

Abstract
Astrocytes are actively involved in a neuroprotective role in the brain, which includes scavenging reactive oxygen species to minimize tissue damage. They also modulate neuroinflammation and reactive gliosis prevalent in several brain disorders like epilepsy, Alzheimer’s, and Parkinson’s disease. In animal models, targeted manipulation of astrocytic function via modulation of their calcium (Ca2+) oscillations by incorporating light-sensitive cation channels like Channelrhodopsin-2 (ChR2) offers a promising avenue in influencing the long-term progression of these disorders. However, using adult animals for Ca2+ imaging poses major challenges, including accelerated deterioration of in situ slice health and age- related changes. Additionally, optogenetic preparations necessitate usage of a red-shifted Ca2+ indicator like Rhod- 2 AM to avoid overlapping light issues between ChR2 and the Ca2+ indicator during simultaneous optogenetic stimulation and imaging. In this unit, we provide an experimental setting that uses live adult murine brain slices (2–5 months) from a knock-in model expressing Channelrhodopsin-2 (ChR2(C128S)) in cortical astrocytes, loaded with Rhod-2 AM to elicit robust Ca2+ response to light stimulation. We have developed and standardized a protocol for brain extraction, sectioning, Rhod- 2 AM loading, maintenance of slice health, and Ca2+ imaging during light stimulation. This has been successfully applied to optogenetically control adult cortical astrocytes, which exhibit synchronous patterns of Ca2+ activity upon light stimulation, drastically different from resting spontaneous activity.
Keywords: Astrocyte, Optogenetics, Calcium imaging, Rhod-2 AM, Channelrhodopsin-2
INTRODUCTION
Astrocytes—key players in the brain, are extensively involved in reducing toxicity in the neuronal environment by scavenging reactive oxygen species, thereby minimizing tissue damage (Devinsky, Vezzani, Najjar, De Lanerolle, & Rogawski, 2013). During brain insults, astrocytes assist microglia in the de-novo synthesis of various cytokines and trophic factors, resulting in the modulation of neuroinflammation (Agulhon et al., 2012; Hanisch, 2002; Vezzani, Friedman, & Dingledine, 2013). This is an essential component of reactive gliosis—a hallmark of a multitude of disorders including epilepsy, stroke, Parkinson’s, and Alzheimer’s disease. Several of these disease models necessitate using adult mice (>2 months) to study disease progression (Maragakis & Rothstein, 2006; Riera, Hatanaka, Uchida, Ozaki, & Kawashima, 2011; Verkhratsky, Rodriguez, & Parpura, 2014).
Calcium serves as a ubiquitous second messenger for intracellular signaling cascades that mediate astrocytic function. Live calcium imaging is utilized to gauge astrocytic activation/excitability and to determine its role in disease and health (Fiacco & McCarthy, 2006; Verkhratsky & Kettenmann, 1996). However, the study of adult (>2 months) and aged (>18 months) animals is challenging due to age-related increase in oxidative stress, glutamatergic excitotoxicity due to higher synaptic spine density and morphofunctional changes disrupting homeostasis, leading to a rapid decline in the viability of acute slices (Matias, Morgado, & Gomes, 2019; Verkhratsky, Zorec, Rodriguez, & Parpura, 2016).
To investigate the role of astrocytic Ca2+ dynamics and its implications in neuroinflammation, it is important to use a non-invasive approach like optogenetics, which provides a platform for targeting and activating specific cell types with high temporal and spatial resolution. Optogenetics also employs the development of transgenic models to genetically target astrocytes, rendering them sensitive to light (Adamantidis, Zhang, de Lecea, & Deisseroth, 2014; Deisseroth, 2015; Fenno, Yizhar, & Deisseroth, 2011). The knock-in mouse model developed by Tanaka et al. (2012) offers robust expression of Channelrhodopsin-2 (ChR2(C128S)) in astrocytes (viz the MlC1 promoter), which is opened by blue light, closed by amber light, and carries a channel fluorescence readout—an Enhanced Yellow Fluorescent Protein (EYFP) tag. Notably, this leaves a narrow excitation spectrum for a fluorescent Ca2+ indicator. Therefore, when using a ChR2 mouse model, it is vital to use a red- shifted Ca2+ indicator like Rhod-2 AM. Several research groups using infant mice brain slices (P14–P20) (He, Linden, & Sapirstein, 2012; Sun et al., 2013; Takano et al., 2014) and Robillard et al (Robillard, Gordon, Choi, Christie, & MacVicar, 2011) employ Rhod-2 AM in murine hippocampal slices. In this unit we provide a protocol for Rhod-2 AM staining to study cortical astrocytes in adult murine brain slices.
In this protocol, we describe a method for utilizing adult (2–5 months) mice brain slices from the above-mentioned transgenic mouse model to assess astrocytic Ca2+ responses to light stimulation in the murine neocortex. We have developed and standardized a protocol for mouse brain extraction, vibratome sectioning, slice recovery, Rhod-2 AM loading, slice health maintenance, and Ca2+ imaging during light stimulation in these adult brain slices. Upon stimulation with light, cortical astrocytes exhibited robust increases in Ca2+ activity qualitatively different from resting spontaneous activity. Simultaneous optogenetics and functional imaging in situ in adult murine cortical astrocytes can provide information on their role in neurodegenerative disorders including Alzheimer’s disease, stroke, and Parkinson’s disease. Notably, many of these diseases exacerbate over time and the protocol provided can be applied to longitudinal studies on mice of various ages. Basic Protocol 1 describes the preparation strategies and setup, prior to beginning the experiment, with strategic overlap of timelines suggested. Support Protocol 1 details the recipes for making the cutting solution and artificial cerebrospinal fluid (aCSF), both of which are critical for maintenance of slice health of the adult brain slices and aimed towards minimizing slice degradation from oxidative stress. Basic Protocol 2 describes the steps crucial for efficient adult murine brain tissue processing—brain extraction, mounting, sectioning, and Rhod-2 AM loading for successful Ca2+ imaging. The vibratome setup with critical sectioning parameters suitable for acquisition of healthy brain slices, experimental checkpoints, and recovery of the adult mice brain slices are discussed. This protocol also elaborates on the suitable Rhod-2 AM concentration, bulk tissue loading and appropriate temperatures, in addition to incubation times. Finally, Basic Protocol 3 discusses the setup for performing simultaneous optogenetic stimulation and calcium imaging of adult murine cortical astrocytes. The outline for time-lapse confocal image acquisition of mice brain slices, data processing and analysis are given in this protocol. Astrocyte specificity of Mlc1-ChR2-EYFP expression in the transgenic model is leveraged to differentiate astrocytes of interest from other Rhod-2 AM loaded cell types, with elaboration on pre-processing, motion correction and fluorescence time-series analysis of astrocytic Ca2+ responses.
BASIC PROTOCOL 1: EXPERIMENTAL PREPARATION, SETUP, SLICE PREPARATION AND RHOD-2 AM STAINING
This protocol has two sections. Section 1A describes what is needed for performing acute slice experiments employing adult mice brains. Section 1B outlines the steps necessary for efficient mouse brain extraction, mounting, settings for vibratome sectioning, incubation parameters, and Rhod-2 AM loading conditions for subsequent Ca2+ imaging. All experimental procedures and animal care in this protocol are approved by the Institutional Animal Care and Use Committee (IACUC) at Florida International University (Approval No. 19-045), IBC exception protocol (18-006) and NIH guidelines. tTA-MlC1-tetO-ChR2(C128S)-EYFP mice (2–5 months old) are housed in standard cages at a 12h-12h light-dark cycle with free access to food and water. Note: A similar protocol must be approved by your IACUC prior to initiating.
