

Abstract
Background
Non-small cell lung carcinoma (NSCLC) is one of the most common human cancers, comprising approximately 80–85% of all lung carcinomas. An estimated incidence of NSCLC is approximately 2 million new cases per year worldwide.
Results
In recent decade, the treatment of NSCLC has made breakthrough progress owing to a large number of targeted therapies which were approved for clinical use. Epidemiology, genetic susceptibility, and molecular profiles in patients are likely to play an important factor in response rates and survival benefits to these targeted treatments and thus warrant further investigation on ethnic differences in NSCLC. In this study, a total number of 1500 Chinese patient samples,1000 formalin fixed paraffin-embedded (FFPE) and 500 blood samples, from patients with NSCLC were analyzed by targeted sequencing to explore mutational landscape in ethnic groups associated with China.
Conclusions
Overall, the data presented here provide a comprehensive analysis of NSCLC mutational landscape in Chinese patients and findings are discussed in the context of similar studies on different ethnic groups.
Keywords: Targeted exome sequencing, Non-small cell lung carcinoma, Chinese patients, Cancer, Disease
Background
Non-small cell lung cancer (NSCLC) represents a heterogeneous group of lung cancer. Two major NSCLC subtypes are distinguished: the adenocarcinoma (AD) and the squamous cell carcinoma (SCC). In general, treatments for NSCLC can include chemotherapy, targeted drug therapy, immunotherapy, surgery, and palliative procedures. Ideal treatment options depend on whether the cancer has already spread and metastasized, what are the genetic changes in the cancer cells, and the patients’ overall health and age. Sequencing of tumor sample may help to screen the patients who may response to and benefit from targeted treatments and help to lower the mortality rate [1]. For instance, if one of the previously identified NSCLC-associated genes, such as EGFR, ALK, ROS1, BRAF, RET, MET, or NTRK, is mutated in the patient’s cancer cells, targeted therapies has to be considered [1]. Accordingly, the National Comprehensive Cancer Network (NCCN) NSCLC guidelines had recommended the routine detection of EGFR or ERBB2 mutations, or ALK, ROS1, or RET fusions prior to treatment. However, previous studies raised the possibility that the distribution of these mutations show a race-dependent pattern, with one study estimating that 10% of Caucasians but as high as 50% of Asians will be found to have drug sensitizing mutations of the EGFR [2]. The observed high variation in mutation frequency in demographic subgroups urges for large-scale studies that systematically investigate mutation landscapes in certain races and offers a better insight what genes has to be tested prior to choosing a targeted therapy [3, 4].
Next-generation sequencing (NGS) has revolutionized the identification process and systematic characterization of genomic alterations, including single nucleotide variations and small insertions/deletions (InDels), and will likely receive recommendations from cancer societies in the very near future about its daily use in clinical oncology practice. Indeed, upfront tumor genotyping is now widely considered as an essential step in guiding treatment decision-making in the management of patients with NSCLC [5].
In this study, a number of 1000 formalin-fixed paraffin-embedded (FFPE) and 500 blood samples with NSCLC were analyzed by NGS-targeted sequencing. This study represents to our knowledge one of the largest efforts so far to systematically characterize mutational landscape in Chinese NSCLC cohort samples.
Results
Clinical features of the patient samples
Discovery and quantification of genetic alterations in NSCLC, from point mutations to large genomic rearrangements, requires a comprehensive genome-wide approach and a large sample cohort. We have collected 1000 formalin-fixed paraffin-embedded (FFPE) tumor samples and 500 blood samples from a total of 1500 patients diagnosed with NSCLC between June 2017 and April 2019. Tissue and blood samples were obtained from independent patient groups. The detailed clinical characteristics of the patients are shown in Table 1. Briefly, lung adenocarcinoma accounted for 84.3% of the FFPE samples (843/1000), squamous cell carcinoma for 14.2% (142/1000), and others for 1.5% (15/1000). As for the blood samples, lung adenocarcinoma accounted for 80.4% (402/500), squamous cell carcinoma for 17% (85/500), and others for 2.6% (13/500). In total, 39 samples were excluded due to not passing quality standards along the sample processing and sequencing.
Table 1.
