

Abstract
Objectives
Obesity is a complex disease associated with a high risk of comorbidities. Gastric bypass surgery, an invasive procedure with low patient eligibility, is currently the most effective intervention that achieves sustained weight loss. This beneficial effect is attributed to alterations in gut hormone signaling. An attractive alternative is to pharmacologically mimic the effects of bariatric surgery by targeting several gut hormonal axes. The G protein-coupled receptor 39 (GPR39) expressed in the gastrointestinal tract has been shown to mediate ghrelin signaling and control appetite, food intake, and energy homeostasis, but the broader effect on gut hormones is largely unknown. A potent and efficacious GPR39 agonist (Cpd1324) was recently discovered, but the in vivo function was not addressed. Herein we studied the efficacy of the GPR39 agonist, Cpd1324, on metabolism and gut hormone secretion.
Methods
Body weight, food intake, and energy expenditure in GPR39 agonist-treated mice and GPR39 KO mice were studied in calorimetric cages. Plasma ghrelin, glucose-dependent insulinotropic polypeptide (GIP), glucagon-like peptide-1 (GLP-1), and peptide YY (PYY) levels were measured. Organoids generated from murine and human small intestine and mouse colon were used to study GLP-1 and PYY release. Upon GPR39 agonist administration, dynamic changes in intracellular GLP-1 content were studied via immunostaining and changes in ion transport across colonic mucosa were monitored in Ussing chambers. The G protein activation underlying GPR39-mediated selective release of gut hormones was studied using bioluminescence resonance energy transfer biosensors.
Results
The GPR39 KO mice displayed a significantly increased food intake without corresponding increases in respiratory exchange ratios or energy expenditure. Oral administration of a GPR39 agonist induced an acute decrease in food intake and subsequent weight loss in high-fat diet (HFD)-fed mice without affecting their energy expenditure. The tool compound, Cpd1324, increased GLP-1 secretion in the mice as well as in mouse and human intestinal organoids, but not in GPR39 KO mouse organoids. In contrast, the GPR39 agonist had no effect on PYY or GIP secretion. Transepithelial ion transport was acutely affected by GPR39 agonism in a GLP-1- and calcitonin gene-related peptide (CGRP)-dependent manner. Analysis of Cpd1324 signaling properties showed activation of Gαq and Gαi/o signaling pathways in L cells, but not Gαs signaling.
Conclusions
The GPR39 agonist described in this study can potentially be used by oral administration as a weight-lowering agent due to its stimulatory effect on GLP-1 secretion, which is most likely mediated through a unique activation of Gα subunits. Thus, GPR39 agonism may represent a novel approach to effectively treat obesity through selective modulation of gastrointestinal hormonal axes.
Keywords: GPR39 agonist, Enteroendocrine cells, Gut hormone secretion, Glucagon-like peptide-1, Obesity
Graphical abstract
Highlights
-
•
Orally active GPR39 agonist decreased food intake and body weight in high-fat diet-fed mice
-
•
Orally administrated GPR39 agonist increased plasma GLP-1 secretion while decreasing plasma ghrelin in mice
-
•
GPR39 agonist induced selective release of GLP-1 from enteroendocrine cells but not the other important L cell product PYY
-
•
GPR39 agonism acutely affected transepithelial ion transport through GLP-1 via CGRP signaling
-
•
GPR39 selective gut hormone release is suggested to be due to activation of the Gαq and Gα i/o signaling pathways
Abbreviations
- BRET
Bioluminescence resonance energy transfer
- BW
Body weight
- CGRP
Calcitonin gene-related peptide
- CCK
Cholecystokinin
- Ex4
Exendin 4
- Ex(9–39)
Exendin (9–39)
- GPR39
G protein-coupled receptor 39
- GPCRs
G protein-coupled receptors
- GI
Gastrointestinal
- GLP-1
Glucagon-like peptide-1
- GIP
Glucose-dependent insulinotropic polypeptide
- HFD
High-fat diet
- KO
Knockout
- NT
Neurotensin
- OGTT
Oral glucose tolerance test
- PYY
Peptide YY
- RER
Respiratory exchange ratio
- TER
Trans-epithelial resistance
- T2D
Type 2 diabetes.
1. Introduction
Obesity is a complex disease that is associated with an increased risk of various comorbidities, such as heart disease, type 2 diabetes (T2D), fatty liver disease, and certain cancers. However, only a few anti-obesity drugs are currently on the market [1,2] and none are sufficiently efficacious to obtain a substantial and sustained weight loss. Currently, the only long-lasting anti-obesity treatment is gastric bypass, which can induce up to 30% weight loss that is sustained at 15 years of follow up, often alongside remission of T2D and fatty liver disease [[3], [4], [5]]. This operation's effectiveness relies to a large extent on metabolic benefits of increased plasma levels of satiety hormones seen after the surgery: glucagon-like peptide 1 (GLP-1), peptide YY (PYY), neurotensin (NT), and cholecystokinin (CCK). The hunger-inducing hormone ghrelin is also reduced following gastric surgery [4]. These changes in hormonal composition are critical for reduced food intake and body weight [6]. Unfortunately, gastric bypass is often associated with post-surgical complications and side effects [7]. Therefore, an attractive alternative to gastric bypass is developing a pharmacological treatment that induces sustained weight loss by mimicking the complex changes in hormone levels characteristic of gastric bypass. However, a major challenge is the lack of mechanistic understanding of how intestinal hormone co-release is controlled and coordinated.
Several G protein-coupled receptors (GPCRs) have been described to regulate hormone secretion from enteroendocrine cells [8,9]. The orphan receptor GPR39 has been suggested to mediate hormone release [10,11]. GPR39 is a member of the ghrelin receptor family and has been implicated in the control of appetite, food intake, and energy homeostasis [12,13]. GPR39 is highly expressed in the gastrointestinal (GI) tract, adipose tissue, pancreas, and liver, with limited distribution in the central nervous system [14,15]. Global knockout studies of GPR39 (GPR39 KO) suggest that receptor activation improves β cell function and survival [16,17] as well as glucose-stimulated insulin secretion. Furthermore, we have previously shown that GPR39 KO animals gained significantly higher fat mass compared to wild-type mice when fed a high-fat diet (HFD). This effect was attributed to increased fat accumulation in combination with a near elimination of diet-induced thermogenesis [18]. Fasting plasma ghrelin levels also increased in these GPR39-deficient animals [10].
The subsequent development of selective GPR39 agonists has permitted direct investigation of the physiological function of GPR39 without potential compensatory mechanisms that may occur in global GPR39 KO mice. However, only a few GPR39 agonists have been developed to date and the selectivity of most ligands remains to be proven in GPR39 KO models [11,[19], [20], [21]]. We recently described the GPR39 agonist, Cpd1324, whose receptor specificity has been verified in knockout models [10]. Cpd1324 caused a decrease in ghrelin secretion from primary gastric mucosa cell cultures, presumably mediated through increased somatostatin secretion [10]. These observations raise the possibility that GPR39 could modulate a broader range of hormone release [10].
Based on the increased sensitivity of the GPR39 KO mice to a HFD and the modulation of ghrelin secretion, in the present study, we characterized the GPR39-mediated impact on body weight regulation, food intake, and secretion of hormones from the gut known to modulate appetite using both the global knockout model and the proven selective GPR39 agonist Cpd1324.
2. Materials and methods
2.1. Chemicals, reagents, and kits
PEG200, HCl, Pefabloc, and acetaminophen were purchased from Sigma Aldrich (Cat. No. P3015, 1.00317, 76,307, and A7085; Soborg, Denmark). BIBO3304 (Y1 antagonist), BIIE0246 (Y2 antagonists), and BIBN4096 (CGRP antagonist) were purchased from Tocris (Cat. No. 2412, 1700 and 4561; Bristol, UK). Somatostatin receptor antagonist was purchased from Bachem (iSST, Cat. No. BIM-23627; Bubendorf, Switzerland). Exendin 4 (Ex4) and Exendin (9–39) (Ex(9–39)) were both purchased from Anaspec (Cat. No. ANA24463 and ANA24467; Cambridge Bioscience, Cambridge, UK). Obestatin was kindly provided by Annette Beck-Sickinger and synthesized as previously described [12]. ELISAs for GLP-1 and insulin were obtained from MesoScale (Cat. No. L4503PA and K152BZC; MesoScale Diagnostics, Maryland, USA). GIP and total ghrelin were acquired from Merck/Millipore (Cat. No. EZRMGIP-55K and EZRGRT-91K; Merck/Millipore Darmstadt, Germany). PYY ELISA was purchased from Crystal Chem (Cat. No. 81501; Zaandam, Netherlands). An acetaminophen kit was obtained from Sekisui Diagnostics (Cat. No. 506–10; Sekisui, Lexington, MA, USA). Cpd1324 was synthesized by InterBioScreen Ltd. as previously described [10] and formulated with a vehicle containing 3.2 μM (−5.5 M) of zinc ions for all of the in vivo experiments.
2.2. Bioluminescence resonance energy transfer (BRET) assays
BRET assays were performed as previously described [[22], [23], [24], [25]]. Activation of specific G proteins was confirmed using BRET sensors that monitored the separation between Gα and Gγ1 subunits in HEK293 cells. The HEK293 cells were seeded in white 96-well plates at a density of 30,000 cells per well and co-transfected with human GPR39, a Gα-RlucII construct, untagged Gβ1, and GFP10-Gγ1 [22] 24 h after seeding.
2.3. Human tissue
Human jejunum tissue samples were obtained from patients undergoing pancreatic surgery (Whipple's procedure) at the Center for Cancer and Organ diseases at Rigshospitalet (Copenhagen, Denmark). All of the patients provided informed consent, and the research protocol was approved by the National Research Ethics Committee (H-18015120). The patients did not receive any anti-cancer treatment prior to surgery.
2.4. In vivo mouse studies
C57BL/6J male mice (Janvier, Le Genest-Saint-Isle, France) were used for all of the studies unless otherwise stated. They were housed in normal 12-h light, 12-h dark cycles and had free access to regular chow (Altromin #1314; Lage, Germany) or a high-fat diet (HFD) (60E% fat, #12492; Research Diets, New Brunswick, NJ, USA) and tap water unless otherwise stated. Germline GPR39-deficient mice (GPR39 KO) [16] and their WT littermates were used to test the housing conditions and acute effects of Cpd1324 on hormone release. All of the studies were performed in male mice aged 9–11 weeks old unless otherwise stated. The animal studies were conducted in accordance with institutional guidelines and approved by the Animal Experiment Inspectorate in Denmark and the Home Office (UK).