SECTION 1A - EXPERIMENTAL PREPARATION AND SETUP
Materials
Vibratome (Model: Vibratome 1000 Plus) Water bath (Polyscience cat. no. WB10A11B) Sonicator (Branson cat. no. 1800)
Anhydrous DMSO (Life Technologies cat. no. D12345) 20% Pluronic (Biotium cat. no. 59004)
Kolliphor EL (Sigma Aldrich cat. no. C5135)
pH meter (Fisher Scientific cat. no. Accumet AB150)
Mixed gas tanks (95% O2, 5% CO2, Airgas cat. no. UN3156)
50 ml tubes (Fisher Scientific cat. no. 1443222) Isopropyl alcohol (Fisher Scientific cat. no. A4591)
Pyrex Heavy-Duty Griffin beakers (Fisher Scientific 2L - 02-555-25H, 1L - 02-555-25F, 600ml - 02-555-25D)
Prepare surfactant stocks once a month. Pluronic: Make 10% Pluronic from the 20% stock in DMSO. Kolliphor EL: Make a 5% solution in DMSO.
Clean all beakers used to prepare and contain aCSF thoroughly using antibacterial soap and water, followed by a final deionized (DI) water rinse.
Rinse the vibratome chamber, specimen holder and blade holder with antibacterial soap and water, followed by an isopropyl alcohol spray and a final DI water rinse. If oil is present on the blade holder, clean with an isopropyl alcohol soaked Kimwipe.
Warm the 10% Pluronic in the water bath at 34°C; this facilitates complete Pluronic dissolution.
Make 1.5L of the ice-cold cutting solution and 0.75L of aCSF according to the recipe given in Reagents and Solutions
Standardize the pH meter and adjust the pH of the solutions in the range of 7.3–7.4 by titration.
Bubble the solutions with mixed gas (95% O2,5% CO2) and recheck pH (Bubbling with mixed gas makes the cutting solution more basic and aCSF more acidic). Titrate the solutions to bring them within the range of 7.3–7.4.
Mix equal parts of the ice-cold cutting solution and aCSF to fill the incubation beaker (~400 mL in the 600 ml beaker), place it in the water bath (34°C), and start bubbling with mixed gas (Bubbling makes this solution more basic). Titrate the pH of the mixed solution so that it ranges from 7.3–7.4. Wait for the mixed solution to come to temperature before use. This solution will be used for the recovery step after slicing.
Freeze a portion of the ice-cold cutting solution (about 200 ml, distributed in 50 ml tubes) at −20°C to be used for brain extraction and temperature control during sectioning (see section 1B, step 6). Freeze the remaining solution at −5 °C to −10°C to be used for filling the vibratome buffer tray to collect sections. The freezing step usually takes about 1.5–2 hours. The ice-cold cutting solution used during extraction should be the consistency of a slush (Complete freezing should be avoided to not drastically alter the osmolality). This solution preserves the enzymatic activity of tissue and slows metabolism while slightly stiffening tissue for optimal sectioning.
SECTION 1B - SLICE PREPARATION AND RHOD-2 AM STAINING
Materials
Double edge safety Razor blades, use one half per preparation (wiped with EtOH) Plastic transfer open pipettes (Globe Scientific Inc. cat. no. 135040)
Custom-designed recovery chamber: made by open trimming of four 50 ml tubes (~ 3–4 cm in height), heat glued together to a fine plastic mesh (200 mesh, ATPWONZ cat. no. 4335511389) (see Figure 1A, Recovery step)
Tissue adhesive (Ted Pella cat. no. 10033)
Agarose gel mount - 2.5% (Agarose-Invitrogen cat. no. 16500 in TAE-Fisher Scientific cat. no. BP13351) Anesthetic (as specified by the approved animal care protocol)
Mice: tTA-MlC1-tetO-ChR2(C128S)-EYFP (2–5 months old, bred in house after obtaining mice lines - RIKEN, Japan RBRC05450 and RBRC05454)
Decapicone (Braintree Scientific cat. no. MDC-200)
Clock or timer Rodent guillotine
Iris Scissors, 14cm super cut curved scissors (World Precision Instruments cat. no. 503262)
Forceps, 25.5.cm (World Precision Instruments cat. no. 500364-G)
Fine spatula (World Precision Instruments cat. no. 504022)
Heavy duty spatula/Lab spoon (US Plastic Corp cat. no. 76236)
Scalpel (Carbon Steel Blades#22, IMS cat.no. CBLD22)
Rhod-2 AM (ThermoFisher Scientific cat. no. R1245MP)
Ultrafree Centrifugal Filters (Millipore cat. no. Duropore - PVDF 0.22um) Mini-Centrifuge (Southwest Sciences cat. no. SC1008-B)
Vortexer (Fisher Scientific cat. no. 02215365) Water bath (Polyscience cat. no. WB10A11B) Sonicator (Branson cat. no. 1800)
Mixed gas tanks (95% O2, 5%CO2, Airgas cat. no. UN3156)
6 well plate (Fisher Scientific cat. no. 0720083)
Custom-designed bubbling probes for the 6-well plate: made using PTFE tubing (PerkinElmer 0.7 mm I.D. cat.no. B0507021) anchored by eppendorf foam slivers and tape (see Figure 1A, orange anchors in staining step)
Isopropyl alcohol (Fisher Scientific cat. no. A4591)
50 ml tubes (Fisher Scientific cat. no. 1443222)
15 ml tubes (Fisher Scientific cat. no. 1495949D)
Anesthesia machine (Vetamac, VAD compact research machine)
Digital scale (Ohaus, cat. no. 30253027)
Induction chamber (Vetequip, cat. no. 941448)
Biohazard bag (Thomas Scientific, cat. no. P410812)
Figure 1. Experimental setup.
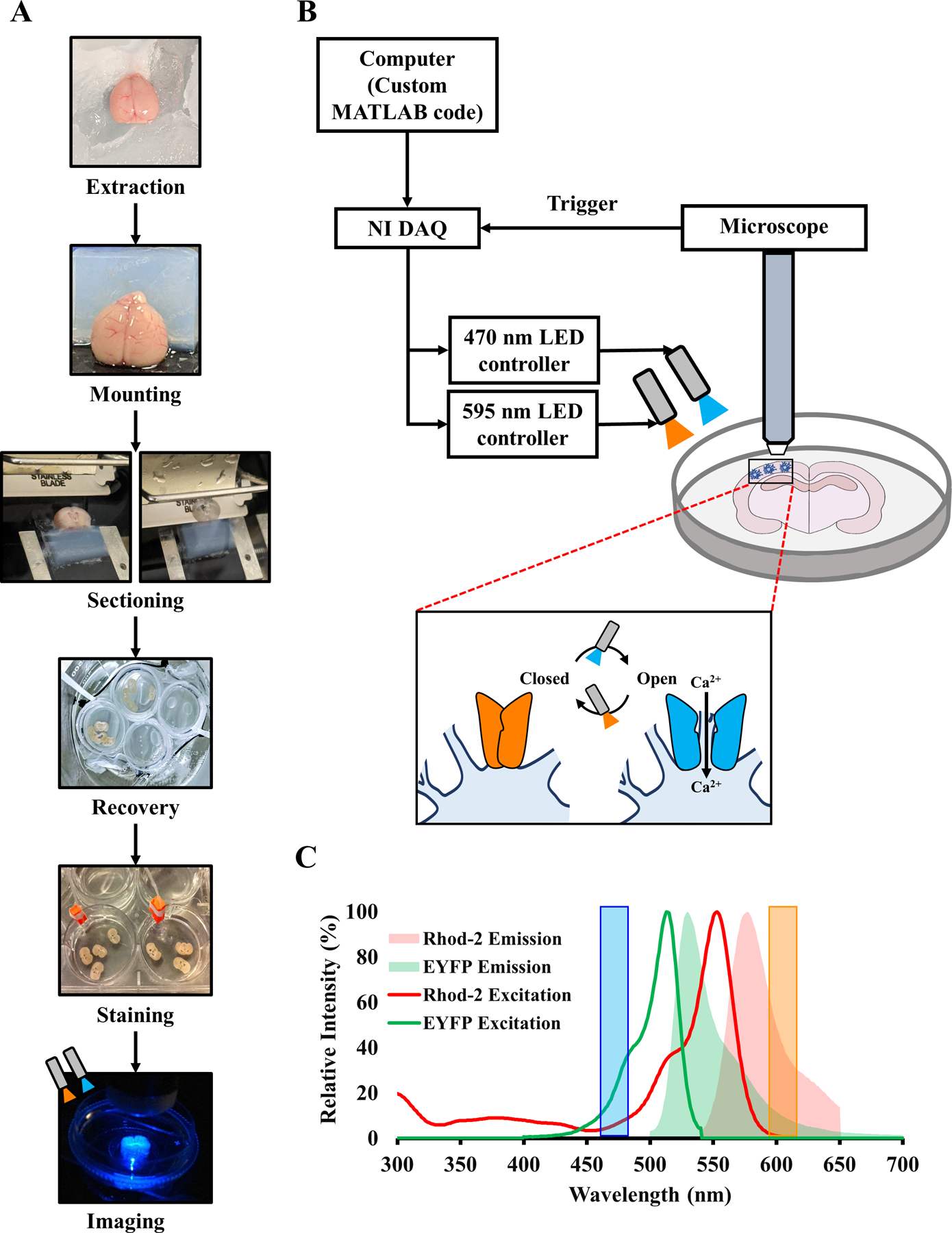
(A) Flowchart of the entire experiment, including extraction, tissue processing, staining, and imaging. (B) Schematic of the setup for simultaneous optogenetic stimulation and confocal imaging of a murine coronal brain slice. Inset shows closed and open configurations of Mlc1-ChR2(C128S)-EYFP on the astrocytic membrane, opened by blue light to allow an influx of Ca2+ and closed by amber light. (C) Excitation-emission spectra of Rhod-2 AM and EYFP. Blue and Amber shaded bars represent wavelengths of LED illumination of the brain slice during light stimulation.