Overview of patient and tumor characteristics in the present study
| Characteristics | Tissues (1000 cases) n (%) |
Blood (500 cases) n (%) |
|---|---|---|
| Sex | ||
| Male | 578 (57.8) | 308 (61.6) |
| Female | 422 (42.2) | 192 (38.4)) |
| Age | ||
| >60 | 392 (39.2) | 164 (32.8) |
| ≤60 | 602 (60.2) | 334 (66.8) |
| Unknown | 6 (0.6) | 2 (0.4) |
| Smoking | ||
| Non-smoking | 945 (94.5) | 477 (95.4) |
| Occasionally | 11 (1.1) | 3 (0.6) |
| Often | 44 (4.4) | 20 (4) |
| Tumor type | ||
| Lung adenocarcinoma | 843 (84.3) | 402 (80.4) |
| Lung squamous carcinoma | 142 (14.2) | 85 (17) |
| Unknown | 15 (1.5) | 13 (2.6) |
Overview of the genomic alterations of 1000 tissue and 500 blood samples of NSCLC patients
The clinical significance of identifying hypermutated tumors has recently been demonstrated in several NSCLC studies [6, 7]. However, there is a large variability in mutation burden within tumor types in NSCLCs [8]. To begin to explore the mutation burden in our cohort, we first identified the overall mutation landscape across the tissue and blood samples. We subclassified mutations into four main types, single mutation (single base variation, insertion or deletion, SM), multiple single mutations (MM), amplification (AMP), and fusion (FUS) (Fig. 1). As for the FFPE NSCLC tissue samples, a total of 968/1000 samples had at least one type of the above-listed mutations, while 387/500 blood NSCLC samples were found to belong to one of the mutation groups. Specifically, there were 127/500 (25.4%) blood samples with single base variation, 224/500 (44.8%) with multiple mutations. Only 36/500 (7.2%) blood samples showed amplification or fusion (Fig. 1). As for tissue samples, there were 113/1000 (11.3%) single base variation, 555/1000 (55.5%) with multiple mutations, and 221/1000 (22.1%) samples had amplification alone or in combination with other mutations. In contrast to 117/500 (22.6%) of blood samples, only 32/1000 tissue samples (3.2%) had not detected mutation within the studied 65 genomic regions (Fig. 1).
Fig. 1.
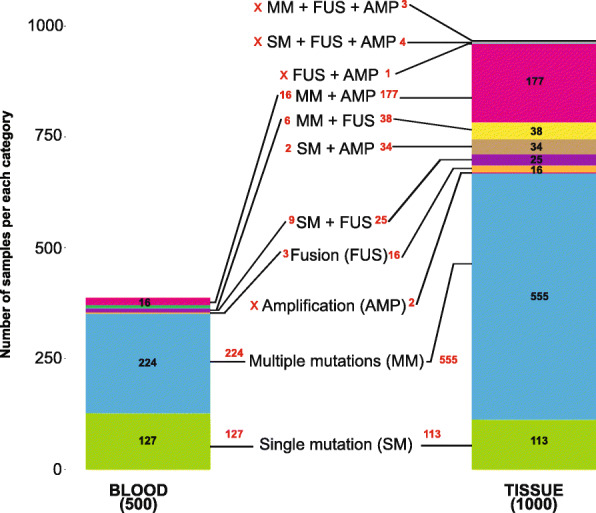
Overview of the genomic alterations of 1000 tissue and 500 blood samples of NSCLC patients. Distribution of tissue and blood samples with single mutation (single base variation, insertion or deletion, SM); multiple single mutations (MM); amplification (AMP), fusion (FUS) or combination of these
Mutation patterns of frequently altered cancer genes
Next, we set out to determine the most common cancer genes enriched for SNV/InDel in our NSCLC patient cohort. We identified many genes previously also found to be mutated in NSCLC, including several tumor suppressor genes TP53 [9], CDKN2A [10], and oncogenes EGFR [11] and KRAS [12]. Notably, we observed highly accumulated TP53 and EGFR mutations in both blood and tissue samples of NSCLC patients (Fig. 2a, b). Co-occurrence of EGFR with the TP53 mutations was remarkable in the tissue samples (>25%). EGFR mutation rate was significantly higher in tissues (~55%) vs. blood (~35%). In addition, we found several other genes that were significantly mutated in our cohort, such as PTCH1 and PIK3CA (Fig. 2a, b). Other, less frequently detected, but previously identified genes included tumor suppressor genes (APC) and tyrosine kinase genes (ERBB2, FGFR, and NTRK genes).
