

Abstract
In malaria, chemical genetics is a powerful method for assigning function to uncharacterized genes. MMV085203 and GNF-Pf-3600 are two structurally related napthoquinone phenotypic screening hits that kill both blood- and sexual-stage P. falciparum parasites in the low nanomolar to low micromolar range. In order to understand their mechanism of action, parasites from two different genetic backgrounds were exposed to sublethal concentrations of MMV085203 and GNF-Pf-3600 until resistance emerged. Whole genome sequencing revealed all 17 resistant clones acquired nonsynonymous mutations in the gene encoding the orphan apicomplexan transporter PF3D7_0312500 (pfmfr3) predicted to encode a member of the major facilitator superfamily (MFS). Disruption of pfmfr3 and testing against a panel of antimalarial compounds showed decreased sensitivity to MMV085203 and GNF-Pf-3600 as well as other compounds that have a mitochondrial mechanism of action. In contrast, mutations in pfmfr3 provided no protection against compounds that act in the food vacuole or the cytosol. A dihydroorotate dehydrogenase rescue assay using transgenic parasite lines, however, indicated a different mechanism of action for both MMV085203 and GNF-Pf-3600 than the direct inhibition of cytochrome bc1. Green fluorescent protein (GFP) tagging of PfMFR3 revealed that it localizes to the parasite mitochondrion. Our data are consistent with PfMFR3 playing roles in mitochondrial transport as well as drug resistance for clinically relevant antimalarials that target the mitochondria. Furthermore, given that pfmfr3 is naturally polymorphic, naturally occurring mutations may lead to differential sensitivity to clinically relevant compounds such as atovaquone.
Keywords: malaria, drug resistance, drug discovery, transporter, mitochondria
With more than 200 million cases and over 400,000 deaths globally, malaria remains a devastating disease and gross burden on public health.1 It is caused by protozoan parasites belonging to the Plasmodium genus and is transmitted by female anophelene mosquitoes. Although substantial effort and resources have been mustered toward the aim of eradicating malaria, the inevitable emergence of drug resistance remains a significant obstacle to complete and lasting malaria control. Not only has the diminished efficacy of available therapeutics necessitated the discovery and development of new candidate antiplasmodial compounds, but also it has underscored the need for a better understanding of the biological correlates of drug resistance in malaria, dubbed the “malaria resistome”. As key components of the malaria resistome, transport proteins are often involved in drug response phenotypes2−4 either as the targets of the drug themselves5,6 or by helping the parasite evade drug action.7 Of the ∼120 members of the P. falciparum transportome, many remain unexplored, pending experimental characterization of their specific function and subcellular localization.4,8 To address this, the forward chemical genetic approach of inducing drug resistance in vitro has revealed a plethora of novel phenotypes associated with drug resistance, which can in turn provide insight into the physiological role of these transport proteins.2,3,9 In this technique, drug resistance is first selected through prolonged exposure to sublethal concentrations of a compound. Next, the genomes of the resistant clones are compared to that of their isogenic parents to discover genetic changes that are likely to be responsible for modulating a drug response.
With the help of in vitro evolution, we were able to identify, characterize, and validate a novel putative transporter as a key mediator of resistance against two structurally similar naphthoquinone derivatives, MMV08520310 (alternative name: GNF-Pf-4450) and GNF-Pf-3600,11 that were identified from previous phenotypic screens (Figure 1a). Not only are both MMV085203 and GNF-Pf-3600 active against the asexual blood stage of the parasite life cycle,11,12 which is responsible for the clinical manifestation of malaria, but also they inhibit the growth of the mature sexual form of the parasite (MMV085203 IC50 = 2.9 μM, GNF-Pf-3600 IC50 = 0.1 μM), which is subsequently transmitted by the mosquito vector across human hosts.13,14 Through cross-resistance profiling of evolved drug-resistant parasites, we were also able to identify the localization for this transporter and elucidate its potential as a facilitator of multidrug resistance in P. falciparum.
Figure 1.

Mutations in pfmfr3 are associated with resistance to MMV085203 and GNF-Pf-3600. (a) Chemical structures of MMV085203 and GNF-Pf-3600, which were used for in vitro selection against Plasmodium falciparum. (b) Protein schematic of PfMFR3 together with the mutations identified by in vitro evolution and whole genome analysis. Predicted transmembrane domains are marked in yellow, and mutations are marked in red stars.
Results
In Vitro Evolution of P. falciparum Resistance to MMV085203 and GNF-Pf-3600
To investigate the mechanism of action and resistance to these two structurally related molecules, we generated resistant parasite lines using in vitro evolution. Parasites from a 3D7 and Dd2 background of P. falciparum were subjected to intermittent treatment with increasing concentrations of GNF-Pf-3600 over the course of four months, resulting in 6 clones each from 3D7 and Dd2 that are up to 5- to 10-fold and 2- to 3-fold less sensitive to GNF-Pf-3600, respectively (Table 2). In a similar fashion, 3D7 strain parasites were also exposed to a stepwise dose progression of MMV085203 from a starting dose of 1 × IC50 up to 6 × IC50 over a period of 6 months. Drug selection yielded five clonal lines that had 4- to 6-fold higher IC50 values against MMV085203 compared to the parent (Table 1).
Table 2. 72 h IC50 Values of GNF-Pf-3600-Resistant Clones Generated from in Vitro Evolution.
| IC50 (nM) | fold shift | |
|---|---|---|
| 3D7 parent | 20.0 ± 1. 5 | |
| 3D7-3B8 | 196.9 ± 7.0 | 10 |
| 3D7-3E3 | 105.5 ± 4.0 | 5 |
| 3D7-3H7 | 158.8 ± 10.4 | 8 |
| 3D7-4A5 | 147.0 ± 21.9 | 7 |
| 3D7-4A8 | 103.3 ± 3.0 | 5 |
| 3D7–4G6 | 166.4 ± 7.8 | 8 |
| Dd2 parent | 34.8 ± 2.2 | |
| Dd2-3D2 | 45.3 ± 5.1 | 1.3 |
| Dd2-4A8 | 79.7 ± 13.1 | 2 |
| Dd2-4B10 | 91.9 ± 8.8 | 3 |
| Dd2-4F4 | 59.1 ± 3.7 | 2 |
| Dd2-3H5 | 74.0 ± 1.2 | 2 |
| Dd2-3C11 | 79.9 ± 2.7 | 2 |
Table 1. 72 h IC50 Values of MMV085203-Resistant Clones Generated from in Vitro Evolution.
| MMV085203 | IC50 (nM) | fold shift |
|---|---|---|
| 3D7 parent | 54.9 ± 27 | |
| 3D7-1F2 | 252.0 ± 114.3 | 5 |
| 3D7-1G5 | 311.6 ± 108.3 | 6 |
| 3D7-2B3 | 289.6 ± 86.1 | 5 |
| 3D7-3B3 | 213.9 ± 85.4 | 4 |
| 3D7-3F3 | 241.8 ± 78.1 | 4 |
To identify possible targets or markers of resistance for the compounds, all 17 resistant clones were subjected to whole genome sequencing to 40–100× coverage. After applying stringent filtering (see the Methods), we detected 13 indels and 21 single nucleotide variants of which 30 were nonsynonymous mutations. Looking only at intragenic mutations that were detected after stringent filtering and not found in the nonselected parental lines, we found 12 variants in 9 genes among the MMV085203-resistant clones. The 12 independent GNF-Pf-3600-resistant clones derived from 3D7 and Dd2 bore nonsynonymous mutations in 15 and 5 genes, respectively. Strikingly, all clonal lines that acquired resistance to either compound contained nonsynonymous mutations in PF3D7_0312500 (pfmfr3), a gene predicted to encode an uncharacterized, putative transporter MFR3 (Tables 3 and 4). Mutations in pfmfr3 were found among all clones that acquired resistance to MMV085203 and GNF-Pf-3600, which demonstrates a significant enrichment of genetic changes in this gene compared to other genes in our data set (p < 2 × 10–16). Additionally, a single nonsynonymous alteration in this gene was sufficient to confer resistance to MMV085203 in clone 3D7-2B3 (Q487E) and GNF-Pf-3600 in clone Dd2-4B10 (D150V). It is noteworthy that mutations in pfmfr3 have not been identified with prior selections using other compounds.3,9,15−19 We also searched for copy number variations (CNVs) by normalizing the mean coverage of coding regions for each clone against their corresponding parental background (3D7 or Dd2) and identifying sections of the genome having two or more contiguous genes that had at least a 2-fold difference in read depth against the parent. On the basis of these parameters, however, we did not detect CNVs in this particular data set.
Table 3. SNVs Obtained from the in Vitro Evolution of a 3D7 Strain of P. falciparum against MMV085203.
| gene name | description | effect | 3D7-1F2 | 3D7-1G5 | 3D7-2B3 | 3D7-3B3 | 3D7-3F3 |
|---|---|---|---|---|---|---|---|
| PF3D7_0221700 | Plasmodium exported protein, unknown function | D63_G64insPKPSTLNP | x | x | |||
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | C401Y | x | x | |||
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | Q487E | x | ||||
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | S519stop | x | ||||
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | N279frameshift | x | ||||
| PF3D7_0718000 | dynein heavy chain, putative | intronic indel | x | ||||
| PF3D7_0812100 | conserved protein, unknown function | T1326_T1335del | x | x | |||
| PF3D7_0823000 | serine/threonine protein kinase VPS15, putative | N830K | x | x | |||
| PF3D7_0918800 | dihydrouridine synthase, putative | N526D | x | x | |||
| PF3D7_1227200 | potassium channel | D1330Y | x | ||||
| PF3D7_1233600 | asparagine- and aspartate-rich protein 1 | D3256_N3262del | x | ||||
| PF3D7_1372200 | histidine-rich protein III | H119_H124del | x | x |
Table 4. SNVs Obtained from the in Vitro Evolution of 3D7 and Dd2 Strains of P. falciparum against GNF-Pf-3600.
