the transcripts with FZD2 and DVL3 showing a marked difference (p < 10-12) in the expression pattern. The expression score is demonstrated by a p-value which denotes the significant difference between two groups of samples viz., normal and HNSCC (Table 3). The functional enrichment analysis showed eleven nodes and 55 edges. The proteins were found to interact more among themselves suggestive of a biologically connected group. The overall PPI enrichment p-value was found to be <1.0 × 10-16. Pathways derived from Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis returned predictions which were more inclined towards other cancer types such as basal cell carcinoma, hepatocellular carcinoma, etc. (Table 4).
Fig. 2.
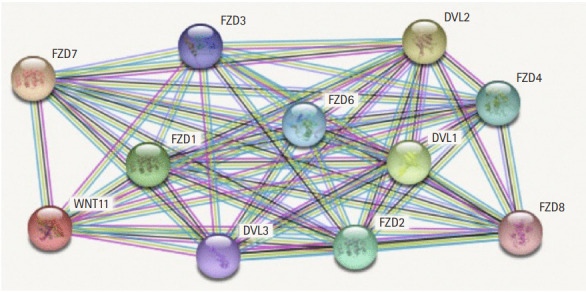
The proteins network interaction of WNT11 gene.
Table 3.
Expression profile of genes interacting with Wnt11 in head and neck squamous cell carcinoma patients
| Gene | Protein | Expression profile | Expression score (p-value) |
|---|---|---|---|
| FZD1 | Frizzled-1; receptor for Wnt proteins | Upregulateda | 2.237 × 10-9 |
| FZD2 | Frizzled-2; receptor for Wnt proteins | Upregulateda | <10-12 |
| FZD3 | Frizzled-3; receptor for Wnt proteins | Upregulated | 1.372 × 10-1 |
| FZD4 | Frizzled-4; receptor for Wnt proteins | Downregulateda | 3.639 × 10-2 |
| FZD6 | Frizzled-6; receptor for Wnt proteins | Upregulateda | 1.624 × 10-12 |
| FZD7 | Frizzled-7; receptor for Wnt proteins | Upregulateda | 8.622 × 10-2 |
| FZD8 | Frizzled-8; receptor for Wnt proteins | Upregulateda | 2.518 × 10-3 |
| DVL1 | Segment polarity protein dishevelled homolog DVL-1 | Upregulateda | 4.600 × 10-2 |
| DVL2 | Segment polarity protein dishevelled homolog DVL-2 | Upregulateda | 1.624 × 10-12 |
| DVL3 | Segment polarity protein dishevelled homolog DVL-3 | Upregulateda | <10-12 |
Differentially expressed genes with a statistically significant gene expression.
Table 4.
Functional enrichment of the Wnt11 interacting genes based on the KEGG pathway analysis
| KEGG pathways | Strength | False discovery rate |
|---|---|---|
| Basal cell carcinoma | 2.49 | 1.76 × 10-26 |
| Melanogenesis | 2.3 | 8.06 × 10-25 |
| Signaling pathways regulating pluripotency stem cells | 2.15 | 1.93 × 10-23 |
| Wnt signaling pathway | 2.14 | 2.11 × 10-23 |
| Gastric cancer | 2.12 | 2.26 × 10-23 |
| Breast cancer | 2.12 | 2.26 × 10-23 |
| mTOR signaling pathway | 2.12 | 2.26 × 10-23 |
| Cushing’s syndrome | 2.11 | 2.26 × 10-23 |
| Hippo signaling pathway | 2.11 | 2.26 × 10-23 |
| Hepatocellular carcinoma | 2.08 | 3.38 × 10-23 |
| Notch signaling pathway | 2.05 | 3.07 × 10-6 |
| HTLV-1 infection | 1.89 | 2.96 × 10-21 |
| Proteoglycans of cancer | 1.86 | 2.27 × 10-14 |
| Human papillomavirus infection | 1.79 | 3.50 × 10-20 |
| Pathways in cancer | 1.58 | 6.22 × 10-18 |
KEGG, Kyoto Encyclopedia of Genes and Genomes; mTOR, mammalian target of rapamycin.