Fig. 2.
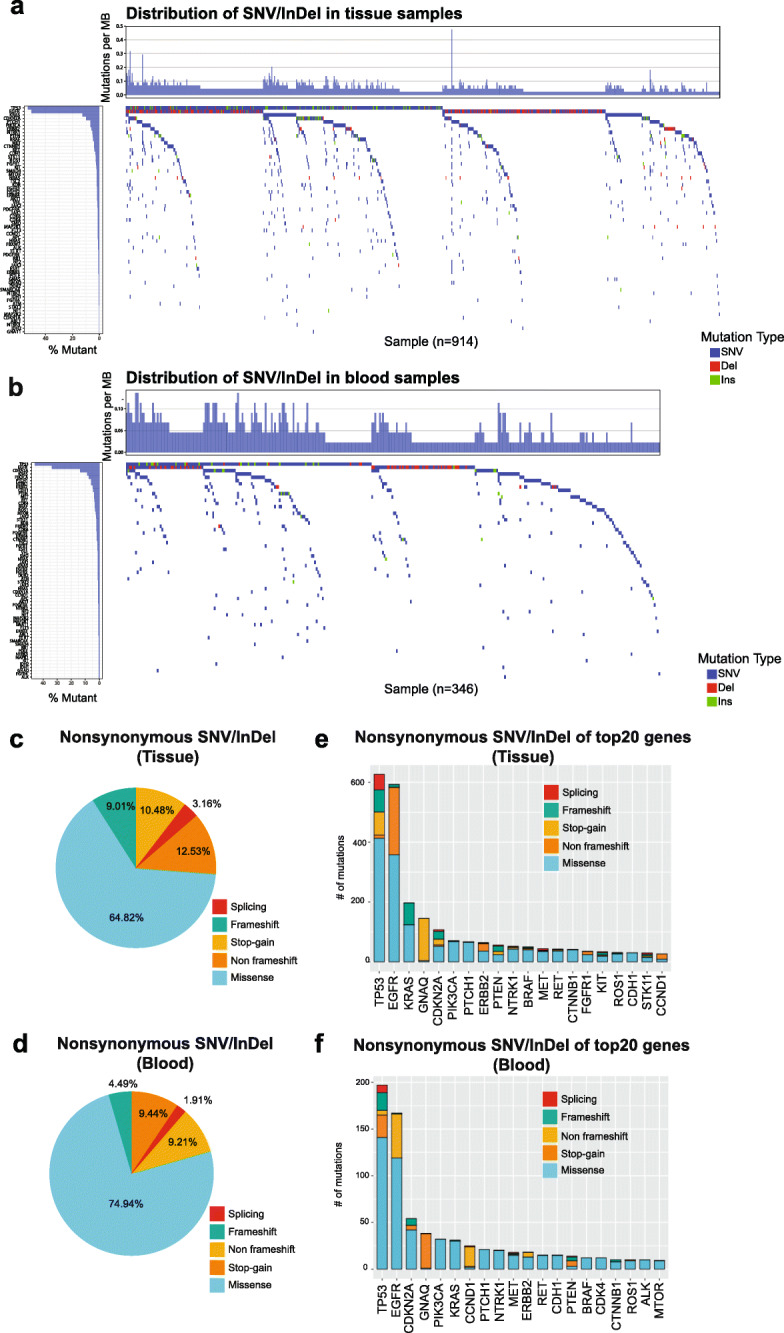
Significantly mutated genes in NSCLC. Waterfall plot of the distribution of SNV/InDel mutations found in tissue (a) and blood (b) patient samples. The top plot show number of mutations per Mb sequenced for a cohort of 914 NSCLC samples. Left plot shows the frequency of samples mutated for the listed gene. The central plot shows the types of mutations (SNV, Insertion, Deletion) in each sample. The distribution of nonsynonymous frameshift insertions and deletions, missense mutations, Stop-gain, and other infrequent alterations (e.g. splicing) in both the tissue (c, e) and blood samples (d, f)
Next, we assessed the distribution of nonsynonymous frameshift insertions and deletions, missense mutations, Stop-gain, and other infrequent alterations (e.g., splicing) in both the tissue and blood samples (Fig. 2c–e). In addition to identifying previously known NSCLC-associated genes, such as TP53, KRAS, EGFR, and CDKN2A, the analysis revealed GNAQ gene, which was previously mostly implicated in melanomas and only a very recent study linked to lung cancer (Fig. 2c–e) [13]. Identified mutations of GNAQ included p.R60G, p.P174R, p.A93D, p.M59L, and p.Q81H.