Prepare the surgical suite with the anesthesia machine, digital scale, induction chamber, dissection tools, biohazard bag, and decapicone (Keep the dissection tools on ice until decapitation).
- Before starting the experiment,
-
aUse the blade angle indicator to adjust the angle of the sectioning blade on the vibratome to 25°.
-
bWipe razor blade half with isopropyl alcohol before inserting into blade holder on vibratome (see Figure 1A).
-
cFill the vibratome half-way with ice-cold cutting solution and begin bubbling with mixed gas.
-
dEnsure that all solutions are saturated with carbogen before use, which ensures stable pH buffering and adequate oxygenation.
-
eCheck that the mixed recovery solution pH is maintained between 7.3–7.4 and immerse the recovery chamber in the mixed solution beaker such that it is half-immersed in solution and held by bent zip ties hanging off of the rim of the beaker (Complete immersion of the recovery chamber will lead to floating of slices, lack of immersion will lead to drying and damage of brain slices). This solution is to be maintained at 34°C in the water bath.
-
fMake a suitable agarose block (see step 5 below) to provide mechanical support to the brain and glue it to the vibratome head using the biocompatible tissue adhesive (Curing time ~ 10 seconds).
-
a
Anesthetize the mouse for initial induction with 5% isoflurane with 1L/min O2, 14.7 PSI. Once the mouse loses righting reflex, change isoflurane to 1.5% for maintenance. Once animal breathing stabilizes (~40–60 bpm), switch off isoflurane, flush the induction box with O2 to prevent inhalation of the anesthetic. Open the box and place the animal inside the decapicone. Decapitate rapidly using the guillotine, while starting the timer.
Extract the brain on the semi-solidified ice-cold cutting solution in under 3 minutes (see Figure 1A). To extract the brain, using the Iris scissors, cut through the scalp and expose the skull. Make a cut in the front between the eyes and the back of the skull where the cerebellum begins, such that only the forebrain remains. Carefully slide the scissors between the skull and the brain to gently cut along the midline of the skull. Exercise caution for the scissors to not scrape the brain to prevent damage. Use the forceps to pry open the pieces of skull to each side and using the fine spatula, gently extract the brain onto frozen cutting solution.
After verifying that no damage is evident on the neocortex, gently dry the brain sufficiently (by removing surrounding liquid using a Kimwipe) to glue the brain’s base to the vibratome head using biocompatible tissue adhesive. Place ventral surface against agarose, ensuring that the brain is pressed against the agarose support block (Figure 1A, Mounting step) and that agarose height does not exceed a couple of mm above the brain, as this will delay initial gross trimming to remove the olfactory bulb.
Fill the vibratome chamber with ice-cold cutting solution, including any chunks of ice that may have formed, until the blade is submerged. Begin sectioning to perform initial trimming and further to achieve 350μm coronal sections. We obtain good sections on a Vibratome 1000Plus at a speed setting of 1.5 (0.3 mm/sec) and an amplitude setting of 8 (1.6 mm). Sectioning of an entire brain takes about 30 minutes; longer sectioning times can lead to deterioration of slice health.
After each slice is obtained, transfer with a plastic transfer pipette (cut the end of the pipette to produce a wide opening) to incubate in the recovery chamber immersed in the mixed solution at 34°C with active bubbling. Incubate at 34°C for 30 minutes once the last slice is collected. At this time, prepare the dye cocktail (see step 8 below) and sonicate for 30 minutes.
Move the recovery chamber containing brain slices to room temperature (RT) in recovery solution and maintain bubbling for an additional 30 minutes.
Rhod-2 AM preparation
We have determined that for 350μm adult mice brain slices, a concentration of 5.7 μM is optimal for Rhod-2 AM staining and imaging in the neocortex. This dye concentration was selected because higher concentrations led to blotches of dye in the slices, observed while imaging.
-
7
Dissolve Rhod-2 AM in 8 μl of 10% Pluronic/ DMSO and subsequently add 8 μl of 5% Kolliphor EL/DMSO. Vortex this dye mix, and after a brief spin down (~5–10 seconds, 2000g (RCF) at 6000 rpm) centrifugation, and then bring up the volume to 250 μl using aCSF.
-
8
Filter dye through the centrifugal filter tube at for ~20–30 seconds at 2000g (RCF)/ 6000 rpm. Add the filtrate to 7.75 ml of aCSF (Total = 8 ml of 5.7 μM Rhod-2 AM). Sonicate for 30 minutes before adding to the slices.
Bulk tissue loading with Rhod-2 AM
-
9
Using an open pipette, place slices in a 6-well plate (3–4 slices/ well).
-
10
Remove excess solution while ensuring that a thin film of liquid remains over the slices (this is to prevent slice health deterioration due to drying).
-
11
Add the Rhod- 2 AM dye solution by gently placing it on each of the slices before filling the well (Total volume/ well ~ 4 ml). Start bubbling mixed gas using the custom-made probes by placing one probe in each well of the 6-well plate containing the slices. Adjust the pressure such that the slices do not swirl in the 6-well plate while getting bubbled.
-
12
Place the 6-well plate at 34°C in a water bath for 45 minutes while bubbling. Fill adjacent wells (not containing brain slices) with aCSF and bubble using extra custom-designed probes (This solution will used in steps 15 and 16 for washing and de-esterification).
-
13
After 45 minutes, remove the bubbling probes and wash slices by gently pipetting out the staining solution followed by immediate addition of freshly bubbled aCSF (from the adjacent wells). Repeat this step 2–3 times.
-
14
Add fresh aCSF (~4 ml) and allow slices to de-esterify in the 6-well plate for 30 minutes at RT, while resuming bubbling.
-
15
After 30 minutes, transfer the slices using an open pipette into a 50 ml beaker containing fresh aCSF, under constant bubbling for imaging afterwards. The entire process of Protocol 1 (Figure 1A) takes approximately 3 hours.