2.5. Food intake, energy expenditure, and body weight
The mice were acclimatized in an indirect calorimetry system (TSE system; Bad Homburg, Germany) for four days. All of the mice were fed regular chow unless otherwise stated. To test phenotypes under basal conditions, the GPR39 KO mice and their WT littermates were placed in a calorimetry system their and food intake and energy expenditure were measured. A stress-free environment was obtained by placing the mice in a calorimetry system within sound-proofed climate chambers with a constant temperature and humidity. To test the effect of Cpd1324, C57BL/6J mice were fed a HFD 4 days prior to dosing. Thirty min before the onset of the dark phase, the mice were orally dosed with either Cpd1324 (30 mg/kg BW) or vehicle 10 ml/kg BW (PEG200/Saline/EtOH) (40/57.5/2.5) with a final concentration of 3.2 μM of ZnCl2. Food intake and energy expenditure were monitored for 24 h. The mice were dosed orally (once daily) for six consecutive days. Body weight was measured before and after the study.
2.6. Pharmacokinetic study of Cpd1324 in mice
The plasma samples (30 μL) were quenched with ice-cold acetonitrile (90 μL) followed by vortexing and centrifugation (15 min, 10.000 g). The supernatants were analyzed by LC-MS using a system consisting of an Agilent 6130 mass spectrometer with electron spray ionization (ESI) coupled to an Agilent 1200 HPLC system with a C18 reverse phase column (Zorbax Eclipse XBD-C18, 4.6 mm × 50 mm), autosampler, diode array detector, and a linear gradient of the binary solvent system of buffer A (MilliQ H2O:acetonitrile:formic acid, 95:5:0.1 v/v%) to buffer B (acetonitrile:formic acid, 100:0.1 v/v%) with a flow rate of 1 mL⁄min. The chromatograms were analyzed based on the selective ion mode (SIM) function and UV absorbance at 214 nm. Compound peaks were integrated and the resulting AUC values were used to quantify the compound concentrations based on a standard curve prepared by spiking compound into blood plasma.
2.7. Gut hormone measurements, glucose tolerance tests, and gastric emptying
The mice were fasted for 16 h (overnight). At 8 a.m., the animals were dosed with 30 mg/kg of Cpd1324 or vehicle. Blood was collected from the orbital sinus vein into EDTA-coated tubes at t = 30, 60, 120 and 240 min after dosing. The plasma was isolated by centrifugation at 3,000 g for 15 min at 4 °C and subsequently stored at −80 °C. For GLP-1 and GIP measurements, DPP-IV inhibitor (valine pyrrolidide kindly provided by Jens Juul Holst) was immediately added to the blood, while Pefabloc (1 mg/ml) and HCl were added to the plasma to a final concentration of 0.05 N for ghrelin measurement. GLP-1, GIP, ghrelin, and PYY were measured using commercially available kits, and neurotensin was measured using an in-house radioimmunoassay [26]. Oral glucose tolerance tests (OGTT) and a gastric emptying test using acetaminophen were carried out in a separate cohort of mice. These mice were fasted for16 h with free access to water before administration of either Cpd1324 (15, 30, 60, or 120 mg/kg) or vehicle 120 min prior to the experiment. At t = 0, the animals were dosed orally with glucose (1.5 g/kg BW) and acetaminophen (100 mg/kg BW). Blood glucose was measured at t = −120, 0, 15, 30, 60, and 120 min in tail blood using a handheld glucometer (Contour Xt Ascensia). Plasma insulin and acetaminophen concentrations were determined at the indicated time points.
2.8. Conditioned taste aversion
Conditioned taste aversion tests were performed in lean mice (n = 7–8) as previously described [27]. During conditioning, the mice received either vehicle, liraglutide (400 μg/kg, s.c.), Cpd1324 (30 mg/kg, p.o.), or Cpd1324 (120 mg/kg, p.o.). Treatment with liraglutide served as a positive control. All of the mice received both an s.c. (vehicle or liraglutide) and oral administration (vehicle or Cpd1324) on the two conditioning days.
2.9. Intestinal organoid studies
Mouse and human intestinal organoids were established and cultured as previously described [27,28]. Fragments of the murine distal ileum (2 cm proximal to the cecum) and proximal colon (1 cm distal to the caecum) were used. GLP-1 secretion experiments were performed as previously described [29,30]. In brief, 15 μL of organoid suspension in Matrigel was plated in each well of a 96-well plate. After 48 h, organoid cultures were washed with DMEM/F12 supplemented with 2 mM of L-glutamine and 10 mM of HEPES and incubated in 50 μL of the same medium with either Cpd1324 or vehicle containing 0.1% DMSO for 2 h at 37 °C. Medium from the wells was collected, centrifuged at 300 g for 2 min at 4 °C, transferred to fresh tubes, and assayed for hormone content. After the experiment, the DNA content was measured in the wells using DNA Quantitation Kit Fluorescence Assays (Cat. No. DNAQF-1 KT; Sigma Aldrich) according to the manufacturer's instructions. The experiments were repeated three times with 3–6 replicates per series. GLP-1 and PYY levels were measured using the previously described kits and values from each well were normalized to the DNA content.
2.10. Measurement of colonic mucosal ion transport (as a short-circuit current, Isc) in vitro
Mucosal preparations with intact submucosal innervation were prepared from adult male C57BL/6J mice as previously described [32,33]. In this study, four adjacent pieces of mucosa from the proximal colon and four pieces from the distal colon were each placed in Ussing chambers. They were voltage-clamped at 0 mV allowing resultant electrogenic ion transport to be measured continuously as Isc (μA.cm−2 and also intermittently checking the transepithelial resistance (TER)). In experiments assessing the pharmacology of Cpd1324 (10 μM in the presence of 10 μM of ZnCl2), the drugs and ZnCl2 were added to the basolateral reservoir. The contributions of endogenous GLP-1, PYY, or αCGRP to Cpd1324 responses were assessed by pretreating tissue with optimal blocking concentrations of either Ex(9–39) (1 μM [32]), a combination of Y1 and Y2 antagonists, BIBO3304 (300 nM), and BIIE0246 (1 μM [33]) or the CGRP antagonist BIBN4096 (1 μM [32]), respectively. Selective antagonism was confirmed by adding Ex4 (100 nM), PYY (10 nM), or αCGRP (10 nM) 30 min after Cpd1324 administration (data not shown). Peak changes in Isc were pooled for each drug and the responses were compared with vehicle controls.
2.11. Immunostaining
Mouse large intestinal organoids were cultured in a 24-well plate (Cat. No. 662892, Greiner sensoplate) as previously described and incubated with either 0.1% DMSO (vehicle) or Cpd1324 (100 μM) in DMEM/F12, 2 mM of L-glutamine, and 10 mM of HEPES media for 30 min at 37 °C. The wells were washed with PBS and fixed with 4% paraformaldehyde (Sigma Aldrich) for 10 min at RT and washed in PBS. The wells were blocked with blocking buffer (2% (w/v) bovine serum albumin in PBS) for 10 min at RT and incubated primary mouse anti-GLP-1 antibodies (Cat. No. ab26278; Abcam) diluted 1:1000 and guinea pig anti-PYY antibodies (Cat. No. EUD5201; Acris) diluted 1:400 in blocking buffer overnight at 4 °C. The wells were then washed with incubated secondary donkey anti-mouse IgG AF568 (Cat. No. ab175700; Abcam) and donkey anti-guinea pig IgG CF488 (Cat. No. 20169; Biotium) antibodies diluted 1:300 in blocking buffer for 1 h at RT before washing and counterstaining with DAPI. The cells were analyzed and images were captured using a confocal microscope (LSM780; Zeiss). L cells were identified based on the presence of PYY-positive granules and the number of cells co-expressing GLP-1 was determined only in cells containing visible nuclei. Quantification of the exact amount of secreted granules after GPR39 agonist treatment was not accessible by this method. Negative control studies revealed no unspecific binding of secondary antibodies.
2.12. Statistical analysis
Data are presented as means ± SEM. Statistical significance was established and graphs were constructed using GraphPad Prism (La Jolla, CA, USA). Student's t tests, one-way ANOVA, and two-way ANOVA with repeated measurements were calculated with appropriate post hoc tests as indicated in the figure legends. For mucosal ion transport measurements, pooled Isc responses are expressed throughout as the mean ± SEM from a minimum of five different specimens. To analyze energy expenditure, the data were corrected for body weight using an ANCOVA model in SAS (SAS Institute Inc., Cary, NC, USA). Single comparisons between data groups were performed using Student's unpaired t test, whereas multiple comparisons used one-way ANOVA with Dunnett's, Bonferroni's or Holm-Šídák's post hoc tests as appropriate. The results were considered significantly different when P < 0.05.
3. Results
3.1. Metabolic phenotype of GPR39 KO mice
Based on the obese phenotype previously observed in GPR39 KO mice [18], we performed a thorough characterization of the metabolic phenotype in calorimetry cages, where we observed significantly higher food intake in the GPR39 KO mice compared to the wild-type littermate control mice. This phenotype was only observed under the climate cabinets’ environmentally stable conditions, presumably due to higher sensitivity to stress when modulating the GPR39 function [18]. In the GPR39 KO mice, food intake was unchanged during the light phase; however, during the dark phase, when the mice are most active, food intake significantly increased, reaching an average intake per day of 3.4 ± 0.2 g compared to 2.7 ± 0.2 g in the WT mice during the three days we monitored the animals (Figure 1A). There was no corresponding increase in the respiratory exchange ratio (RER) (Figure 1B) or energy expenditure in the GPR39 KO mice compared to the control mice (Figure 1C and Figs. S1A–D).
Figure 1.

GPR39 KO mice displayed increased food intake. (A) The average food intake over a 3-day period (B), energy expenditure, and (C) respiratory exchange ratio (RER) of GPR39-deficient (open columns; n = 7) and WT littermate (gray columns, n = 8) mice were measured under stable environmental conditions in climate cabinets. The data were collected during a three-day period and divided into light and dark phases. Statistical significance was tested by Student's unpaired t test. Data are shown as means ± SEM. ∗P ≤ 0.05.