Gene expression and survival analysis
The gene expression profile of WNT11 between normal and primary tumor samples revealed a significant difference with a p-value of 3.043 × 10-3. The relative expression profile of WNT11 gene in different grades of HNSCC also returned significant values between normal vs. grade 2, normal vs. grade 4, grade 1 vs. grade 4, grade 2 vs. grade 4, and grade 3 vs. grade 4 (Fig. 3A). The expression pattern of WNT11 gene produced significant difference between normal and female HNSCC subjects (p = 2.169 × 10-2). Significant difference was not observed between groups normal vs. male and male vs. female (Fig. 3B). Although the present observation do not confirm sex predilection of WNT11 gene expression with HNSCC, a significant difference in the survival probability between male and female subjects was observed with low/medium level expression (p = 0.021). Furthermore, female subjects who presented with a low/medium level expression exhibited low survival probability when compared to male subjects. A p-value less than 0.05 was considered to be significant (Fig. 4).
Fig. 3.

(A) Box-Whisker plot showing relative expression profile of WNT11 gene in different grades of head and neck squamous cell carcinoma (HNSCC). The X axis denotes The Cancer Genome Atlas (TCGA) samples and Y axis denotes the transcripts per million values. The comparison of gene expression patterns between different grades of HNSCC returned significant values between normal vs grade 2 (p = 3.8 × 10-2), normal vs. grade 4 (p = 4.3 × 10-3), grade 1 vs. grade 4 (p = 7.11 × 10-3), grade 2 vs. grade 4 (p = 2.54 × 10-11) and grade 3 vs. grade 4 (p = 1.9 × 10-3). A p-value less than 0.05 was considered to be significant. (B) Box-Whisker plot showing relative expression profile of WNT11 gene in male and female HNSCC subjects. The X axis denotes the TCGA samples and Y axis denotes the transcripts per million values. The comparison of gene expression patterns between male and female viz., normal vs. male (p = 9.923 × 10-2), normal vs. female (p = 2.169 × 10-2), male vs. female (p = 2.12 × 10-1). A p-value less than 0.05 was considered to be significant.
Fig. 4.
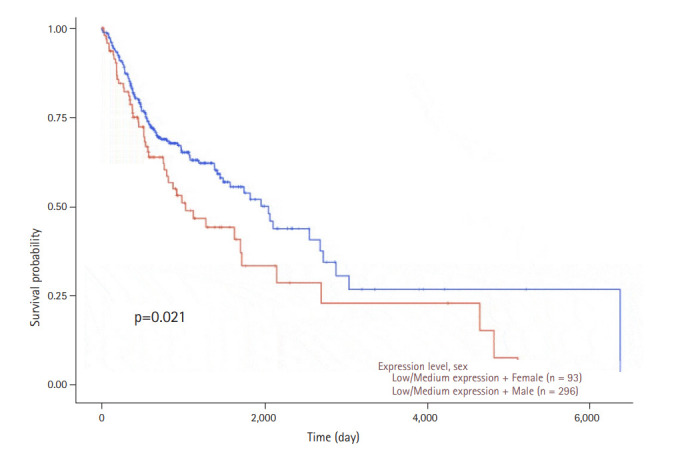
Kaplan-Meier plots showing the association of WNT11 gene expression in combination with the sex with head and neck squamous cell carcinoma patient’s survival. The x-axis represents time in days and y-axis shows the survival probability. The blue line indicates low/medium expression in male patients and the red line indicates low/medium level expression of the WNT11 gene in female patients. A significant difference in the survival probability was observed between the two groups (p = 0.021). Female subjects with a low/medium level expression presented with a low survival probability when compared to male subjects. A p-value less than 0.05 was considered to be significant.
The expression profile of WNT11 was checked in other types of squamous cell carcinoma such as lung (LUSC) and esophageal cancers (ESCC). Although both LUSC and ESCC produced a significant difference with respect to normal and primary tumors, WNT11 expression was upregulated in ESCC cases which was in consonance with HNSCC type. In both the cancer types, there was upregulation of WNT11 gene, whereas in case of LUSC WNT11 expression was downregulated. The receptor proteins interacting with WNT11 have also shown significant differences based on the levels of gene expression in different sex groups (data not shown). These results add to the association of WNT11 gene alterations with HNSCC.