BASIC PROTOCOL 2: IMAGE ACQUISITION AND ANALYSIS
In this unit, we validate Rhod-2 AM loading of adult murine cortical astrocytes and the ability to perform simultaneous optogenetics and Ca2+ imaging. We discuss an example setup (block stimulation paradigm) for performing optogenetic stimulation and provide an outline for confocal image acquisition, the corresponding data processing and analysis are also given in this protocol. It is noteworthy that the analysis of Ca2+ imaging data can be done by using other equivalent software and implementing the suggested software is not a limiting factor in performing simultaneous Rhod-2 imaging and optogenetic stimulation.
Materials
Imaging chamber – 35 mm dish superglued on a slide (Southern Labware cat. no. 706011) Tissue restraining harp – flattened inert metal (e.g. silver) wire and nylon fibers, tissue adhesive Kimwipes
Confocal microscope (Model: Olympus FV 1200 with filter cubes cat. no. U-N49002 and U-N49004) 10X Water objective (Olympus cat. no. UMPlanFLN)
Microscope TTL cable (Olympus cat. no. FV-TTLCA) Data Acquisition (DAQ) System – NI USB 6259 BNC Power Meter (Thorlabs cat. no. PM100A)
Silicon Power Head (Thorlabs cat. no. S120C)
Fiber Coupled LED – 470nm (Thorlabs cat. no. M470F3) Fiber Coupled LED – 595nm (Thorlabs cat. no. M595F2) LED driver (Thorlabs cat. no. LEDD1B)
Pigtail Rotary Joint, 200μm, 1.25 Ferrule (Thorlabs cat. no. RJPSL2)
Pigtail Rotary Joint, 400μm, 2.5 Ferrule (Thorlabs cat. no. RJPSF4)
Analysis software (such as FIJI, MATLAB (Mathworks, R2018a), GraphPad Prism)
Preparation for data acquisition
-
1
For optogenetic stimulation, LED power and position are checked to assure consistency across experiments. Fiber optic LEDs are pointed at a 45-degree angle to illuminate the slice area under the objective. The microscope objective is moved to the side to allow for placement of power meter probe incident to the LEDs. LED power is adjusted to be 500μW for the 470nm fiber-coupled LED (blue), and 100μW for the 595nm fiber-coupled LED (amber). The entire setup for optogenetic stimulation and Ca2+ imaging is shown in Figure 1B.
-
2For optogenetic stimulation of the slice, the field of view is illuminated using the fiber optic LEDs and synced to image acquisition. An analog trigger signal sent every time an image is acquired is read using a TTL cable connecting the NI-DAQ to the microscope box. The desired stimulation paradigm is programmed in MATLAB using the Data Acquisition Toolbox and synchronized to the confocal acquisition trigger signal on the DAQ. The program uses the DAQ digital output channels to trigger the T- cube LED drivers via a BNC connector according to the stimulation paradigm.The example recording shown in Figure 3C is 12 minutes long, with the blue LEDs beginning at 100s into the recording. The blue LED is on for 20 seconds, followed by a 5s amber pulse, repeating every 100 seconds.
-
3
Construct an imaging chamber by fixing the lid of a 35mm dish onto a glass slide with cyanoacrylate glue.
-
4
Cut a piece of Kimwipe to fit the bottom of the imaging chamber, fill with enough aCSF to soak it, and position the brain slice in the center of the Kimwipe using an open transfer pipette. The Kimwipe beneath the slice creates friction between the harp and the plastic dish needed to minimize slice motion.
-
5
With minimal liquid covering slice, position the tissue restraining harp over the slice to cover the area evenly and avoid folding or wrinkling of tissue. Once positioned, fill the chamber with enough aCSF to image using the 10X water immersion objective.
Figure 3.
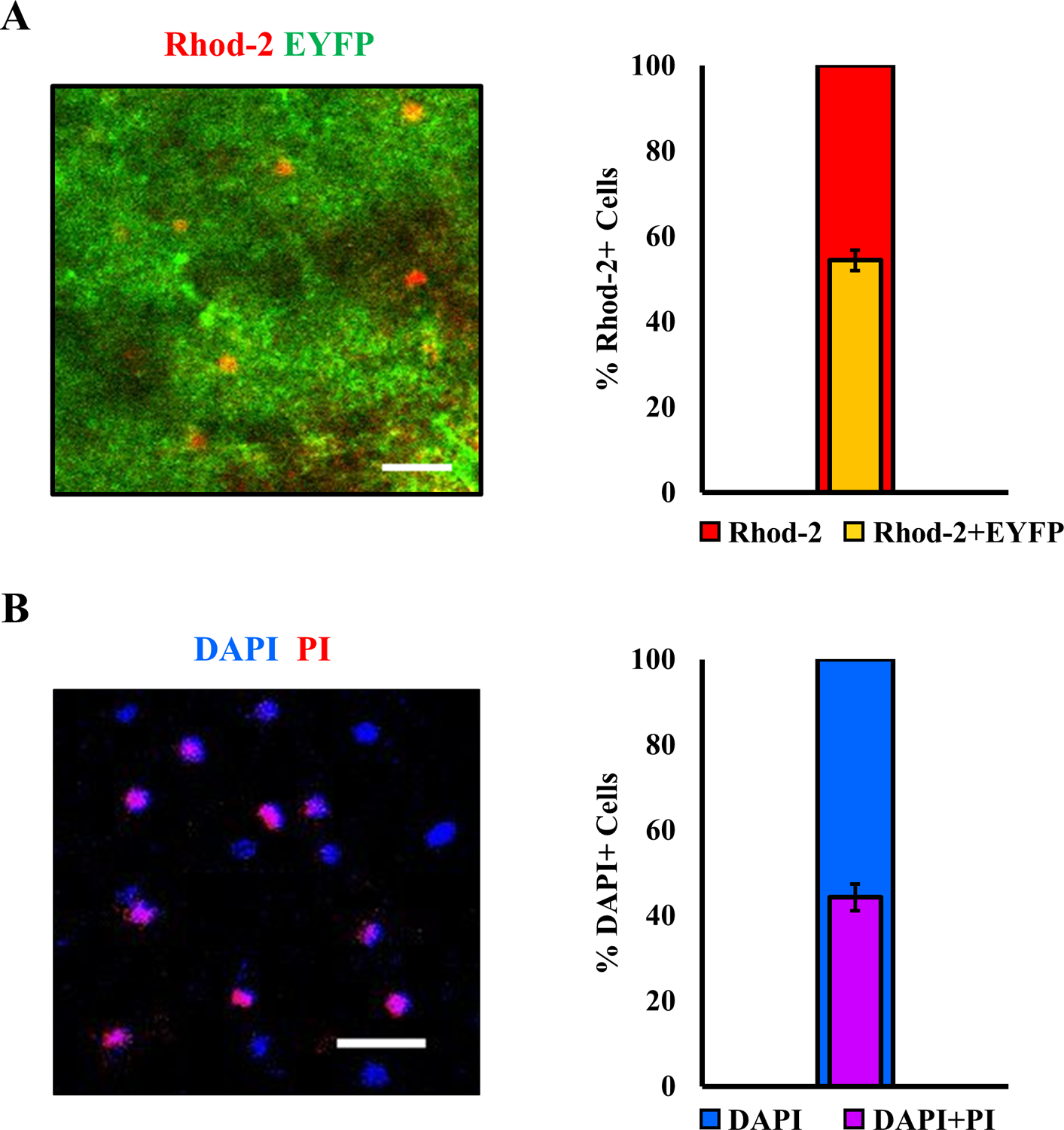
Quantification of astrocytic staining and cell viability. (A) Left: representative image of a cortical field of view from mice expressing Mlc1-ChR2(C128S)-EYFP (green), showing overlap with Rhod-2 AM staining (red). Right: a fraction of Rhod-2 AM positive cells expressing Mlc1-ChR2(C128S)-EYFP in the neocortex, indicating astrocytes (n = 8 mice, 10 sections, mean ± std). (B) Left: representative image of Propidium Iodide (PI) stained cells (red) showing overlap with DAPI stained cells (blue). Cells that are positive for DAPI and PI are considered dead. Right: a fraction of DAPI positive cells also stained by PI in the neocortex (n = 2 mice, 4 sections, mean ± std). Scale bar – 30 μm.