3.2. Effects of Cpd1324 on food intake, body weight, and energy expenditure
We previously described an orally active selective GPR39 agonist, compound TM-N1324 (herein called Cpd1324), which was developed as a tool compound to explore the physiological function of GPR39 [10]. Since zinc is known to modulate the function of Cpd1324 [10] and enhance GPR39 activity, we included zinc in the vehicle for the in vivo and ex vivo experiments. To evaluate the bioavailability after a single oral dose of Cpd1324 at respectively 30 and 120 mg/kg, we monitored plasma levels of Cpd1324 after 2, 4, and 6 h. As shown in Figs. S2A and a 30 mg/kg dosage resulted in robust plasma concentrations above 7.5 μM for up to 6 h after gavage, a concentration that is more than sufficient to obtain full activation of the GPR39 receptor [10]. The optimal treatment dose for food intake was identified in a pilot experiment in which we followed food intake after a single oral bolus of 15, 30, 60, or 120 mg/kg of Cpd1324 in the HFD-treated mice after four days of habituation to the diet (Figs. S2B–I), suggesting 30 mg/kg as an optimal dose. The mice treated with 30 mg/kg of Cpd1324 displayed a lower food intake compared to the vehicle-treated mice (Figure 2A,D). The food intake decreased to 76% in the vehicle group (Figure 2D) during the first dark phase following Cpd1324 dosing, whereas there was no significant difference in energy expenditure (Figure 2B,E, and S2J-M). However, a trend toward a lower RER was present during the dark phase (Figure 2C,F, P = 0.078), potentially due to the decrease in food intake (Figure 2D) as no effect on activity levels was observed (Fig. S3A). Importantly, oral administration of 30 mg/kg Cpd1324 to the GPR39 KO mice on the HFD did not cause any change in food intake (Figure 2G), indicating that the food intake reducing effect was dependent on the receptor GPR39. Daily dosing of Cpd1324 for six consecutive days on the HFD resulted in a small but significant body weight reduction of 4.5% (Figure 2H) compared to the vehicle.
Figure 2.

GPR39 agonist Cpd1324 led to an acute reduction in food intake and body weight. C57BL/6J mice were fed a HFD 4 days prior to dosing of Cpd1324 or vehicle. The 24 h accumulation: (A) food intake, (B) energy expenditure, and (C) respiratory exchange ratio (RER) in C57BL/6J mice after a single oral bolus of Cpd1324 (30 mg/kg) (black dots) or vehicle (gray dots). Food intake was reduced by 24%. (D) The 24 h food intake, (E) energy expenditure, and (F) RER divided into dark and light phases in Cpd1324 (gray columns) and vehicle-treated mice (open columns). (G) The 24 h accumulated food intake in GPR39 KO mice after a single oral bolus of Cpd1324 (30 mg/kg). (H) Body weight after 6 days of once-daily dosing of Cpd1324 (30 mg/kg) or vehicle to C57BL/6J mice. (I) Plasma acetaminophen (μg/ml) following oral gavage of acetaminophen. Significance of the difference was tested by 2-way ANOVA followed by Sidak's test (A, B, C, D, F, and G) or Student's unpaired t test (E and H). n = 6–8. All of the animals were 9–11 weeks of age. Data are shown as means ± SEM. ∗P ≤ 0.05.
3.3. Effects of Cpd1324 on gastric emptying and glucose tolerance
A decrease in food intake may result from a decreased rate of gastric emptying, which in humans may translate into nausea [34]. To investigate the effect on gastric emptying, we administrated Cpd1324 orally 2 h before an oral gavage of acetaminophen and no difference in gastric emptying was observed between the mice treated with Cpd1324 and vehicle, indicating that Cpd1324 did not modulate gastric motility (Figure 2I). We also examined conditioned taste aversion using administration of 400 μg/kg of liraglutide (s.c.), the dose used in several weight-reducing studies [35] as a positive control and observed a 6-fold decrease in saccharine intake compared to the control group. In comparison, a single oral bolus of 30 or 120 mg/kg of Cpd1324 (p.o.) caused only a 2- or 3-fold reduction in saccharin intake, respectively (Fig. S3B). To evaluate the effect of Cpd1324 on glucose homeostasis, we performed a glucose tolerance test 2 h after a moderate (30 mg/kg) and high (120 mg/kg) oral dose of Cpd1324 on lean and diet-induced obese (DIO) mice. In the lean mice, no difference was observed between the Cpd1324-treated and vehicle-treated mice in the glucose clearance rate (Figure 3A,B) or insulin levels during the test (Figure 3C,D) at either 30 mg/kg or 120 mg/kg. However, in the DIO mice, while the glucose clearance rate was unchanged (Figure 3E,F), we observed increased plasma insulin after 30 mg/kg 15 min after a moderate dose of 30 mg/kg (Figure 3G). Using a high dose of Cpd1324 (120 mg/kg) in the DIO mice, we observed a further enhanced insulin secretion that was significantly better than the vehicle-treated mice at the basal level (P = 0.01) and very close to significant after 15 min (P = 0.0505) (Figure 3H).
Figure 3.

OGTT and plasma insulin levels after Cpd1324 administration in lean and diet-induced obese mice. (A, B, E, and F) Blood glucose and (C, D, G, and H) plasma insulin 15 min after oral glucose (2 g/kg) challenge in lean (A, B, C, and D) or DIO (E, F, G, and H) mice treated with a single oral dose of 30 mg/kg (A, C, E, and G), 120 mg/kg (B, D, F, and H), Cpd1324 (black dots), or vehicle (gray dots) 2 h prior to the experiment. All of the animals were 9–11 weeks of age. Significance of the difference was tested by Student's unpaired t test (C, D, G, and H). n = 7–8. Data are shown as means ± SEM. ∗P ≤ 0.05.
3.4. Effects of Cpd1324 on hormone release
To study whether appetite regulatory gut hormones were responsible for the decreases in food intake and body weight, we measured a panel of gut hormones in plasma samples at 30, 60, 120, and 240 min following oral administration of 30 mg/kg of Cpd1324 in the lean mice. Based on our previously published ex vivo data [10] from gastrointestinal preparations, a decrease in the plasma level of the gastric hormone ghrelin could be expected following Cpd1324 administration. A decrease in ghrelin was indeed observed after 2 and 4 h (P = 0.0012 and P = 0.002, respectively, Figure 4A) when performed with a low level of environmental stress. GIP secreted from K cells located in the upper small intestine has been demonstrated to be efficiently regulated by nutrient receptors such as short- and long-chain free fatty acids [36,37]. However, Cpd1324 did not significantly affect GIP secretion at any investigated time point (Figs. 4B and S4D), although a slight attenuation in the plasma levels was observed after 30 min (P = 0.08, Fig. S4D). Secretion of NT, which is expressed in N cells and co-expressed with GLP-1 and PYY in L cells [38,39], only increased to a minor degree by Cpd1324 (Figure 4C). PYY and GLP-1 are secreted from L cells in the small and large intestine and both hormones have been shown to regulate appetite and food intake [40]. We observed a 2.2-fold increase in total GLP-1 levels 2 h and 4 h after Cpd1324 administration (Figure 4D), with no effect at earlier time points (Fig. S4A). Surprisingly, PYY levels were not affected by administration of the GPR39 agonist (Figure 4E), although it is well established that PYY and GLP-1 granules are clustered together within the same enteroendocrine L cells and that peptides are often co-secreted [38,39,[41], [42], [43]] We then investigated whether the effect of Cpd1324 on GLP-1 secretion was mediated specifically through GPR39 receptors. We found that Cpd1324 had no effect on GLP-1 secretion in the global GPR39 KO mice (Figure 4F), suggesting GLP-1 secretion was dependent on GPR39. However, it was not possible to determine the exact tissue origin of the Cpd1324-mediated increase in plasma GLP-1 levels.
Figure 4.

Satiety hormone levels in mice upon GPR39 activation. Plasma gut hormone levels following a single oral dose of Cpd1324 (30 mg/kg) in overnight-fasted C57BL/6J mice. Total plasma (A) active ghrelin (the data from two separate cohorts were pooled), n = 20–23 in each group, (B) GIP, (C) neurotensin (NT), (D) GLP-1, and (E) PYY were measured 2 (gray columns) and 4 (dark gray columns) h after dosing and compared to vehicle (open columns). (F) Plasma GLP-1 levels measured in GPR39 KO mice after dosing. Statistical significance was tested by Student's unpaired t test or one-way ANOVA followed by Tukey's post hoc test. n = 7–8 in each group unless otherwise stated. All of the animals were 9–11 weeks of age. For the GPR39 KO, WT littermates were used. Data are shown as means ± SEM. ∗∗P < 0.01 and ∗∗∗P < 0.001.
3.5. Ex vivo characterization of GLP-1 and PYY secretion following GPR39 agonism
To further elucidate the apparent selective secretion pattern induced by the GPR39 agonist in vivo, we used organoids cultured from the murine ileum and colon as an ex vivo model. The organoids are generated by self-renewing cryptal stem cells, which produce differentiated cell types in the intestinal epithelium, including enteroendocrine cells. They are characterized by a three-dimensional structure similar to the in vivo situation, where hormone-producing cells are maintained by the extracellular matrix [31,44]. Using ileal organoids from wild-type mice incubated with 10 μM or 50 μM of Cpd1324, we observed a significant concentration-dependent increase in GLP-1 release (Figure 5A). In contrast, Cpd1324-induced GLP-1 secretion was absent in organoids derived from the GPR39 KO mice (Figure 5B), indicating that GPR39-expressing intestinal cells were responsible for GLP-1 release. G-protein coupled bile acid receptor (GPBAR or TGR5) agonist L3740 [45] was used a positive control to demonstrate an increased GLP-1 secretion in ileal organoids from the wild-type and GPR39 KO mice. Since we did not detect any PYY secretion in response to either the TGR5 agonist or Cpd1324 (data not shown), suggesting that PYY was not expressed in these small intestinal preparations.
Figure 5.