| gene name | description | effect | 3D7-3B8 | 3D7-3E3 | 3D7-3H7 | 3D7-4A5 | 3D7-4A8 | 3D7-4G6 |
|---|---|---|---|---|---|---|---|---|
| PF3D7_0305500 | conserved Plasmodium protein, unknown function | D1228_D1229del | x | x | x | x | x | |
| PF3D7_0307900 | conserved Plasmodium protein, unknown function | L2968F | x | x | x | x | x | x |
| PF3D7_0307900 | conserved Plasmodium protein, unknown function | D1648_Q1650dup | x | x | x | x | x | x |
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | G146R | x | x | x | x | x | x |
| PF3D7_0501400 | interspersed repeat antigen | Q127Q | x | |||||
| PF3D7_0614300 | organic anion transporter | intron variant | x | x | x | x | ||
| PF3D7_0929000 | conserved Plasmodium protein, unknown function | intron variant | x | |||||
| PF3D7_1106600 | DEAD/DEAH box helicase, putative | N90_N91dup | x | |||||
| PF3D7_1132400 | conserved Plasmodium membrane protein, unknown function | D1030_N1031del | x | x | x | x | x | x |
| PF3D7_1118500 | box C/D snoRNP rRNA 2′-O-methylation factor, putative | H545Y | x | x | ||||
| PF3D7_1222600 | transcription factor with AP2 domain(s) | S2162R | x | |||||
| PF3D7_1222800 | conserved Plasmodium protein, unknown function | intron variant | x | x | x | x | x | x |
| PF3D7_1314300 | conserved Plasmodium protein, unknown function | S197S | x | x | x | x | x | x |
| PF3D7_1363400 | polyubiquitin binding protein, putative | N220_N221dup | x | x | ||||
| PF3D7_1408200 | transcription factor with AP2 domain(s) | N900_N901del | x | x | x | x | x | |
| PF3D7_1470100 | conserved Plasmodium protein, unknown function | L2246S | x | x |
| gene name | description | effect | Dd2-3C11 | Dd2-3D2 | Dd2-3H5 | Dd2-4A8 | Dd2-4B10 | Dd2-4F4 |
|---|---|---|---|---|---|---|---|---|
| PF3D7_0310200 | phd finger protein, putative | V2606frameshift | x | x | x | x | x | |
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | S16R | x | x | x | |||
| PF3D7_0312500 | major facilitator superfamily related transporter, putative | D150V | x | x | x | |||
| PF3D7_0402300 | reticulocyte binding protein homologue 1 | K1244N | x | |||||
| PF3D7_0515400 | conserved Plasmodium protein, unknown function | C174S | x | x | x | |||
| PF3D7_1254500 | rifin|PIR protein | Q4H | x | x |
An orphan, previously uncharacterized transporter, MFR3, bears very little overall sequence similarity to any other known protein in current databases. On the basis of its general topology, this 579-amino acid protein is classified under the major facilitator superfamily (MFS) of transporters20 due to the presence of an MFS-like motif, which is characterized by 12 transmembrane helices divided into 2 distinct domains on the N and C terminal ends of the protein, with each domain consisting of 6 consecutive transmembrane segments20−22 (Figure 1b). Six out of the 7 nonsynonymous variants identified among the clones are located in the predicted transmembrane regions of the protein (S16R, G146R, D150V, C401Y, Q487E, and S519stop), while one frameshift mutation (N279 fs) is found right after the sixth transmembrane segment. All of these findings indicate that this novel parasite MFS transporter is crucial to regulating parasite sensitivity against MMV085203 and GNF-Pf-3600.
pfmfr3 Mediates Resistance to MMV085203, GNF-Pf-3600, and Atovaquone
To further validate the link between pfmfr3 and the resistance phenotype we observed in the in vitro-evolved parasite lines, we introduced the N279 frameshift mutation into a wild-type Dd2 background using CRISPR-Cas9 (Figure 2a). Originally identified in a clone (3D7-3B3) that was 4-fold more resistant to MMV085203, this frameshift event results in a N279I change as well as a premature stop codon at position 281, effectively truncating the gene product from 579 to 280 amino acids, resulting in the loss of the last six transmembrane domains and likely disrupting its function. Although we were able to confirm editing in all clones transfected with both the N279 fs plasmid and the silent control plasmid, all attempts at PCR amplification of the entire gene were not successful. Alternatively, PCR genotyping was performed in order to detect the possible recombination of the entire plasmid into the parasite genome, resulting from CRISPR-induced double-strand break and homologous-directed repair. We found that all transgenic clones underwent disruption of endogenous pfmfr3 by plasmid integration into the genome, which is detected by p1277+p282 and p283+p1281 primer pairs (Figure 2a). Nevertheless, Sanger sequencing of the targeted segment of pfmfr3 revealed that the intact donor sequence was integrated into the gene as expected, thereby still resulting in a premature stop codon at position 281 effectively shortening the gene product and presumably leading to the loss of protein function.
Figure 2.

Disruption of pfmfr3 confers resistance to MMV085203 and GNF-Pf-3600. (a) Map of plasmid used for CRISPR-Cas9 editing of endogenous pfmfr3 accompanied by the evidence of complete donor plasmid recombination into the genomic pfmfr3 locus. Integration on the 5′ and 3′ ends of the gene is demonstrated by the p1277+p282 and p1281+p283 amplicons, respectively. Dd2/N279 fs represents clones transfected with the donor containing the mutation, while Dd2/silent represents clones transfected with a silent control donor plasmid. (b) The sensitivity of the CRISPR-edited pfmfr3 mutant expressing the truncated form of the protein (MFR3-KO) was evaluated against MMV085203, GNF-Pf-3600, and other antimalarial compounds with known mechanisms of action with wild-type Dd2 (Dd2) as a control; additional data can be found in Table 5. The sensitivity of the evolved (c) MMV085203-resistant mutant (pfmfr3 Q487E) and the (d) GNF-Pf-3600-resistant mutant (pfmfr3 D150V) was also evaluated against the same set of compounds as MFR3-KO, in comparison to their respective wild-type 3D7 and Dd2 parent lines; additional data can be found in Table 6. Bars represent the mean ± SD IC50 values from at least three independent biological replicates. Pairwise comparisons between parasite lines were performed using the Student’s t test.
Comparing the drug sensitivity of the edited pfmfr3 mutant line against wild-type Dd2, we observed that the clone expressing the truncated protein was 2.5- and 4-fold less sensitive to both MMV085203 (p = 0.002) and GNF-Pf-3600 (p = 0.003), respectively (Figure 2b, Table 5), demonstrating the role of this putative transporter as an important modulator of drug response to both compounds. Additionally, we found clone 3D7-2B3, which was specifically evolved against MMV085203 and bore a single Q487E mutation in pfmfr3, to also be cross-resistant against GNF-Pf-3600 (Figure 2c, Table 6). Likewise, resistant clone Dd2-4B10, which was specifically exposed to GNF-Pf-3600 and contained a single D150V mutation in pfmfr3, was also significantly less sensitive to MMV085203 (Figure 2d, Table 6).
Table 5. 72 h IC50 Values of Dd2 vs MFR3-KO Strains against Antimalarial Compounds.
| IC50 | Dd2 | MFR3-KO |
|---|---|---|
| MMV085203 (nM) | 90.0 ± 39.7 | 213.9 ± 68.6 |
| GNF-Pf-3600 (nM) | 148.5 ± 32.5 | 563.5 ± 58.6 |
| atovaquone (nM) | 0.550 ± 0.4 | 1.31 ± 1.10 |
| artemisinin (nM) | 15.9 ± 1.2 | 16.2 ± 0.5 |
| chloroquine (nM) | 59.4 ± 32.5 | 47.7 ± 41.6 |
| epoxomicin (nM) | 7.19 ± 1.00 | 6.55 ± 0.8 |
| brefeldin A (μM) | 1.48 ± 0.00 | 1.66 ± 0.2 |
| KDU691 (nM) | 43.2 ± 3.80 | 54.4 ± 5.4 |
| KAF156 (nM) | 15.7 ± 2.30 | 13.3 ± 2.8 |
| cycloheximide (nM) | 143.9 ± 24.0 | 140.2 ± 32.7 |
| actinomycin D (nM) | 0.310 ± 0.10 | 0.340 ± 0.00 |
Table 6. 72 h IC550 Values of 3D7 vs MFR3-Q487E (3D7 Background) and Dd2 vs MFR3-D150V (Dd2 Background) against Antimalarial Compounds.
| IC50 | 3D7 | MFR3-Q487E | Dd2 | MFR3-D150V |
|---|---|---|---|---|
| MMV085203 (nM) | 152.7 ± 25.8 | 438.4 ± 40.8 | 47.6 ± 6.8 | 78.6 ± 21.9 |
| GNF-Pf-3600 (nM)a | 96.1 | 476.6 | 21.7 ± 8.6 | 60.8 ± 15.9 |
| atovaquone (nM) | 0.585 ± 0.11 | ±0.09 | 0.546 ± 0.11 | 0.497 ± 0.21 |
| artemisinin (nM) | 21.7 ± 4.3 | 19.5 ± 0.95 | 15.4 ± 2.8 | 12.9 ± 5.6 |
| chloroquine (nM) | 70.2 ± 51.2 | 71.4 ± 54.9 | 556.8 ± 197.6 | 556.1 ± 224.2 |
| epoxomicin (nM) | 10.1 ± 1.8 | 9.82 ± 1.8 | 10.1 ± 2.6 | 8.17 ± 0.15 |
| brefeldin A (μM) | 1.71 ± 0.09 | 1.63 ± 0.11 | 2.13 ± 0.25 | 1.94 ± 0.26 |
| KDU691 (nM) | 45.0 ± 5.74 | 44.4 ± 2.19 | 41.7 ± 3.0 | 42.8 ± 7.2 |
| KAF156 (nM) | 6.68 ± 0.38 | 7.60 ± 0.10 | 10.7 ± 0.51 | 9.6 ± 0.73 |
| cycloheximide (nM) | 202.9 ± 53.2 | 174.4 ± 34.3 | 152.2 ± 80.3 | 135.3 ± 47.5 |
| actinomycin D (nM) | 3.74 ± 0.67 | 3.58 ± 0.55 | 1.98 ± 0.2 | 1.73 ± 0.3 |
Only one biological replicate.
We also sought to gain some insight into the parasiticidal mechanism of these two structurally related compounds by assaying the sensitivity of pfmfr3-mutated clones against nine compounds that have antimalarial activity and exhibit different modes of drug action. In addition to the functional knockout line, which bears the nonsense mutation, we also tested the evolved clones 3D7-2B3 and Dd2-4B10 against the same panel of antimalarial drugs. The truncated-MFR3 clone showed differential decreased sensitivity to atovaquone (p = 0.04) compared to wild-type Dd2, while remaining similarly sensitive to artemisinin, chloroquine, epoxomicin, brefeldin A, KDU691, KAF156, cycloheximide, and actinomycin D (Figure 2a, Table 5). On the other hand, no other cross-resistance phenotypes were observed in the case of the Q487E and D150V mutants (Figure 2c,d, Table 6). The significant decrease in sensitivity to atovaquone, which also contains a 1,4-naphthoquinone scaffold like MMV085203 and GNF-Pf-3600, observed in the functional knockout line suggests a possible role for MFR3 in the transport of this frontline antimalarial drug. On the other hand, the lack of resistance observed in the Q487E and D150V point mutants against atovaquone suggests differences in which specific transmembrane domains are responsible for the binding and transport among these three compounds.
MMV085203 and GNF-Pf-3600 Do Not Inhibit Cytochrome bc1
On the basis of the shared structural features across atovaquone, MMV085203, and GNF-Pf-3600 as well as the alteration in sensitivity for all three compounds resulting from the disruption of pfmfr3, we explored the possibility of MMV085203 and GNF-Pf-3600 having the same mechanism of action as atovaquone, a clinically relevant antiparasitic drug that targets the cytochrome bc1 complex in Plasmodium parasites through competitive inhibition of ubiquinol.23,24 Not only does blockage of cytochrome bc1 by atovaquone disrupt the mitochondrial electron transport chain (mETC), but also it triggers the downstream inhibition of pyrimidine biosynthesis due to the loss in production of ubiquinone (CoQ), a molecule that is regenerated through the mETC and is in turn the substrate of Pf dihydroorotate dehydrogenase (DHODH), the enzyme responsible for the conversion of dihydroorotate to orotate, a pyrimidine precursor25 (Figure 3a). The parasiticidal action of atovaquone, therefore, is driven by the inhibition of these essential biological processes.
Figure 3.