Discussion
Genetic variations such as single nucleotide variants and copy number variants have long been associated with oral cancer and other devastating diseases. Wnt signaling is vital for a plethora of cellular function ranging from homeostasis to the development of mature tissue. Embryonic development also requires Wnt-mediated canonical signaling [7–9]. Moreover, Wnt signaling is inevitable for regulating cellular proliferation, apoptosis, metastasis, and migration of cells [10]. Of note, Wnt operates through either canonical or non-canonical pathways which are differentiated by β-catenin involvement [11]. Both pathways are activated by the binding of Wnt protein to the Frizzled (Fzd) seven transmembrane receptor. The fundamental difference between these two pathways is the involvement of β-catenin [18,19].
Non-canonical Wnt signaling pathways, which are independent of β-catenin, rely on the signal transduction of Wnt through Fzd as well as its co-receptors such as receptor tyrosine kinase-like orphan receptor 2 or receptor-like tyrosine kinase [20]. On the other hand, the canonical Wnt signaling pathway, also known as Wnt/β-catenin signaling pathway, involves the activation of cytoplasmic β-catenin signaling cascades upon Wnt signal transduction at the cell membrane [21]. Altered Wnt/β-catenin signaling has been considered a promoting event for various types of cancers. Canonical Wnt signaling pathway acts as a master regulator for a wide range of biological effects through up- or down-regulation of genes that act as direct effectors, transcription regulators, or other signaling pathway regulators. Thus, Wnt target gene expression can either directly or indirectly activate cell cycle progression, cell proliferation, cell differentiation, cell migration, inhibit apoptosis, and regulate embryonic development.
The involvement of canonical Wnt signaling pathway in the formation of HNSCC has also been examined in several experimental studies. Evidence of the association between the pathway and HNSCC was first discovered through cDNA arrays on patient samples. The study found that Fzd, Fzd homolog 3, and Dvl homolog genes, which are functionally important in the canonical Wnt signaling pathway, were highly expressed in HNSCC by two to five fold when compared to normal tissues [22]. Furthermore, another study has also demonstrated that the gene expression levels of Wnts, particularly Wnt11 and Wnt10b, were markedly higher by 17 and 3-fold, respectively, in HNSCC cells compared to normal oral squamous epithelial cells [23]. Over-expression of these major components in HNSCC cells compared to normal cells clearly signifies the involvement of abnormal activation of Wnt signaling cascades in HNSCC. These reports were in agreement with the observation made in the present study wherein the up-regulation of WNT11 correlated with concomitant increase in the gene expression level of interacting genes. Recently, the role of canonical Wnt signaling pathway in regulating self-renewal of HNSCC cancer stem cells is also being emphasized in several studies. Abnormal activation of the pathway has been correlated with increased proliferation and thus self-renewal of cancer stem cells in HNSCC [24]. These observations were confirmed by our computational analysis wherein the KEGG pathways arising out of functional enrichment analysis revealed involvement of WNT11 in multiple pathways viz., pathways associated with cancer, infections, syndrome, and signaling process regulating pluripotency of stem cells.
Although gene expression profiling is well documented in case of WNT family of genes, the effect of gene alterations such as mutations, deletions, and copy number variations upon the expression of the genes in the family is scarce or limited. The present study throws light on those alterations which might act as putative drivers in establishing tumorigenesis. Numerous novel variants and reported variants were identified in HNSCC patients. The potential role of these variants in the disease process is yet to be explored. Similar studies have already been carried out to unravel the potential markers with putative association with HNSCC. The survival curve analysis provides cues on the prognostic significance of these markers in relation to the disease [25,26].
Computational tools provide a cost-effective alternative for analyzing the genetic alterations especially in a complex disorder such as cancer. Although the crosstalk between multiple signaling pathways play a vital role in the development of cancer, further understanding of one important molecular mechanism, which is the canonical Wnt signaling pathway, is critical as targeting the pathway could be a promising approach in eradicating the treatment failure and relapse in HNSCC. With all the pros addressed, the study design also suffers certain drawbacks such as population bias and reproducibility in other ethnic groups. The identification of polymorphic variants and chromosomal abnormalities would aid us in preparing a panel of markers intended for use as early diagnostic leads. These markers can be further validated using genotyping methods or next-generation sequencing approaches to derive a strong association with the disease phenotype.
Acknowledgments
The authors are grateful to all the consorts and groups involved in the compilation of data from patients for public use. Our sincere thanks also go to all the patients who have indirectly contributed to the scientific community through providing consent for sharing their data for research use.