GPR39 induced GLP-1 secretion in vitro. (A) Cpd1324-induced GLP-1 secretion in mouse ileal organoids (n = 6). TGR5 agonist (TGR5-ago; L-3740) was used as a positive control. (B) GLP-1 secretion in GPR39 KO mouse ileal organoids in the presence of Cpd1324 and L-3740 (n = 3). (C) GLP-1 secretion in human jejunum organoids stimulated with Cpd1324 or L-3740 (n = 3). (D) Cpd1324-induced GLP-1 secretion in mouse colon organoids (n = 4). (E) Cpd1324-induced PYY secretion in mouse colon organoids (n = 4). Data are shown as means ± SEM. Data involving more than two groups were assessed by ANOVA with Bonferroni's post hoc test using GraphPad Prism 8 software. ∗P < 0.05, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001. (F) Representative confocal microscopy images of GLP-1 (red) and PYY (green) immunofluorescence-stained enteroendocrine cells in colonic organoids stimulated with 100 μM of Cpd1324 or vehicle. Counterstained with DAPI (blue). Bar = 5 μm. (G) Quantification of colonic organoid enteroendocrine cell populations containing GLP-1 and PYY or only PYY staining following dosing.
To examine whether Cpd1324-mediated GLP-1 secretion was dependent on a decreasing somatostatin tone, we stimulated ileal organoids with Cpd1324, a broad-spectrum somatostatin receptor (SSTR) antagonist (BIM-23627) or a combination of the two. As shown in Fig. S5A, while administration of the SSTR antagonist to the organoids resulted in secretion of GLP-1 similar to that of Cpd1324-stimulated organoids, a combination of the two resulted in an additive effect, suggesting that GLP-1 secretion was not dependent on the somatostatin tone. Thus, GPR39 activation resulted in elevated plasma concentrations of GLP-1, which could contribute to the anorectic effect of Cpd1324. Other groups have reported that the gastric hormone obestatin can interact with GPR39 [46,47]; however, obestatin had no effect on GLP-1 secretion in our organoids (Fig. S5B).
Cpd1324 also increased GLP-1 secretion in organoid preparations from the human jejunum (Figure 5C) in which the samples were obtained from patients undergoing surgery, suggesting translation of the GPR39-mediated hormone signatures from rodents to humans (Figure 5C).
In mouse colon organoids, Cpd1324 increased GLP-1 secretion to a similar extent as observed in the small intestine (Figure 5D). In accordance with our observations in vivo, Cpd1324 had no effect on PYY secretion in organoids derived from mouse colon, where the PYY secretion is highest in the GI tract [[48], [49], [50]], while the positive control TGR5 agonist promoted a 3-fold increase in PYY secretion (Figure 5E).
To visually confirm the expression of PYY and GLP-1 in the murine colonic organoids, we performed immunostaining for GLP-1 and PYY and examined the preparations via confocal microscopy (Figure 5F and Fig. S6). In the vehicle-treated colonic organoids, co-expression of GLP-1 and PYY in the same enteroendocrine cells was observed in 96% of the 52 identified cells, with only 4% of the cells staining for PYY only (Figure 5G). In accordance with the secretion data, after 30 min of Cpd1324 treatment, the fraction of enteroendocrine cells co-storing GLP-1 and PYY decreased to 77%, while the PYY-only cells increased to 23% of the total 57 identified cells. These data confirmed the presence of enteroendocrine cells in the colonic organoids that co-stored GLP-1 and PYY and supported the notion that a subpopulation of enteroendocrine cells appears to respond to Cpd1324 by selectively secreting GLP-1 while retaining PYY granules.
3.6. Endogenous GLP-1 was responsible for GPR39-mediated colonic mucosal responses
To investigate the potential effects of GPR39 agonism on native colonic mucosa, we measured the acute effects of Cpd1324 on the transepithelial ion transport in murine proximal colon placed in Ussing chambers. A pair of representative recordings of transepithelial changes in Isc from adjacent colonic pieces shows that the presence of Cpd1324 rapidly increased the basal Isc (upper trace) (Figure 6A). This response was sensitive to pretreatment with the GLP-1 antagonist Ex(9–39) (added to the lower trace only), and the antagonist further abolished the subsequent response induced by the GLP-1 receptor agonist Ex4, yet had no effect on the hypersecretory response induced by subsequent addition of forskolin or the anti-secretory effect of α2 agonist UK14,304 (Figure 6A). The pooled data of Cpd1324 responses (from 10 mice) is shown in Figure 6B. Importantly, the Cpd1324-induced increase in Isc was not attenuated by administration of two antagonists specific for PYY Y1 and Y2 receptors (BIBO3304 and BIIE0246, respectively), indicating that endogenous PYY did not mediate Cpd1324 responses in the proximal colon mucosa. Because CGRP has been shown to mediate colonic GLP-1 responses [32], the selective CGRP antagonist BIBN4096 was used to determine whether the GPR39-induced increase in Isc depended on this paracrine sensory neuropeptide. BIBN4096 significantly attenuated Cpd1324 responses (Figure 6D) and abolished subsequent αCGRP responses (data not shown).
Figure 6.

GPR39 induced GLP-1-dependent and GLP-1-independent changes in epithelial ion transport. (A–B) Representative traces showing Cpd1324 (10 μM, basolaterally) increased Isc in vehicle-exposed proximal colon mucosa (upper trace/open column, n = 10) in the presence of 10 μM of ZnCl2. This effect was attenuated by pretreating with the GLP-1R antagonist Ex(9–39) (1 μM; lower trace/gray column, n = 10). (C) Cpd1324 responses were not affected by pretreatment with a combination of PYY-Y1, BIBO3304 (300 nM), and Y2 antagonist BIIE0246 (1 μM, n = 4) but were significantly inhibited by (D) the CGRP antagonist BIBN4096 (1 μM, n = 5). (E) Schematic illustration depicting the potential modes of action of the GPR39 agonist in the mouse proximal colon. Cpd1324 caused release of GLP-1 from L cells, which then acted on GLP-1R-positive submucosal neurons inducing the release of CGRP. This sensory neuropeptide then directly activated epithelial ion transport via Gαs-coupled CGRP receptors, leading to an increase in apical Cl− secretion through cAMP-sensitive CFTR channels. Residual responses to Cpd1324 (in B and D) were likely to be epithelial in origin and Gαq-mediated. Pooled data are the means ± SEM. Statistical significance was tested using Student's unpaired t test (in B, C, and D). ∗P < 0.05 and ∗∗∗P < 0.001.9–39: Ex(9–39), Veh: vehicle, BIBO: BIBO3304, BIIE: BIIE0246.
These data suggest, in accordance with our observations from hormone-secretion studies, that the Cpd1324 mucosal response was mediated via preferential secretion of GLP-1 and provide additional details on the endogenous mechanisms of GPR39 activity crucially demonstrated in this study using native tissue (Figure 6E).
3.7. Signaling properties of the small molecule agonist for GPR39
To understand the molecular determinants responsible for the Cpd1324 (Figure 7A)-induced selective GLP-1 secretion, we next characterized the signaling properties of GPR39 in a heterologous expression system. GPR39 is known to signal through the Gαq pathway, where accumulation of inositol trisphosphate (InsP3) has been measured as a second messenger [51]. To gain further insight into the pharmacological properties of GPR39, we tested the effects of Cpd1324 on the full spectrum of G protein activation. We assessed the direct activation of G proteins using bioluminescence resonance energy transfer (BRET)-based biosensors. These assays monitored the separation of the Gα and Gβγ subunits that occurred during G protein activation by measuring BRET between the Gα subunits fused to the energy donor RlucII and Gγ1 fused to the energy acceptor GFP10 in the presence of untagged Gβ1. The activation of the G protein resulted in a decrease in BRET signal, where the amplitude of this signal was used as an index of the G protein activation [22]. As shown in Figure 7B, in addition to activating Gαq, Cpd1324 activated all of the tested Gαi/o proteins and the Gα13 subunit with a high potency similar to Gαq. In contrast, the ligand activated Gαs with a low potency in the μM range. In general, 10 μM of Zn2+ is known to activate GPR39 independently and accordingly Zn2+ has been suggested to be the endogenous ligand for GPR39. The addition of 10 μM Zn2+ to Cpd1324 increased the potency of Cpd1324 to activate Gαq, Gαi1-3, and Gα13, whereas Gαs activation remained largely unchanged. These data indicate that Cpd1324 displays a unique signaling pattern with high Gαq signaling and strong concomitant activation of Gαi/o subunits that collectively may oppose potential downstream signals induced via the Gαs pathway.
Figure 7.

Cpd1324 structure and G protein signaling ability. (A) Molecular structure of the GPR39 agonist Cpd1324. (B) Measurements of G protein activation and using bioluminescence resonance energy transfer (BRET) technology following Cpd1324 administration. The data are shown as -BRET since the BRET sensors measured the decrease in BRET signals upon receptor activation. n = 3. Data are shown as means ± SEM.
4. Discussion
In the present study, we exploited a recently developed, highly potent, and efficacious GPR39 agonist, Cpd1324, as a tool compound to uncover the physiological effects of GPR39 on body weight development and hormone secretion from the gut. In this process, we revealed that the orally active agonist Cpd1324 decreased food intake acutely and led to significantly decreased body weight in the HFD-fed mice after 6 days of treatment. Importantly, GPR39 agonism led to decreased secretion of ghrelin and interestingly a selective release in GLP-1 but not PYY. The selective secretion pattern from the L cells appeared to be mediated through a unique G protein activation pattern.
4.1. GPR39-mediated modulation of food intake, energy expenditure, and body weight
GPR39 KO mice have previously been shown to have an increased susceptibility to a HFD [18], which can be partly attributed to increased food intake as we observed in this study. Thus, to probe for GPR39-mediated changes in food intake and energy metabolism, lean C57BL/6J mice were fed a HFD prior to administration of the GPR39 agonist Cpd1324 and were kept on the HFD throughout the study. The observed decrease in acute food intake was accompanied by a 4.5% body weight loss after 6 days of once-daily oral treatment. Based on the conditioned taste aversion study that demonstrated significantly less taste aversion compared to liraglutide, Cpd1324 is seemingly a well-tolerated compound; the decreased body weight is rather proposed to be the result of a combined GLP-1 secretion and decreased ghrelin secretion. As the most pronounced effect on food intake was observed within the first 2 h after administration of Cpd1324, we suggest that the hormones modulating feeding behavior during the acute phase may have been acting via the afferent vagal nerves. As the afferent vagal nerve endings are in close proximity to the site of gut hormone release, it is possible that even minor secretion modulation of ghrelin and possibly other hormones could exert this effect via the nerve terminals before being diluted in the systemic circulation. This could also explain the discrepancy in timing between the decreased food intake and observed changes in plasma concentrations of ghrelin and GLP-1.