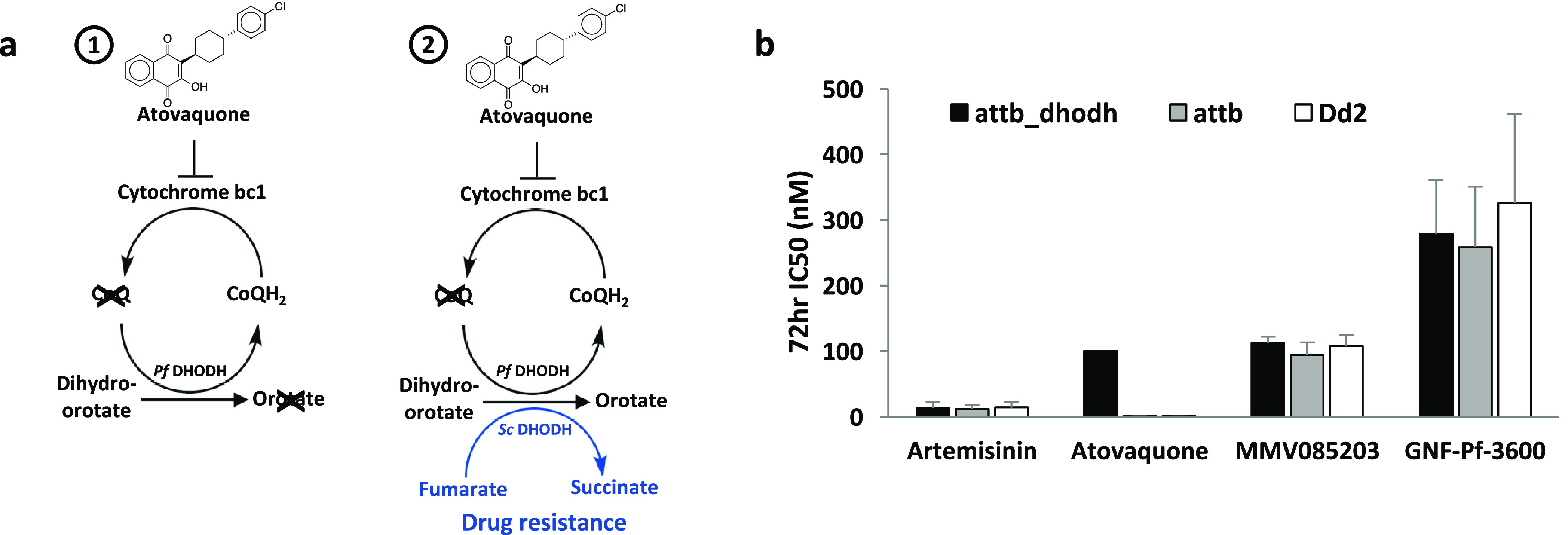
MMV085203 and GNF-Pf-3600 do not target the mitochondrial electron transport chain. (a) Simplified schematic of the mitochondrial electron transport chain (mETC) and pyrimidine synthesis in P. falciparum in the (1) absence and (2) presence of ScDHODH. The loss of ubiquinone (CoQ) due to the inhibition of cytochrome bc1 by atovaquone results in the downstream obstruction of the CoQ-mediated conversion of dihydroorotate to orotate by PfDHODH. However, genetic supplementation of CoQ-independent ScDHODH is able to bypass this blockage, rendering the parasite resistant against cytochrome bc1 inhibition. (b) The sensitivity against artemisinin (noncytochrome bc1 inhibitor), atovaquone (cytochrome bc1 inhibitor), MMV085203, and GNF-Pf-3600 was measured across three parasite lines: the transgenic P. falciparum line overexpressing yeast DHODH generated using a Dd2 line bearing an attb recombination site (attb_dhodh), P. falciparum bearing an attB recombination site on a Dd2 background (attb), and a wild-type Dd2 strain (Dd2). Additional data can be found in Table 7. Bars represent mean ± SD IC50 values from three independent biological replicates.
To investigate, we conducted a genetic supplementation assay that utilizes a transgenic parasite line that overexpresses the S. cerevisiae-derived DHODH enzyme (attb_dhodh).25 Unlike the parasite DHODH (PfDHODH), the yeast-derived DHODH (ScDHODH) is able to catalyze orotate production even in the absence of ubiquinone, effectively bypassing the mETC.25,26 Parasites that also express yeast DHODH, in addition to their own, are therefore refractory to compounds that act by disrupting the mETC through cytochrome bc1 inhibition, such as atovaquone (Figure 3a). Accordingly, we compared the 72 h IC50 for MMV085203 and GNF-Pf-3600 from a parasite line expressing ScDHODH (attb_dhodh) against the Dd2_attb parent (attb) from which it was derived, which only expresses the parasite version of the enzyme (PfDHODH). Both strains are derived from a wild-type Dd2 background and contain an attB site designed for chromosomal integration.27 We also included a wild-type Dd2 as a control to rule out any possible phenotypic interference that could be due to the presence of the attB sequence. As expected, genetic supplementation of ScDHODH in the attb_dhodh parasites rendered them over 100-fold resistant against atovaquone relative to the attb and wild-type Dd2 parasites. In contrast, IC50 values for MMV085203 and GNF-Pf-3600 were comparable between the three parasite lines. The same outcome was likewise observed in the case of artemisinin, a potent antimalarial that does not target cytochrome bc128−30 (Figure 3b, Table 7). This finding demonstrates that, despite sharing a 1,4-naphthoquinone scaffold with atovaquone, the mechanism of action of MMV085203 and GNF-Pf-3600 does not involve the specific inhibition of cytochrome bc1.
Table 7. 72 h IC50 Values of Dd2-attb_dhodh, Dd2-attb, and Dd2 Strains.
| IC50 (nM) | Dd2-attb_dhodh | Dd2-attb | Dd2 |
|---|---|---|---|
| artemisinin | 13.3 ± 8.7 | 12.1 ± 6.3 | 13.8 ± 8.7 |
| atovaquone | >100a | 0.27 ± 0.1 | 0.25 ± 0.1 |
| MMV085203 | 112.3 ± 9.7 | 94.6 ± 18.7 | 107.9 ± 16.1 |
| GNF-Pf-3600 | 278.8 ± 82.3 | 258.6 ± 92.3 | 326 ± 135.4 |
Incomplete curve fitting. 50% inhibition observed at >100 nM.
PfMFR3 Is a Putative Mitochondrial Transporter
Finally, we sought to determine the cellular localization of MFR3 through the episomal overexpression of a green fluorescent protein (GFP)-tagged species of MFR3. The full-length coding sequence of this gene was generated through gene-specific PCR amplification of pfmfr3 from total cDNA derived from a wild-type Dd2 clone and then inserted into a pDC2-cam-mrfp-2A–gfp31 vector backbone from which the mRFP-2A segment had been removed upstream of the GFP tag. Transfection of wild-type Dd2 with this construct leads to a parasite line that expresses MFR3 bearing a C-terminal GFP tag under the control of a constitutive calmodulin (CAM) promoter (Dd2_mfr3over) (Figure 4a). As a control, we also transfected a Dd2 parent with the empty vector, which was grown alongside the tagged MFR3 overexpression line (Dd2_empty). PCR genotyping of total DNA extracted from the resulting transfectants confirms the presence of the correct episome in both the empty vector and MFR3-GFP lines (Figure 4b), while quantitative PCR shows 3-fold overexpression of pfmfr3 in the parasites transfected with the MFR3-GFP plasmid compared to the control line bearing only the empty vector (Table 8). Crucially, we were also able to observe a slight but reproducible increase in susceptibility against MMV05203 (p = 0.02) and GNF-Pf-3600 (p = 0.04) accompanying overexpression of MFR3 (Figure 4c, Table 9).
Figure 4.

PfMF3 localizes to the parasite mitochondrion. (a) Map of plasmid used for episomal overexpression of GFP-tagged pfmfr3. An empty vector (no pfmfr3 insert) was used for transfection of a control (Dd2_empty) parasite line. (b) PCR amplification of the blasticidin-resistance (BSD Deaminase) marker in both parasite lines confirms the successful transfection of both control and overexpression/tagging parasite lines, while PCR amplification of the chimeric MFR3-GFP template confirms the presence of the MFR3-GFP episome in the overexpression line (Dd2_mfr3over) but not in the control (Dd2_empty). (c) The sensitivity of parasites overexpressing pfmfr3 (Dd2-MFR3_over) and the corresponding control line (Dd2_empty) was evaluated against MMV085203, GNF-Pf-3600, and other antimalarial compounds with known mechanisms of action. Bars represent mean ± SD IC50 values from at least three independent biological replicates. Pairwise comparisons between parasite lines were performed using the Student’s t test. Additional data can be found in Table 9. (d) Blood-stage parasites expressing a GFP-tagged version of PfMFR3 were costained with MitoTracker Red (200 nM) and DAPI and then imaged using confocal microscopy. The blue signal pertains to the DAPI-stained parasite nuclei; the red signal represents the parasite mitochondrion, and the green signal, GFP-tagged PfMFR3.
Table 8. Checking for pfmfr3 Overexpression Using Quantitative PCR.
| Dd2_MFR3over | Dd2_empty | |
|---|---|---|
| endogenous + episomal pfmfr3 (Ct) | 22 ± 0.14 | 24.9 ± 0.34 |
| arginyl tRNA synthetase (control) (Ct) | 18.4 ± 0.02 | 19.8 ± 0.02 |
| fold change pfmfr3 | 2.96 ± 0.89 |
Table 9. 72 h IC50 Values of Dd2 vs MFR3_over.
| IC50 | Dd2_empty | MFR3_over |
|---|---|---|
| MMV085203 (nM) | 163.5 ± 21.9 | 118.2 ± 15.9 |
| GNF-Pf-3600 (nM) | 181.8 ± 24.9 | 166.2 ± 29.9 |
| atovaquone (nM) | 0.468 ± 0.072 | 0.200 ± 0.016 |
| artemisinin (nM) | 19.8 ± 1.91 | 18.1 ± 2.58 |
| chloroquine (nM) | 377.8 ± 40.9 | 341.7 ± 29.1 |
| epoxomicin (nM) | 9.26 ± 2.04 | 8.52 ± 1.95 |
| brefeldin A (μM) | 1.33 ± 0.02 | 1.02 ± 0.61 |
| KDU691 (nM) | 34.5 ± 7.87 | 37.5 ± 3.64 |
| KAF156 (nM) | 11.5 ± 0.91 | 12.8 ± 3.23 |
| cycloheximide (nM) | 108.5 ± 9.40 | 96.5 ± 6.67 |
| actinomycin D (nM) | 1.29 ± 0.39 | 1.12 ± 0.41 |
As with the functional knockout (MFR3-KO) line, we also evaluated the sensitivity of the MFR3-overexpression line against other drugs with established antimalarial activities and varying mechanisms of action. We found that overexpressing PfMFR3 rendered the parasite significantly more sensitive to atovaquone (p = 0.0003), demonstrating the opposite phenotype as the line expressing the truncated form of the protein. On the other hand, the control parasite line that had been transfected with the empty vector was similarly susceptible as the MFR3-overexpression line against the other antimalarials that do not target the mitochondrion.
Using confocal microscopy, we then visualized unfixed, thin smears of different stages of intraerythrocytic Dd2_mfr3over parasites to track the location of the GFP-tagged species of MFR3; smears were also stained with DAPI and MitoTracker Red to visualize the parasite nuclei and mitochondria, respectively. Microscopy-based imaging revealed perinuclear distribution of the GFP signal corresponding to the tagged MFR3 transporter. Strikingly, this GFP signal also appeared to colocalize with the MitoTracker Red signal across different stages of parasite blood-stage development, indicating that this orphan transporter localizes to the parasite mitochondrion (Figure 4d). Interestingly, matching the predicted peptide sequence of MFR3 using position-specific iterated alignment (PSI-BLAST), which allows for the identification of more distantly related proteins, we found that this parasite protein is related to the yeast protein FMP42, an uncharacterized integral membrane protein that localizes to the mitochondrion and vacuole. Our observation that MFR3 is located in the parasite mitochondrion corroborates the link between MFR3 and atovaquone sensitivity given that its drug target is cytochrome bc1. Additionally, it also brings to light the possibility of this subcellular organelle being a site of action of both MMV085203 and GNF-Pf-3600 in P. falciparum.