Recurrent SNV mutations in NSCLC
Next, we explored the positional distribution and recurrence of SNV mutations in the genes with most frequent mutations, focusing on the most frequently mutated genes, TP53, EGFR, KRAS, CDKN2A, PTCH1, and PIK3CA (Fig. 3).
Fig. 3.

Recurrent SNV mutations in TP53, EGFR, KRAS, CDKN2A, PTCH1, and PIK3CA. Positional distribution of SNV mutations across blood and tissue NSCLC samples. SNV mutations detected by exome sequencing are depicted on lolliplot and mapped to the structure of the corresponding gene
Most clinical studies suggest that lung cancer with alterations detected in TP53 carries an overall worse prognosis and such cases are more resistant to chemotherapy and radiation [14]. Indeed, as it was shown in Fig. 2, mutations of the TP53 gene occurred in over 50% of NSCLC samples in our cohort. In our cohort, only 8 samples showed mutations at codons 157, 6 samples at codon 158, 11 samples at codon 179, and 27 samples at codon 248 of TP53. These codons are typically mutated in lung cancer from smokers and uncommonly observed in lung cancer from nonsmokers [15].
Previous analysis of the TK domain of the EGFR by Shigematsu et al. identified that all mutations in lung cancer specimens occurred within exons 18–21, with a prevalence of 21% [11, 16, 17]. Consistent with these previous reports, EGFR mainly had three subtype of mutation (p.L858R, Exon 19del, p.T790M). EGFR p.L858R and Exon 19del were the most common EGFR active mutant, which may be sensitive to EGFR-TKI inhibitors such as gefitinib, erlotinib, or afatinib. We found the percentage of these mutation in FFPE and blood sample were similar. There were 42.4% p.L858R in blood sample and 44.4% in FFPE samples. Similarly, there were 38.5.4% Exon 19del in blood sample and 34.2% in FFPE samples. Interestingly, there was significantly different percent of p.T790M in FFPE and blood sample. The percent of p.T790M in FFPE and blood sample were 24.8% and 2.4%, respectively.
We found that mutations in KRAS were mostly detected at amino acid positions 12, 13, 61, in regions which are considered mutational hotspots (Fig. 3). Recurrent mutations included p.G12C, p.G12V, p.G13D, and p.Q61H. In addition, we have also found pA146T in two tissue samples.
In addition to the previously described mutations involving TP53, EGFR, and KRAS genes, our analysis in this large cohort revealed several other recurrent point mutations in NSCLC. For instance, recurrent point mutations (E545K) in the PIK3CA gene were identified. In fact, somatic mutations of the PIK3CA gene have been also described NSCLC [18, 19].
CDKN2A gene mutation was detected in ~10% of the analyzed NSCLC tissue samples. CDKN2A is a well-known tumor suppressor, which regulates cell cycle progression by inhibiting cyclinD-CDK4 and cyclinD-CDK6 complexes responsible for initiating the G1/S phase transition. Recurrent mutations included p.A68V, p.R80X, p.A85P, p.D108Y, p.E120X, and p.V115E.
Recently, the PTCH1 gene mutations were also identified in NSCLC. Previous studies found that the most common genetic alterations in PTCH1 are missense mutations (2.17%), frameshift (0.46%), nonsense mutations (0.17%), and S1203Afs*52 (0.15%) [20]. We found p.A741V, p.D898N recurrent mutations (Fig. 3).
Structural rearrangement signatures and overview of aberration frequencies identified in our NSCLC patient cohort
Previous studies have been able to detect significant copy number alteration in lung adenocarcinomas [21, 22]. Sequencing of the coding exons of the 65 pre-selected candidate cancer genes in our study identified gene amplifications in both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) (Fig. 4a, b). Similarly to previous reports, we have found both EGFR and KRAS gene copy number gains to occur frequently in NSCLC [23, 24].
Fig. 4.