Currently available orally active pharmaceutical interventions for obesity are rather limited and may be associated with an increased cardiovascular risk as observed in psychostimulatory drugs [2]. Other drugs with mild side effects, such as orlistat or lorcaserin, have limited efficacy and produce only a 3–6% decrease in body weight after 6 months of treatment [2]. Thus, there is a substantial need for novel, orally active, and safe pharmaceutical interventions with good efficacy for obesity. The observed effects of the GPR39 agonist on body weight and food intake revealed GPR39 as a novel potential anti-obesity target. To date, the only pharmaceutical intervention for obesity that leads to a substantial and sustained decrease in body weight is GLP-1-based pharmaceuticals, which are peptides with no or very little oral bioavailability [52,53]. To circumvent the challenges of oral bioavailability for GLP-1 mimetics, an alternative approach could be to develop a drug that increases the endogenous secretion of GLP-1 potentially in combination with modulation of a number of other appetite regulatory hormones, such as ghrelin.
4.2. GPR39-mediated GLP-1 secretion
In this study, we determined in murine in vivo setting and ex vivo preparations that Cpd1324 agonism was able to induce GLP-1 secretion in a GPR39-dependent manner since the response was eliminated in the absence of the receptor. However, since GPR39 is expressed in several metabolically important tissue such as the liver, pancreas, and brain, the effect on body weight regulation could be due to Cpd1324's action in these tissues in addition to the intestine. Thus, we cannot exclude that the Cpd1324-mediated increase in plasma GLP-1 levels may also be attributed to other sources such as central nervous system neurons and the intestinal L cells.
Regarding the intestine, Cpd1324 increased GLP-1 secretion in ex vivo preparations from healthy human organoids, indicating that GPR39 could constitute a relevant therapeutic target in human obesity. The increase in GLP-1 levels induced by GPR39 activation was consistently 2- to 3-fold above basal levels in the human and mouse preparations and in vivo and ex vivo models. Despite the higher increase in GLP-1 secretion observed for the TGR5 agonist, none of these ligands have been pursued in further clinical trials due to lack of efficacy in glucose metabolism and unwanted side effects related to the pancreatic and biliary tracts [54,55]. Although Cpd1324 significantly increased plasma GLP-1 and the insulin level in the DIO mice, we did not observe an improvement in glucose homeostasis during an OGTT. The lack of improved glucose metabolism even in the DIO mice that displayed increased insulin secretion was in accordance with previous observations [21] and may have resulted from a yet-undefined insulin resistance in the periphery or via hepatic glucose production. Furthermore, the GPR39 protein expression patterns were different in the DIO/T2D rats compared to the healthy controls [56].
4.3. GPR39-mediated regulation of other gut hormones
Other nutrients and metabolite receptors have been considered as therapeutic targets to increase endogenous GLP-1 levels including the lipid receptors GPR40 (free fatty acid receptor 1, FFAR1), GPR120 (free fatty acid receptor 4, FFAR4), and GPR119 as well as amino acid receptors or transporters such as GPR142 [30,36,[57], [58], [59]]. Stimulation of these receptors is characterized by secretion of a broad range of gut hormones, including GIP secretion from K cells in the upper small intestine. In contrast, GPR39 activation induced a decrease in ghrelin secretion and a selective secretion of GLP-1 with no significant changes to PYY, NT, or GIP release.
We previously found that GPR39 activation in vivo and ex vivo decreased ghrelin secretion [10] via an indirect effect mediated by somatostatin secretion from D cells in the gastric epithelium, leading to inhibition of ghrelin secretion. In the present study, in which the GPR39 agonist was administered orally, we observed a significant inhibition of plasma ghrelin levels, but this was apparently not dependent on somatostatin.
4.4. GPR39-mediated selective secretion of GLP-1 from L cells
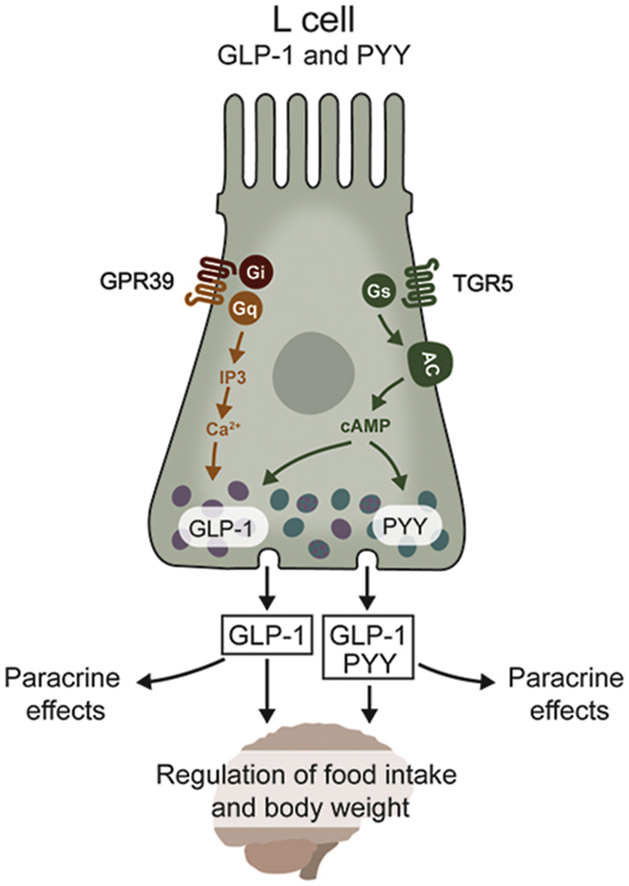
Surprisingly, GPR39 activation induced a consistent, robust increase in GLP-1 secretion without a concomitant secretion of the other L cell product, PYY. This may prove advantageous as elevated plasma levels of PYY3–36 have been associated with severe nausea and abdominal discomfort, which may mask the therapeutic application of PYY-based pharmaceutical interventions [60]. As PYY is not consistently expressed in the murine upper small intestine [8,48,50], we focused on the distal part of the small intestine and colon to study PYY's potential release. In these distal GI segments, the majority of L cells co-synthesize GLP-1 and PYY [38,48,61,62]; however, the expression pattern changes as the enteroendocrine cells mature and migrate from the crypt to the villi/surface [38,39]. When we examined murine colonic organoid cultures, the TGR5 agonist was able to induce a 4-fold increase in PYY secretion, whereas no detectable GPR39 agonist-induced PYY secretion was observed.
It remains to be determined whether GLP-1 and PYY are co-stored in the same vesicles or if significant proportions of L cells store these two important hormones in separate populations of secretory granules as described by Cho et al. [43]. A recent study by Billing et al. focused on colonic L cells expressing INSL-5 identified a striking overlap between the three major L cell products PYY, GLP-1, and INSL-5 in the secretory vesicles and that a broad range of stimuli produced the co-release of all three hormones concomitantly [63]. This notion is supported by prior studies that showed vesicular co-storage of PYY and pro-glucagon products [62,64,65], whereas other studies found that enteroendocrine cells appear to store the individual hormones in separate granules [38,43,61,66,67].
Since PYY is apparently not co-secreted with GLP-1 after Cpd1324 exposure in vivo and ex vivo, this supports the notion that GLP-1 and PYY can be stored separately and could be released independently from the same enteroendocrine cell. Importantly, we observed by immunostaining that GPR39 activation selectively induced the removal of GLP-1 from L cells while retaining secretory vesicles containing PYY. We cannot exclude the possibility that a proportion of the GLP-1 output in vivo derived from a subpopulation of GPR39-positive L cells storing only GLP-1 and not PYY. However, our immunohistochemical data confirmed that GLP-1 and PYY were co-localized within the vast majority of enteroendocrine L cells under basal and stimulated conditions in the colonic preparations.
We previously demonstrated that PYY predominantly mediates the effects of GPR119 agonism [68] and FFA2 agonism [69]. Using a similar in vitro design, we determined that GPR39 agonism in the proximal colon was independent of PYY since antagonists targeting Y1 and Y2 receptors were ineffective, while the GLP-1 receptor antagonist Ex(9–39) successfully attenuated the GPR39 mucosal response. The CGRP antagonist was also able to attenuate the GPR39 agonist-mediated mucosal response. These data indicated that GPR39 activation caused endogenous GLP-1 secretion in the proximal colon and that subsequent activation of GLP-1 receptors on submucosal sensory neurons led to CGRP release and activation of epithelial ion transport (see schematic; Figure 6E). It is notable that our expression data from FACS-sorted L cells indicated that the surrounding enterocytes also expressed the GPR39 receptor at significant levels (Fig. S7). Thus, it is also possible that GPR39 acts directly on enterocytes, inducing epithelial Cl− secretion and elevating Isc levels in the mouse proximal colon (Figure 6E). Thus, direct epithelial and indirect neuroendocrine mechanisms may be co-activated by the GPR39 agonist Cpd1324 in native colonic tissue.
Our data consistently suggest that GPR39 activation induces GLP-1 secretion independent of PYY despite the two peptides being co-localized. One explanation for this observation is that GPR39 agonism initiates a signaling pathway in L cells that favors exocytosis of GLP-1-positive granules. Cpd1324 is characterized by a unique activation pattern of the GPR39 receptor since we observed a highly potent activation of both Gαq and Gαi/o signaling but only stimulates the Gαs pathway with low potency. Furthermore, the second messenger activation that may happen downstream of Gαs is most likely neutralized by the concomitant more potent activation of Gαi coupling. In contrast, the TGR5 agonist used as a positive control in the present study induces highly potent activation of Gαs coupling that may facilitate the strong activation of L cells to release the “full package” of secretory granules, including PYY and GLP-1.
5. Conclusion
Our data showed that an orally active GPR39 agonist acutely decreased food intake and led to decreased body weight in mice fed a HFD after 6 days of treatment. This effect was most likely due to secretion of a specific signature of gut hormones including robust GLP-1 secretion and decreased ghrelin secretion. Importantly, the other major L cell product, PYY, was not released by GPR39 agonism, suggesting that PYY and GLP-1 were responsive to different activation pathways to engage in exocytosis.