PfMFR3 Modulates Sensitivity to Compounds that Target the Mitochondrion
Given its localization, we also investigated whether the loss of function of this transporter would alter the parasite’s sensitivity against other compounds that, like atovaquone, target the mitochondrion, even without the 1,4-naphthoquinone scaffold. To test this, we chose six compounds (MMV1271410, MMV1042937, MMV1425891, MMV1451822, MMV1432711, and MMV1427995) with antimalarial activity that also inhibit cytochrome bc1 while being structurally dissimilar to atovaquone, MMV085203, and GNF-Pf-3600 (Figure 5a). A comparison of the metabolomic profiles of trophozoite-stage parasites exposed to the six compounds with those of other clinically relevant antimalarial drugs32,33 revealed that they all cocluster with atovaquone and demonstrate an increase in levels of N-carbamoyl-l-aspartate and dihydroorotate, a metabolic signature that is specifically attributed to inhibitors of cytochrome bc132 (Figure 5b, Supplementary Data 1). Furthermore, we found that all six demonstrated a profound (20- to 3000-fold) rightward shift in IC50 in the transgenic P. falciparum lines expressing yeast DHODH relative to the attb parent line (Figure 5b), indicating a mechanism of action that involves inhibition of the mitochondrial electron transport chain. Unsurprisingly, the metabolomic profile of MMV085203 did not cocluster with atovaquone or any of the other clinically relevant antimalarials evaluated and shows a completely different pattern of dysregulated metabolites compared to the six other MMV compounds (Figure 5b). This outcome is to be expected given that the DHODH rescue assay has ruled it out as a cytochrome bc1 inhibitor. Interestingly, the metabolic signature for MMV085203 involves a distinct upregulation (>2-fold) of aconitate and fumarate (Supplementary Data 1), both intermediates of the tricarboxylic acid (TCA) cycle, which is a process that takes place in the mitochondrion, further reinforcing the likelihood of MMV085203 engaging a cellular target that is located in this specific organelle.
Figure 5.
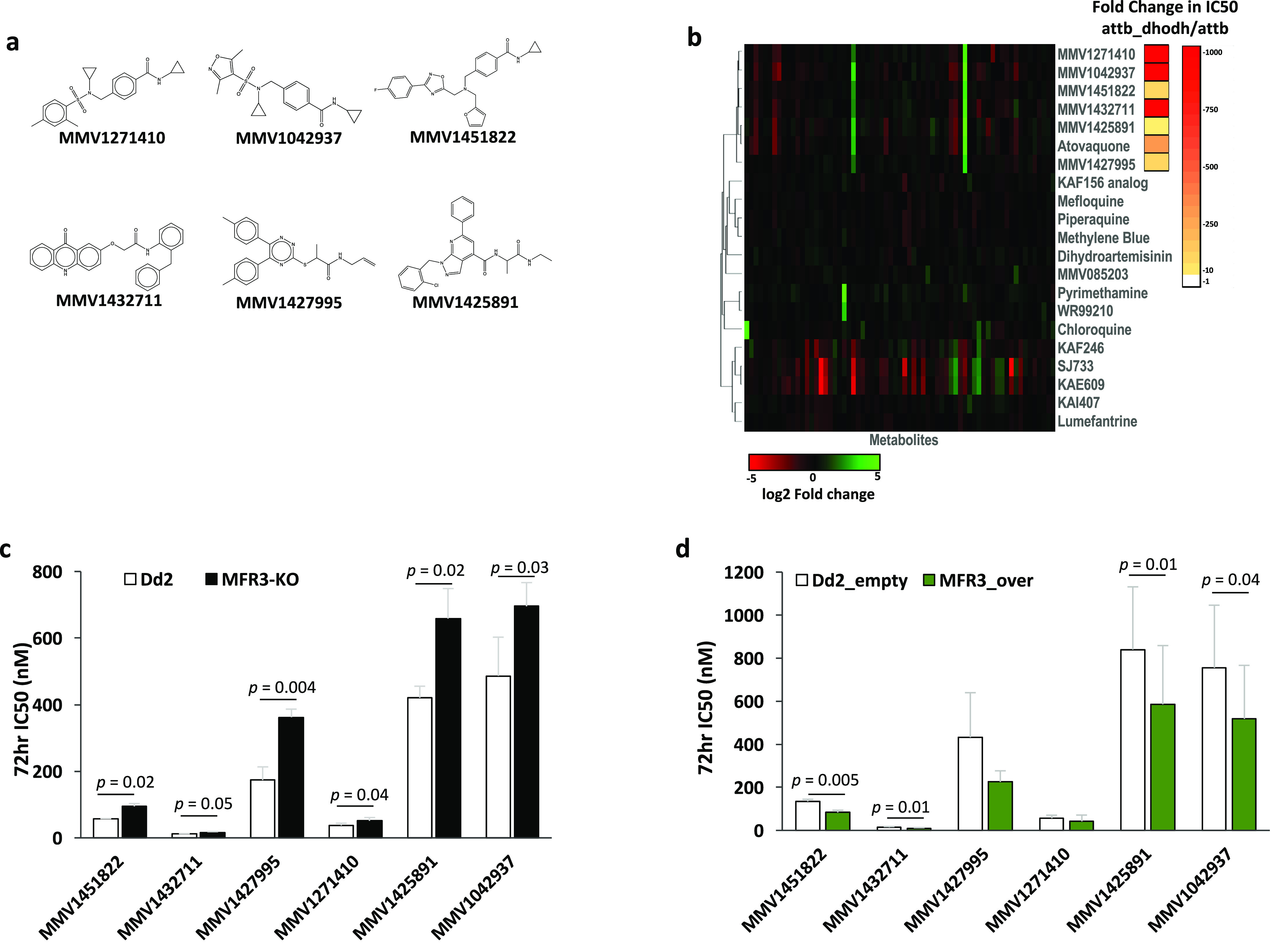
Disruption and overexpression of pfmfr3 modulates sensitivity against inhibitors of cytochrome bc1. (a) Chemical structures of six structurally diverse compounds (MMV1271410, MMV1042937, MMV1425891, MMV1451822, MMV1432711, and MMV1427995) that target cytochrome bc1 as predicted by metabolomic profiling and the ScDHODH rescue assay. (b) Metabolomic profiles of parasites exposed to the six MMV compounds were coclustered with other clinically relevant antimalarials, including a known inhibitor of cytochrome bc1, atovaquone. Fold-differences in each metabolite can be found in Supplementary Data 1. Each compound was also evaluated for resistance conferred by ScDHODH supplementation, indicating cytochrome bc1 inhibition. Sensitivity of the (c) CRISPR-edited pfmfr3 mutant expressing the truncated form of the protein (MFR3-KO) as well as the (d) parasite line overexpressing MFR3 was measured against the above-mentioned cyctochrome bc1 inhibitors. Additional data can be found in Table 10. Bars represent the mean ± SD IC50 values from at least three independent biological replicates. Pairwise comparisons between parasite lines were performed using the Student’s t test.
Evaluating our functional MFR3 knockout line against MMV1271410, MMV1042937, MMV1425891, MMV1451822, MMV1432711, and MMV1427995, we found that disruption of this putative mitochondrial transporter rendered the parasite significantly less sensitive to all six inhibitors of cytochrome bc1 (Figure 5c, Table 10), just as we previously observed with atovaquone. Inversely, overexpression of MFR3 led to diminished 72 h IC50 values for all six compounds tested with the increase in sensitivity coming up to significance for MMV1451822, MMV1432711, MMV1425891, and MMV1042937 (Figure 5d, Table 10), and although we have yet to identify the specific function of this novel putative transporter within the parasite, the fact that the disruption and overexpression of this protein leads to a significant alteration in drug sensitivity across a set of structurally diverse molecules that target the mitochondrion reveals the potential of MFR3 to be a relevant multidrug-resistance factor in malaria.
Table 10. 72 h IC50 Values of Dd2 vs MFR3-KO and Dd2_empty vs MFR_over Strains against Mitochondrial Inhibitors.
| IC50(nM) | Dd2 | MFR3-KO | Dd2_empty | MFR3_over |
|---|---|---|---|---|
| MMV1451822 | 56.9 ± 0.9 | 94.57 ± 8.5 | 134.43 ± 9.9 | 83.10 ± 10.6 |
| MMV1432711 | 11.35 ± 1.3 | 16.50 ± 2.2 | 13.78 ± 3.9 | 9.40 ± 3.3 |
| MMV1427995 | 173.63 ± 39.5 | 362.17 ± 24.4 | 432.53 ± 207.2 | 225.33 ± 51.4 |
| MMV1271410 | 37.00 ± 7.0 | 51.10 ± 9.9 | 56.22 ± 14.4 | 41.80 ± 29.5 |
| MMV1425891 | 420.60 ± 34.9 | 658.30 ± 90.4 | 840.25 ± 291.2 | 585.58 ± 272.9 |
| MMV1042937 | 486.40 ± 116.3 | 695.53 ± 71.1 | 755.02 ± 291.1 | 518.03 ± 248.5 |
Discussion
Directed evolution of drug resistance has long been a staple technique for identifying drug targets and mechanisms of resistance in the human malaria parasite.3,9 Using this method, we were able to identify and validate a novel mitochondrial transporter in Plasmodium falciparum, pfmfr3 (PF3D7_0312500),20 as a mediator of resistance against a number of known mitochondrial inhibitors as well two structurally related compounds with unknown mechanisms of action, MMV085203 and GNF-Pf-3600. While pfmfr3 is expressed throughout the blood (sexual and asexual), liver, and mosquito stages of the life cycle of P. falciparum,34,35 genome-wide mutagenesis and knockout screens in Plasmodium falciparum(36) and Plasmodium berghei(37) demonstrate ready mutability of pfmfr3, indicating that it is not essential to the intraerythrocytic development of the parasite. Despite the observation that nonsynonymous mutations in this gene are overrepresented in MMV085203/GNF-Pf-3600-resistant parasite lines derived from two genetically distinct backgrounds, the nonessentiality of this particular gene product, coupled with its function as a transporter, suggests that it is not an actual drug target but rather a shared resistance mechanism.
The involvement of MFS transporters in mediating resistance against MMV085203 is also substantiated by directed evolution experiments in a yeast model38 where a genetically modified strain of S. cerevisiae was subjected to increasing concentrations of the antimalarial drug. Notably, three of the seven clones that demonstrated diminished susceptibility to MMV085203 bore mutations in ScARN1, which encodes an iron siderophore transporter, with one of the resistant lines having only a single polymorphism in this gene, while two resistant clones gained mutations in ScAFT1, a transcription factor that modulates expression of a cluster of genes, including ScARN1,39 that are involved in iron homeostasis.40 One of four homologous genes in the yeast genome (ARN1–4) that are regulated by AFT1, ScARN1, encodes a transport protein that, like PfMFR3, belongs to the MFS superfamily of transporters. Its predicted topology reveals the presence of 14 transmembrane domains with the segment spanning the first 12 transmembrane helices exhibiting homology to other MFS transporters found in bacteria41,42 (Figure 6). Depending on extracellular concentration of its siderophore iron substrate, ARN1 is trafficked between the plasma membrane and endosomal compartments in the cytoplasm.43,44 Five out of the seven MMV085203-resistant yeast lines acquired mutations that are likely to alter the functionality of this transporter, which points to the involvement of ARN1 in modulating sensitivity to this molecule. Furthermore, outcomes from both yeast and parasite resistance models reinforce the role of these MFS transporters as possibly driving cellular uptake or efflux of MMV085203 and other structurally related molecules.