Data acquisition
-
6
While Rhod-2 AM loaded cells are distributed throughout the depth of the slice, astrocytes at the surface of the slice (0 to 20μm deep) often display a reactive phenotype (visualized as dye aggregates and hypertrophy of primary processes). It is advisable to avoid the superficial layer and select the field of view such that the cells imaged are >20μm deep into the slice.
-
7
Acquire an image with the EYFP (astrocytic marker due to Mlc1-ChR2(C128S-EYFP)) and Rhod-2 AM filter sets of the chosen field of view. This image will serve as the reference image during processing to identify astrocytes stained with Rhod-2 AM, refer to step 13.
-
8
Depending on the stimulation setup protocol selected, the user will need to run the custom-MATLAB script which will wait for a trigger from the microscope to initiate stimulation (refer to supplementary section; NIDAQ_trigger code). This will also serve to synchronize Ca2+ imaging with the optogenetic stimulation.
-
9
Begin time series data collection for simultaneous optogenetic stimulation and Ca2+ imaging on the Olympus Fluoview or equivalent microscope software.
Data processing and analysis
In our experiment, we acquire raw Ca2+ imaging data (time series, step 10) and a static image overlap of EYFP and Rhod-2 AM (step 8). After acquisition using the Olympus Fluoview software, or analogous acquisition software, the steps for data processing and analysis are given in Figure 4. These data processing steps suggested, account for common artifacts encountered in single-photon fluorescence (confocal) data acquisition.
Figure 4.
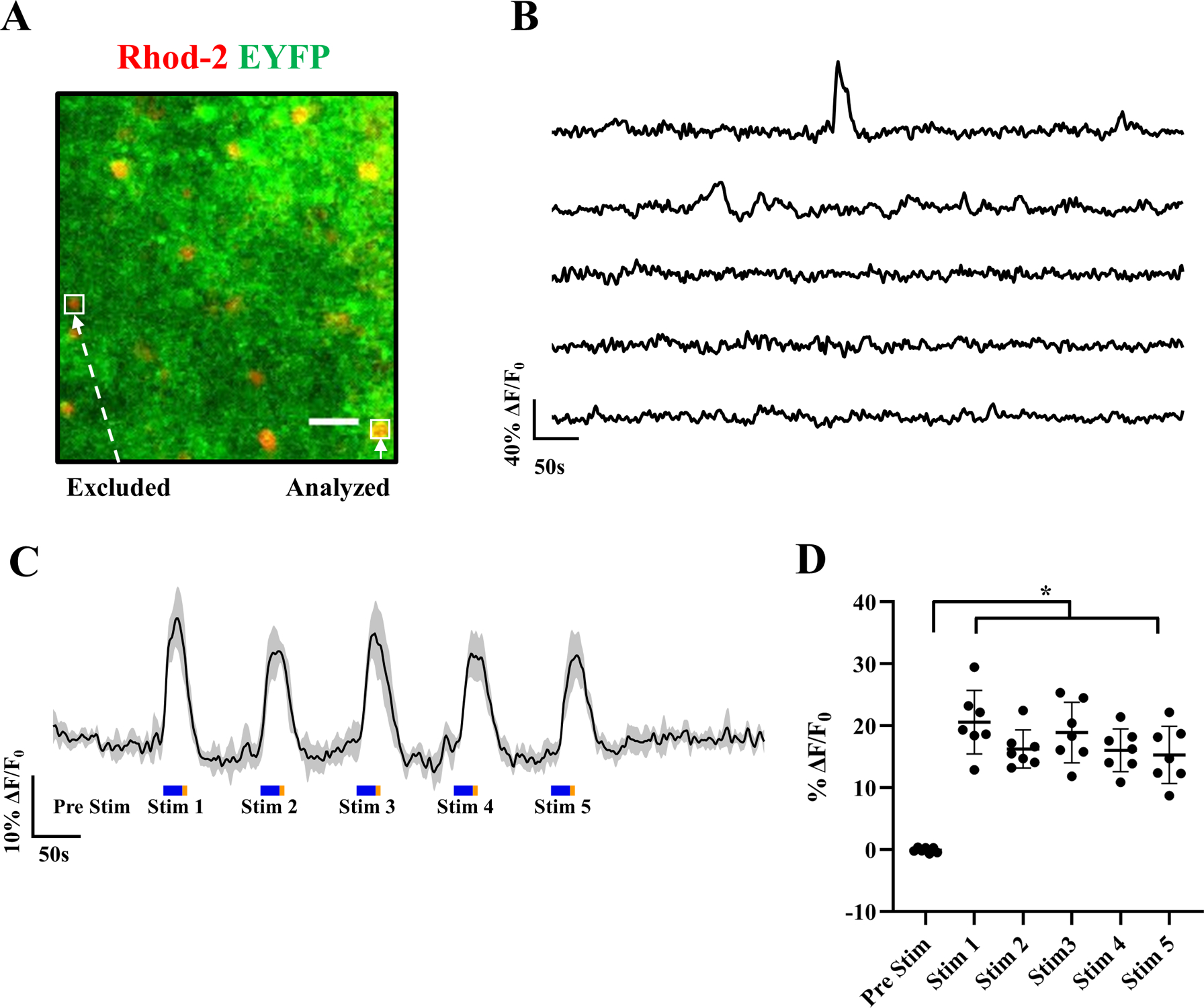
Spontaneous and light-evoked Rhod-2 AM Ca2+ responses from adult murine cortical astrocytes. (A) Representative image of a cortical section from mice expressing Mlc1-ChR2(C128S)-EYFP (green), showing overlap with Rhod-2 AM (red). Examples of an analyzed astrocyte (Rhod-2 AM+/Mlc1-ChR2(C128S)-EYFP+) and excluded cell (only Rhod-2 AM+) are bound by white boxes. (B) Representative ΔF/F0 traces of spontaneous calcium responses from cortical astrocytes expressing Mlc1-ChR2(C128S)-EYFP. (C) Average ΔF/F0 (central dark trace) of 7 astrocytes from the same brain slice subjected to repeated light stimulation over the recording period, shaded region depicts the standard deviation (n = 7 astrocytes). Light stimuli: Blue-20s/500 μW and Amber-5s/100 μW pulses were produced using LED light sources to open and close ChR2(C128S) channels, respectively. (D) Scatter plot of the median ΔF/F0 of the pre-stimulation period along with maximum ΔF/F0 astrocytic responses during each blue light stimulation window, corresponding to plot C. Average data are represented as mean ± std. 1-way ANOVA, p < 0.0001, 95% CI. Dunnett’s multiple comparisons test reveals the significance of pairwise comparisons to be: Pre Stim versus Stim 1 (***), Pre Stim versus Stim 2 (****), Pre Stim versus Stim 3 (***), Pre Stim versus Stim 4 (***), Pre Stim versus Stim 5 (**). Scale bar – 30 μm.
-
10
The raw data is exported to MATLAB for motion correction to improve image registration using NoRMCorre (Pnevmatikakis & Giovannucci, 2017)—a piecewise nonrigid motion correction algorithm implemented in MATLAB (Step 1, Figure 4; refer to supplementary codes – NoRMCorre section).
-
11
Successfully registered videos are then exported as 16-bit Tiff stacks for analysis using FIJI ROI Analyzer (Schindelin et al., 2012). ROI selection is based on the inclusion criteria, as illustrated in Figure 3A. A Z project image is generated from the time series, which demarcates active Rhod-2 AM labeled cells with dynamic changes in fluorescence (Step 2, Figure 4). This directs resources to analyze the most active or responsive cells from each slice.