Amplifications and gene fusion signatures identified in our NSCLC patient cohort. Structural rearrangement signatures identified in Chinese NSCLC patients. Rearrangement hotspots identified in NSCLC patients. (a) Gene amplificaitons in tissue samples; (b) Gene amplificaitons in blood samples; (c) Gene fusions in tissue samples; (d) Gene fusions in blood samples; (e) ALK fusions in tissue samples; (f) RET fusions in tissue samples
The recent discovery of a fusion involving the echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) genes in tumor specimens from a subset of patients with NSCLC (mostly adenocarcinoma) and the quite effective treatment of these cases by ALK kinase inhibitors have reinvigorated efforts to identify additional genomic rearrangements that could be therapeutic targets [8, 25]. Thus, we also analyzed the tumor genomes for fusion genes and were able to systematically identify fusion genes (Fig. 4c). ALK fusion mutation was very common in our NSCLC cohort. We found that among the samples which had any type of genomic rearrangements, ~61% FFPE and ~74% blood samples had rearrangements related to ALK. The most common rearrangement of ALK in tissue samples was EML4-ALK (88.9%), and the other subtypes included GPC6-ALK (1.9%), LTBP1-ALK (1.9%), GPATCH8-ALK (1.9%), DIS3L2-ALK (1.9%), HIP1-ALK (1.9%), and LPIN1-ALK (1.9%) (Fig. 4e). The most common rearrangement of RET in tissue samples was KIF5B-RET (64.7.9%), and the other subtypes included MPP7-RET (5.8%), CCNYL2-RET (5.8%), KIAA1468-RET (5.8%), CCDC-RET (5.8%), and YME1L1-RET (5.8%) (Fig. 4e).
Combination of SNV, amplification, and fusion of significantly mutated genes
Finally, to further explore the mutations in the most common cancer genes involved in Chinese NSCLC patients, we also assessed the co-occurrence of single nucleotide variations with other mutational events. Strikingly, majority of samples (~90%) carrying KRAS mutations were not containing any other type of mutations (Fig. 5). In contrast, EGFR has often co-occurred with other mutations.
Fig. 5.

An overview of significantly mutated genes. Assessment of single mutations (SNVs and InDels), multiple mutations, and amplifications across the top most frequently mutated genes, excluding TP53. Genes were depicted according to aberration frequencies
Discussion
In this study, we analyzed genomic events in a large set of FFPE and blood samples from patients with NSCLC. Specifically, we used targeted sequencing of selected candidate genes to identify most common mutations in a large cohort of Chinese NSCLC patients. The vast amount of genomic information generated in this and similar studies is expected to transform our current understanding of lung cancer and advance personalized lung cancer therapy. We also anticipate that our study along with other studies implementing tumor mutation landscape analysis using targeted and genome-wide NGS across different ethnic groups in lung cancer will enormously expand our knowledge base in lung cancer biology, treatment strategy, new drug target development, and NSCLC outcome.
In fact, recent discoveries made based on previous mutational analysis already significantly improved and expanded the availability of targeted therapies. Development of new receptor kinase inhibitors, such as erlotinib and gefitinib (against EGFR) and most recently crizotinib (against rearranged ALK), and antibodies such as cetuximab (against EGFR) are all great examples how NGS can help to improve personalized medicine [26]. However, while these drugs are effective in a subset of patients, our analysis and other studies clearly suggest a very complex mutational landscape in NSCLC and warrant for even more targeted drug development to be able to further decrease the still high mortality rate of NSCLC.
An interesting target that came out from our analysis is GNAQ (Fig. 2). GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) is known as a subunit of one of the heterotrimeric guanine nucleotide binding proteins (G proteins) that is involved in multiple processes of mammary cells including hormonal signal transduction, metabolism, development, cell survival, and sensory functions. Previous studies mostly implicated its mutations in melanoma, and GNAQ mutations have not been documented in NSCLC. We found several nonsynonymous SNV (Stop-gain) in GNAQ both in blood and tissue samples, though none of the identified mutations were shown recurrence across the samples.
Another interesting candidate for follow-up studies was the tumor suppressor Patched 1 (PTCH1), a multi-pass transmembrane protein which is over-expressed in many metastatic cancers. In an unbound inactive state, PTCH1 acts as a negative regulator of smoothened (SMO), while upon activation it leads to activation of GLI1 proto-oncoprotein. Since PTCH1 is a multidrug transporter, it contributes to chemotherapy resistance by the efflux of chemotherapeutic agents such as doxorubicin [27]. PTCH1-altered tumors can be now targeted with three different FDA-approved SMO inhibitors, namely sonidegib, vismodegib, and glasdegib [27].