Figure 6.
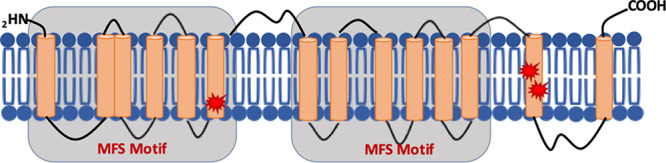
Predicted structure of S. cerevisiae ARN1. Protein schematic of yeast ARN1 together with the mutations identified by in vitro evolution and whole genome analysis.38 Predicted transmembrane domains are marked in orange, and mutations are marked in red stars.
Denoted as an “orphan” transporter, the mechanism of transport and substrate specificity of MFR3 in Plasmodium falciparum is still unknown. The major facilitator superfamily is one of the largest family of transporters and demonstrates immense sequence and functional diversity, and the fact that MFR3 does not strongly resemble any other prokaryotic or eukaryotic protein in the current databases makes it difficult to speculate on its biological function within the parasite. On the other hand, pbmfr3, the rodent ortholog of pfmfr3, has been shown to be important in sporozoite formation and male gamete exflagellation.45 Interestingly, RNA-seq of male and female gametocytes in P. berghei revealed that pbmfr3 is significantly overexpressed in male gametocytes versus female gametocytes,46 while in the case of P. falciparum, MFR3 is 2-fold more abundant among females versus males.47 This differential level of pfmfr3 expression might also play into the differential susceptibility of P. falciparum gametocytes to MMV085203. A prior dual gamete formation screen on the malaria box demonstrated that, while MMV085203 effectively inhibits stage V male gametocytes, it shows no activity against females.14 This sex-specific expression pattern could suggest a role for pfmfr3 in the development of transmission stages in P. falciparum. Given its possible function as a mitochondrial transporter, this is not surprising as the parasite mitochondrion is known to undergo significant expansion and activation during gametocytogenesis,48 and gametocytes display higher levels of glucose utilization and TCA function,49 reflecting increased energy demands for the subsequent stages of the life cycle.
We also determined that, despite the structural similarity across MMV085203, GNF-Pf-3600, and atovaquone, neither MMV085203 nor GNF-Pf-3600 targets the mETC. This result is also supported by the discrepancy in susceptibility of sexual stages of the parasite against these three drugs, wherein atovaquone has been shown to be inactive against late stage P. falciparum gametocytes,50,51 while both MMV085203 and GNF-Pf-3600 remain efficacious at inhibiting stage V gametocytes.13 Additionally, a previous malaria box screen for inhibitors of the parasite enzyme thioredoxin reductase (TrxR) identified MMV085203 as demonstrating the highest level of TrxR inhibition, in contrast with atovaquone, which proved inactive against the enzyme.52 An essential component of redox homeostasis maintenance, two isoforms of TrxR, is produced by the parasite from the same pftrxr locus with one isoform located in the cytosol and the other localizing to the parasite mitochondrion.53 Interestingly, metabolomic profiling of parasites treated with MMV085203 demonstrated a distinct upregulation of aconitate, which is an intermediate formed as citrate is being converted to isocitrate in the tricarboxylic acid cycle. This reaction is catalyzed by aconitate hydratase, or aconitase, an enzyme encoded by PF3D7_1342100 or pfirp that has also been found to be located in the mitochondrion.54 While not essential for asexual blood-stage growth, PfIRP is important to sexual-stage development, since knocking out this gene renders the parasites unable to form mature gametocytes.55 The gametocyte-specific activity of MMV085203, coupled with its effect on aconitate levels in treated parasites, suggests that PfIRP could be another attractive candidate for further investigation as a drug target for MMV085203. Taken together with our results showing localization of MFR3 to the mitochondrion, all of these findings further bolster the possibility of this organelle as one of the sites of action for MMV085203/GNF-Pf-3600.
For certain compounds containing a 1,4-naphthoquinone moiety, their cytotoxic effect is tied to their activity as “subversive substrates” of NADPH-dependent disulfide reductases (such as thioredoxin reductase and glutathione reductase), which leads to inhibition of the physiological reaction catalyzed by these enzymes while also resulting in the production of reactive oxygen species and subsequent disruption of hemoglobin digestion;56,57 it is possible that this is one of the mechanisms through which MMV085203 and GNF-Pf-3600 are acting against the parasite.
Our observation that the genetic changes in pfmfr3 alters the parasite’s drug response against multiple mitochondrial targeting compounds bearing dissimilar chemical scaffolds and likely involving different mechanisms of action demonstrates the potential of MFR3 as a significant mediator of antimalarial multidrug resistance. Essential biological pathways occurring in the mitochondrion such as the mitochondrial electron transport chain58,59 and the tricarboxylic acid cycle49,55 involve key players that are attractive druggable targets for developing clinical antimalarials. Cytochrome bc1, for example, has been shown to be inhibited by a wide variety of chemotypes60−63 and can be targeted for prophylactic, therapeutic, and transmission-blocking purposes.64 In addition, the shared resistance caused by disruption of MFR3 against three compounds bearing a 1,4-naphthoquinone scaffold could also speak to a general involvement of this protein in transporting compounds having a similar structure. In addition to MMV085203, GNF-Pf-3600, and atovaquone, the pfmfr3 Q487E single point mutant also showed 3-fold resistance against another compound with a naphthoquinone group, GNF-Pf-3703 (data not shown). If this is indeed the case, MFR3 activity could be a significant correlate of resistance against a multitude of candidate antimalarials given that naphthoquinone derivatives have immense potential as antiplasmodial leader molecules and have been widely used for the development of many compound series.56,65−71 Importantly, polymorphisms in pfmfr3 have been found to naturally exist in parasite populations in the field with over 50% occurring in transmembrane domains.72 Although none of the genetic alterations that were identified in this study have been documented in clinical isolates, one cannot rule out the possibility of these natural mutations leading to modifications in transporter activity and specificity and eventually contributing to clinical drug resistance.
While a number of plasmodium transport proteins have the potential to be attractive therapeutic targets, the fact that PfMFR3 is not essential for parasite growth and replication in the asexual blood stage precludes it from being an ideal antimalarial drug target. Furthermore, it is naturally polymorphic among field isolates, which suggests a higher likelihood of drug resistance emerging against inhibitors of this putative transporter. Overall, these two important considerations impart only a modest clinical impact to the therapeutic inhibition of this protein.
Nevertheless, its role as a transporter in an important and highly druggable organelle make it more interesting as a possible multidrug resistance factor. Further investigation, therefore, into the function of this as-yet-uncharacterized transporter could provide new insight into general parasite biology and lead to a better understanding of what drives resistance in malaria.
Methods
In Vitro Culture of P. falciparum
Two strains (Dd2 and 3D7) of P. falciparum were used for in vitro drug selection. Continuous cultivation was performed under standard conditions as previously described.73 Parasites were grown in human O-positive (O+) whole blood obtained from the Blood Bank of The Scripps Research Institute (TSRI) (La Jolla, CA). Leukocyte-free erythrocytes are washed and then stored at 50% hematocrit in RPMI 1640. The evaluation of parasitemia and parasite morphology was performed using a microscopic evaluation of thin blood smears that were first fixed with methanol (Merck) and then stained with Giemsa (Sigma).
In Vitro Selection of Drug-Resistant P. falciparum
In vitro selection for MMV085203-resistant parasites was performed on a 3D7 background of P. falciparum. Parasites were exposed to a progressively increasing drug concentration of MMV085203 starting at 20 nM and eventually culminating at a final exposure concentration of 6× the starting dose (120 nM) after 6 months of selection. In the case of GNF-Pf-3600, in vitro selection for resistance was performed on two parental strains, 3D7 and Dd2. Parasites were exposed to stepwise-increasing concentrations of GNF-Pf-3600 starting from 20 and 35 nM for 3D7 and Dd2, respectively. Over the course of 4 months, the drug concentration used for continuous exposure was increased up to 6× the starting dose (125 nM) for 3D7 and 4× the starting dose for Dd2 (150 nM). Once a detectable rightward shift in IC50 was detected using the 72 h drug sensitivity assay, MMV085203- and GNF-Pf-3600-resistant clones were obtained through limiting dilution. Clones were then cultivated and phenotyped to confirm resistance, and their genomic DNA was subsequently sent out for whole genome sequencing. Nontreated control parasite lines for 3D7 and Dd2 were maintained in parallel throughout the course of in vitro selection.
72 h Drug Sensitivity Assay Using SYBR Green I
Drug sensitivities of Plasmodium falciparum were evaluated using a SYBR green I-based fluorescence assay.74 Briefly, ring-stage parasites growing in a synchronous culture were synchronized by treatment with 5% (w/v) sorbitol. They were then incubated for 72 h in 96-well plates in a 12-dose titration of each drug at a final parasitemia of 0.6% and 2% hematocrit. After 72 h, a 1:1000 mixture of SYBR Green I (Invitrogen) in lysis buffer (0.16% Saponin, 1.6% Triton X-100, 5 mM EDTA, and 20 mM Tris-HCl) was added to each well, and plates were incubated in the dark overnight. Parasite viability was quantified on the basis of a fluorescence readout using a Synergy HTX Multi-Mode Microplate Reader (BioTek). Assays were performed with at least three independent biological replicates with each replicate consisting of technical duplicates. IC50’s were then calculated using the drc package in R.
Whole Genome Sequencing and Variant Calling
To obtain genomic DNA (gDNA) from clonal parasite samples, infected RBCs were washed with 0.05% saponin and gDNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen) according to the standard protocols. Sequencing libraries were prepared with the Nextera XT kit (Cat. No. FC-131-1024, Illumina) via the standard dual index protocol and sequenced on the Illumina HiSeq 2500 in RapidRun mode to generate paired-end reads 100 bp in length. Reads were aligned to the P. falciparum 3D7 reference genome (PlasmoDB v13.0) using the previously described Platypus pipeline.75 A total of 20 clones were sequenced to an average coverage of 83× with an average of 98.5% of reads mapping to the reference genome. Following alignment, SNVs and INDELs were called using GATK HaplotypeCaller and filtered according to GATK’s best practice recommendations.76 Variants were annotated using SnpEff77 and further filtered by comparing those from resistant clones to the parent clone, such that only a mutation present in the resistant clone but not the sensitive parent clone would be retained. Since all parasite lines were cloned before sequencing, only variant calls with >90% reads mapped to the alternate allele were considered for resistance conferral. No CNVs were detected in any of the samples following a previously described analysis protocol.58
Disruption of pfmfr3 Using CRISPR/Cas9
The CRISPR-Cas9 plasmid pDC2-coCas9-U6.2-hdhfr78 was used to introduce the pfmfr3 N279 fs mutation (originally detected the MMV085203-resistant clone 3D7-3B3) into P. falciparum Dd2 (Figure 2a). The codon for asparagine at residue 279 has a single adenosine (A) deletion resulting in a frameshift and premature termination (N279 fs). The protein size is thus shortened from 579 to 280 amino acid residues. A donor fragment of 577 bp, centered on the frameshift mutation, was synthesized along with silent “shield” mutations at the binding site for the gRNA (TGATAATCAGCTTGTATCAG) located 29 bp upstream of the desired mutation. PCR genotyping of the target locus in cloned transfectants revealed that the entire plasmid recombined into the genome possibly as a result of the double-strand break and homologous-directed repair events by the CRISPR-Cas9 system. However, sequencing of this genomic region confirmed that this recombination event still resulted in a truncated, nonfunctional version of pfmfr3. The resulting transgenic parasites were subjected to a 72 h drug sensitivity assay against MMV085203 to confirm the drug resistance phenotype. All primers used in this study are listed in Table 11.