-
12
We leverage astrocyte specificity of Mlc1-ChR2-EYFP expression in our model to differentiate astrocytes of interest from other Rhod-2 AM loaded cell types. Cells with explicit somatic Rhod-2 AM loading overlapping with EYFP labeling are chosen for fluorescence signal extraction and further analysis.
-
13
Using processed data from steps 12 and 13, regions of interest (ROI) encompassing astrocytes for fluorescent signal extraction are manually selected using the ROI manager (Step 3, Figure 4). The time series data for the selected ROIs is generated on FIJI (Step 4, Figure 4) and saved as an excel file.
-
14
Fluorescence time-series data from selected ROIs is detrended to correct for the effect of fluorophore bleaching on the baseline of traces in MATLAB (Step 5, Figure 4).
-
15
The traces are smoothed using a gaussian filter to remove imaging artifacts (Step 6, Figure 4).
-
16
Finally, traces are normalized and visualized as a percentage of ΔF/F0, where baseline fluorescence (F0) is taken to be the median fluorescence intensity of the pre-stimulus imaging window (Step 7, Figure 4). The custom-written MATLAB code (dFF processing code) for steps 15–17 is available in the supplementary section.
-
17
Statistical tests are run using GraphPad Prism version 8.4.3 (Step 8, Figure 4). For the dataset in Figure 3, we confirm the normal distribution of the data using the Shapiro-Wilkins test, following which we perform a one-way ANOVA and Dunnett’s T3 multiple comparisons test with significance declared at p<0.05. We present summary data as mean ± std and do not exclude any experimental data points. Sample size is defined as the number of mice, number of slices or number of cells, mentioned in each figure legend.
REAGENTS AND SOLUTIONS
Preparation of fresh cutting solution and aCSF
Detailed recipes for the preparation of ice-cold cutting solution and aCSF in DI water are given in this section. The solutions are recommended to be made fresh, on the day of the experiment to avoid bacterial contamination.
Materials
Sucrose (Fisher Scientific cat. no. S25590)
Glucose (Fisher Scientific cat. no. BP350-1)
Sodium Chloride (Fisher Scientific cat. no. S271)
Sodium Phosphate, Monobasic (Ward’s Science cat. no. 470302-666)
Potassium Chloride (Fisher Scientific cat. no. BP366)
Sodium Bicarbonate (Fisher Scientific cat. no. S233)
Calcium Chloride Dihydrate (Fisher Scientific cat. no. C70)
Sodium Ascorbate (Fisher Scientific cat. no. AC352681000)
Magnesium Sulphate Heptahydrate (Fisher Scientific cat. no. M63)
Deionized water
COMMENTARY
Background information
Longitudinal studies for investigating neurodegenerative disorders like Alzheimer’s disease, stroke, and Parkinson’s disease employ mice across ages—young (<2 months), adults (2–14 months) and aged mice (>18 months) (Riera et al., 2011; Rodriguez-Arellano, Parpura, Zorec, & Verkhratsky, 2016; Verkhratsky et al., 2014). The study of brain slices from adult mice is challenging due to astrocytic vulnerability pertaining to age-related toxicity and morphological factors. This is attributed to atrophy and high levels of oxidative stress, mainly due to the rapid depletion of cellular antioxidants, including ascorbate and reduced glutathione. Brain slices from these older mice are also more prone to insults during dissection leading to lower viability (Brahma, Forman, Stewart, Nicholson, & Rice, 2000; Matias et al., 2019). While this manuscript focuses on slice preparations from mice aged 2–5 months, the authors encourage adopters to perceive this method as work in progress towards imaging in even older mice. We believe that our study can serve as a guideline towards methodological improvements to curb deterioration and prolong slice viability when using mice > 5 months.
It is crucial for studies that combine the use of ChR2 and Ca2+ imaging to choose a red-shifted Ca2+ indicator due to limited spectral availability, without a confounding effect between light stimulation and Ca2+ indicator excitation (refer to Figure 1C). To evaluate astrocytic Ca2+ responses, we utilize Rhod-2 AM—a robust red-shifted indicator. Rhod-2 AM has been extensively characterized in acute brain slice studies probing several cell types across young mice cohorts, predominantly juvenile and young adults <2 months (He et al., 2012; Sun et al., 2013). It is noteworthy that recently, a highly sensitive Ca2+ indicator in the red spectrum—Calbryte 590 AM has been used in vivo to detect neuronal dendritic calcium spikes (Suzuki & Larkum, 2017). Although this is pending optimization for use in acute brain slice studies, it holds the potential for obtaining Ca2+ responses with enhanced sensitivity and higher signal-to-noise ratio. Other tools like genetically encoded calcium indicators, specifically R-Camps, can be further engineered to target astrocytes to facilitate simultaneous optogenetic stimulation and Ca2+ imaging (Akerboom et al., 2013; Inoue et al., 2015). Also, it is noteworthy to mention that the protocol discussed in this unit can also be employed in adult mice brain slice preparations which have been virally injected for delivery of ChR2 to cortical astrocytes.
It is important to note that usage of a 2-photon microscope would aid in demarcation of Ca2+ responses from astrocytic soma versus processes and would facilitate recording for longer periods of time with reduced photobleaching. The authors would like to mention that the analysis and recording setup employed in this unit can be implemented using alternative software like AQuA and Python, for quantification of astrocytic activity and LabVIEW, or an Arduino for signal synchronization and trigger setup.
Critical parameters and troubleshooting
Several steps in our optimized protocol, including holding temperatures, incubation, and vibratome slicing parameters, were determined by assessing the health of the brain slices. A robust methodology employed by Buskila et al (Buskila et al., 2014), was used to co-stain DAPI, the nuclear stain, and propidium iodide (PI), a marker for identification of dead cells, thereby giving us an estimate of live/ dead cells (Figure 2B). Table 2 below highlights several challenges that one might encounter during the experiments, along with possible causes and solutions.
Figure 2.
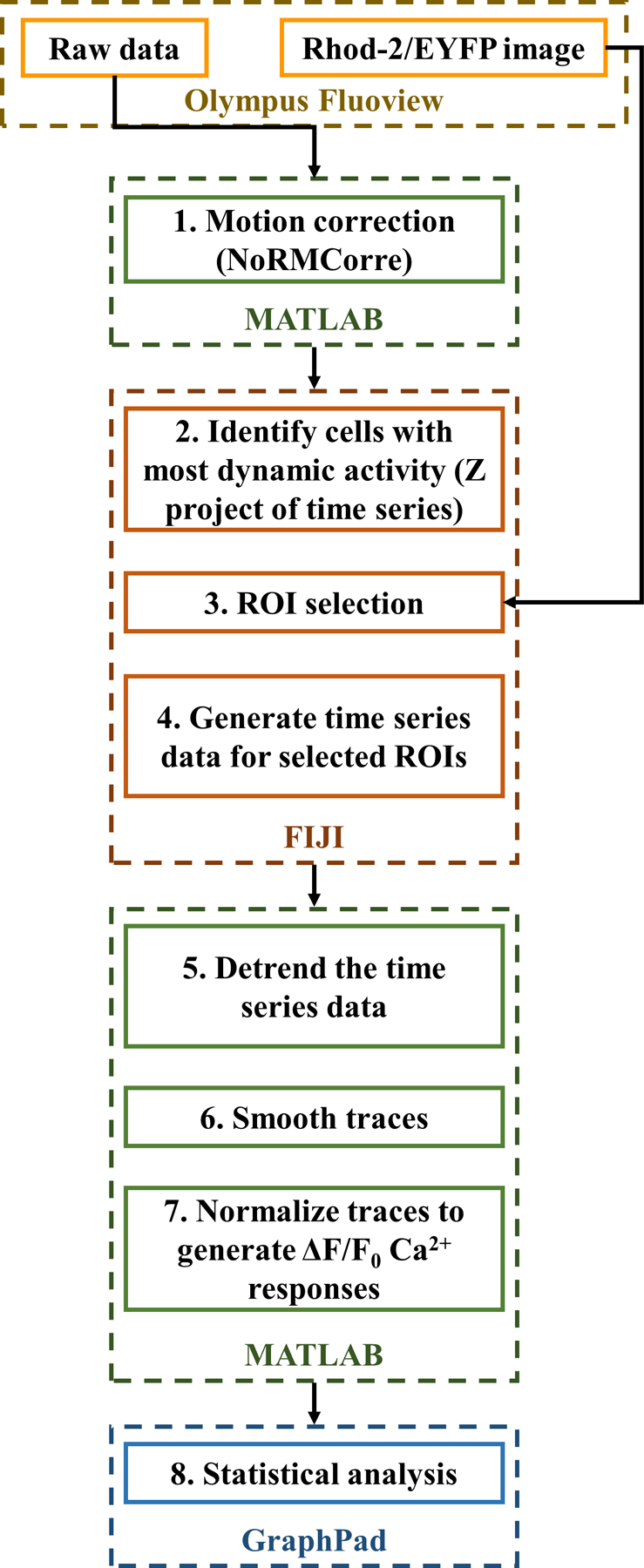
Flowchart for processing 1-photon Ca2+ imaging data. Suggestive data processing pipeline for Ca2+ imaging time-lapse data. After acquiring data using the microscope software (e.g., Olympus Fluoview), the data is motion corrected in MATLAB, time series plots are generated on FIJI and further detrended, smoothed and normalized on MATLAB. GraphPad was employed to perform statistical analysis of the data.