An important context to discuss is related to health disparities, which are a recognized and well-documented phenomenon on the cancer field but has not yet been addressed in case of NSCLC. Socioeconomic and cultural differences across ethnic groups undoubtedly account for some of the disparities, namely that certain groups may bear a disproportionate burden of cancer compared with other groups. Our study specifically aimed to collect and explore data of a well-defined group of patients based on geographic location. Our data collection and/or exploration did not yet include gathering information on income, education, disabilities, and other possibly relevant characteristics. Nevertheless, it is important to highlight that the analyzed samples are all representing non-smoker patients and we gathered information on gender that will be further correlated with mutational landscapes in follow-up studies.
While a number of cancer centers have already begun to integrate molecular profiling and even clinical next-generation sequencing (NGS) into the pipeline of routine cancer diagnosis in order to increase accuracy and efficiency of treatments, it is important to recognize and discuss the limitations of the targeted therapy in the treatment of NSCLC. For instance, EGFR inhibitors, such as gefitinib, erlotinib, or afatinib, can effectively shrink tumors for several months; these drugs eventually stop working for most patients, usually because the cancer cells within the tumor develop additional mutation(s) in the EGFR gene. Studies investigating the clinicopathological factors influencing post-recurrence survival and the effect of post-recurrence therapy in NSCLC will be critical to further advance therapies.
Conclusions
In summary, using targeted whole exome sequencing, we have identified mutations in a large cohort of Chinese NSCLC blood and tissue samples for 65 genes and provide an overview of the mutational landscape by analyzing CNVs, fusions, and SNV/InDel in details.
Methods
Samples
The study was conducted in accordance with the Helsinki Declaration and was approved by the institute’s Ethics Committee. All the patients enrolled had been informed about the content and purposes of this study and signed the consents. In this study, we have collected and processed a total of 1000 formalin-fixed paraffin-embedded (FFPE) tumor samples and 500 blood samples of patients diagnosed with NSCLC between June 2017 and April 2019. Patient samples were collected from The First Affiliated Hospital of Nanchang University (Nanchang, Jiangxi, China), PLA General Hospital (Beijing, China), Jingdezhen First People’s Hospital (Jingdezhen, Jiangxi, China), 334 Affiliated Hospital of Nanchang University (Nanchang, Jiangxi, China), and The First Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China). Tissue and blood samples were collected from independent patient groups.
DNA extraction and Next-Generation Sequencing
Genomic profiling was performed in a College of American Pathologists (CAP)-accredited lab at OrigiMed (Shanghai, China) according to standard procedures. Briefly, genomic DNA was extracted from tissue and plasma samples were tested for cell-free DNA (cfDNA). DNA was extracted from tissue and liquid blood biopsies using standard DNA Extraction Kit (QIAamp DNA FFPE Tissue Kit; Qiagen, Hilden, Germany) and MagMAX Cell-free DNA isolation kit (Thermo, Cat#A29319), respectively, according to manufacturer’s recommendations. A total of 3.6–35 ng of DNA was used as input to prepare barcoded libraries for each sample. The exon regions of 65 cancer driver genes were tested using the IDT (Integrated DNA Technologies, Coralville, IA, USA) custom-designed panel. The genes included in this panel are ABL1, AKT1, ALK, APC, AR, ATM, BRAF, CCND1, CDK4, CDK6, CDKN1A, CDKN2A, CTNNB1, DDR2, EGFR, ERBB2, ERBB3, ERBB4, ESR1, FBXW7, FGFR1, FGFR2, FGFR3, FGFR4, FLT3, GNA11, GNAQ, GNAS, HRAS, IDH1, IDH2, JAK1, JAK2, JAK3, KDR, KIT, KRAS, MEK1, MET, MTOR, NF1, NF2, NRAS, NTRK1, NTRK2, NTRK3, PDGFRA, PDGFRB, PIK3CA, POLE, PTCH1, PTEN, RB1, RET, ROS1, SATA3, SMAD4, SMARCA4, SMO, STK11, TERT, TP53, TSC1, TSC2, and VHL.