Table 11. Primers and Oligonucleotides Used in This Study.
| name | sequence | description |
|---|---|---|
| p282 | AACATATGTTAAATATTTATTTCTC | for genotyping of recombinant pfmfr3 locus |
| p283 | AGGGTTATTGTCTCATGAGCGG | for genotyping of recombinant pfmfr3 locus |
| p1277 | TGACAGATATCCTGTGGAAGATATCG | for genotyping of recombinant pfmfr3 locus |
| p1281 | GTGAGGCAAATGTATTTATTATACC | for genotyping of recombinant pfmfr3 locus |
| F1_mfr3_avrII | ATCGCCTAGGATGAAAAAAGTAAAGG | for amplification of full length pfmfr3 cDNA |
| R1_mfr3_mfeI | ATCGCAATTGTTACATTTGCTGTAG | for amplification of full length pfmfr3 cDNA |
| F2_mfr3_mfeI | ATCGCAATTGGATATATATCTTTAGTG | for amplification of full length pfmfr3 cDNA |
| R2_mfr3_nheI | ATCGGCTAGCCTTTGAAGAAGGAAGGG | for amplification of full length pfmfr3 cDNA |
| BSD_F | ATCAACAGCATCCCCATCTC | for amplification of the BSD resistance cassette |
| BSD_R | ATGCAGATCGAGAAGCACCT | for amplification of the BSD resistance cassette |
| mfr3_GFP_F | TCACCTTCACCCTCTCCACT | for amplification of the pfmfr3-GFP junction on the overexpression/tagging episome |
| mfr3_GFP_R | CCAAAGGCAATAGCTCAAGG | for amplification of the pfmfr3-GFP junction on the overexpression/tagging episome |
| rrs_qpcr_F | GAGTACCCCAATCACCTACA | for qPCR (ΔΔCt), reference |
| rrs_qpcr_R | AAGAGATGCATGTTGGTCATTT | for qPCR (ΔΔCt), reference |
| mfr3_qpcr_F | CCAAAGGCAATAGCTCAAGG | for qPCR (ΔΔCt), target |
| mfr3_qpcr_R | TTGAAGAAGGAAGGGAAATCA | for qPCR (ΔΔCt), target |
Generation of pfmfr3-GFP Overexpressing Lines
A Dd2 strain was made to episomally overexpress a GFP-tagged version of PfMFR3 using the pDC2-cam-mrfp-2A–gfp plasmid31 (Figure 4a). The RFP-2A segment was excised, and the full-length coding sequence of pfmfr3, generated from gene-specific PCR amplification of total cDNA extracted from wild-type Dd2, was inserted into the vector upstream of the GFP tag. A parasite line transfected with the empty plasmid was also generated and grown in 2.5 μg/mL blasticidin (BSD) alongside the overexpression lines to serve as a vector control. PCR genotyping confirmed the presence of the empty vector and MFR3-GFP-containing episome in each respective parasite line (Figure 4b).
To confirm overexpression, real-time quantitative PCR (relative quantification) was also performed using gene-specific primers that are able to interrogate both the endogenous and episomal pfmfr3 transcripts and using PF3D7_1218600 (Arginyl-tRNA synthetase, pfrrs) as the reference gene. The parasite line transfected with the empty plasmid was used as the control for ΔΔCt.
Total RNA was extracted from synchronized trophozoite-stage parasite cultures using the TRIZol-Chloroform method.79 First strand cDNA synthesis was first carried out on 50 ng of each transfectant line using oligo-dT-primed reverse transcription using SuperScript II Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. The resulting first strand product was then used as template for real-time qPCR using Power SYBR Green Master Mix (Applied Biosystems) and using pfmr3 (target gene) and pfrrs (reference gene primers). The ΔΔCt method was used to analyze the relative changes in the expression level of pfmfr3, where ΔΔCt = [(Ct of sample pfmfr3 – Ct of sample pfrrs) – (Ct of control pfmfr3 – Ct of control pfrrs)] and 2–ΔΔCt is the fold-difference in gene expression. All primers used in this study are listed in Table 11.
Imaging of pfmfr3-GFP Overexpressing Parasites
Asynchronous blood-stage parasites episomally expressing MFR3-GFP were incubated with 200 nM MitoTracker Red (Invitrogen) for 30 min and subsequently washed three times with warm (37 °C), 1× PBS. Thin blood smears using the blood (2–4 μL) were then generated and mounted with Vectashield with DAPI (Vector Laboratories) and then sealed with glass coverslips. Images were acquired using a Zeiss LSM880 with Airyscan confocal microscope (63× oil immersion lens); diode laser power was set to 2% for 405, 488, and 561 nm. The images were captured and processed using the confocal ZEN software (Black edition, Zeiss).
Metabolomic Profiling of P. falciparum Blood-Stage Parasites
The metabolite profiling of drug-treated, trophozoite-stage parasites was performed as previously described.32,33 Briefly, highly synchronized, MACS-purified parasites aged 24–36 h post-invasion were treated with compound at a dose of 10× IC50 for 2.5 h alongside an untreated control. Parasitized red blood cells were extracted with 90% methanol containing 0.5 mM 13C15N-labeled aspartate as an internal standard and stored at −80 °C prior to downstream processing. Samples were subsequently resuspended in HPLC-grade water mixed with 1 mM chlorpropamide as an additional internal standard and analyzed by ultrahigh-performance liquid chromatography mass spectrometry (UHPLC-MS). Hierarchical clustering of metabolite profiles was performed using Cluster 3.0 and visualized using Treeview 3.0.
Acknowledgments
This work was supported by the Bill and Melinda Gates Foundation (OPP1054480 - Target Discovery for Antimalarials). M.R.L. and K.C. are supported in part by a Ruth L. Kirschstein Institutional National Research Award from the National Institute for General Medical Sciences (T32 GM008666).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00676.
Table of parasite metabolites profiled after drug treatment (XLSX)
Author Contributions
Drug selections using MMMV085203 were performed by P.G. with help from E.S., while GNF-Pf-3600 selections were done by E.S.I. The MFR3-KO line was generated by M.C.S.L., E.F.C., and K.K., while the MFR3 tagged/overexpression line was generated by F.R. with help from K.C. Parasite drug assays on the MFR3-KO and MFR3-tagged/overexpression lines were conceptualized by F.R., E.A.W., S.O., M.C.S.L., and D.E.G. and were performed by F.R. Assays involving ScDHODH supplementation were performed by F.R. with help from N.M. Metabolomics experiments were designed by M.L. and performed by E.O. Microscopy was carried out by F.R. with help from J.C. Whole-genome sequencing was performed by the UCSD Institute for Genomic Medicine Core Facility and analyzed by M.R.L. and F.R. The manuscript was written by F.R. All authors read and approved this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization (2019) World Malaria Report 2019, World Health Organization, Geneva, Switzerland, https://apps.who.int/iris/handle/10665/330011.
- Cowell A.; Winzeler E. (2018) Exploration of the Plasmodium falciparum Resistome and Druggable Genome Reveals New Mechanisms of Drug Resistance and Antimalarial Targets. Microbiol. Insights 11, 117863611880852. 10.1177/1178636118808529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell A. N.; Istvan E. S.; Lukens A. K.; Gomez-Lorenzo M. G.; Vanaerschot M.; Sakata-Kato T.; Flannery E. L.; Magistrado P.; Owen E.; Abraham M.; LaMonte G.; Painter H. J.; Williams R. M.; Franco V.; Linares M.; Arriaga I.; Bopp S.; Corey V. C.; Gnadig N. F.; Coburn-Flynn O.; Reimer C.; Gupta P.; Murithi J. M.; Moura P. A.; Fuchs O.; Sasaki E.; Kim S. W.; Teng C. H.; Wang L. T.; Akidil A.; Adjalley S.; Willis P. A.; Siegel D.; Tanaseichuk O.; Zhong Y.; Zhou Y.; Llinas M.; Ottilie S.; Gamo F. J.; Lee M. C. S.; Goldberg D. E.; Fidock D. A.; Wirth D. F.; Winzeler E. A. (2018) Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 359 (6372), 191–199. 10.1126/science.aan4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. E. (2020) The transportome of the malaria parasite. Biol. Rev. Camb Philos. Soc. 95 (2), 305–332. 10.1111/brv.12565. [DOI] [PubMed] [Google Scholar]
- Spillman N. J.; Kirk K. (2015) The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int. J. Parasitol.: Drugs Drug Resist. 5 (3), 149–62. 10.1016/j.ijpddr.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golldack A.; Henke B.; Bergmann B.; Wiechert M.; Erler H.; Blancke Soares A.; Spielmann T.; Beitz E. (2017) Substrate-analogous inhibitors exert antimalarial action by targeting the Plasmodium lactate transporter PfFNT at nanomolar scale. PLoS Pathog. 13 (2), e1006172 10.1371/journal.ppat.1006172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valderramos S. G.; Fidock D. A. (2006) Transporters involved in resistance to antimalarial drugs. Trends Pharmacol. Sci. 27 (11), 594–601. 10.1016/j.tips.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. E.; Ginsburg H.; Kirk K. (2009) Membrane transport proteins of the malaria parasite. Mol. Microbiol. 74 (3), 519–28. 10.1111/j.1365-2958.2009.06863.x. [DOI] [PubMed] [Google Scholar]
- Luth M. R.; Gupta P.; Ottilie S.; Winzeler E. A. (2018) Using in Vitro Evolution and Whole Genome Analysis To Discover Next Generation Targets for Antimalarial Drug Discovery. ACS Infect. Dis. 4 (3), 301–314. 10.1021/acsinfecdis.7b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T.; Burrows J. N.; Kowalczyk P.; McDonald S.; Wells T. N.; Willis P. (2013) The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 8 (6), e62906 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. (2008) In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U. S. A. 105 (26), 9059–64. 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirawurah J. D.; Ansah F.; Nyarko P. B.; Duodu S.; Aniweh Y.; Awandare G. A. (2017) Antimalarial activity of Malaria Box Compounds against Plasmodium falciparum clinical isolates. Int. J. Parasitol.: Drugs Drug Resist. 7 (3), 399–406. 10.1016/j.ijpddr.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D. M.; Wree M.; Du A. Y.; Meister S.; Li F.; Patra K.; Lubar A.; Okitsu S. L.; Flannery E. L.; Kato N.; Tanaseichuk O.; Comer E.; Zhou B.; Kuhen K.; Zhou Y.; Leroy D.; Schreiber S. L.; Scherer C. A.; Vinetz J.; Winzeler E. A. (2016) High-Throughput Assay and Discovery of Small Molecules that Interrupt Malaria Transmission. Cell Host Microbe 19 (1), 114–26. 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruecker A.; Mathias D. K.; Straschil U.; Churcher T. S.; Dinglasan R. R.; Leroy D.; Sinden R. E.; Delves M. J. (2014) A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob. Agents Chemother. 58 (12), 7292–302. 10.1128/AAC.