Table 2.
Troubleshooting critical parameters during the experiment.
| Problem | Possible cause | Solution |
|---|---|---|
| Compression of the mounted brain during sectioning or lack of intact sections | Effect of incorrect: 1. amplitude |
1. Sectioning at a very low amplitude (below 1.2 mm) causes compression of the brain tissue, while at a very high amplitude (2 mm) leads to the shearing of the tissue. |
| 2. speed of the vibratome | 2. Sectioning at a higher speed (greater than 0.4 mm/sec) leads to compromised tissue integrity and very low cell viability. At lower speeds (lesser than 0.2 mm/sec), sectioning of the brain is challenging. | |
| The amplitude and speed used for sectioning in this study were 1.6 mm and 0.3 mm/sec corresponding to settings 8 and 1.5 on the Vibratome 1000 Plus, respectively. | ||
| Dye aggregates in the slice visualized during imaging | Lack of sonication and filtration of the dye during preparation | Filter the dye mixture and sonicate for a minimum of 20 minutes before adding to the brain slices. |
| Improper function of the vibratome head/ motor | Water leak to the inner base of the vibratome | Seal the back of the vibratome chamber using paper towels to absorb excess water while sectioning, preventing solution leak. |
| Rapid deterioration of health of brain slices (within 6 hours of sectioning), reflected as bad Rhod-2 AM signal-to-noise ratio while imaging | 1. Stoppage of bubbling during the experiment | 1. Rigorous monitoring of bubbling is imperative for continuous buffering of HCO3- present in the aCSF. This avoids drastic changes in pH (Outside the 7.3–7.4 range). |
| 2. Lack of fresh Pluronic/ Kolliphor EL | 2. Make fresh Pluronic and Kolliphor EL once a month. | |
| 3. The pH of solution turning extremely acidic/ basic (outside the desired 7.3–7.4 range) | 3. Recheck the pH of the solutions periodically during the experiment. | |
| 4. Buildup of toxins | 4. Holding in a large beaker after de-esterification keeps slices healthier, making them last longer. Avoid using a 6-well plate for holding. | |
| 5. Incorrect DMSO/ Pluronic concentrations | 5. Usage of recommended DMSO and Pluronic concentrations (Hamad, Krause, & Wahle, 2015). | |
| 6. Increased temperature of water bath | 6. Maintain water bath at 34°C. | |
| Repeated issues with staining, visualized as cells being too bright or too dim (unloaded) | Issue with a specific lot number of the dye | Order a fresh batch of dye. |
Understanding results
These protocols enable the generation of viable adult cortical murine brain slices stained with Rhod-2 AM, which are suitable for simultaneous optogenetic stimulation and Ca2+ imaging experiments. The experimenter can expect to have about 8–10 brain slices with a thickness of 350μm from an adult mouse. The viability of the slices generally deteriorates about 6–7 hours after decapitation. Cell counts of Rhod-2 loaded astrocytes, and cellular viability in the Mlc1-ChR2(C128S)-EYFP transgenic mice slices are shown in Figure 2. The overlap between ChR2(C128S)-EYFP and Rhod-2 AM channels is used to determine astrocytes loaded with Rhod-2 AM to subsequently study Ca2+ responses ((Figure 2A, n = 8 mice, 10 slices). Approximately half of all Rhod-2 AM loaded cells are astrocytes expressing the optogenetic construct (right panel), as per the inclusion criteria delineated in Figure 3A. We report this yield in the neocortex as an average of percentages across slices. Cellular viability is roughly 55%, quantified as the number of DAPI+ cells only. Those which have also taken up PI—indicating membrane permeability and cell death are expressed as percentages (Figure 2B, n = 2 mice, 4 slices). This yield was achieved at optimized vibratome sectioning and processing steps.
Time-lapse imaging of Mlc1-ChR2(C128S)-EYFP mice brain slices loaded with Rhod-2 AM is performed to evaluate astrocytic Ca2+ responses (Figure 3). A representative image with examples of cells excluded or analyzed is shown in Figure 3A. Refer to steps 13 and 14 of data processing and analysis section (basic protocol 2) for elaboration on the inclusion/ exclusion criteria. Spontaneous Ca2+ traces (depicted as ΔF/F0) demonstrate random transients when no stimulus is applied (Figure 3B, n = 5 astrocytes). Figure 3C shows robust synchronized increases in Rhod-2 AM fluorescence upon repeated pulsed blue light stimulation from 7 astrocytes (plotted as mean ± std), indicative of sharp Ca2+ fluctuations in these cells. These gradually return to basal fluorescence upon a brief amber light pulse, followed by cessation of all light. Figure 3D shows the quantification of astrocytic responses to multiple pulsed light stimulation windows. After confirming normality using the Shapiro-Wilkins test, a one-way ANOVA and Dunnett’s T3 multiple comparisons test between ΔF/F0 of the astrocytes during pre-stim and stimulation periods were conducted. Statistically significant increases in ΔF/F0 during each one of the stimulation pulses are observed with respect to the fluorescence in the pre-stimulation period.
Time considerations
The outlined protocol typically takes about 11 hours from the preparation of solutions until the end of imaging for 6–7 brain slices (~12-minute recordings). Pluronic should be warmed in the water bath (34°C) at the beginning of the experiment so that it completely dissolves and is ready for dye preparation. After about 2 hours of warming in the water bath, Pluronic and Kolliphor EL need to be sonicated for 1– 2 hours. The dye mixture comprising of Rhod-2 AM, Pluronic, and Kolliphor EL must be prepared immediately post sectioning while the slices are incubated at 34°C, to allow adequate time for sonication. Calibration of LED power to ensure consistency between experiments can be done during the slice staining step.
Supplementary Material
Table 1.
Recipes for ice-cold cutting solution and aCSF.
| Reagent | Cutting Solution Conc. (mM) | aCSF Conc. (mM) |
|---|---|---|
| Sucrose | 110 | - |
| Glucose | 5 | 10 |
| Sodium Chloride | 60 | 124 |
| Sodium Phosphate, Monobasic | 1.25 | 1.25 |
| Potassium Chloride | 3 | 5 |
| Sodium Bicarbonate | 28 | 22 |
| Calcium Chloride Dihydrate | 0.5 | 2 |
| Sodium Ascorbate | 0.6 | - |
| Magnesium Sulphate Heptahydrate | - | 2 |
SIGNIFICANCE STATEMENT.