Author contributions
B.H. conceptualized and designed the study. T.A.D., K·V.G., N·P., I.R.T., L.J.S., C.J., F.M., M.P.A., L.M.E.S., S.L.J., B.H. L.L., J.D.A.B., and R.B.S. performed and analyzed the experiments. B.H., H·C., A.B., and M.B. supervised the experiments. K.V.G. and B.H. drafted the manuscript. H.C., N·P., C.J., T.A.D., L.J.S., F.M., M.P.A., R.B·S., M.B., S.L.J., S.A.H., and T.M.F. edited the manuscript. All of the authors approved the final version of the manuscript.
Grants
This study was supported by Carlsberg Foundation and Novo Nordisk Foundation Challenge grants NNF14OC0016798, project grants in Endocrinology and Metabolism Nordic Region 2019 (#0057417), and NNF15OC0013655. The Novo Nordisk Foundation Center for Basic Metabolic Research (www.metabol.ku.dk) is supported by an unconditional grant (NNF10CC1016515) from the Novo Nordisk Foundation to the University of Copenhagen. I.R.T. is supported by a BBSRC grant (BB/N006763/1) to H.C. M.B. is supported by a CIHR foundation grant (#148431).
Conflicts of interest
M.B. is the chairman of the scientific advisory board of Domain Therapeutics to which some of the biosensors used in this study are licensed for commercial use. The biosensors are freely available for academic research from M.B. The rest of the authors declare no conflicts of interest.
Acknowledgments
We greatly appreciate Bianca Plouffe, Anette Bjerregaard, and Lisbeth Meyer Petersen for technical assistance and Anna Sofie Husted for the graphical abstract. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101207.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Energy expenditure in GPR39 KO mice compared to WT mice. VO2 consumption for individual WT (black dots, n = 8) and GPR39 KO (open dots, n = 7) mice during (A) the dark phase and (B) light phase with a correlation trend line for WT (straight) and GPR39 KO mice (dashed). Energy expenditure during (C) dark and (D) light phases was corrected for body mass using an ANCOVA model. n = 7–8.
Multimedia component 2Bioavailability, food intake, and energy expenditure following different doses of Cpd1324. (A) Plasma concentration of Cpd1324 in C57Bl/6J mice 0, 2, 4, and 6 h after administration of 30 mg/kg (red dots, n = 4) or 120 mg/kg (blue dots, n = 4). (B, D, F, and H) The 24 h accumulated food intake in C57BL/6J mice after a single oral bolus of (B and C) 15, (D and E) 30, (F and G) 60, or (H and I) 120 mg/kg of Cpd1324 (black dots) or vehicle (open dots) and (C, E, G, and I) 24 h accumulated food intake divided into dark and light phases in Cpd1324 (black columns) and vehicle-treated mice (open columns, n = 6–8). VO2 consumption of individual vehicle-treated (black dots, n = 6) and Cpd1324-treated (30 mg/kg, open dots, n = 8) mice during (J) dark and (K) light phases with a correlation trend line for vehicle-treated (straight) and Cpd1324-treated mice (dashed). Energy expenditure during (L) dark and (M) light phases was corrected for body mass using an ANCOVA model following administration of vehicle (black column) or Cpd1324 (30 mg/kg, open column). Significance of the difference was tested by 2-way ANOVA using Sidak's test (A, C, E, G, I). Data are shown as means ± SEM. ∗P ≤ 0.05, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001.
Multimedia component 3Activity levels and conditioned taste aversion following oral administration of Cpd1324. (A) Activity counts per 20 min following Cpd1324 (black dots) or vehicle (gray dots) dosing (n = 8). (B) Conditioned taste aversion following administration of vehicle, 400 μg/kg liraglutide (s.c.), 30- or 120 mg/kg Cpd1324 (p.o.) (n = 7–8). Statistical significance was tested by Student's unpaired t test (B). Data are shown as means ± SEM. ∗∗P ≤ 0.01 and ∗∗∗∗P ≤ 0.0001.
Multimedia component 4Acute effect (30–60 min) on gut satiety hormones following administration of Cpd1324. Plasma gut hormone levels following a single oral dose of Cpd1324 (30 mg/kg) in overnight-fasted C57BL/6J mice. Total plasma (A), total GLP-1, (B) glucagon, (C) insulin, (D) GIP, and (E) active ghrelin measurements 30 min (light gray columns, n = 13) and 60 min (dark gray columns, n = 13) h after dosing and compared to vehicle (open columns, n = 10). Statistical significance was tested by one-way ANOVA. All of the animals were 9–11 weeks of age. Data are shown as means ± SEM.
Multimedia component 5The effect of somatostatin receptor antagonism following GPR39 agonism or obestatin on organoid GLP-1 secretion. GLP-1 secretion from (A) ileal organoids stimulated with vehicle, Cpd1324, a broad spectrum somatostatin receptor (SSTR) antagonist (iSST; BIM-23627), or both (n = 4–21). (B) Ileal organoids stimulated for 2 h with vehicle, 1 μM of TGR5 agonist (L3740), 100 nM of obestatin, or 500 nM of obestatin (n = 4). Statistical significance was tested by one-way ANOVA with either a Dunnett's (A) or Holm-Šídák's multiple comparisons test (B).
Multimedia component 6GLP-1 and PYY immunocytofluorescence staining in colonic organoids following Cpd1324 administration. Representative confocal microscopy images of GLP-1 (red) and PYY (green) immunofluorescence-stained enteroendocrine cells in colonic organoids stimulated with 100 μM of Cpd1324 or vehicle (DMSO). Counterstained with DAPI (blue). Bar = 5 μm.
Multimedia component 7GPR39 expression in intestinal L cells. Gpr39 mRNA levels in FACS-purified L cells (gray columns) and surrounding cells (black columns) in small and large intestine segments. Mean ± SEM of 3 independent studies in which each study was comprised of L cells collected from 10 male mice.
References
- 1.Srivastava G., Apovian C.M. Current pharmacotherapy for obesity. Nature Reviews Endocrinology. 2018;14(1):12–24. doi: 10.1038/nrendo.2017.122. [DOI] [PubMed] [Google Scholar]
- 2.Müller T.D., Clemmensen C., Finan B., Dimarchi R.D., Tschöp M.H. Anti-obesity therapy: from rainbow pills to polyagonists. Pharmacological Reviews. 2018;70(4):712–746. doi: 10.1124/pr.117.014803. [DOI] [PubMed] [Google Scholar]
- 3.Cummings D.E., Overduin J., Foster-Schubert K.E. Gastric bypass for obesity: mechanisms of weight loss and diabetes resolution. Journal of Clinical Endocrinology & Metabolism. 2004;89:2608–2615. doi: 10.1210/jc.2004-0433. [DOI] [PubMed] [Google Scholar]
- 4.Svane M., Madsbad S. Bariatric surgery - effects on obesity and related co-morbidities. Current Diabetes Reviews. 2014;10(3):208–214. doi: 10.2174/1573399810666140616144141. [DOI] [PubMed] [Google Scholar]
- 5.Dirksen C., Jørgensen N.B., Bojsen-Møller K.N., Jacobsen S.H., Hansen D.L., Worm D., et al. Mechanisms of improved glycaemic control after Roux-en-Y gastric bypass. Diabetologia. 2012;55(7):1890–1901. doi: 10.1007/s00125-012-2556-7. [DOI] [PubMed] [Google Scholar]
- 6.Al-Najim W., Docherty N.G., Le Roux C.W. Food intake and eating behavior after bariatric surgery. Physiological Reviews. 2018;98(3):1113–1141. doi: 10.1152/physrev.00021.2017. [DOI] [PubMed] [Google Scholar]
- 7.Ma I.T., Madura J.A. Gastrointestinal complications after bariatric surgery. Gastroenterology and Hepatology. 2015;11(8):526–535. [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts G.P., Larraufie P., Richards P., Kay R.G., Galvin S.G., Miedzybrodzka E.L., et al. Comparison of human and murine enteroendocrine cells by transcriptomic and peptidomic profiling. Diabetes. 2019;68(5):1062–1072. doi: 10.2337/db18-0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Husted A.S., Trauelsen M., Rudenko O., Hjorth S.A., Schwartz T.W. GPCR-mediated signaling of metabolites. Cell Metabolism. 2017;25(4):777–796. doi: 10.1016/j.cmet.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Frimurer T.M., Mende F., Graae A.S., Engelstoft M.S., Egerod K.L., Nygaard R., et al. Model-based discovery of synthetic agonists for the Zn2+-sensing G-protein-coupled receptor 39 (GPR39) reveals novel biological functions. Journal of Medicinal Chemistry. 2017;60(3):886–898. doi: 10.1021/acs.jmedchem.6b00648. [DOI] [PubMed] [Google Scholar]
- 11.Peukert S., Hughes R., Nunez J., He G., Yan Z., Jain R., et al. Discovery of 2-pyridylpyrimidines as the first orally bioavailable GPR39 agonists. ACS Medicinal Chemistry Letters. 2014;5(10):1114–1118. doi: 10.1021/ml500240d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holst B., Egerod K.L., Schild E., Vickers S.P., Cheetham S., Gerlach L.O., et al. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology. 2007;148(1):13–20. doi: 10.1210/en.2006-0933. [DOI] [PubMed] [Google Scholar]
- 13.Müller T.D., Nogueiras R., Andermann M.L., Andrews Z.B., Anker S.D., Argente J., et al. Ghrelin. Molecular Metabolism. 2015;4(6):437–460. doi: 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egerod K.L., Holst B., Petersen P.S., Hansen J.B., Mulder J., Hökfelt T., et al. GPR39 splice variants versus antisense gene LYPD1: expression and regulation in gastrointestinal tract, endocrine pancreas, liver, and white adipose tissue. Molecular Endocrinology (Baltimore, Md. 2007;21(7):1685–1698. doi: 10.1210/me.2007-0055. [DOI] [PubMed] [Google Scholar]
- 15.Młyniec K., Gaweł M., Librowski T., Reczyński W., Bystrowska B., Holst B. Investigation of the GPR39 zinc receptor following inhibition of monoaminergic neurotransmission and potentialization of glutamatergic neurotransmission. Brain Research Bulletin. 2015;115:23–29. doi: 10.1016/j.brainresbull.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Holst B., Egerod K.L., Jin C., Petersen P.S., Østergaard M.V., Hald J., et al. G protein-coupled receptor 39 deficiency is associated with pancreatic islet dysfunction. Endocrinology. 2009;150(6):2577–2585. doi: 10.1210/en.2008-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tremblay F., Richard A.M.T., Will S., Syed J., Stedman N., Perreault M., et al. Disruption of G protein-coupled receptor 39 impairs insulin secretion in vivo. Endocrinology. 2009;150(6):2586–2595. doi: 10.1210/en.2008-1251. [DOI] [PubMed] [Google Scholar]