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman R. T.; Dharia N. V.; Winzeler E. A.; Fidock D. A. (2011) Piperaquine resistance is associated with a copy number variation on chromosome 5 in drug-pressured Plasmodium falciparum parasites. Antimicrob. Agents Chemother. 55 (8), 3908–16. 10.1128/AAC.01793-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariey F.; Witkowski B.; Amaratunga C.; Beghain J.; Langlois A. C.; Khim N.; Kim S.; Duru V.; Bouchier C.; Ma L.; Lim P.; Leang R.; Duong S.; Sreng S.; Suon S.; Chuor C. M.; Bout D. M.; Menard S.; Rogers W. O.; Genton B.; Fandeur T.; Miotto O.; Ringwald P.; Le Bras J.; Berry A.; Barale J. C.; Fairhurst R. M.; Benoit-Vical F.; Mercereau-Puijalon O.; Menard D. (2014) A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505 (7481), 50–5. 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocamora F.; Zhu L.; Liong K. Y.; Dondorp A.; Miotto O.; Mok S.; Bozdech Z. (2018) Oxidative stress and protein damage responses mediate artemisinin resistance in malaria parasites. PLoS Pathog. 14 (3), e1006930 10.1371/journal.ppat.1006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas A. R.; Sharma A. I.; Wong W.; Early A. M.; Redmond S.; Bopp S.; Neafsey D. E.; Volkman S. K.; Hartl D. L.; Wirth D. F. (2018) Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc. Natl. Acad. Sci. U. S. A. 115 (50), 12799–12804. 10.1073/pnas.1812317115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandt R. E. K.; Lafuente-Monasterio M. J.; Sakata-Kato T.; Luth M. R.; Segura D.; Pablos-Tanarro A.; Viera S.; Magan N.; Ottilie S.; Winzeler E. A.; Lukens A. K.; Gamo F. J.; Wirth D. F. (2019) In vitro selection predicts malaria parasite resistance to dihydroorotate dehydrogenase inhibitors in a mouse infection model. Sci. Transl Med. 11 (521), eaav1636. 10.1126/scitranslmed.aav1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. E.; Henry R. I.; Abbey J. L.; Clements J. D.; Kirk K. (2005) The ‘permeome’ of the malaria parasite: an overview of the membrane transport proteins of Plasmodium falciparum. Genome Biol. 6 (3), R26. 10.1186/gb-2005-6-3-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy V. S.; Shlykov M. A.; Castillo R.; Sun E. I.; Saier M. H. Jr. (2012) The major facilitator superfamily (MFS) revisited. FEBS J. 279 (11), 2022–35. 10.1111/j.1742-4658.2012.08588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N. (2013) Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 38 (3), 151–9. 10.1016/j.tibs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Fry M.; Pudney M. (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43 (7), 1545–53. 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- Kessl J. J.; Lange B. B.; Merbitz-Zahradnik T.; Zwicker K.; Hill P.; Meunier B.; Palsdottir H.; Hunte C.; Meshnick S.; Trumpower B. L. (2003) Molecular basis for atovaquone binding to the cytochrome bc1 complex. J. Biol. Chem. 278 (33), 31312–8. 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]
- Painter H. J.; Morrisey J. M.; Mather M. W.; Vaidya A. B. (2007) Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446 (7131), 88–91. 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- Nagy M.; Lacroute F.; Thomas D. (1992) Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl. Acad. Sci. U. S. A. 89 (19), 8966–70. 10.1073/pnas.89.19.8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkrumah L. J.; Muhle R. A.; Moura P. A.; Ghosh P.; Hatfull G. F.; Jacobs W. R. Jr.; Fidock D. A. (2006) Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat. Methods 3 (8), 615–21. 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshnick S. R. (2002) Artemisinin: mechanisms of action, resistance and toxicity. Int. J. Parasitol. 32 (13), 1655–60. 10.1016/S0020-7519(02)00194-7. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhang C. J.; Chia W. N.; Loh C. C.; Li Z.; Lee Y. M.; He Y.; Yuan L. X.; Lim T. K.; Liu M.; Liew C. X.; Lee Y. Q.; Zhang J.; Lu N.; Lim C. T.; Hua Z. C.; Liu B.; Shen H. M.; Tan K. S.; Lin Q. (2015) Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 6, 10111. 10.1038/ncomms10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail H. M.; Barton V.; Phanchana M.; Charoensutthivarakul S.; Wong M. H.; Hemingway J.; Biagini G. A.; O’Neill P. M.; Ward S. A. (2016) Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc. Natl. Acad. Sci. U. S. A. 113 (8), 2080–5. 10.1073/pnas.1600459113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J.; Lee M. C.; Lee A. H.; Zeitler B.; Williams A. E.; Pearl J. R.; Zhang L.; Rebar E. J.; Gregory P. D.; Llinas M.; Urnov F. D.; Fidock D. A. (2012) Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat. Methods 9 (10), 993–8. 10.1038/nmeth.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman E. L.; Painter H. J.; Samra J.; Carrasquilla M.; Llinas M. (2016) Metabolomic Profiling of the Malaria Box Reveals Antimalarial Target Pathways. Antimicrob. Agents Chemother. 60 (11), 6635–6649. 10.1128/AAC.01224-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murithi J. M.; Owen E. S.; Istvan E. S.; Lee M. C. S.; Ottilie S.; Chibale K.; Goldberg D. E.; Winzeler E. A.; Llinas M.; Fidock D. A.; Vanaerschot M. (2020) Combining Stage Specificity and Metabolomic Profiling to Advance Antimalarial Drug Discovery. Cell Chem. Biol. 27 (2), 158–171.E3. 10.1016/j.chembiol.2019.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barragan M. J.; Lemieux J.; Quinones M.; Williamson K. C.; Molina-Cruz A.; Cui K.; Barillas-Mury C.; Zhao K.; Su X. Z. (2011) Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics 12, 587. 10.1186/1471-2164-12-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanghi G.; Vembar S. S.; Baumgarten S.; Ding S.; Guizetti J.; Bryant J. M.; Mattei D.; Jensen A. T. R.; Renia L.; Goh Y. S.; Sauerwein R.; Hermsen C. C.; Franetich J. F.; Bordessoulles M.; Silvie O.; Soulard V.; Scatton O.; Chen P.; Mecheri S.; Mazier D.; Scherf A. (2018) A Specific PfEMP1 Is Expressed in P. falciparum Sporozoites and Plays a Role in Hepatocyte Infection. Cell Rep. 22 (11), 2951–2963. 10.1016/j.celrep.2018.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Wang C.; Otto T. D.; Oberstaller J.; Liao X.; Adapa S. R.; Udenze K.; Bronner I. F.; Casandra D.; Mayho M.; Brown J.; Li S.; Swanson J.; Rayner J. C.; Jiang R. H. Y.; Adams J. H. (2018) Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360 (6388), eaap7847. 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell E.; Gomes A. R.; Sanderson T.; Anar B.; Girling G.; Herd C.; Metcalf T.; Modrzynska K.; Schwach F.; Martin R. E.; Mather M. W.; McFadden G. I.; Parts L.; Rutledge G. G.; Vaidya A. B.; Wengelnik K.; Rayner J. C.; Billker O. (2017) Functional Profiling of a Plasmodium Genome Reveals an Abundance of Essential Genes. Cell 170 (2), 260–272.E8. 10.1016/j.cell.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottilie S., Luth M. R., Hellemann E., Goldgof G. M., Vigil E., Kumar P., Cheung A. L., Song M., Godinez-Macias K. P., Carolino K., Yang J., Lopez G., Abraham M., Tarsio M., LeBlanc E., Whitesell L., Schenken J., Gunawan F., Patel R., Smith J., Love M. S., Williams R. M., McNamara C. W., Gerwick W. H., Ideker T., Suzuki Y., Wirth D. F., Lukens A. K., Kane P. M., Cowen L. E., Durrant J. D., and Winzeler E. A. (2021) Defining the Yeast Resistome through in vitro Evolution and Whole Genome Sequencing, submitted for publication, 10.1101/2021.02.17.430112. [DOI] [Google Scholar]
- Yun C. W.; Ferea T.; Rashford J.; Ardon O.; Brown P. O.; Botstein D.; Kaplan J.; Philpott C. C. (2000) Desferrioxamine-mediated iron uptake in Saccharomyces cerevisiae. Evidence for two pathways of iron uptake. J. Biol. Chem. 275 (14), 10709–15. 10.1074/jbc.275.14.10709. [DOI] [PubMed] [Google Scholar]
- Rutherford J. C.; Ojeda L.; Balk J.; Muhlenhoff U.; Lill R.; Winge D. R. (2005) Activation of the iron regulon by the yeast Aft1/Aft2 transcription factors depends on mitochondrial but not cytosolic iron-sulfur protein biogenesis. J. Biol. Chem. 280 (11), 10135–40. 10.1074/jbc.M413731200. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Lampert S. M.; Philpott C. C. (2005) A receptor domain controls the intracellular sorting of the ferrichrome transporter, ARN1. EMBO J. 24 (5), 952–62. 10.1038/sj.emboj.7600579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A.; Park J.; Paulsen I. T.; Jonniaux J. L.; Dinh T.; Mordant P.; Saier M. H. Jr. (1997) Multidrug-resistant transport proteins in yeast: complete inventory and phylogenetic characterization of yeast open reading frames with the major facilitator superfamily. Yeast 13 (1), 43–54. . [DOI] [PubMed] [Google Scholar]
- Kim Y.; Yun C. W.; Philpott C. C. (2002) Ferrichrome induces endosome to plasma membrane cycling of the ferrichrome transporter, Arn1p, in Saccharomyces cerevisiae. EMBO J. 21 (14), 3632–42. 10.1093/emboj/cdf382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C. W.; Tiedeman J. S.; Moore R. E.; Philpott C. C. (2000) Siderophore-iron uptake in saccharomyces cerevisiae. Identification of ferrichrome and fusarinine transporters. J. Biol. Chem. 275 (21), 16354–9. 10.1074/jbc.M001456200. [DOI] [PubMed] [Google Scholar]