Astrocytes modulate neuroinflammation and reactive gliosis prevalent in several brain disorders including epilepsy and Alzheimer’s disease. Optogenetic control (viz incorporation of light-sensitive cation channels like Channelrhodopsin-2 i.e., ChR2) of astrocytic Ca2+ oscillations can aid in controlling the long-term progression of these disorders. Here, we provide an experimental setting that uses live adult murine brain slices (2–5 months) from a knock-in model expressing ChR2 in cortical astrocytes, loaded with Rhod-2 AM to elicit robust Ca2+ response to light stimulation, with protocols for efficient sectioning, Rhod-2 AM loading, maintenance of slice health, and Ca2+ imaging during light stimulation. The methodology described in this study can be employed to seamlessly perform simultaneous optogenetics and functional imaging in situ of adult murine cortical astrocytes.
Acknowledgements
This study was supported by the Wallace Coulter Foundation BME SEED grant at Florida International University (FIU). Karla Alejandra Montejo was supported by the National Institutes of Health NIGMS R25 GM75148 and NINDS R01 NS107336–02S1. The authors would like to sincerely thank Mr. Anders Asp, Dr. Shaohua Wang, and Mr. Chavier Laffitte for insightful discussions and feedback on the calibration of calcium imaging analysis. The authors would also like to thank Dr. James Schummers, Dr. Ranu Jung, Dr. Timothy Allen and Dr. Kalai Mathee for their help with equipment employed in this study and RIKEN, Japan, and JAX Labs, USA for their assistance with establishing the transgenic mice lines in the United States.
Contributor Information
Lakshmini Balachandar, NMD Laboratory at Florida International University Engineering Center, Miami, FL 33174 USA.
Karla A. Montejo, NMD Laboratory at Florida International University Engineering Center, Miami, FL 33174 USA and the Department of Neurologic Surgery, Mayo Clinic, Rochester, MN 55902 USA
Eleane Castano, NMD Laboratory at Florida International University Engineering Center, Miami, FL 33174 USA.
Melissa Perez, NMD Laboratory at Florida International University Engineering Center, Miami, FL 33174 USA.
Carolina Moncion, NMD Laboratory at Florida International University Engineering Center, Miami, FL 33174 USA.
Jeremy W. Chambers, Department of Environmental Health Sciences at Florida International University, Robert Stempel College of Public Health & Social Work, Miami, FL 33199 USA
J. Luis Lujan, Department of Neurologic Surgery, Mayo Clinic, Rochester, MN 55902 USA and the Department of Physiology and Biomedical Engineering, Mayo Clinic, Rochester, MN 55905 USA.
Jorge Riera Diaz, NMD Laboratory at Florida International University Engineering Center, Miami, FL 33174 USA.
Literature Cited
- Adamantidis AR, Zhang F, de Lecea L, & Deisseroth K (2014). Optogenetics: opsins and optical interfaces in neuroscience. Cold Spring Harb Protoc, 2014(8), 815–822. 10.1101/pdb.top083329 [DOI] [PubMed] [Google Scholar]
- Agulhon C, Sun MY, Murphy T, Myers T, Lauderdale K, & Fiacco TA (2012). Calcium Signaling and Gliotransmission in Normal vs. Reactive Astrocytes. Front Pharmacol, 3, 139. 10.3389/fphar.2012.00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Carreras Calderon N, Tian L, Wabnig S, Prigge M, Tolo J, Looger LL (2013). Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci, 6, 2. 10.3389/fnmol.2013.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buskila Y, Breen PP, Tapson J, van Schaik A, Barton M, & Morley JW (2014). Extending the viability of acute brain slices. Sci Rep, 4(1), 5309. 10.1038/srep05309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K (2015). Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci, 18(9), 1213–1225. 10.1038/nn.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, & Rogawski MA (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci, 36(3), 174–184. 10.1016/j.tins.2012.11.008 [DOI] [PubMed] [Google Scholar]
- Fenno L, Yizhar O, & Deisseroth K (2011). The development and application of optogenetics. Annu Rev Neurosci, 34, 389–412. 10.1146/annurev-neuro-061010-113817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, & McCarthy KD (2006). Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia, 54(7), 676–690. 10.1002/glia.20396 [DOI] [PubMed] [Google Scholar]
- Hamad MI, Krause M, & Wahle P (2015). Improving AM ester calcium dye loading efficiency. J Neurosci Methods, 240, 48–60. 10.1016/j.jneumeth.2014.11.010 [DOI] [PubMed] [Google Scholar]
- Hanisch UK (2002). Microglia as a source and target of cytokines. Glia, 40(2), 140–155. 10.1002/glia.10161 [DOI] [PubMed] [Google Scholar]
- He L, Linden DJ, & Sapirstein A (2012). Astrocyte inositol triphosphate receptor type 2 and cytosolic phospholipase A2 alpha regulate arteriole responses in mouse neocortical brain slices. PLoS One, 7(8), e42194. 10.1371/journal.pone.0042194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Takeuchi A, Horigane S, Ohkura M, Gengyo-Ando K, Fujii H, … Bito H (2015). Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nat Methods, 12(1), 64–70. 10.1038/nmeth.3185 [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, & Rothstein JD (2006). Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol, 2(12), 679–689. 10.1038/ncpneuro0355 [DOI] [PubMed] [Google Scholar]
- Matias I, Morgado J, & Gomes FCA (2019). Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front Aging Neurosci, 11, 59. 10.3389/fnagi.2019.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pnevmatikakis EA, & Giovannucci A (2017). NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J Neurosci Methods, 291, 83–94. 10.1016/j.jneumeth.2017.07.031 [DOI] [PubMed] [Google Scholar]
- Riera J, Hatanaka R, Uchida T, Ozaki T, & Kawashima R (2011). Quantifying the uncertainty of spontaneous Ca2+ oscillations in astrocytes: particulars of Alzheimer’s disease. Biophys J, 101(3), 554–564. 10.1016/j.bpj.2011.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robillard JM, Gordon GR, Choi HB, Christie BR, & MacVicar BA (2011). Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS One, 6(5), e20676. 10.1371/journal.pone.0020676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Arellano JJ, Parpura V, Zorec R, & Verkhratsky A (2016). Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience, 323, 170–182. 10.1016/j.neuroscience.2015.01.007 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, … Cardona A (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods, 9(7), 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare J-F, Xu Q, Chen M, Peng W, … Nedergaard M (2013). Glutamate- dependent neuroglial calcium signaling differs between young and adult brain. Science, 339(6116), 197–200. 10.1126/science.1226740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, & Larkum ME (2017). Dendritic calcium spikes are clearly detectable at the cortical surface. Nature Communications, 8(1), 1–11. 10.1038/s41467-017-00282-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, He W, Han X, Wang F, Xu Q, Wang X, … Nedergaard M (2014). Rapid manifestation of reactive astrogliosis in acute hippocampal brain slices. Glia, 62(1), 78–95. 10.1002/glia.22588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka KF, Matsui K, Sasaki T, Sano H, Sugio S, Fan K, Yamanaka A (2012). Expanding the repertoire of optogenetically targeted cells with an enhanced gene expression system. Cell Rep, 2(2), 397–406. 10.1016/j.celrep.2012.06.011 [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, & Kettenmann H (1996). Calcium signalling in glial cells. Trends Neurosci, 19(8), 346–352. 10.1016/0166-2236(96)10048-5 [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Rodriguez JJ, & Parpura V (2014). Neuroglia in ageing and disease. Cell Tissue Res, 357(2), 493–503. 10.1007/s00441-014-1814-z [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Zorec R, Rodriguez JJ, & Parpura V (2016). Astroglia dynamics in ageing and Alzheimer’s disease. Curr Opin Pharmacol, 26, 74–79. 10.1016/j.coph.2015.09.011 [DOI] [PubMed] [Google Scholar]
- Vezzani A, Friedman A, & Dingledine RJ (2013). The role of inflammation in epileptogenesis. Neuropharmacology, 69, 16–24. 10.1016/j.neuropharm.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.