The authors Dr. Vijayashree Priyadharsini J. and Dr. Paramasivam A. thank the Science and Engineering Research Board (SERB), Government of India for the financial support rendered through the core research grant (CRG) (No. CRG/2019/003756) and (EEQ) (No.EEQ/2019/000411).
Footnotes
Authors’ Contribution
Conceptualization: JVP, AP. Data curation: JVP, AP. Formal analysis: JVP, AP. Funding acquisition: JVP, AP. Writing - original draft: JA, ASSG. Writing - review & editing: JVP, AP.
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
References
- 1.Sengupta A. Recent advances in head and neck cancer. Apollo Med. 2012;9:96–103. [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Szyfter K, Kiwerska K, Wierzbicka M. HPV-related HNC: new challenge and hope for head and neck cancer subjects. J Med Sci. 2018;87:112–116. [Google Scholar]
- 4.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–548. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta B, Johnson NW, Kumar N. Global epidemiology of head and neck cancers: a continuing challenge. Oncology. 2016;91:13–23. doi: 10.1159/000446117. [DOI] [PubMed] [Google Scholar]
- 6.Yan K, Agrawal N, Gooi Z. Head and neck masses. Med Clin North Am. 2018;102:1013–1025. doi: 10.1016/j.mcna.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 9.Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer: a review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol. 2015;309:C511–C521. doi: 10.1152/ajpcell.00117.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noguti J, CF DEM, Hossaka TA, Franco M, Oshima CT, Dedivitis RA, et al. The role of canonical WNT signaling pathway in oral carcinogenesis: a comprehensive review. Anticancer Res. 2012;32:873–878. [PubMed] [Google Scholar]
- 11.Ozbey U, Attar R, Romero MA, Alhewairini SS, Afshar B, Sabitaliyevich UY, et al. Apigenin as an effective anticancer natural product: spotlight on TRAIL, WNT/beta-catenin, JAK-STAT pathways, and microRNAs. J Cell Biochem. 2018;120:1060–1067. doi: 10.1002/jcb.27575. [DOI] [PubMed] [Google Scholar]
- 12.Javed Z, Muhammad Farooq H, Ullah M, Zaheer Iqbal M, Raza Q, Sadia H, et al. Wnt signaling: a potential therapeutic target in head and neck squamous cell carcinoma. Asian Pac J Cancer Prev. 2019;20:995–1003. doi: 10.31557/APJCP.2019.20.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrashekar DS, Bashel B, Balasubramanya SA, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 19.Katoh M, Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 2007;13:4042–4045. doi: 10.1158/1078-0432.CCR-06-2316. [DOI] [PubMed] [Google Scholar]
- 20.Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010;106:1798–1806. doi: 10.1161/CIRCRESAHA.110.219840. [DOI] [PubMed] [Google Scholar]
- 21.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 22.Leethanakul C, Patel V, Gillespie J, Pallente M, Ensley JF, Koontongkaew S, et al. Distinct pattern of expression of differentiation and growth-related genes in squamous cell carcinomas of the head and neck revealed by the use of laser capture microdissection and cDNA arrays. Oncogene. 2000;19:3220–3224. doi: 10.1038/sj.onc.1203703. [DOI] [PubMed] [Google Scholar]
- 23.Rhee CS, Sen M, Lu D, Wu C, Leoni L, Rubin J, et al. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene. 2002;21:6598–6605. doi: 10.1038/sj.onc.1205920. [DOI] [PubMed] [Google Scholar]
- 24.Felthaus O, Ettl T, Gosau M, Driemel O, Brockhoff G, Reck A, et al. Cancer stem cell-like cells from a single cell of oral squamous carcinoma cell lines. Biochem Biophys Res Commun. 2011;407:28–33. doi: 10.1016/j.bbrc.2011.02.084. [DOI] [PubMed] [Google Scholar]
- 25.J VP, A P. Virtual screening of mutations in antioxidant genes and its putative association with HNSCC: an in silico approach. Mutat Res. 2020;821:111710. doi: 10.1016/j.mrfmmm.2020.111710. [DOI] [PubMed] [Google Scholar]
- 26.Anita R, Paramasivam A, Priyadharsini JV, Chitra S. The m6A readers YTHDF1 and YTHDF3 aberrations associated with metastasis and predict poor prognosis in breast cancer patients. Am J Cancer Res. 2020;10:2546–2554. [PMC free article] [PubMed] [Google Scholar]