- 18.Petersen P.S., Jin C., Madsen A.N., Rasmussen M., Kuhre R., Egerod K.L., et al. Deficiency of the GPR39 receptor is associated with obesity and altered adipocyte metabolism. The FASEB Journal. 2011;25(11):3803–3814. doi: 10.1096/fj.11-184531. [DOI] [PubMed] [Google Scholar]
- 19.Bassilana F., Carlson A., Dasilva J.A., Grosshans B., Vidal S., Beck V., et al. Target identification for a Hedgehog pathway inhibitor reveals the receptor GPR39. Nature Chemical Biology. 2014;10(5):343–349. doi: 10.1038/nchembio.1481. [DOI] [PubMed] [Google Scholar]
- 20.Sato S., Huang X.P., Kroeze W.K., Roth B.L. Discovery and characterization of novel GPR39 agonists allosterically modulated by zinc. Molecular Pharmacology. 2016;90(6):726–737. doi: 10.1124/mol.116.106112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fjellström O., Larsson N., Yasuda S.I., Tsuchida T., Oguma T., Marley A., et al. Novel Zn2+ modulated GPR39 receptor agonists do not drive acute insulin secretion in rodents. PloS One. 2015;10(12) doi: 10.1371/journal.pone.0145849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galés C., Van Durm J.J.J., Schaak S., Pontier S., Percherancier Y., Audet M., et al. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nature Structural & Molecular Biology. 2006;13(9):778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- 23.Paradis J.S., Ly S., Blondel-Tepaz É., Galan J.A., Beautrait A., Scott M.G.H., et al. Receptor sequestration in response to β-arrestin-2 phosphorylation by ERK1/2 governs steady-state levels of GPCR cell-surface expression. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(37):E5160–E5168. doi: 10.1073/pnas.1508836112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quoyer J., Janz J.M., Luo J., Ren Y., Armando S., Lukashova V., et al. Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(52):E5088–E5097. doi: 10.1073/pnas.1312515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mende F., Hundahl C., Plouffe B., Skov L.J., Sivertsen B., Madsen A.N., et al. Translating biased signaling in the ghrelin receptor system into differential in vivo functions. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(43):E10255–E10264. doi: 10.1073/pnas.1804003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ratner C., He Z., Grunddal K.V.K.V., Skov L.J.L.J., Hartmann B., Zhang F., et al. Long-acting neurotensin synergizes with liraglutide to reverse obesity through a melanocortin-dependent pathway. Diabetes. 2019;68(6):1329–1340. doi: 10.2337/db18-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratner C., Skov L.J., Raida Z., Bächler T., Bellmann-Sickert K., Foll C. Le., et al. Effects of peripheral neurotensin on appetite regulation and its role in gastric bypass surgery. Endocrinology. 2016;157(9):3482–3492. doi: 10.1210/en.2016-1329. [DOI] [PubMed] [Google Scholar]
- 28.Sato T., Stange D.E., Ferrante M., Vries R.G.J., Van Es J.H., Van Den Brink S., et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 29.Sato T., Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 30.Rudenko O., Shang J., Munk A., Ekberg J.P., Petersen N., Engelstoft M.S., et al. The aromatic amino acid sensor GPR142 controls metabolism through balanced regulation of pancreatic and gut hormones. Molecular Metabolism. 2019;19:49–64. doi: 10.1016/j.molmet.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersen N., Frimurer T.M., Terndrup Pedersen M., Egerod K.L., Wewer Albrechtsen N.J., Holst J.J., et al. Inhibiting RHOA signaling in mice increases glucose tolerance and numbers of enteroendocrine and other secretory cells in the intestine. Gastroenterology. 2018;155(4):1164–1176. doi: 10.1053/j.gastro.2018.06.039. e2. [DOI] [PubMed] [Google Scholar]
- 32.Tough I.R., Moodaley R., Cox H.M. Mucosal glucagon-like peptide 1 (GLP-1) responses are mediated by calcitonin gene-related peptide (CGRP) in the mouse colon and both peptide responses are area-specific. Neuro-Gastroenterology and Motility. 2018;30(1) doi: 10.1111/nmo.13149. [DOI] [PubMed] [Google Scholar]
- 33.Tough I.R., Forbes S., Tolhurst R., Ellis M., Herzog H., Bornstein J.C., et al. Endogenous peptide YY and neuropeptide y inhibit colonic ion transport, contractility and transit differentially via Y 1 and Y 2 receptors. British Journal of Pharmacology. 2011;164(2 B):471–484. doi: 10.1111/j.1476-5381.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jelsing J., Vrang N., Hansen G., Raun K., Tang-Christensen M., Bjerre Knudsen L. Liraglutide: short-lived effect on gastric emptying-long lasting effects on body weight. Diabetes, Obesity and Metabolism. 2012;14(6):531–538. doi: 10.1111/j.1463-1326.2012.01557.x. [DOI] [PubMed] [Google Scholar]
- 35.Adams J.M., Pei H., Sandoval D.A., Seeley R.J., Chang R.B., Liberles S.D., et al. Liraglutide modulates appetite and body weight through glucagon-like peptide 1 receptor-expressing glutamatergic neurons. Diabetes. 2018;67:1538–1548. doi: 10.2337/db17-1385. American Diabetes Association Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekberg J.H., Hauge M., Kristensen L.V., Madsen A.N., Engelstoft M.S., Husted A.S., et al. GPR119, a major enteroendocrine sensor of dietary triglyceride metabolites coacting in synergy with FFA1 (GPR40) Endocrinology. 2016;157(12):4561–4569. doi: 10.1210/en.2016-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adriaenssens A.E., Biggs E.K., Darwish T., Tadross J., Sukthankar T., Girish M., et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metabolism. 2019;30(5):987–996. doi: 10.1016/j.cmet.2019.07.013. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grunddal K.V., Ratner C.F., Svendsen B., Sommer F., Engelstoft M.S., Madsen A.N., et al. Neurotensin is coexpressed, coreleased, and acts together with GLP-1 and PYY in enteroendocrine control of metabolism. Endocrinology. 2016;157(1):176–194. doi: 10.1210/en.2015-1600. [DOI] [PubMed] [Google Scholar]
- 39.Billing L.J., Larraufie P., Lewis J., Leiter A., Li J., Lam B., et al. Single cell transcriptomic profiling of large intestinal enteroendocrine cells in mice – identification of selective stimuli for insulin-like peptide-5 and glucagon-like peptide-1 co-expressing cells. Molecular Metabolism. 2019;29(September):158–169. doi: 10.1016/j.molmet.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holst J.J., Madsbad S., Bojsen-Møller K.N., Svane M.S., Jørgensen N.B., Dirksen C., et al. Mechanisms in bariatric surgery: gut hormones, diabetes resolution, and weight loss. Surgery for Obesity and Related Diseases. 2018;14(5):708–714. doi: 10.1016/j.soard.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ullmer C., Alvarez Sanchez R., Sprecher U., Raab S., Mattei P., Dehmlow H., et al. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. British Journal of Pharmacology. 2013;169(3):671–684. doi: 10.1111/bph.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun L., Goh H.J., Govindharajulu P., Leow M.K.S., Henry C.J. Differential effects of monounsaturated and polyunsaturated fats on satiety and gut hormone responses in healthy subjects. Foods. 2019;8(12) doi: 10.3390/foods8120634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho H.J., Robinson E.S., Rivera L.R., McMillan P.J., Testro A., Nikfarjam M., et al. Glucagon-like peptide 1 and peptide YY are in separate storage organelles in enteroendocrine cells. Cell and Tissue Research. 2014;357(1):63–69. doi: 10.1007/s00441-014-1886-9. [DOI] [PubMed] [Google Scholar]
- 44.Simon-Assmann P., Turck N., Sidhoum-Jenny M., Gradwohl G., Kedinger M. In vitro models of intestinal epithelial cell differentiation. Cell Biology and Toxicology. 2007;23(4):241–256. doi: 10.1007/s10565-006-0175-0. [DOI] [PubMed] [Google Scholar]
- 45.Lund M.L., Sorrentino G., Egerod K.L., Kroone C., Mortensen B., Knop F.K., et al. L-cell differentiation is induced by bile acids through GPBAR1 and paracrine GLP-1 and serotonin signaling. Diabetes. 2020;69(4):614–623. doi: 10.2337/db19-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J.V., Jahr H., Luo C.W., Klein C., Van Kolen K., Ver Donck L., et al. Obestatin induction of early-response gene expression in gastrointestinal and adipose tissues and the mediatory role of G protein-coupled receptor, GPR39. Molecular Endocrinology. 2008;22(6):1464–1475. doi: 10.1210/me.2007-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J.V., Ren P.G., Avsian-Kretchmer O., Luo C.W., Rauch R., Klein C., et al. Medicine: obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science. 2005;310(5750):996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- 48.Svendsen B., Pedersen J., Albrechtsen N.J.W., Hartmann B., Toräng S., Rehfeld J.F., et al. An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology. 2015;156(3):847–857. doi: 10.1210/en.2014-1710. [DOI] [PubMed] [Google Scholar]