The FFPE and blood samples were sequenced by Illumina Nova seq. As for the FFPE samples, the mean sequencing depth was nearly 1200x, the coverage rate was 99.99%, and fraction of bases mapped to target region was between 40 and 70%. At least 200x nucleic acid coverage and 1% of mutation allele fraction were used as the standard cutoff to make the final variant call. As for the blood samples, the mean sequencing depth was nearly 10000x, the coverage rate was 99.99%, and fraction of bases mapped to target region was between 4 and 70%. At least 2000x nucleic acid coverage and 0.5% of mutation allele fraction were used as the cutoff for the final variant call.
Bioinformatics analysis
Our initial analysis aimed to explore genomic alterations, including gene rearrangements, copy number variations (CNVs), single nucleotide variants (SNVs), and short and long insertions/deletions (InDels). Raw sequencing reads were aligned to the human reference genome (hg19) using Burrows-Wheeler Aligner (BWA). Consensus reads were generated for error suppressing and PCR duplicates were removed using in-house software ECR. Read depth and coverage of the targeted regions were calculated by in-house software LibraryQC. The log-ratio per region of each target genes was calculated, and customized algorithms were used to detect copy number variations. Focal amplifications were characterized as genes with thresholds ≥4 copies. Gene rearrangements and long indels were detected using CREST [28] and Manta [29]. SNVs and short indels were identified by MuTect [30] and Pindel [31].
Acknowledgements
Not applicable
Abbreviations
- NSCLC
Non-small cell lung carcinoma
- FFPE
Formalin-fixed paraffin-embedded
- AD
Adenocarcinoma
- SCC
Squamous cell carcinoma
- NCCN
National Comprehensive Cancer Network
- NGS
Next-generation sequencing
Authors’ contributions
YJZ and WZ wrote the manuscript; QHZ, YY, and CW collected the data; CF, CJL, LN, and LMW collected the data. All authors read and approved the final manuscript.
Funding
This study was supported by the Science and Technology Plan Fund Project of Science and Technology Department of Jiangxi Province (20181BBG78023).
Availability of data and materials
All data generated or analyzed during this study are included in this published article. The sequence data will be provided upon request.
Declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Helsinki Declaration and was approved by the institute’s Ethics Committee. All the patients enrolled had been informed about the content and purposes of this study and signed the consents.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ya-jun Zhou and Wei Zheng contributed equally to this work.
Contributor Information
Li Niu, Email: niul@cheerlandgroup.com.
Li-ming Wu, Email: 304088889@qq.com.
References
- 1.Suster DI, Mino-Kenudson M. Molecular pathology of primary non-small cell lung cancer. Arch Med Res. 2020. [DOI] [PubMed]
- 2.Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER) J Thorac Oncol. 2014;9(2):154–162. doi: 10.1097/JTO.0000000000000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee J, Sun JM, Lee SH, Ahn JS, Park K, Ahn MJ. Are there any ethnic differences in the efficacy and safety of immune checkpoint inhibitors for treatment of lung cancer? J Thorac Dis. 2020;12(7):3796–3803. doi: 10.21037/jtd.2019.08.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng L, Wu YL. Immunotherapy in the Asiatic population: any differences from Caucasian population? J Thorac Dis. 2018;10(Suppl 13):S1482–S1S93. doi: 10.21037/jtd.2018.05.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, Normanno N, Scarpa A, Robson M, Meric-Bernstam F, Wagle N, Stenzinger A, Bonastre J, Bayle A, Michiels S, Bièche I, Rouleau E, Jezdic S, Douillard JY, Reis-Filho JS, Dienstmann R, André F. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31(11):1491–1505. doi: 10.1016/j.annonc.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 6.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 7.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, Bose R, Ornitz D, Xiong D, You M, Dooling DJ, Watson M, Mardis ER, Wilson RK. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150(6):1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar A, Minna J. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246(4929):491–494. doi: 10.1126/science.2554494. [DOI] [PubMed] [Google Scholar]