- Parker K. E. R.; Fairweather S. J.; Rajendran E.; Blume M.; McConville M. J.; Broer S.; Kirk K.; van Dooren G. G. (2019) The tyrosine transporter of Toxoplasma gondii is a member of the newly defined apicomplexan amino acid transporter (ApiAT) family. PLoS Pathog. 15 (2), e1007577 10.1371/journal.ppat.1007577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh L. M.; Goodman C. D.; Mollard V.; McFadden G. I.; Ralph S. A. (2017) Comparative transcriptomics of female and male gametocytes in Plasmodium berghei and the evolution of sex in alveolates. BMC Genomics 18 (1), 734. 10.1186/s12864-017-4100-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasonder E.; Rijpma S. R.; van Schaijk B. C.; Hoeijmakers W. A.; Kensche P. R.; Gresnigt M. S.; Italiaander A.; Vos M. W.; Woestenenk R.; Bousema T.; Mair G. R.; Khan S. M.; Janse C. J.; Bartfai R.; Sauerwein R. W. (2016) Integrated transcriptomic and proteomic analyses of P. falciparum gametocytes: molecular insight into sex-specific processes and translational repression. Nucleic Acids Res. 44 (13), 6087–101. 10.1093/nar/gkw536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N.; Spurck T. P.; Goodman C. D.; McFadden G. I. (2009) Apicoplast and mitochondrion in gametocytogenesis of Plasmodium falciparum. Eukaryotic Cell 8 (1), 128–32. 10.1128/EC.00267-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae J. I.; Dixon M. W.; Dearnley M. K.; Chua H. H.; Chambers J. M.; Kenny S.; Bottova I.; Tilley L.; McConville M. J. (2013) Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol. 11, 67. 10.1186/1741-7007-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre J.; Almela M. J.; Lozano S.; Miguel C.; Franco V.; Leroy D.; Herreros E. (2012) Activity of clinically relevant antimalarial drugs on Plasmodium falciparum mature gametocytes in an ATP bioluminescence “transmission blocking” assay. PLoS One 7 (4), e35019 10.1371/journal.pone.0035019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit-Vical F.; Lelievre J.; Berry A.; Deymier C.; Dechy-Cabaret O.; Cazelles J.; Loup C.; Robert A.; Magnaval J. F.; Meunier B. (2007) Trioxaquines are new antimalarial agents active on all erythrocytic forms, including gametocytes. Antimicrob. Agents Chemother. 51 (4), 1463–72. 10.1128/AAC.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari N. K.; Reynolds P. J.; Calderon A. I. (2016) Preliminary LC-MS Based Screening for Inhibitors of Plasmodium falciparum Thioredoxin Reductase (PfTrxR) among a Set of Antimalarials from the Malaria Box. Molecules 21 (4), 424. 10.3390/molecules21040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehr S.; Sturm N.; Rahlfs S.; Przyborski J. M.; Becker K. (2010) Compartmentation of redox metabolism in malaria parasites. PLoS Pathog. 6 (12), e1001242 10.1371/journal.ppat.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges M.; Yikilmaz E.; Patterson G.; Kasvosve I.; Rouault T. A.; Gordeuk V. R.; Loyevsky M. (2005) An iron regulatory-like protein expressed in Plasmodium falciparum displays aconitase activity. Mol. Biochem. Parasitol. 143 (1), 29–38. 10.1016/j.molbiopara.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Ke H.; Lewis I. A.; Morrisey J. M.; McLean K. J.; Ganesan S. M.; Painter H. J.; Mather M. W.; Jacobs-Lorena M.; Llinas M.; Vaidya A. B. (2015) Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 11 (1), 164–74. 10.1016/j.celrep.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller T.; Johann L.; Jannack B.; Bruckner M.; Lanfranchi D. A.; Bauer H.; Sanchez C.; Yardley V.; Deregnaucourt C.; Schrevel J.; Lanzer M.; Schirmer R. H.; Davioud-Charvet E. (2011) Glutathione reductase-catalyzed cascade of redox reactions to bioactivate potent antimalarial 1,4-naphthoquinones--a new strategy to combat malarial parasites. J. Am. Chem. Soc. 133 (30), 11557–71. 10.1021/ja201729z. [DOI] [PubMed] [Google Scholar]
- Morin C.; Besset T.; Moutet J. C.; Fayolle M.; Bruckner M.; Limosin D.; Becker K.; Davioud-Charvet E. (2008) The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioredoxin and glutathione reductases and of human erythrocyte glutathione reductase. Org. Biomol. Chem. 6 (15), 2731–42. 10.1039/b802649c. [DOI] [PubMed] [Google Scholar]
- Antonova-Koch Y.; Meister S.; Abraham M.; Luth M. R.; Ottilie S.; Lukens A. K.; Sakata-Kato T.; Vanaerschot M.; Owen E.; Jado J. C.; Maher S. P.; Calla J.; Plouffe D.; Zhong Y.; Chen K.; Chaumeau V.; Conway A. J.; McNamara C. W.; Ibanez M.; Gagaring K.; Serrano F. N.; Eribez K.; Taggard C. M.; Cheung A. L.; Lincoln C.; Ambachew B.; Rouillier M.; Siegel D.; Nosten F.; Kyle D. E.; Gamo F. J.; Zhou Y.; Llinas M.; Fidock D. A.; Wirth D. F.; Burrows J.; Campo B.; Winzeler E. A. (2018) Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 362 (6419), eaat9446. 10.1126/science.aat9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon G. L.; Pidathala C.; Shone A. E.; Antoine T.; Fisher N.; O’Neill P. M.; Ward S. A.; Biagini G. A. (2013) Targeting the mitochondrial electron transport chain of Plasmodium falciparum: new strategies towards the development of improved antimalarials for the elimination era. Future Med. Chem. 5 (13), 1573–91. 10.4155/fmc.13.121. [DOI] [PubMed] [Google Scholar]
- Barton V.; Fisher N.; Biagini G. A.; Ward S. A.; O’Neill P. M. (2010) Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr. Opin. Chem. Biol. 14 (4), 440–6. 10.1016/j.cbpa.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Dong C. K.; Urgaonkar S.; Cortese J. F.; Gamo F. J.; Garcia-Bustos J. F.; Lafuente M. J.; Patel V.; Ross L.; Coleman B. I.; Derbyshire E. R.; Clish C. B.; Serrano A. E.; Cromwell M.; Barker R. H. Jr.; Dvorin J. D.; Duraisingh M. T.; Wirth D. F.; Clardy J.; Mazitschek R. (2011) Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum cytochrome bc1 inhibitors. Chem. Biol. 18 (12), 1602–10. 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A. M.; Noviyanti R.; Sinden R. E.; Kocken C. H. M.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jimenez-Diaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F. J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. (2013) Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl Med. 5 (177), 177ra37. 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallieres C.; Fisher N.; Antoine T.; Al-Helal M.; Stocks P.; Berry N. G.; Lawrenson A. S.; Ward S. A.; O’Neill P. M.; Biagini G. A.; Meunier B. (2012) HDQ, a potent inhibitor of Plasmodium falciparum proliferation, binds to the quinone reduction site of the cytochrome bc1 complex. Antimicrob. Agents Chemother. 56 (7), 3739–47. 10.1128/AAC.00486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickles A. M.; Ting L. M.; Morrisey J. M.; Li Y.; Mather M. W.; Meermeier E.; Pershing A. M.; Forquer I. P.; Miley G. P.; Pou S.; Winter R. W.; Hinrichs D. J.; Kelly J. X.; Kim K.; Vaidya A. B.; Riscoe M. K.; Nilsen A. (2015) Inhibition of cytochrome bc1 as a strategy for single-dose, multi-stage antimalarial therapy. Am. J. Trop. Med. Hyg. 92 (6), 1195–201. 10.4269/ajtmh.14-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranchi D. A.; Cesar-Rodo E.; Bertrand B.; Huang H. H.; Day L.; Johann L.; Elhabiri M.; Becker K.; Williams D. L.; Davioud-Charvet E. (2012) Synthesis and biological evaluation of 1,4-naphthoquinones and quinoline-5,8-diones as antimalarial and schistosomicidal agents. Org. Biomol. Chem. 10 (31), 6375–87. 10.1039/c2ob25812a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Barrantes P. M.; Lamoureux G. V.; Perez A. L.; Garcia-Sanchez R. N.; Martinez A. R.; San Feliciano A. (2013) Synthesis and biological evaluation of novel ferrocene-naphthoquinones as antiplasmodial agents. Eur. J. Med. Chem. 70, 548–57. 10.1016/j.ejmech.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Pingaew R.; Prachayasittikul V.; Worachartcheewan A.; Nantasenamat C.; Prachayasittikul S.; Ruchirawat S.; Prachayasittikul V. (2015) Novel 1,4-naphthoquinone-based sulfonamides: Synthesis, QSAR, anticancer and antimalarial studies. Eur. J. Med. Chem. 103, 446–59. 10.1016/j.ejmech.2015.09.001. [DOI] [PubMed] [Google Scholar]
- de Sena Pereira V. S.; da Silva Emery F.; Lobo L.; Nogueira F.; Oliveira J. I. N.; Fulco U. L.; Albuquerque E. L.; Katzin A. M.; de Andrade-Neto V. F. (2018) In vitro antiplasmodial activity, pharmacokinetic profiles and interference in isoprenoid pathway of 2-aniline-3-hydroxy-1.4-naphthoquinone derivatives. Malar. J. 17 (1), 482. 10.1186/s12936-018-2615-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck D. C.; Ferreira S. B.; Cruz L. N.; da Rocha D. R.; Moraes M. S.; Nakabashi M.; Rosenthal P. J.; Ferreira V. F.; Garcia C. R. (2013) Biological evaluation of hydroxynaphthoquinones as anti-malarials. Malar. J. 12, 234. 10.1186/1475-2875-12-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira D. R.; de Sa M. S.; Macedo T. S.; Menezes M. N.; Reys J. R.; Santana A. E.; Silva T. L.; Maia G. L.; Barbosa-Filho J. M.; Camara C. A.; da Silva T. M.; da Silva K. N.; Guimaraes E. T.; dos Santos R. R.; Goulart M. O.; Soares M. B. (2015) Evaluation of naphthoquinones identified the acetylated isolapachol as a potent and selective antiplasmodium agent. J. Enzyme Inhib. Med. Chem. 30 (4), 615–21. 10.3109/14756366.2014.958083. [DOI] [PubMed] [Google Scholar]
- Vale V. V.; Cruz J. N.; Viana G. M. R.; Póvoa M. M.; Brasil D. d. S. B.; Dolabela M. F. (2020) Naphthoquinones isolated from Eleutherine plicata herb: in vitro antimalarial activity and molecular modeling to investigate their binding modes. Med. Chem. Res. 29 (29), 487–494. 10.1007/s00044-019-02498-z. [DOI] [Google Scholar]
- MalariaGEN (2016) The Pf3K Project: Pf3k pilot data release 5, https://www.malariagen.net/data/pf3k-5.
- Trager W.; Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193 (4254), 673–5. 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Johnson J. D.; Dennull R. A.; Gerena L.; Lopez-Sanchez M.; Roncal N. E.; Waters N. C. (2007) Assessment and continued validation of the malaria SYBR green I-based fluorescence assay for use in malaria drug screening. Antimicrob. Agents Chemother. 51 (6), 1926–33. 10.1128/AAC.01607-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manary M. J.; Singhakul S. S.; Flannery E. L.; Bopp S. E.; Corey V. C.; Bright A. T.; McNamara C. W.; Walker J. R.; Winzeler E. A. (2014) Identification of pathogen genomic variants through an integrated pipeline. BMC Bioinf. 15, 63. 10.1186/1471-2105-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A.; Hanna M.; Banks E.; Sivachenko A.; Cibulskis K.; Kernytsky A.; Garimella K.; Altshuler D.; Gabriel S.; Daly M.; DePristo M. A. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P.; Platts A.; Wang le L.; Coon M.; Nguyen T.; Wang L.; Land S. J.; Lu X.; Ruden D. M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6 (2), 80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpiyevich M.; Adjalley S.; Mol M.; Ascher D. B.; Mason B.; van der Heden van Noort G. J.; Laman H.; Ovaa H.; Lee M. C. S.; Artavanis-Tsakonas K. (2019) Nedd8 hydrolysis by UCH proteases in Plasmodium parasites. PLoS Pathog. 15 (10), e1008086 10.1371/journal.ppat.1008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdech Z.; Mok S.; Gupta A. P. (2012) DNA microarray-based genome-wide analyses of Plasmodium parasites. Methods Mol. Biol. 923, 189–211. 10.1007/978-1-62703-026-7_13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.