- 49.Kuhre R.E., Christiansen C.B., Saltiel M.Y., Wewer Albrechtsen N.J., Holst J.J. On the relationship between glucose absorption and glucose-stimulated secretion of GLP-1, neurotensin, and PYY from different intestinal segments in the rat. Physiological Reports. 2017;5(23) doi: 10.14814/phy2.13507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wewer Albrechtsen N.J., Kuhre R.E., Toräng S., Holst J.J. The intestinal distribution pattern of appetite- and glucose regulatory peptides in mice, rats and pigs. BMC Research Notes. 2016;9(1):60. doi: 10.1186/s13104-016-1872-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holst B., Holliday N.D., Bach A., Elling C.E., Cox H.M., Schwartz T.W. Common structural basis for constitutive activity of the ghrelin receptor family. Journal of Biological Chemistry. 2004;279(51):53806–53817. doi: 10.1074/jbc.M407676200. [DOI] [PubMed] [Google Scholar]
- 52.Knudsen L.B., Lau J. The discovery and development of liraglutide and semaglutide. Frontiers in Endocrinology. 2019;10:155. doi: 10.3389/fendo.2019.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buckley S.T., Bækdal T.A., Vegge A., Maarbjerg S.J., Pyke C., Ahnfelt-Rønne J., et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Science Translational Medicine. 2018;10(468) doi: 10.1126/scitranslmed.aar7047. [DOI] [PubMed] [Google Scholar]
- 54.Pols T.W.H., Nomura M., Harach T., Lo Sasso G., Oosterveer M.H., Thomas C., et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metabolism. 2011;14(6):747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hodge R.J., Nunez D.J. Therapeutic potential of takeda-G-protein-receptor-5 (TGR5) 2016;vol. 5:439–443. doi: 10.1111/dom.12636. [DOI] [PubMed] [Google Scholar]
- 56.Kolodziejski P.A., Pruszynska-Oszmalek E., Sassek M., Kaczmarek P., Szczepankiewicz D., Billert M., et al. Changes in obestatin gene and GPR39 receptor expression in peripheral tissues of rat models of obesity, type 1 and type 2 diabetes. Journal of Diabetes. 2017;9(4):353–361. doi: 10.1111/1753-0407.12417. [DOI] [PubMed] [Google Scholar]
- 57.Diakogiannaki E., Pais R., Tolhurst G., Parker H.E., Horscroft J., Rauscher B., et al. Oligopeptides stimulate glucagon-like peptide-1 secretion in mice through proton-coupled uptake and the calcium-sensing receptor. Diabetologia. 2013;56(12):2688–2696. doi: 10.1007/s00125-013-3037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin H.V., Efanov A.M., Fang X., Beavers L.S., Wang X., Wang J., et al. GPR142 controls tryptophan-induced insulin and incretin hormone secretion to improve glucose metabolism. PloS One. 2016;11(6) doi: 10.1371/journal.pone.0157298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hirasawa A., Tsumaya K., Awaji T., Katsuma S., Adachi T., Yamada M., et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nature Medicine. 2005;11(1):90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 60.Sloth B., Holst J.J., Flint A., Gregersen N.T., Astrup A. Effects of PYY1-36 and PYY3-36 on appetite, energy intake, energy expenditure, glucose and fat metabolism in obese and lean subjects. American Journal of Physiology - Endocrinology and Metabolism. 2007;292(4) doi: 10.1152/ajpendo.00450.2006. [DOI] [PubMed] [Google Scholar]
- 61.Fothergill L.J., Ringuet M.T., Sioras E., Hunne B., Fazio Coles T.E., Martins P.R., et al. Cellular and sub-cellular localisation of oxyntomodulin-like immunoreactivity in enteroendocrine cells of human, mouse, pig and rat. Cell and Tissue Research. 2019;375(2):359–369. doi: 10.1007/s00441-018-2921-z. [DOI] [PubMed] [Google Scholar]
- 62.Nilsson O., Bilchik A.J., Goldenring J.R., Ballantyne G.H., Adrian T.E., Modlin I.M. Distribution and immunocytochemical colocalization of peptide YY and enteroglucagon in endocrine cells of the Rabbit Colon. Endocrinology. 1991;129(1):139–148. doi: 10.1210/endo-129-1-139. [DOI] [PubMed] [Google Scholar]
- 63.Billing L.J., Smith C.A., Larraufie P., Goldspink D.A., Galvin S., Kay R.G., et al. Co-storage and release of insulin-like peptide-5, glucagon-like peptide-1 and peptideYY from murine and human colonic enteroendocrine cells. Molecular Metabolism. 2018;16(July):65–75. doi: 10.1016/j.molmet.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Böttcher G., Sjölund K., Ekblad E., Håkanson R., Schwartz T.W., Sundler F. Coexistence of peptide YY and glicentin immunoreactivity in endocrine cells of the gut. Regulatory Peptides. 1984;8(4):261–266. doi: 10.1016/0167-0115(84)90034-X. [DOI] [PubMed] [Google Scholar]
- 65.Ali-Rachedi A., Varndell I.M., Adrian T.E., Gapp D.A., Van Noorden S., Bloom S.R., et al. Peptide YY (PYY) immunoreactivity is co-stored with glucagon-related immunoreactants in endocrine cells of the gut and pancreas. Histochemistry. 1984;80(5):487–491. doi: 10.1007/BF00495439. [DOI] [PubMed] [Google Scholar]
- 66.Stengel A., Goebel M., Yakubov I., Wang L., Witcher D., Coskun T., et al. Identification and characterization of nesfatin-1 immunoreactivity in endocrine cell types of the rat gastric oxyntic mucosa. Endocrinology. 2009;150(1):232–238. doi: 10.1210/en.2008-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fothergill L.J., Callaghan B., Hunne B., Bravo D.M., Furness J.B. Costorage of enteroendocrine hormones evaluated at the cell and subcellular levels in male mice. Endocrinology. 2017;158(7):2113–2123. doi: 10.1210/en.2017-00243. [DOI] [PubMed] [Google Scholar]
- 68.Cox H.M., Tough I.R., Woolston A.M., Zhang L., Nguyen A.D., Sainsbury A., et al. Peptide YY is critical for acylethanolamine receptor Gpr119-induced activation of gastrointestinal mucosal responses. Cell Metabolism. 2010;11(6):532–542. doi: 10.1016/j.cmet.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Forbes S., Stafford S., Coope G., Heffron H., Real K., Newman R., et al. Selective FFA2 agonism appears to act via intestinal PYY to reduce transit and food intake but does not improve glucose tolerance in mouse models. Diabetes. 2015;64(11):3763–3771. doi: 10.2337/db15-0481. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Energy expenditure in GPR39 KO mice compared to WT mice. VO2 consumption for individual WT (black dots, n = 8) and GPR39 KO (open dots, n = 7) mice during (A) the dark phase and (B) light phase with a correlation trend line for WT (straight) and GPR39 KO mice (dashed). Energy expenditure during (C) dark and (D) light phases was corrected for body mass using an ANCOVA model. n = 7–8.
Multimedia component 2Bioavailability, food intake, and energy expenditure following different doses of Cpd1324. (A) Plasma concentration of Cpd1324 in C57Bl/6J mice 0, 2, 4, and 6 h after administration of 30 mg/kg (red dots, n = 4) or 120 mg/kg (blue dots, n = 4). (B, D, F, and H) The 24 h accumulated food intake in C57BL/6J mice after a single oral bolus of (B and C) 15, (D and E) 30, (F and G) 60, or (H and I) 120 mg/kg of Cpd1324 (black dots) or vehicle (open dots) and (C, E, G, and I) 24 h accumulated food intake divided into dark and light phases in Cpd1324 (black columns) and vehicle-treated mice (open columns, n = 6–8). VO2 consumption of individual vehicle-treated (black dots, n = 6) and Cpd1324-treated (30 mg/kg, open dots, n = 8) mice during (J) dark and (K) light phases with a correlation trend line for vehicle-treated (straight) and Cpd1324-treated mice (dashed). Energy expenditure during (L) dark and (M) light phases was corrected for body mass using an ANCOVA model following administration of vehicle (black column) or Cpd1324 (30 mg/kg, open column). Significance of the difference was tested by 2-way ANOVA using Sidak's test (A, C, E, G, I). Data are shown as means ± SEM. ∗P ≤ 0.05, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001.
Multimedia component 3Activity levels and conditioned taste aversion following oral administration of Cpd1324. (A) Activity counts per 20 min following Cpd1324 (black dots) or vehicle (gray dots) dosing (n = 8). (B) Conditioned taste aversion following administration of vehicle, 400 μg/kg liraglutide (s.c.), 30- or 120 mg/kg Cpd1324 (p.o.) (n = 7–8). Statistical significance was tested by Student's unpaired t test (B). Data are shown as means ± SEM. ∗∗P ≤ 0.01 and ∗∗∗∗P ≤ 0.0001.
Multimedia component 4Acute effect (30–60 min) on gut satiety hormones following administration of Cpd1324. Plasma gut hormone levels following a single oral dose of Cpd1324 (30 mg/kg) in overnight-fasted C57BL/6J mice. Total plasma (A), total GLP-1, (B) glucagon, (C) insulin, (D) GIP, and (E) active ghrelin measurements 30 min (light gray columns, n = 13) and 60 min (dark gray columns, n = 13) h after dosing and compared to vehicle (open columns, n = 10). Statistical significance was tested by one-way ANOVA. All of the animals were 9–11 weeks of age. Data are shown as means ± SEM.
Multimedia component 5The effect of somatostatin receptor antagonism following GPR39 agonism or obestatin on organoid GLP-1 secretion. GLP-1 secretion from (A) ileal organoids stimulated with vehicle, Cpd1324, a broad spectrum somatostatin receptor (SSTR) antagonist (iSST; BIM-23627), or both (n = 4–21). (B) Ileal organoids stimulated for 2 h with vehicle, 1 μM of TGR5 agonist (L3740), 100 nM of obestatin, or 500 nM of obestatin (n = 4). Statistical significance was tested by one-way ANOVA with either a Dunnett's (A) or Holm-Šídák's multiple comparisons test (B).
Multimedia component 6GLP-1 and PYY immunocytofluorescence staining in colonic organoids following Cpd1324 administration. Representative confocal microscopy images of GLP-1 (red) and PYY (green) immunofluorescence-stained enteroendocrine cells in colonic organoids stimulated with 100 μM of Cpd1324 or vehicle (DMSO). Counterstained with DAPI (blue). Bar = 5 μm.
Multimedia component 7GPR39 expression in intestinal L cells. Gpr39 mRNA levels in FACS-purified L cells (gray columns) and surrounding cells (black columns) in small and large intestine segments. Mean ± SEM of 3 independent studies in which each study was comprised of L cells collected from 10 male mice.