- 10.Packenham JP, Taylor JA, White CM, Anna CH, Barrett JC, Devereux TR. Homozygous deletions at chromosome 9p21 and mutation analysis of p16 and p15 in microdissected primary non-small cell lung cancers. Clin Cancer Res. 1995;1(7):687–690. [PubMed] [Google Scholar]
- 11.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97(5):339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 12.Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, et al. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res. 1988;48(20):5738–5741. [PubMed] [Google Scholar]
- 13.Choi JY, Lee YS, Shim DM, Seo SW. Effect of GNAQ alteration on RANKL-induced osteoclastogenesis in human non-small-cell lung cancer. Bone Joint Res. 2020;9(1):29–35. doi: 10.1302/2046-3758.91.BJR-2019-0085.R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mogi A, Kuwano H. TP53 mutations in nonsmall cell lung cancer. J Biomed Biotechnol. 2011;2011:583929. doi: 10.1155/2011/583929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hainaut P, Olivier M, Pfeifer GP. TP53 mutation spectrum in lung cancers and mutagenic signature of components of tobacco smoke: lessons from the IARC TP53 mutation database. Mutagenesis. 2001;16(6):551–553. doi: 10.1093/mutage/16.6.551. [DOI] [PubMed] [Google Scholar]
- 16.Prabhakar CN. Epidermal growth factor receptor in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4(2):110–118. doi: 10.3978/j.issn.2218-6751.2015.01.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 18.Scheffler M, Bos M, Gardizi M, Konig K, Michels S, Fassunke J, et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget. 2015;6(2):1315–1326. doi: 10.18632/oncotarget.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Yang Y, Li H, Chen Z, Jiang G, Fei K. Assessment of the clinical application of detecting EGFR, KRAS, PIK3CA and BRAF mutations in patients with non-small cell lung cancer using next-generation sequencing. Scand J Clin Lab Invest. 2016;76(5):386–392. doi: 10.1080/00365513.2016.1183813. [DOI] [PubMed] [Google Scholar]
- 20.Consortium APG AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818–831. doi: 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, Barletta JA, Borecki IB, Broderick S, Chang AC, Chiang DY, Chirieac LR, Cho J, Fujii Y, Gazdar AF, Giordano T, Greulich H, Hanna M, Johnson BE, Kris MG, Lash A, Lin L, Lindeman N, Mardis ER, McPherson JD, Minna JD, Morgan MB, Nadel M, Orringer MB, Osborne JR, Ozenberger B, Ramos AH, Robinson J, Roth JA, Rusch V, Sasaki H, Shepherd F, Sougnez C, Spitz MR, Tsao MS, Twomey D, Verhaak RGW, Weinstock GM, Wheeler DA, Winckler W, Yoshizawa A, Yu S, Zakowski MF, Zhang Q, Beer DG, Wistuba II, Watson MA, Garraway LA, Ladanyi M, Travis WD, Pao W, Rubin MA, Gabriel SB, Gibbs RA, Varmus HE, Wilson RK, Lander ES, Meyerson M. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450(7171):893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Modrek B, Ge L, Pandita A, Lin E, Mohan S, Yue P, Guerrero S, Lin WM, Pham T, Modrusan Z, Seshagiri S, Stern HM, Waring P, Garraway LA, Chant J, Stokoe D, Cavet G. Oncogenic activating mutations are associated with local copy gain. Mol Cancer Res. 2009;7(8):1244–1252. doi: 10.1158/1541-7786.MCR-08-0532. [DOI] [PubMed] [Google Scholar]
- 24.Broet P, Dalmasso C, Tan EH, Alifano M, Zhang S, Wu J, et al. Genomic profiles specific to patient ethnicity in lung adenocarcinoma. Clin Cancer Res. 2011;17(11):3542–3550. doi: 10.1158/1078-0432.CCR-10-2185. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, Nakagawa K, Mano H, Ishikawa Y. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 26.Choong NW, Ma PC, Salgia R. Therapeutic targeting of receptor tyrosine kinases in lung cancer. Expert Opin Ther Targets. 2005;9(3):533–559. doi: 10.1517/14728222.9.3.533. [DOI] [PubMed] [Google Scholar]
- 27.Hasanovic A, Mus-Veteau I. Targeting the multidrug transporter Ptch1 potentiates chemotherapy efficiency. Cells. 2018;7(8). [DOI] [PMC free article] [PubMed]
- 28.Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, Holmfeldt L, Payne-Turner D, Fan X, Wei L, Zhao D, Obenauer JC, Naeve C, Mardis ER, Wilson RK, Downing JR, Zhang J. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods. 2011;8(8):652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 30.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article. The sequence data will be provided upon request.